
Chemoenzymatic Synthesis of Optically Active Alcohols Possessing 1,2,3,4-Tetrahydroquinoline Moiety Employing Lipases or Variants of the Acyltransferase from Mycobacterium smegmatis †

 , , , and
, , , and 
Abstract
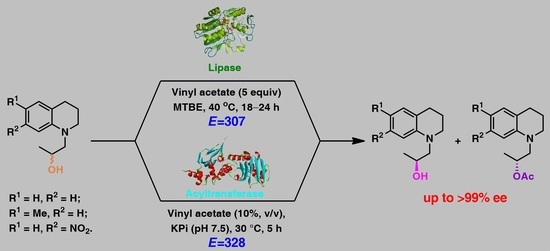
:
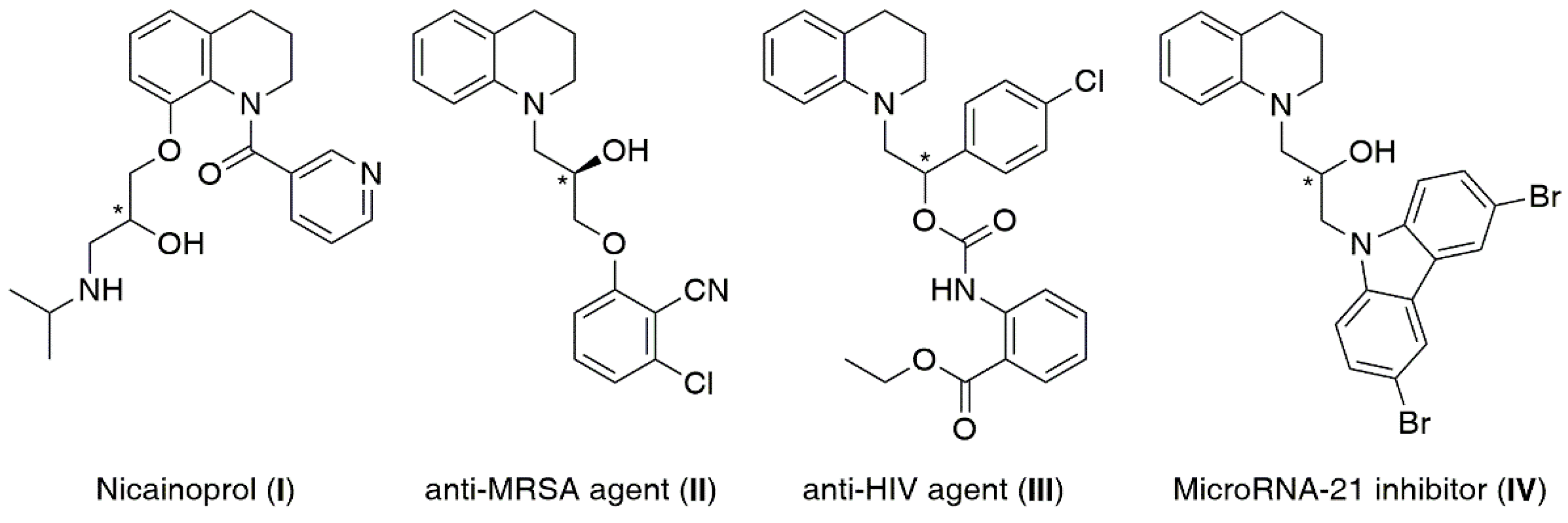
1. Introduction
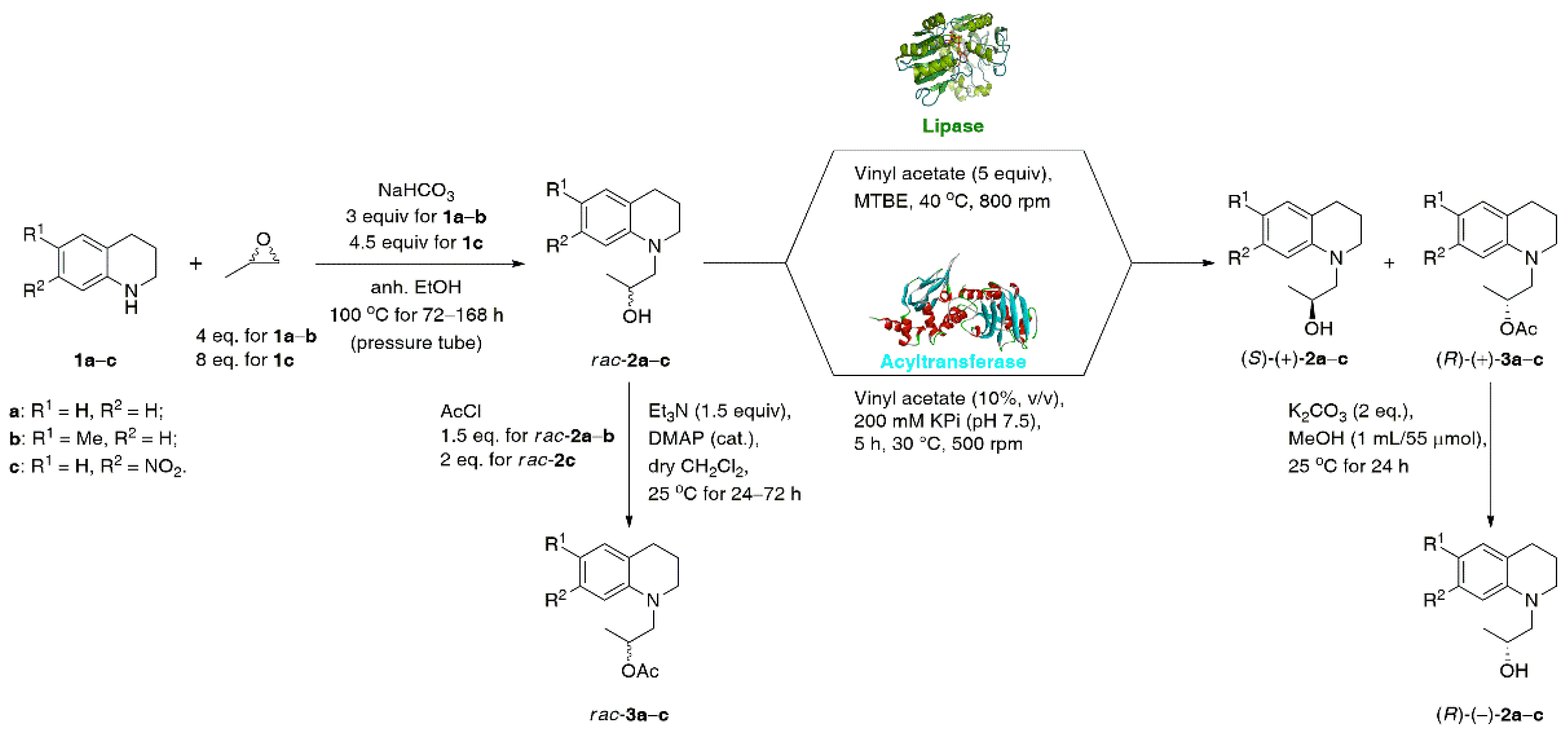
2. Results and Discussion
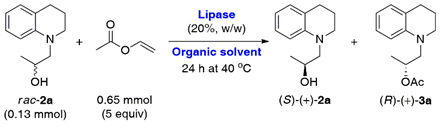
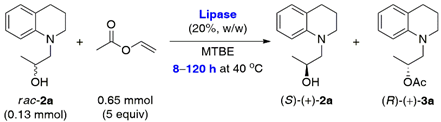
2.1. Lipase-Catalyzed KR of Racemic 1,2,3,4-Tetrahydroquinoline-Based Alcohols rac-2a–c Using Vinyl Acetate in Organic Solvents
2.2. Acyltransfarase-Catalyzed KR of Racemic 1,2,3,4-Tetrahydroquinoline-Based Alcohols rac-2a–c Using Vinyl Acetate in Water
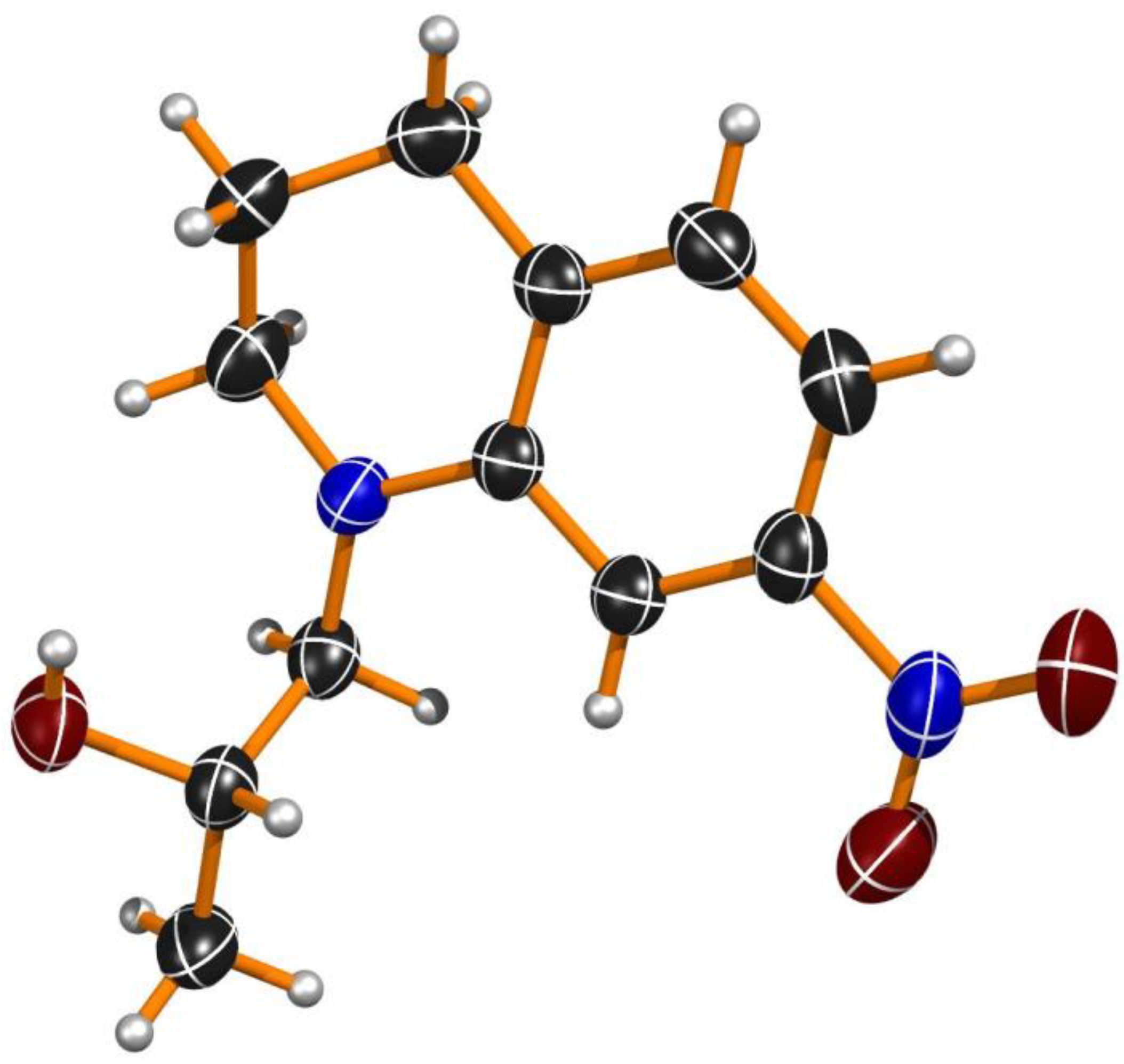
2.3. The Assignment of the Absolute Configuration of Enantiomeric Products 2a–c and 3a–c
3. Materials and Methods
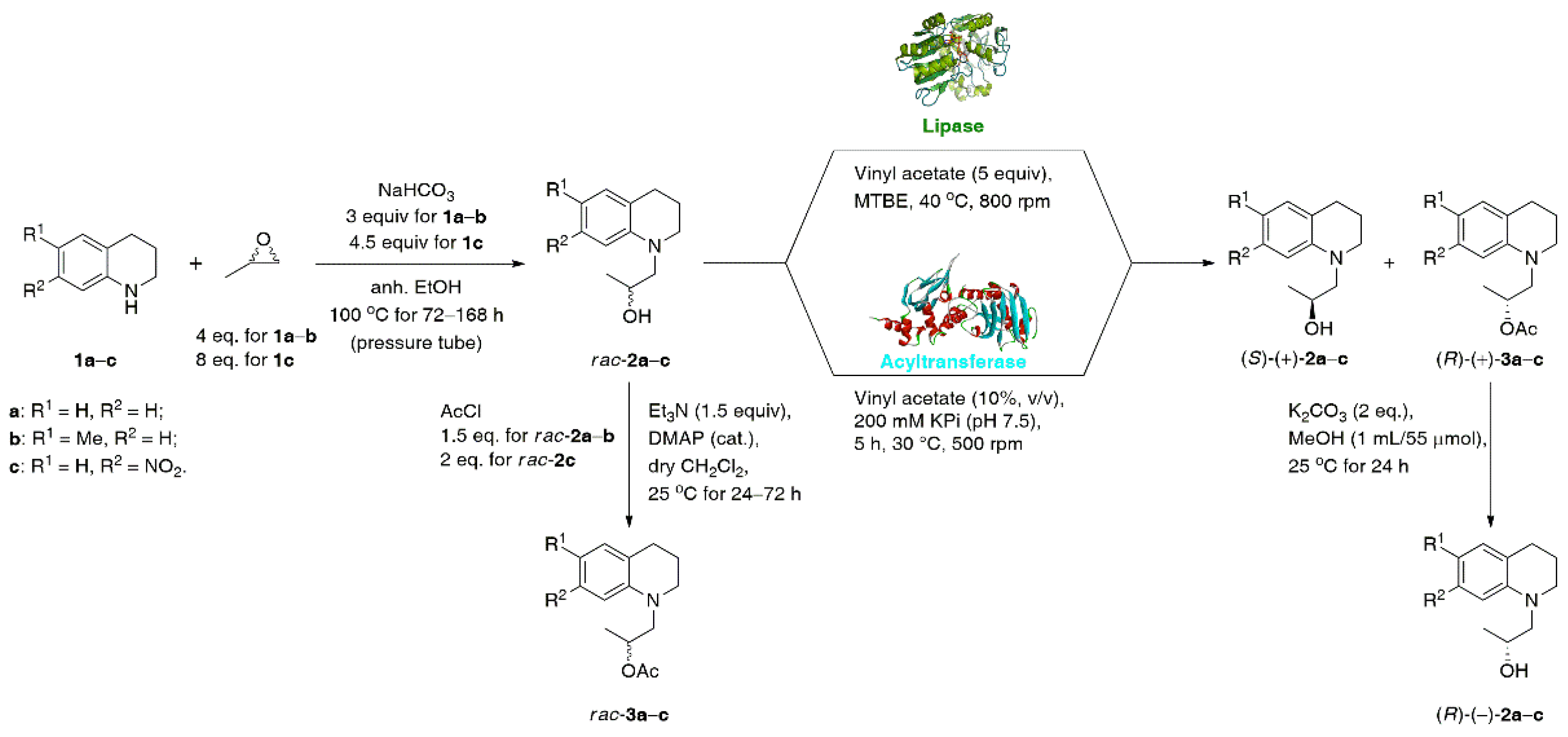
3.1. General Procedure for the Synthesis of Racemic Alcohols rac-2a–c
3.2. General Procedure for the Synthesis of Racemic Esters rac-3a–c
3.3. General Procedure for Analytical Scale Lipase-Catalyzed KR of rac-2a—Enzyme Screening
3.4. General Procedure for Analytical Scale Lipase-Catalyzed KR of rac-2a—Co-Solvent Screening
3.5. General Procedure for Analytical Scale Lipase-Catalyzed KR of rac-2a—Effect of Temperature
3.6. General Procedure for Analytical Scale Lipase-Catalyzed KR of rac-2a—Effect of Time
3.7. General Procedure for Preparative Scale Lipase-Catalyzed KR of rac-2a–c
3.8. General Procedure for K2CO3-Catalyzed Methanolysis of (R)-(+)-3a–c
3.9. General Procedure for the Synthesis of (R)-(–)-2a–c Using Commercially Available (R)-(+)-Propylene Oxide (99% ee)
3.10. Screening Conditions for MsAcT-Catalyzed KR of rac-2a–c Using Vinyl Acetate in Water
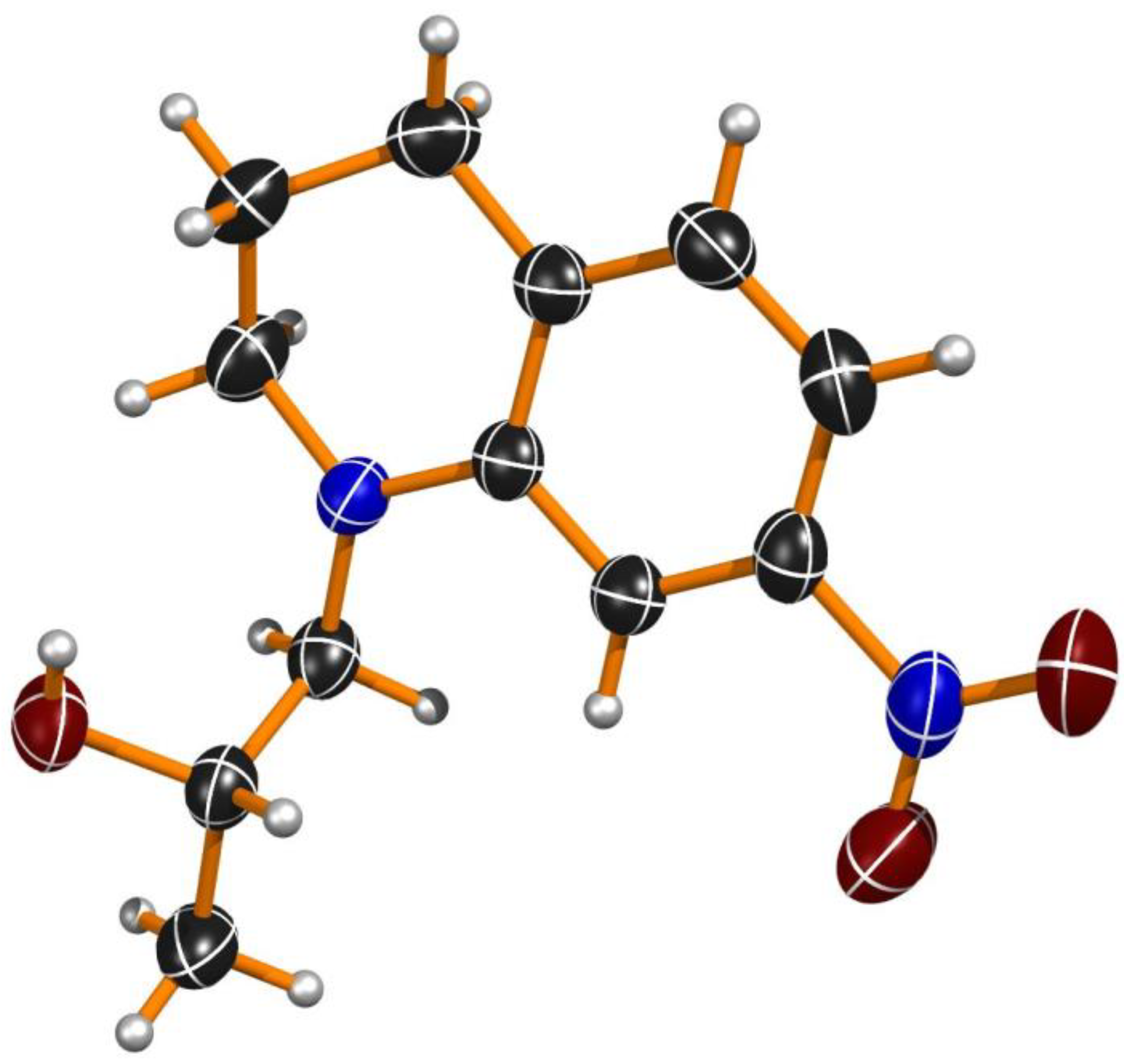
3.11. XRD Analyses
3.11.1. Conditions for Crystal Growth of (2S)-1-(7-Nitro-3,4-dihydroquinolin-1(2H)-yl)propan-2-ol ((S)-(+)-2c)
3.11.2. Crystal Structure Determination of (S)-(+)-2c
3.11.3. Crystal Data for (S)-(+)-2c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.K.; Schrittwieser, J.H.; Kroutil, W. Power of Biocatalysis for Organic Synthesis. ACS Central Sci. 2021, 7, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. [Google Scholar] [CrossRef]
- Kar, S.; Sanderson, H.; Roy, K.; Benfenati, E.; Leszczynski, J. Green chemistry in the synthesis of pharmaceuticals. Chem. Rev. 2022, 122, 3637–3710. [Google Scholar] [CrossRef] [PubMed]
- Hauer, B. Embracing nature’s catalysts: A viewpoint on the future of biocatalysis. ACS Catal. 2020, 10, 8418–8427. [Google Scholar] [CrossRef]
- Faber, K. (Ed.) Biotransformations in Organic Chemistry: A Textbook, 7th ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Abdelraheem, E.M.M.; Busch, H.; Hanefeld, U.; Tonin, F. Biocatalysis explained: From pharmaceutical to bulk chemical production. React. Chem. Eng. 2019, 4, 1878–1894. [Google Scholar] [CrossRef] [Green Version]
- Simic, S.; Zukic, E.; Schmermund, L.; Faber, K.; Winkler, C.K.; Kroutil, W. Shortening Synthetic Routes to Small Molecule Active Pharmaceutical Ingredients Employing Biocatalytic Methods. Chem. Rev. 2022, 122, 1052–1126. [Google Scholar] [CrossRef]
- Kinner, A.; Nerke, P.; Siedentop, R.; Steinmetz, T.; Classen, T.; Rosenthal, K.; Nett, M.; Pietruszka, J.; Lutz, S. Recent Advances in Biocatalysis for Drug Synthesis. Biomedicines 2022, 10, 964. [Google Scholar] [CrossRef]
- Slagman, S.; Fessner, W.D. Biocatalytic routes to anti-viral agents and their synthetic intermediates. Chem. Soc. Rev. 2021, 50, 1968–2009. [Google Scholar] [CrossRef]
- Fryszkowska, A.; Devine, P.N. Biocatalysis in drug discovery and development. Curr. Opin. Chem. Biol. 2020, 55, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Devine, P.N.; Howard, R.M.; Kumar, R.; Thompson, M.P.; Truppo, M.D.; Turner, N.J. Extending the application of biocatalysis to meet the challenges of drug development. Nature Rev. Chem. 2018, 2, 409–421. [Google Scholar] [CrossRef]
- Hoyos, P.; Pace, V.; Alcántara, A. Biocatalyzed Synthesis of Statins: A Sustainable Strategy for the Preparation of Valuable Drugs. Catalysts 2019, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- De María, P.; de Gonzalo, G.; Alcántara, A. Biocatalysis as Useful Tool in Asymmetric Synthesis: An Assessment of Recently Granted Patents (2014–2019). Catalysts 2019, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, P.; Starikov, E.B. Agricultural Biocatalysis: Enzymes in Agriculture and Industry, 1st ed.; Jenny Stanford Publishing: Dubai, United Arab Emirates, 2022. [Google Scholar]
