
Electrocatalytic Reduction of CO2 to C1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
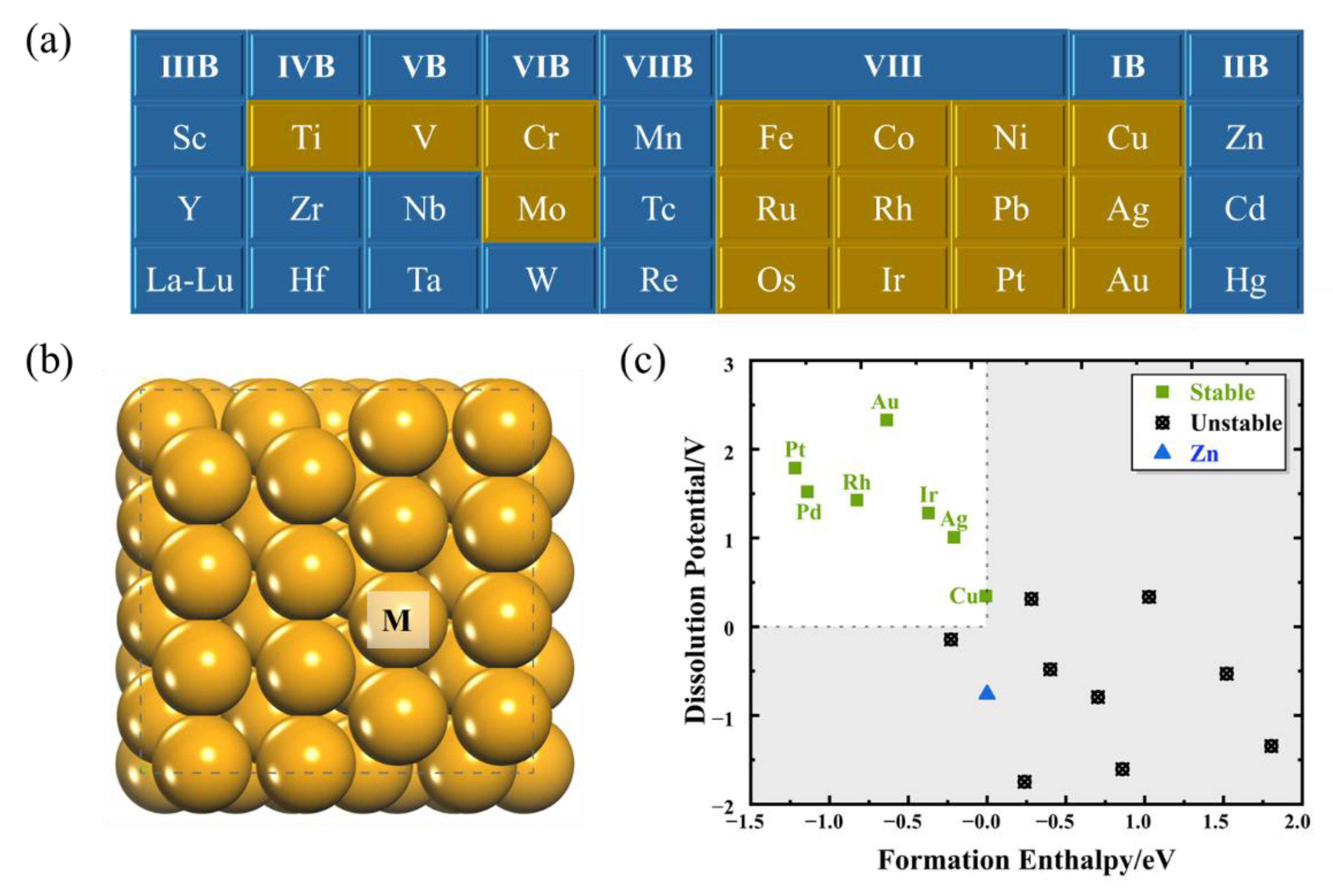
2.1. Screening Stable SAAs
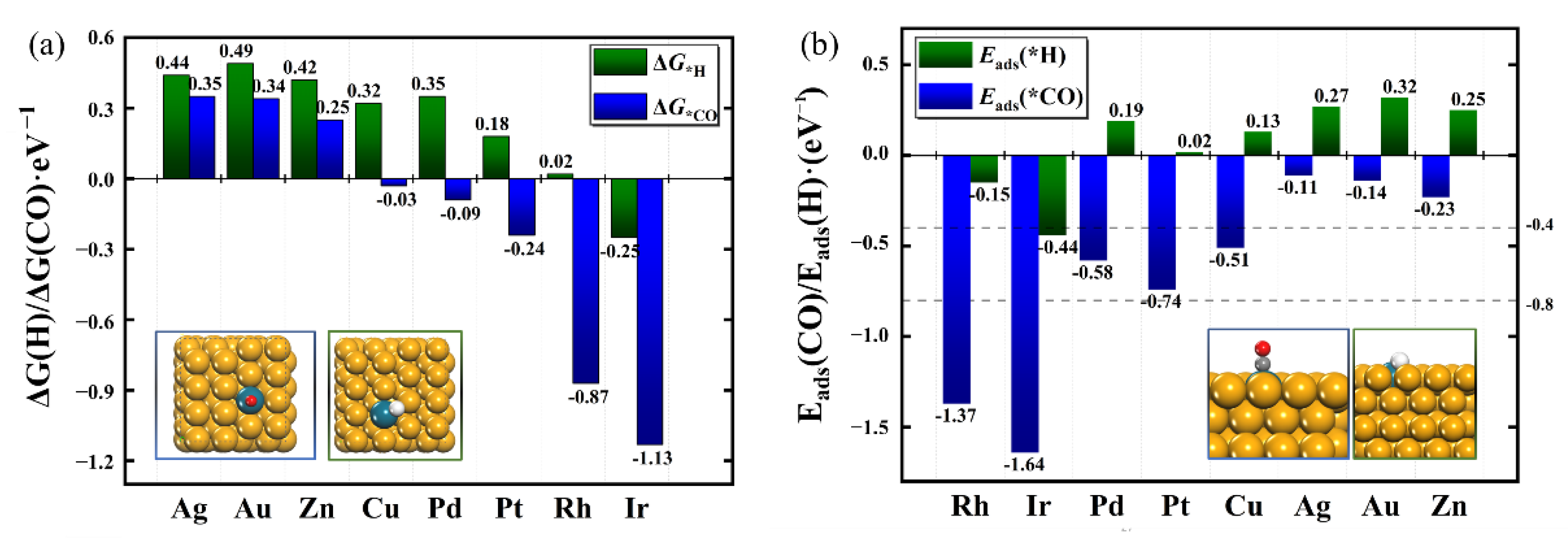
2.2. Identification of Catalysts with High Selectivity for CORR
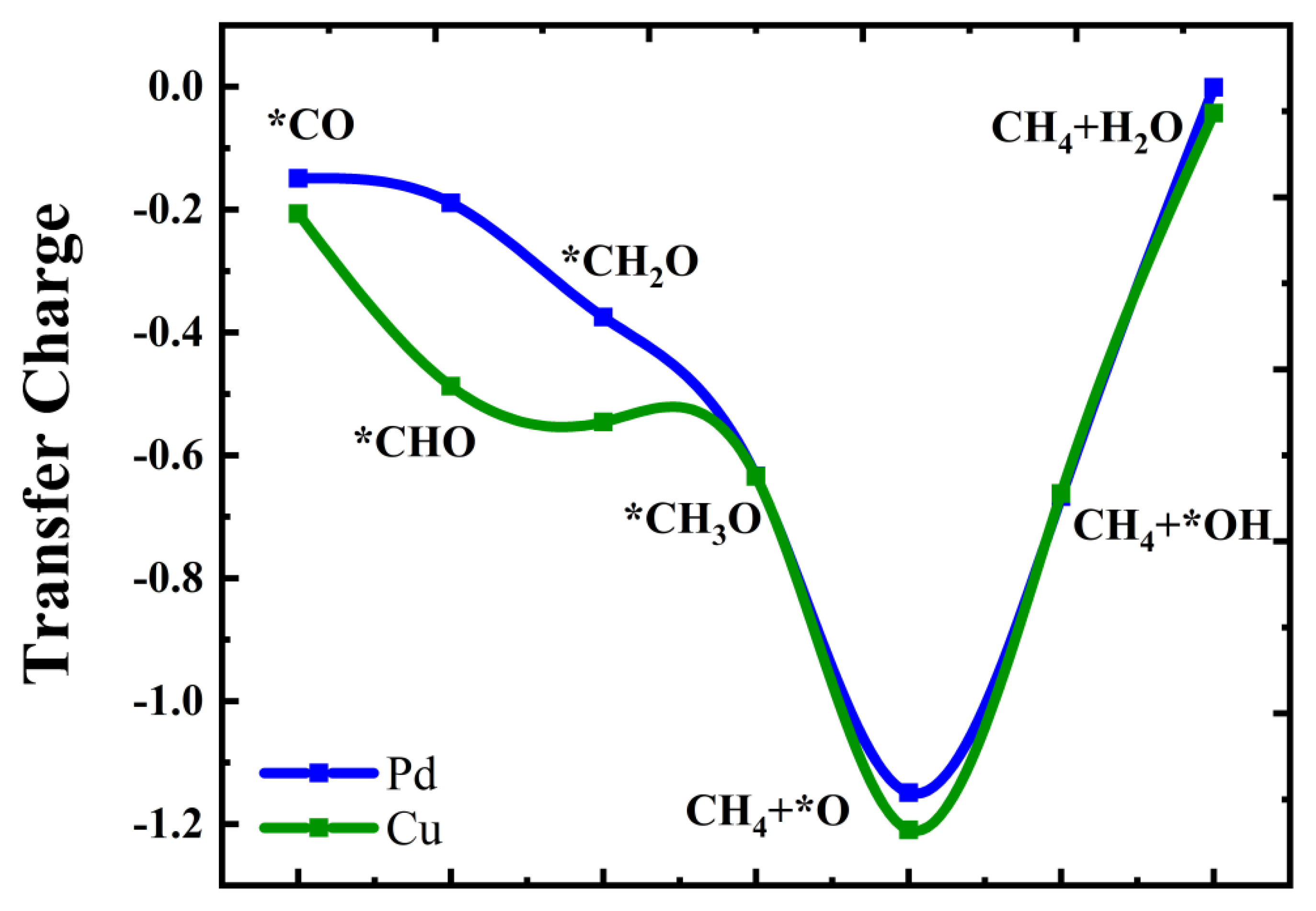
2.3. Mechanisms of CO Reduction to Methane Catalyst by Cu@Zn (101) and Pd@Zn (101)
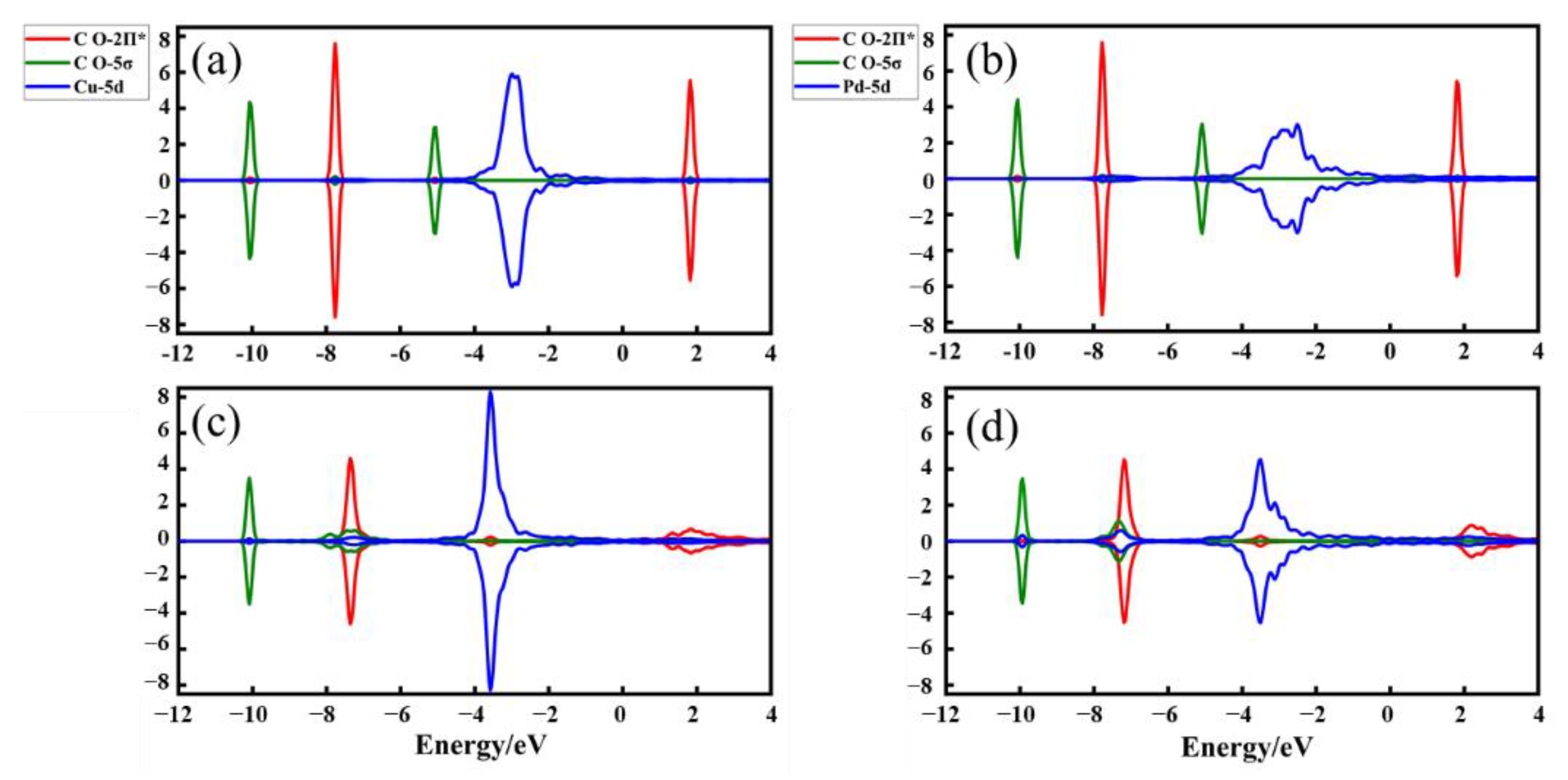
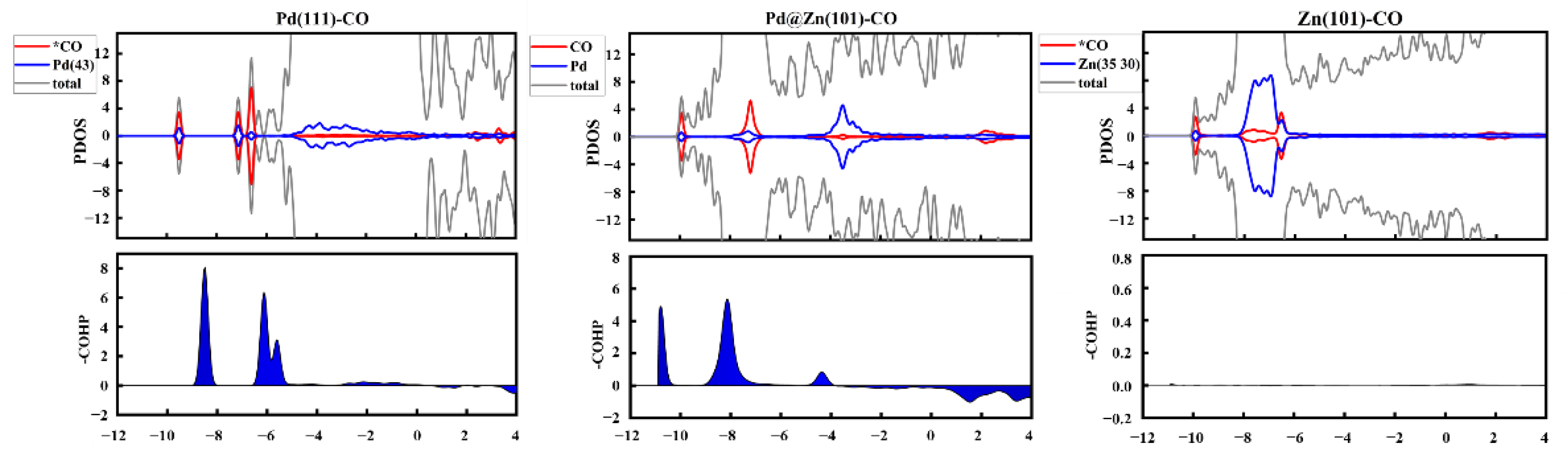
2.4. The Source of Catalytic Activity
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gao, D.; Arán-Ais, R.M.; Jeon, H.S.; Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2019, 2, 198–210. [Google Scholar] [CrossRef]
- Kowalec, I.; Kabalan, L.; Catlow, C.R.A.; Logsdail, A.J. A computational study of direct CO2 hydrogenation to methanol on Pd surfaces. Phys. Chem. Chem. Phys. 2022, 24, 9360–9373. [Google Scholar] [CrossRef]
- Zheng, M.; Zhou, X.; Zhou, Y.; Li, M. Theoretical insights into mechanisms of electrochemical reduction of CO2 to ethylene catalyzed by Pd3Au. Appl. Surf. Sci. 2022, 572, 151474. [Google Scholar] [CrossRef]
- Tierney, H.L.; Baber, A.E.; Kitchin, J.R.; Sykes, E.C.H. Hydrogen dissociation and spillover on individual isolated palladium atoms. Phys. Rev. Lett. 2009, 103, 246102. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, G.; Boucher, M.B.; Jewell, A.D.; Lewis, E.A.; Lawton, T.J.; Baber, A.E.; Tierney, H.L. Isolated Metal Atom Geometries as a Strategy for Selective Heterogeneous Hydrogenations. Science 2012, 335, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowski, M.D.; Darby, M.T.; Liu, J.; Wimble, J.M.; Lucci, F.R.; Lee, S.; Michaelides, A.; Flytzani-Stephanopoulos, M.; Stamatakis, M.; Sykes, E.C.H. Pt/Cu single-atom alloys as coke-resistant catalysts for efficient C–H activation. Nat. Chem. 2018, 10, 325–332. [Google Scholar] [CrossRef]
- Cheng, M.-J.; Clark, E.L.; Pham, H.H.; Bell, A.T.; Head-Gordon, M. Quantum mechanical screening of single-atom bimetallic alloys for the selective reduction of CO2 to C1 hydrocarbons. ACS Catal. 2016, 6, 7769–7777. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lucci, F.R.; Yang, M.; Lee, S.; Marcinkowski, M.D.; Therrien, A.J.; Williams, C.T.; Sykes, E.C.H.; Flytzani-Stephanopoulos, M. Tackling CO poisoning with single-atom alloy catalysts. J. Am. Chem. Soc. 2016, 138, 6396–6399. [Google Scholar] [CrossRef]
- Boucher, M.B.; Zugic, B.; Cladaras, G.; Kammert, J.; Marcinkowski, M.D.; Lawton, T.J.; Sykes, E.C.H.; Flytzani-Stephanopoulos, M. Single atom alloy surface analogs in Pd0.18Cu15 nanoparticles for selective hydrogenation reactions. Phys. Chem. Chem. Phys. 2013, 15, 12187–12196. [Google Scholar] [CrossRef]
- Yang, K.; Yang, B. Surface restructuring of Cu-based single-atom alloy catalysts under reaction conditions: The essential role of adsorbates. Phys. Chem. Chem. Phys. 2017, 19, 18010–18017. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Yang, B. Identification of the active and selective sites over a single Pt atom-alloyed Cu catalyst for the hydrogenation of 1,3-Butadiene: A combined DFT and microkinetic modeling study. J. Phys. Chem. C 2018, 122, 10883–10891. [Google Scholar] [CrossRef]
- Kruppe, C.M.; Krooswyk, J.D.; Trenary, M. Selective hydrogenation of acetylene to ethylene in the presence of a carbonaceous surface layer on a Pd/Cu(111) single-atom alloy. ACS Catal. 2017, 7, 8042–8049. [Google Scholar] [CrossRef]
- Da Won, H.; Shin, H.; Koh, J.; Chung, J.; Lee, H.S.; Kim, H.; Woo, S.I. Highly efficient, selective, and stable CO2 electroreduction on a hexagonal Zn catalyst. Angew. Chem. Int. Ed. 2016, 55, 9297–9300. [Google Scholar] [CrossRef]
- Qin, B.; Li, Y.; Fu, H.; Wang, H.; Chen, S.; Liu, Z.; Peng, F. Electrochemical reduction of CO2 into tunable syngas production by regulating the crystal facets of earth-abundant Zn catalyst. ACS Appl. Mater. Interfaces 2018, 10, 20530–20539. [Google Scholar] [CrossRef]
- Guo, M.; Li, X.; Huang, Y.; Li, L.; Li, J.; Lu, Y.; Xu, Y.; Zhang, L. CO2-induced fibrous Zn catalyst promotes electrochemical reduction of CO2 to CO. Catalysts 2021, 11, 477. [Google Scholar] [CrossRef]
- Low, Q.H.; Loo NW, X.; Calle-Vallejo, F.; Yeo, B.S. Enhanced electroreduction of carbon dioxide to methanol using zinc dendrites pulse-deposited on silver foam. Angew. Chem. Int. Ed. 2019, 58, 2256–2260. [Google Scholar] [CrossRef]