- Aleu, J.; Bustillo, A.J.; Hernandez-Galan, R.; Collado, I.G. Biocatalysis applied to the synthesis of agrochemicals. Curr. Org. Chem. 2006, 10, 2037–2054. [Google Scholar] [CrossRef]
- Heath, R.S.; Ruscoe, R.E.; Turner, N.J. The beauty of biocatalysis: Sustainable synthesis of ingredients in cosmetics. Natural Prod. Rep. 2022, 39, 335–388. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa; Singh, R.; Arora, P.K. Microbial lipases and their industrial applications: A comprehensive review. Microb. Cell Fact. 2020, 19, 169. [Google Scholar] [CrossRef]
- Guerrand, D. Lipases industrial applications: Focus on food and agroindustries. OCL 2017, 24, D403. [Google Scholar] [CrossRef]
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Nat. Acad. Sci. USA 1985, 82, 3192–3196. [Google Scholar] [CrossRef] [Green Version]
- Carrea, G.; Riva, S. Properties and Synthetic Applications of Enzymes in Organic Solvents. Angew. Chem. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Klibanov, A.M. Improving enzymes by using them in organic solvents. Nature 2001, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Salihu, A.; Alam, M.Z. Solvent tolerant lipases: A review. Process. Biochem. 2015, 50, 86–96. [Google Scholar] [CrossRef]
- Kumar, A.; Dhar, K.; Kanwar, S.S.; Arora, P.K. Lipase catalysis in organic solvents: Advantages and applications. Biol. Proc. Online 2016, 18, 2. [Google Scholar] [CrossRef] [Green Version]
- Subileau, M.; Jan, A.H.; Drone, J.; Rutyna, C.; Perrier, V.; Dubreucq, E. What makes a lipase a valuable acyltransferase in water abundant medium? Catal. Sci. Technol. 2017, 7, 2566–2578. [Google Scholar] [CrossRef]
- Godoy, C.A.; Pardo-Tamayo, J.S.; Barbosa, O. Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks. Int. J. Mol. Sci. 2022, 23, 9933. [Google Scholar] [CrossRef]
- Verma, S.; Choudhary, R.N.; Kanadje, A.P.; Banerjee, U.C. Diversifying Arena of Drug Synthesis: In the Realm of Lipase Mediated Waves of Biocatalysis. Catalysts 2021, 11, 1328. [Google Scholar] [CrossRef]
- Contesini, F.J.; Davanço, M.G.; Borin, G.P.; Vanegas, K.G.; Cirino, J.P.G.; Melo, R.R.d.; Mortensen, U.H.; Hildén, K.; Campos, D.R.; Carvalho, P.d.O. Advances in Recombinant Lipases: Production, Engineering, Immobilization and Application in the Pharmaceutical Industry. Catalysts 2020, 10, 1032. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; González-Martínez, D.; Gotor-Fernández, V. Stereoselective biocatalysis: A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotrans. 2017, 36, 102–130. [Google Scholar] [CrossRef] [Green Version]
- Borowiecki, P.; Milner-Krawczyk, M.; Brzezińska, D.; Wielechowska, M.; Plenkiewicz, J. Synthesis and Antimicrobial Activity of Imidazolium and Triazolium Chiral Ionic Liquids. Eur. J. Org. Chem. 2013, 2013, 712–720. [Google Scholar] [CrossRef]
- Ríos-Lombardía, N.; Porcar, R.; Busto, E.; Alfonso, I.; Montejo-Bernardo, J.; García-Granda, S.; Gotor, V.; Luis, S.V.; García-Verdugo, E.; Gotor-Fernández, V. Enantiopure Triazolium Salts: Chemoenzymatic Synthesis and Applications in Organocatalysis. ChemCatChem 2011, 3, 1921–1928. [Google Scholar] [CrossRef]
- Borowiecki, P.; Poterała, M.; Maurin, J.; Wielechowska, M.; Plenkiewicz, J. Preparation and thermal stability of optically active 1,2,4-triazolium-based ionic liquids. Arkivoc 2012, 2012, 262–281. [Google Scholar] [CrossRef]
- Borowiecki, P.; Milner-Krawczyk, M.; Plenkiewicz, J. Chemoenzymatic synthesis and biological evaluation of enantiomerically enriched 1-(β-hydroxypropyl)imidazolium- and triazolium-based ionic liquids. Beilstein J. Org. Chem. 2013, 9, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucher, O.V.; Kolodiazhnaya, A.O.; Smolii, O.B.; Prisuazhnyk, D.V.; Tolmacheva, K.A.; Zaporozhets, O.A.; Moroz, Y.S.; Mykhailiuk, P.K.; Tolmachev, A.A. Enzyme-Catalyzed Kinetic Resolution of 2,2,2-Trifluoro-1-(heteroaryl)ethanols: Experimental and Docking Studies. Eur. J. Org. Chem. 2014, 2014, 7692–7698. [Google Scholar] [CrossRef]
- De Amici, M.; De Micheli, C.; Carrea, G.; Spezia, S. Chemoenzymatic synthesis of chiral isoxazole derivatives. J. Org. Chem. 2002, 54, 2646–2650. [Google Scholar] [CrossRef]
- Hapău, D.; Brem, J.; Zaharia, V. Heterocycles 30: Lipase catalyzed kinetic resolution of racemic 1-(2-aryl-4-methyl-thiazol-5-yl)ethanols. Tetrahedron Asymm. 2011, 22, 2165–2171. [Google Scholar] [CrossRef]
- Varga, A.; Naghi, M.A.; Füstös, M.; Katona, G.; Zaharia, V. Heterocycles 35. CaL-B mediated synthesis of enantiomerically pure (R)- and (S)-ethyl 3-(2-arylthiazol-4-yl)-3-hydroxypropanoates. Tetrahedron Asymm. 2014, 25, 298–304. [Google Scholar] [CrossRef]
- Łukowska-Chojnacka, E.; Bernaś, U.; Plenkiewicz, J. Lipase-catalyzed enantioseparation of alcohols containing a tetrazole ring. Tetrahedron Asymm. 2012, 23, 136–143. [Google Scholar] [CrossRef]
- Łukowska-Chojnacka, E.; Mierzejewska, J. Enzymatic hydrolysis of esters containing a tetrazole ring. Chirality 2014, 26, 811–816. [Google Scholar] [CrossRef]
- Brem, J.; Liljeblad, A.; Paizs, C.; Toşa, M.I.; Irimie, F.-D.; Kanerva, L.T. Lipases A and B from Candida antarctica in the enantioselective acylation of ethyl 3-heteroaryl-3-hydroxypropanoates: Aspects on the preparation and enantiopreference. Tetrahedron Asymm. 2011, 22, 315–322. [Google Scholar] [CrossRef]
- Brem, J.; Naghi, M.; Toşa, M.-I.; Boros, Z.; Poppe, L.; Irimie, F.-D.; Paizs, C. Lipase mediated sequential resolution of aromatic β-hydroxy esters using fatty acid derivatives. Tetrahedron Asymm. 2011, 22, 1672–1679. [Google Scholar] [CrossRef]
- Kim, B.M.; Lee, H.-Y.; Munson, P.M.; Guare, J.P.; McDonough, C. A short synthesis of 3(R)-hydroxy-2(R)-isopropyltetrahydrothiophene: A precursor to a high-affinity P2-ligand of HIV-1 protease inhibitors. Tetrahedron Lett. 1993, 34, 6517–6520. [Google Scholar] [CrossRef]
- Martin-Matute, B.; Edin, M.; Bogar, K.; Kaynak, F.B.; Backvall, J.E. Combined ruthenium(II) and lipase catalysis for efficient dynamic kinetic resolution of secondary alcohols. Insight into the racemization mechanism. J. Am. Chem. Soc. 2005, 127, 8817–8825. [Google Scholar] [CrossRef] [PubMed]
- Ball, A.J.; Corr, S.; Micklefield, J. Lipase-catalysed kinetic resolutions of secondary alcohols in pressurised liquid hydrofluorocarbons. Tetrahedron Lett. 2009, 50, 3543–3546. [Google Scholar] [CrossRef]
- Hara, P.; Turcu, M.-C.; Sundell, R.; Toşa, M.; Paizs, C.; Irimie, F.-D.; Kanerva, L.T. Lipase-catalyzed asymmetric acylation in the chemoenzymatic synthesis of furan-based alcohols. Tetrahedron Asymm. 2013, 24, 142–150. [Google Scholar] [CrossRef]
- Ferreira, D.S.P.; Ferreira, J.G.; Filho, E.F.S.; Princival, J.L. Tuning lipase-catalysed kinetic resolution of 2-substituted thiophenes and furans: A scalable chemoenzymatic route to masked γ-bis-oxo-alcohols. J. Mol. Catal. B Enzym. 2016, 126, 37–45. [Google Scholar] [CrossRef]
- Cheedrala, R.K.; Sachwani, R.; Krishna, P.R. Lipase mediated kinetic resolution of benzimidazolyl ethanols. Tetrahedron Asymm. 2008, 19, 901–905. [Google Scholar] [CrossRef]
- Łukowska-Chojnacka, E.; Staniszewska, M.; Bondaryk, M.; Maurin, J.K.; Bretner, M. Lipase-Catalyzed Kinetic Resolution of Novel Antifungal N-Substituted Benzimidazole Derivatives. Chirality 2016, 28, 347–354. [Google Scholar] [CrossRef]
- Pchełka, B.K.; Loupy, A.; Petit, A. Preparation of various enantiomerically pure (benzotriazol-1-yl)- and (benzotriazol-2-yl)-alkan-2-ols. Tetrahedron Asymm. 2006, 17, 2516–2530. [Google Scholar] [CrossRef]
- Wawro, A.M.; Wielechowska, M.; Bretner, M. Synthesis of new optically pure tetrabromobenzotriazole derivatives via lipase-catalyzed transesterification. J. Mol. Catal. B Enzym. 2013, 87, 44–50. [Google Scholar] [CrossRef]
- Borowiecki, P.; Fabisiak, M.; Ochal, Z. Lipase-catalyzed kinetic resolution of 1-(1,3-benzothiazol-2-ylsulfanyl)propan-2-ol with antifungal activity: A comparative study of transesterification versus hydrolysis. Tetrahedron 2013, 69, 4597–4602. [Google Scholar] [CrossRef]
- Naghi, M.A.; Bencze, L.C.; Brem, J.; Paizs, C.; Irimie, F.D.; Toşa, M.I. Sequential enzymatic procedure for the preparation of enantiomerically pure 2-heteroaryl-2-hydroxyacetic acids. Tetrahedron Asymm. 2012, 23, 181–187. [Google Scholar] [CrossRef]
- Büyükadalı, N.N.; Aslan, N.; Gümüş, S.; Gümüş, A. Stereoselective synthesis of benzofuran and benzothiophene substituted dihydropyran derivatives via ring closing metathesis. Tetrahedron Asymm. 2016, 27, 954–959. [Google Scholar] [CrossRef]