- Tang, Q.-L.; Duan, X.-X.; Liu, B.; Wei, A.-Q.; Liu, S.-L.; Wang, Q.; Liang, Y.-P.; Ma, X.-H. A density functional study on properties of a Cu3Zn material and CO adsorption onto its surfaces. Appl. Surf. Sci. 2016, 363, 128–139. [Google Scholar] [CrossRef]
- Moreno-Garcia, P.; Schlegel, N.; Zanetti, A.; Cedeno Lopez, A.; Galvez-Vazquez, M.J.; Dutta, A.; Rahaman, M.; Broekmann, P. Selective electrochemical reduction of CO2 to CO on Zn-based foams produced by Cu2+ and template-assisted electrodeposition. ACS Appl. Mater. Interfaces 2018, 10, 31355–31365. [Google Scholar] [CrossRef]
- Hussain, J.; Jónsson, H.; Skúlason, E. Calculations of product selectivity in electrochemical CO2 reduction. ACS Catal. 2018, 8, 5240–5249. [Google Scholar] [CrossRef]
- Wang, Y.; Papanikolaou, K.G.; Hannagan, R.T.; Patel, D.A.; Balema, T.A.; Cramer, L.A.; Kress, P.L.; Stamatakis, M.; Sykes, E.C.H. Surface facet dependence of competing alloying mechanisms. J. Chem. Phys. 2020, 153, 244702. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Esopi, M.R.; Janik, M.J.; Asthagiri, A. Selectivity of CO2 reduction on copper electrodes: The role of the kinetics of elementary steps. Angew. Chem. Int. Ed. 2013, 52, 2459–2462. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M. Accounting for bifurcating pathways in the screening for CO2 reduction catalysts. ACS Catal. 2017, 7, 7346–7351. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, J.; Peng, H.; Hong, X.; Chan, K.; Nørskov, J.K. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 2017, 8, 15438. [Google Scholar] [CrossRef] [Green Version]