- Paizs, C.; Toşa, M.; Majdik, C.; Moldovan, P.; Novák, L.; Kolonits, P.; Marcovici, A.; Irimie, F.-D.; Poppe, L. Optically active 1-(benzofuran-2-yl)ethanols and ethane-1,2-diols by enantiotopic selective bioreductions. Tetrahedron Asymm. 2003, 14, 1495–1501. [Google Scholar] [CrossRef]
- Nagy, B.; Dima, N.; Paizs, C.; Brem, J.; Irimie, F.D.; Tosa, M.I. New chemo-enzymatic approaches for the synthesis of (R)- and (S)-bufuralol. Tetrahedron Asymm. 2014, 25, 1316–1322. [Google Scholar] [CrossRef]
- Büyükadalı, N.N.; Seven, S.; Aslan, N.; Yenidede, D.; Gümüş, A. Chemoenzymatic synthesis of novel 1,4-disubstituted 1,2,3-triazole derivatives from 2-heteroaryl substituted homopropargyl alcohols. Tetrahedron Asymm. 2015, 26, 1285–1291. [Google Scholar] [CrossRef]
- Villar-Barro, Á.; Gotor, V.; Brieva, R. Enzymatic resolution of five membered heterocyclic bromohydrins. Tetrahedron Asymm. 2013, 24, 694–698. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernández, V.; Gotor, V. Chemoenzymatic synthesis of chiral 4-(N,N-dimethylamino)pyridine derivatives. Tetrahedron Asymm. 2005, 16, 3427–3435. [Google Scholar] [CrossRef]
- López-Iglesias, M.; González-Martínez, D.; Rodríguez-Mata, M.; Gotor, V.; Busto, E.; Kroutil, W.; Gotor-Fernández, V. Asymmetric Biocatalytic Synthesis of Fluorinated Pyridines through Transesterification or Transamination: Computational Insights into the Reactivity of Transaminases. Adv. Synth. Catal. 2017, 359, 279–291. [Google Scholar] [CrossRef]
- Solares, L.F.; Lavandera, I.; Gotor-Fernández, V.; Brieva, R.; Gotor, V. Biocatalytic preparation of enantioenriched 3,4-dihydroxypiperidines and theoretical study of Candida antarctica lipase B enantioselectivity. Tetrahedron 2006, 62, 3284–3291. [Google Scholar] [CrossRef]
- Banoth, L.; Narayan, T.K.; Banerjee, U.C. New chemical and chemo-enzymatic routes for the synthesis of (R,S)- and (S)-enciprazine. Tetrahedron Asymm. 2012, 23, 1272–1278. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, J.A.; Quijada, F.J.; Brieva, R.; Rebolledo, F.; Gotor, V. Chemoenzymatic synthesis of orthogonally protected (3R,4R)- and (3S,4S)-trans-3-amino-4-hydroxypyrrolidines. Tetrahedron 2013, 69, 5407–5412. [Google Scholar] [CrossRef]
- Van den Heuvel, M.; Cuiper, A.D.; van der Deen, H.; Kellogg, R.M.; Feringa, B.L. Optically active 6-acetyloxy-2H-pyran-3(6H)-one obtained by lipase catalyzed transesterification and esterification. Tetrahedron Lett. 1997, 38, 1655–1658. [Google Scholar] [CrossRef] [Green Version]
- Alatorre-Santamaría, S.; Gotor-Fernández, V.; Gotor, V. Stereoselective synthesis of optically active cyclic α- and β-amino esters through lipase-catalyzed transesterification or interesterification processes. Tetrahedron Asymm. 2010, 21, 2307–2313. [Google Scholar] [CrossRef]
- Paizs, C.; Tähtinen, P.; Toşa, M.; Majdik, C.; Irimie, F.-D.; Kanerva, L.T. Biocatalytic enantioselective preparation of phenothiazine-based cyanohydrin acetates: Kinetic and dynamic kinetic resolution. Tetrahedron 2004, 60, 10533–10540. [Google Scholar] [CrossRef]
- Brem, J.; Toşa, M.-I.; Paizs, C.; Munceanu, A.; Matković-Čalogović, D.; Irimie, F.-D. Lipase-catalyzed kinetic resolution of racemic 1-(10-alkyl-10H-phenothiazin-3-yl)ethanols and their butanoates. Tetrahedron Asymm. 2010, 21, 1993–1998. [Google Scholar] [CrossRef]
- Brem, J.; Toşa, M.I.; Paizs, C.; Vass, E.; Irimie, F.D. Enzyme-catalyzed synthesis of (R)- and (S)-3-hydroxy-3-(10-alkyl-10H-phenothiazin-3-yl)propanoic acids. Tetrahedron Asymm. 2010, 21, 365–373. [Google Scholar] [CrossRef]
- Brem, J.; Pilbák, S.; Paizs, C.; Bánóczi, G.; Irimie, F.-D.; Toşa, M.-I.; Poppe, L. Lipase-catalyzed kinetic resolutions of racemic 1-(10-ethyl-10H-phenothiazin-1,2, and 4-yl)ethanols and their acetates. Tetrahedron Asymm. 2011, 22, 916–923. [Google Scholar] [CrossRef]
- Borowiecki, P.; Paprocki, D.; Dranka, M. First chemoenzymatic stereodivergent synthesis of both enantiomers of promethazine and ethopropazine. Beilstein J. Org. Chem. 2014, 10, 3038–3055. [Google Scholar] [CrossRef] [Green Version]
- Schoffers, E.; Kohler, L. Efficient alcoholysis of 5,6-dihydro-1,10-phenanthroline-5,6-epoxide with ytterbium(III) triflate and subsequent enantioselective transesterification with lipases. Tetrahedron Asymm. 2009, 20, 1879–1902. [Google Scholar] [CrossRef] [Green Version]
- Borowiecki, P.; Paprocki, D.; Dudzik, A.; Plenkiewicz, J. Chemoenzymatic Synthesis of Proxyphylline Enantiomers. J. Org. Chem. 2016, 81, 380–395. [Google Scholar] [CrossRef]
- Borowiecki, P.; Mlynek, M.; Dranka, M. Chemoenzymatic synthesis of enantiomerically enriched diprophylline and xanthinol nicotinate. Bioorg. Chem. 2021, 106, 104448. [Google Scholar] [CrossRef] [PubMed]
- Cvetovich, R.J.; Chartrain, M.; Hartner, F.W., Jr.; Roberge, C.; Amato, J.S.; Grabowski, E.J. An Asymmetric Synthesis of L-694,458, a Human Leukocyte Elastase Inhibitor, via Novel Enzyme Resolution of beta-Lactam Esters. J. Org. Chem. 1996, 61, 6575–6580. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, T.; Kitano, K.; Matsubara, J.; Kawano, Y.; Komatsu, M.; Uchida, M.; Tabusa, F.; Nagao, Y. Efficient Synthesis of Optically Active 4,5-Dihydroxy-2,3,4,5-tetrahydro-1H-1-benzazepine Derivatives Utilizing Lipase-Catalyzed Transesterification. Heterocycles 2006, 69, 333. [Google Scholar] [CrossRef]
- Gotor, V.; Limeres, F.; García, R.; Bayod, M.; Brieva, R. Enzymatic resolution of (±)-6-(5-chloropyridin-2-yl)-6-vinyloxy-carbonyloxy-6,7-dihydro[5H]pyrrolo[3,4-b]pyrazin-5-one. Synthesis of (+)-zopiclone. Tetrahedron Asymm. 1997, 8, 995–997. [Google Scholar] [CrossRef]
- Palomo, J.M.; Mateo, C.; Fernández-Lorente, G.; Solares, L.F.; Diaz, M.; Sánchez, V.c.M.; Bayod, M.; Gotor, V.; Guisan, J.M.; Fernandez-Lafuente, R. Resolution of (±)-5-substituted-6-(5-chloropyridin-2-yl)-7-oxo-5,6-dihydropyrrolo[3,4b]pyrazine derivatives-precursors of (S)-(+)-Zopiclone, catalyzed by immobilized Candida antarctica B lipase in aqueous media. Tetrahedron Asymm. 2003, 14, 429–438. [Google Scholar] [CrossRef]
- Gelo, M.; Šunjić, V. Lipase Catalyzed Enantioselective Hydrolysis of (±)-2-Acetoxy-1-chloro-3-phthalimidopropane. Synthesis 1993, 1993, 855–857. [Google Scholar] [CrossRef]
- Borowiecki, P.; Zdun, B.; Popow, N.; Wiklinska, M.; Reiter, T.; Kroutil, W. Development of a novel chemoenzymatic route to enantiomerically enriched beta-adrenolytic agents. A case study toward propranolol, alprenolol, pindolol, carazolol, moprolol, and metoprolol. RSC Adv. 2022, 12, 22150–22160. [Google Scholar] [CrossRef]
- Imarah, A.O.; Silva, F.M.W.G.; Tuba, L.; Malta-Lakó, Á.; Szemes, J.; Sánta-Bell, E.; Poppe, L. A Convenient U-Shape Microreactor for Continuous Flow Biocatalysis with Enzyme-Coated Magnetic Nanoparticles-Lipase-Catalyzed Enantiomer Selective Acylation of 4-(Morpholin-4-yl)butan-2-ol. Catalysts 2022, 12, 1065. [Google Scholar] [CrossRef]
- Borowiecki, P. Chemoenzymatic Synthesis of Optically Active Ethereal Analog of iso-Moramide—A Novel Potentially Powerful Analgesic. Int. J. Mol. Sci. 2022, 23, 11803. [Google Scholar] [CrossRef]
- Borowiecki, P.; Dranka, M.; Ochal, Z. Lipase-Catalyzed Kinetic Resolution of N-Substituted 1-(β-Hydroxypropyl)indoles by Enantioselective Acetylation. Eur. J. Org. Chem. 2017, 2017, 5378–5390. [Google Scholar] [CrossRef]
- Borowiecki, P.; Justyniak, I.; Ochal, Z. Lipase-catalyzed kinetic resolution approach toward enantiomerically enriched 1-(β-hydroxypropyl)indoles. Tetrahedron Asymm. 2017, 28, 1717–1732. [Google Scholar] [CrossRef]
- Gotor-Fernández, V.; Fernández-Torres, P.; Gotor, V. Chemoenzymatic preparation of optically active secondary amines: A new efficient route to enantiomerically pure indolines. Tetrahedron Asymm. 2006, 17, 2558–2564. [Google Scholar] [CrossRef]
- Alatorre-Santamaria, S.; Rodriguez-Mata, M.; Gotor-Fernandez, V.; de Mattos, M.C.; Sayago, F.J.; Jimenez, A.I.; Cativiela, C.; Gotor, V. Efficient access to enantiomerically pure cyclic alpha-amino esters through a lipase-catalyzed kinetic resolution. Tetrahedron Asymm. 2009, 19, 1714–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Iglesias, M.; Busto, E.; Gotor, V.; Gotor-Fernandez, V. Stereoselective synthesis of 2,3-disubstituted indoline diastereoisomers by chemoenzymatic processes. J. Org. Chem. 2012, 77, 8049–8055. [Google Scholar] [CrossRef]
- López-Iglesias, M.; Arizpe, A.; Sayago, F.J.; Gotor, V.; Cativiela, C.; Gotor-Fernández, V. Lipase-catalyzed dynamic kinetic resolution of dimethyl (1,3-dihydro-2H-isoindol-1-yl)phosphonate. Tetrahedron 2016, 72, 7311–7316. [Google Scholar] [CrossRef]
- Busto, E.; Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V. Chemoenzymatic Asymmetric Synthesis of Serotonin Receptor Agonist (R)-Frovatriptan. Eur. J. Org. Chem. 2013, 2013, 4057–4064. [Google Scholar] [CrossRef]