- Durand, W.J.; Peterson, A.A.; Studt, F.; Abild-Pedersen, F.; Nørskov, J.K. Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces. Surf. Sci. 2011, 605, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Peterson, A.A.; Nørskov, J.K. Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Norgren, S.; Ågren, J. Enthalpies of formation of transition-metal lanthanide laves phases with the MgCu2 structure. J. Phase Equilibria 1997, 18, 441. [Google Scholar] [CrossRef]
- Ulissi, Z.W.; Tang, M.T.; Xiao, J.; Liu, X.; Torelli, D.A.; Karama, M.; Cummins, K.; Hahn, C.; Lewis, N.S.; Jaramillo, T.F.; et al. Machine-learning methods enable exhaustive searches for active bimetallic facets and reveal active site motifs for CO2 reduction. ACS Catal. 2017, 7, 6600–6608. [Google Scholar] [CrossRef] [Green Version]
- Nellaiappan, S.; Katiyar, N.K.; Kumar, R.; Parui, A.; Malviya, K.D.; Pradeep, K.G.; Singh, A.K.; Sharma, S.; Tiwary, C.S.; Biswas, K. High-entropy alloys as catalysts for the CO2 and CO reduction reactions: Experimental realization. ACS Catal. 2020, 10, 3658–3663. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef] [PubMed]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skúlason, E.; Bligaard, T.; Nørskov, J.K. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys. Rev. Lett. 2007, 99, 016105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed AG, A.; Zhou, E.; Zeng, Z.; Xie, J.; Gao, D.; Wang, Y. Asymmetric Oxo-bridged ZnPb bimetallic electrocatalysis boosting CO2-to-HCOOH reduction. Adv. Sci. 2022, 9, 2104138. [Google Scholar] [CrossRef]