- Mathews, I.; Soltis, M.; Saldajeno, M.; Ganshaw, G.; Sala, R.; Weyler, W.; Cervin, M.A.; Whited, G.; Bott, R. Structure of a novel enzyme that catalyzes acyl transfer to alcohols in aqueous conditions. Biochemistry 2007, 46, 8969–8979. [Google Scholar] [CrossRef]
- Müller, H.; Terholsen, H.; Godehard, S.P.; Badenhorst, C.P.S.; Bornscheuer, U.T. Recent Insights and Future Perspectives on Promiscuous Hydrolases/Acyltransferases. ACS Catal. 2021, 11, 14906–14915. [Google Scholar] [CrossRef]
- Cannazza, P.; Donzella, S.; Pellis, A.; Contente, M.L. Mycobacterium smegmatis acyltransferase: The big new player in biocatalysis. Biotechnol. Adv. 2022, 59, 107985. [Google Scholar] [CrossRef]
- Szymańska, K.; Odrozek, K.; Zniszczoł, A.; Torrelo, G.; Resch, V.; Hanefeld, U.; Jarzębski, A.B. MsAcT in siliceous monolithic microreactors enables quantitative ester synthesis in water. Catal. Sci. Technol. 2016, 6, 4882–4888. [Google Scholar] [CrossRef]
- Mestrom, L.; Claessen, J.G.R.; Hanefeld, U. Enzyme-Catalyzed Synthesis of Esters in Water. ChemCatChem 2019, 11, 2004–2010. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, F.; Contente, M.L.; Pinna, C.; Tamborini, L.; Pinto, A. Biocatalyzed Flow Oxidation of Tyrosol to Hydroxytyrosol and Efficient Production of Their Acetate Esters. Antioxidants 2021, 10, 1142. [Google Scholar] [CrossRef] [PubMed]
- Contente, M.L.; Tamborini, L.; Molinari, F.; Paradisi, F. Aromas flow: Eco-friendly, continuous, and scalable preparation of flavour esters. J. Flow Chem. 2020, 10, 235–240. [Google Scholar] [CrossRef]
- Finnveden, M.; Semlitsch, S.; He, O.; Martinelle, M. Mono-substitution of symmetric diesters: Selectivity of Mycobacterium smegmatis acyltransferase variants. Catal. Sci. Technol. 2019, 9, 4920–4927. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, N.; Torrelo, G.; Bisterfeld, C.; Resch, V.; Mestrom, L.; Straulino, E.; van der Weel, L.; Hanefeld, U. Ester Synthesis in Water: Mycobacterium smegmatis Acyl Transferase for Kinetic Resolutions. Adv. Synth. Catal. 2018, 360, 242–249. [Google Scholar] [CrossRef]
- Jost, E.; Kazemi, M.; Mrkonjić, V.; Himo, F.; Winkler, C.K.; Kroutil, W. Variants of the Acyltransferase from Mycobacterium smegmatis Enable Enantioselective Acyl Transfer in Water. ACS Catal. 2020, 10, 10500–10507. [Google Scholar] [CrossRef]
- Sridharan, V.; Suryavanshi, P.A.; Menendez, J.C. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, S.; Gupta, G.; Chaturvedi, V.; Sinha, S.; Srivastava, R. Natural product inspired diversity oriented synthesis of tetrahydroquinoline scaffolds as antitubercular agent. ACS Comb. Sci. 2011, 13, 65–71. [Google Scholar] [CrossRef]
- Nammalwar, B.; Bunce, R.A. Recent syntheses of 1,2,3,4-tetrahydroquinolines, 2,3-dihydro-4(1H)-quinolinones and 4(1H)-quinolinones using domino reactions. Molecules 2013, 19, 204–232. [Google Scholar] [CrossRef] [Green Version]
- Chander, S.; Ashok, P.; Zheng, Y.T.; Wang, P.; Raja, K.S.; Taneja, A.; Murugesan, S. Design, synthesis and in-vitro evaluation of novel tetrahydroquinoline carbamates as HIV-1 RT inhibitor and their antifungal activity. Bioorg. Chem. 2016, 64, 66–73. [Google Scholar] [CrossRef]
- Connelly, C.M.; Boer, R.E.; Moon, M.H.; Gareiss, P.; Schneekloth, J.S., Jr. Discovery of Inhibitors of MicroRNA-21 Processing Using Small Molecule Microarrays. ACS Chem. Biol. 2017, 12, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ju, Y.; Li, C.; Yang, T.; Deng, Y.; Luo, Y. Design, synthesis, and antibacterial evaluation of novel derivatives of NPS-2143 for the treatment of methicillin-resistant S. aureus (MRSA) infection. J. Antibiot. 2019, 72, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Borowiecki, P.; Zdun, B.; Dranka, M. Chemoenzymatic enantioselective and stereo-convergent syntheses of lisofylline enantiomers via lipase-catalyzed kinetic resolution and optical inversion approach. Mol. Catal. 2021, 504, 111451. [Google Scholar] [CrossRef]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 1982, 104, 7294–7299. [Google Scholar] [CrossRef]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef]
- Janseen, A.E.; Van der Padt, A.; Van Sonsbeek, H.M.; Van’t Riet, K. The effect of organic solvents on the equilibrium position of enzymatic acylglycerol synthesis. Biotechnol. Bioeng. 1993, 41, 95–103. [Google Scholar] [CrossRef]
- Kvittingen, L. Some aspects of biocatalysis in organic solvents. Tetrahedron 1994, 50, 8253–8274. [Google Scholar] [CrossRef]
- Sakurai, T.; Margolin, A.L.; Russell, A.J.; Klibanov, A.M. Control of enzyme enantioselectivity by the reaction medium. J. Am. Chem. Soc. 1988, 110, 7236–7237. [Google Scholar] [CrossRef]
- Parida, S.; Dordick, J.S. Substrate Structure and Solvent Hydrophobicity Control Lipase Catalysis and Enantioselectivity in Organic Media. J. Am. Chem. Soc. 1991, 113, 2253–2259. [Google Scholar] [CrossRef]
- Nakamura, K.; Takebe, Y.; Kitayama, T.; Ohno, A. Effect of solvent structure on enantioselectivity of lipase-catalyzed transesterification. Tetrahedron Lett. 1991, 32, 4941–4944. [Google Scholar] [CrossRef]
- Fitzpatrick, P.A.; Klibanov, A.M. How Can the Solvent Affect Enzyme Enantioselectivity. J. Am. Chem. Soc. 1991, 113, 3166–3171. [Google Scholar] [CrossRef]
- Wescott, C.R.; Noritomi, H.; Klibanov, A.M. Rational control of enzymatic enantioselectivity through solvation thermodynamics. J. Am. Chem. Soc. 1996, 118, 10365–10370. [Google Scholar] [CrossRef]
- Janssen, A.E.; Van der Padt, A.; Riet, K.V. Solvent effects on lipase-catalyzed esterification of glycerol and fatty acids. Biotechnol. Bioeng. 1993, 42, 953–962. [Google Scholar] [CrossRef]
- Ke, T.; Wescott, C.R.; Klibanov, A.M. Prediction of the solvent dependence of enzymatic prochiral selectivity by means of structure-based thermodynamic calculations. J. Am. Chem. Soc. 1996, 118, 3366–3374. [Google Scholar] [CrossRef]
- Hirata, H.; Higuchi, K.; Yamashina, T. Lipase-catalyzed transesterification in organic solvent: Effects of water and solvent, thermal stability and some applications. J. Biotechnol. 1990, 14, 157–167. [Google Scholar] [CrossRef]
- Hirose, Y.; Kariya, K.; Sasaki, I.; Kurono, Y.; Ebiike, H.; Achiwa, K. Drastic solvent effect on lipase-catalyzed enantioselective hydrolysis of prochiral 1,4-dihydropyridines. Tetrahedron Lett. 1992, 33, 7157–7160. [Google Scholar] [CrossRef]
- Franken, B.; Eggert, T.; Jaeger, K.E.; Pohl, M. Mechanism of acetaldehyde-induced deactivation of microbial lipases. BMC Biochem. 2011, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Marsden, S.R.; Mestrom, L.; McMillan, D.G.G.; Hanefeld, U. Thermodynamically and Kinetically Controlled Reactions in Biocatalysis–from Concepts to Perspectives. ChemCatChem 2019, 12, 426–437. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Berbasov, D.O. Self-disproportionation of enantiomers of (R)-ethyl 3-(3,5-dinitrobenzamido)-4,4,4-trifluorobutanoate on achiral silica gel stationary phase. J. Fluor. Chem. 2006, 127, 597–603. [Google Scholar] [CrossRef]
- Soloshonok, V.A. Remarkable amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. 2006, 45, 766–769. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Borkowski, M.; Strzelecka, D.; Kielar, K.; Kicińska, W.; Karevych, S.; Jasiński, R.; Demchuk, O.M. A General Phenomenon of Spontaneous Amplification of Optical Purity under Achiral Chromatographic Conditions. Symmetry 2019, 11, 680. [Google Scholar] [CrossRef] [Green Version]
- Amabilino, D.B.; Kellogg, R.M. Spontaneous Deracemization. Israel J. Chem. 2011, 51, 1034–1040. [Google Scholar] [CrossRef]
- Godehard, S.P.; Badenhorst, C.P.S.; Müller, H.; Bornscheuer, U.T. Protein Engineering for Enhanced Acyltransferase Activity, Substrate Scope, and Selectivity of the Mycobacterium smegmatis Acyltransferase MsAcT. ACS Catal. 2020, 10, 7552–7562. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Weissfloch, A.N.E.; Rappaport, A.T.; Cuccia, L.A. A rule to predict which enantiomer of a secondary alcohol reacts faster in reactions catalyzed by cholesterol esterase, lipase from Pseudomonas cepacia, and lipase from Candida rugosa. J. Org. Chem. 2002, 56, 2656–2665. [Google Scholar] [CrossRef]
- CRYSALISPRO Software System; Rigaku: Oxford, UK, 2022.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Sec. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sec. C Struct. Chem. 2015, 71, 3–8. [Google Scholar]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Cryst. Sec. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. Sec. D, Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Crystallogr. 2008, 41, 96–103. [Google Scholar] [CrossRef]
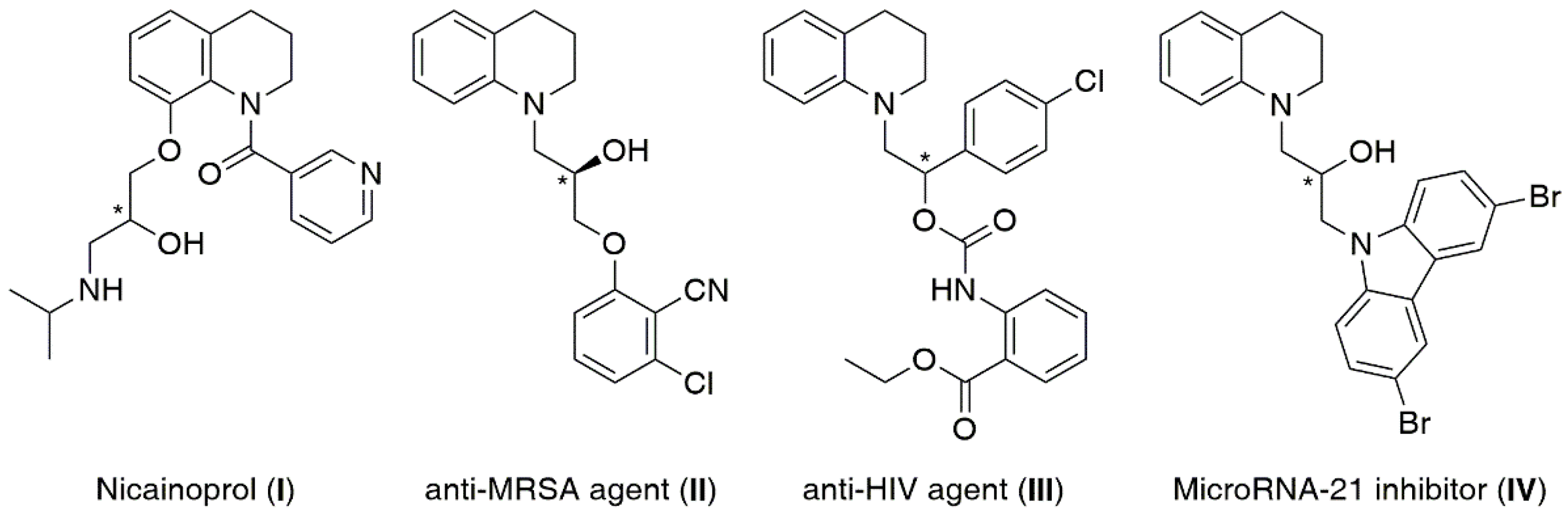

{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Lipase Preparation a | Conv. (%) b | ees (%) c | eep (%) c | E d |
| 1 | Novozym 435 | 66 | >99 | 51 | 20 |
| 2 | Novozym 435-STREM | 75 | >99 | 34 | 12 |