- Feng, Y.; An, W.; Wang, Z.; Wang, Y.; Men, Y.; Du, Y. Electrochemical CO2 reduction reaction on M@Cu(211) bimetallic single-atom surface alloys: Mechanism, kinetics, and catalyst screening. ACS Sustain. Chem. Eng. 2019, 8, 210–222. [Google Scholar] [CrossRef]
- Xie, H.; Wan, Y.; Wang, X.; Liang, J.; Lu, G.; Wang, T.; Chai, G.; Adil, N.M.; Priest, C.; Huang, Y.; et al. Boosting Pd-catalysis for electrochemical CO2 reduction to CO on Bi-Pd single atom alloy nanodendrites. Appl. Catal. B Environ. 2021, 289, 119783. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, G. Cu-based single-atom catalysts boost electroreduction of CO2 to CH3OH: First-principles predictions. J. Phys. Chem. C 2019, 123, 4380–4387. [Google Scholar] [CrossRef]
- Li, M.; Hua, B.; Wang, L.-C.; Zhou, Z.; Stowers, K.J.; Ding, D. Discovery of single-atom alloy catalysts for CO2-to-methanol reaction by density functional theory calculations. Catal. Today 2022, 388–389, 403–409. [Google Scholar] [CrossRef]
- Shi, C.; Hansen, H.A.; Lausche, A.C.; Nørskov, J.K. Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 2014, 16, 4720–4727. [Google Scholar] [CrossRef]
- Nie, X.; Luo, W.; Janik, M.J.; Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. J. Catal. 2014, 312, 108–122. [Google Scholar] [CrossRef]
- Tsai, A.P.; Kameoka, S.; Nozawa, K.; Shimoda, M.; Ishii, Y. Intermetallic: A pseudoelement for catalysis. Acc. Chem. Res. 2017, 50, 2879–2885. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Hafner, J. Ab-initio simulations of materials using vasp: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmann, S.N.; Michel, C. How to gain atomistic insights on reactions at the water/solid interface? ACS Catal. 2022, 12, 6294–6301. [Google Scholar] [CrossRef]
- Mathew, K.; Kolluru VS, C.; Mula, S.; Steinmann, S.N.; Hennig, R.G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zheng, M.; Wang, X.; Zhou, X. Electrocatalytic Reduction of CO2 to C1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation. Catalysts 2022, 12, 1617. https://doi.org/10.3390/catal12121617
Wang Y, Zheng M, Wang X, Zhou X. Electrocatalytic Reduction of CO2 to C1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation. Catalysts. 2022; 12(12):1617. https://doi.org/10.3390/catal12121617
Chicago/Turabian StyleWang, Yixin, Ming Zheng, Xin Wang, and Xin Zhou. 2022. "Electrocatalytic Reduction of CO2 to C1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation" Catalysts 12, no. 12: 1617. https://doi.org/10.3390/catal12121617
APA StyleWang, Y., Zheng, M., Wang, X., & Zhou, X. (2022). Electrocatalytic Reduction of CO2 to C1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation. Catalysts, 12(12), 1617. https://doi.org/10.3390/catal12121617