| 3 | Lipozyme 435 | 71 | >99 | 40 | 15 |
| 4 | Chirazyme L-2, C-2 | 61 | >99 | 65 | 33 |
| 5 | Chirazyme L-2, C-3 | 54 | >99 | 87 | 107 |
| 6 | Lipozyme TL IM | 35 | 48 | 89 | 28 |
| 7 | Lipozyme RM IM | 20 | 25 | 98 | 126 |
| 8 | Amano PS-IM | 45 | 79 | 98 | 240 |
| 9 | Amano PS | <10 | N.D. e | N.D. e | N.D. e |
| 10 | Amano AK | 0 | N.D. e | N.D. e | N.D. e |
| 11 | Lipase from Candida rugosa Type VII | 0 | N.D. e | N.D. e | N.D. e |
| 12 | Lipase from wheat germ Type I | 0 | N.D. e | N.D. e | N.D. e |
| 13 | Lipase from Rhizopus niveus | 0 | N.D. e | N.D. e | N.D. e |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Lipase Preparation a | Solvent (log P) b | Conv. (%) c | ees (%) d | eep (%) d | E e |
| 1 | Chirazyme L-2, C-3 | Acetone (0.20) | 23 | 29 | 97 | 87 |
| 2 | EtOAc (0.29) | 46 | 81 | 94 | 81 | |
| 3 | Vinyl acetate (0.54) | 35 | 52 | 97 | 111 | |
| 4 | 2-MeTHF (0.72) | 50 | 93 | 93 | 94 | |
| 5 | MTBE (0.96) | 48 | 88 | 96 | 143 | |
| 6 | tert-Amyl alcohol (1.09) | 50 | 94 | 96 | 175 | |
| 7 | PhCH3 (2.52) | 22 | 27 | 96 | 64 | |
| 8 | Amano PS-IM | Acetone (0.20) | 5 | 5 | 98 | 104 |
| 9 | EtOAc (0.29) | 12 | 13 | >99 | 226 | |
| 10 | Vinyl acetate (0.54) | 16 | 19 | >99 | 240 | |
| 11 | 2-MeTHF (0.72) | 10 | 11 | >99 | 222 | |
| 12 | MTBE (0.96) | 31 | 44 | >99 | 307 | |
| 13 | tert-Amyl alcohol (1.09) | 17 | 20 | >99 | 242 | |
| 14 | PhCH3 (2.52) | 21 | 26 | >99 | 257 | |
 | |||||
|---|---|---|---|---|---|
| Entry | Temperature (°C) a | Conv. (%) b | ees (%) c | eep (%) c | E d |
| 1 | 40 | 47 | 87 | 98 | 283 |
| 2 | 50 | 49 | 94 | 97 | 235 |
| 3 | 60 | 48 | 89 | 97 | 198 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Lipase Preparation a | Time (h) | Conv. (%) b | ees (%) c | eep (%) c | E d |
| 1 | Novozym 435 | 8 | 43 | 70 | 93 | 58 |
| 2 | 12 | 52 | 95 | 87 | 53 | |
| 3 | 18 | 57 | >99 | 74 | 34 | |
| 4 | Amano PS-IM | 24 | 31 | 44 | >99 | 307 |
| 5 | 72 | 47 | 87 | 98 | 283 | |
| 6 | 120 | 50 | 97 | 97 | 278 | |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate a | Lipase | Time (h) | Conv. (%) b | ees (%) c/Yield (%) d | eep (%) c/Yield (%) d | E e |
| 1 | rac-2a | Novozym 435 | 18 | 54 | 98/39 | 82/48 | 46 |
| 2 | Amano PS-IM | 24 | 28 | 36/59 | 99/25 | 283 | |
| 3 | rac-2b | Novozym 435 | 18 | 62 | >99/29 | 62/49 | 21 |
| 4 | Amano PS-IM | 24 | 26 | 35/51 | 98/23 | 140 | |
| 5 | rac-2c | Novozym 435 | 18 | 60 | >99/35 | 67/52 | 24 |
| 6 | Amano PS-IM | 24 | 46 | 79/49 | 94/40 | 78 | |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate a | MsAcT | Conv. (%) b | ees (%) c | eep (%) c | E d |
| 1 | rac-2a | F154V | 16 | 16 | 84 | 13 |
| 2 | F154A | 29 | 39 | 95 | 57 | |
| 3 | F150A/F154A | 51 | 98 | 94 | 149 | |
| 4 | F150V/F154V | 37 | 52 | 88 | 26 | |
| 5 | F154V/F174V | 44 | 75 | 94 | 73 | |
| 6 | rac-2b | F154V | 13 | 14 | 93 | 32 |
| 7 | F154A | 34 | 50 | 99 | 328 | |
| 8 | F150A/F154A | 48 | 89 | 97 | 198 | |
| 9 | F150V/F154V | 17 | 19 | 93 | 33 | |
| 10 | F154V/F174V | 29 | 38 | 95 | 57 | |
| 11 | rac-2c | F154V | 16 | 14 | 76 | 8 |
| 12 | F154A | 22 | 26 | 91 | 27 | |
| 13 | F150A/F154A | 58 | 76 | 55 | 8 | |
| 14 | F150V/F154V | 39 | 7 | 11 | 1 | |
| 15 | F154V/F174V | 32 | 17 | 36 | 2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zdun, B.; Kopińska, I.; Dranka, M.; Reiter, T.; Kroutil, W.; Borowiecki, P. Chemoenzymatic Synthesis of Optically Active Alcohols Possessing 1,2,3,4-Tetrahydroquinoline Moiety Employing Lipases or Variants of the Acyltransferase from Mycobacterium smegmatis . Catalysts 2022, 12, 1610. https://doi.org/10.3390/catal12121610
Zdun B, Kopińska I, Dranka M, Reiter T, Kroutil W, Borowiecki P. Chemoenzymatic Synthesis of Optically Active Alcohols Possessing 1,2,3,4-Tetrahydroquinoline Moiety Employing Lipases or Variants of the Acyltransferase from Mycobacterium smegmatis . Catalysts. 2022; 12(12):1610. https://doi.org/10.3390/catal12121610
Chicago/Turabian StyleZdun, Beata, Izabela Kopińska, Maciej Dranka, Tamara Reiter, Wolfgang Kroutil, and Paweł Borowiecki. 2022. "Chemoenzymatic Synthesis of Optically Active Alcohols Possessing 1,2,3,4-Tetrahydroquinoline Moiety Employing Lipases or Variants of the Acyltransferase from Mycobacterium smegmatis " Catalysts 12, no. 12: 1610. https://doi.org/10.3390/catal12121610
APA StyleZdun, B., Kopińska, I., Dranka, M., Reiter, T., Kroutil, W., & Borowiecki, P. (2022). Chemoenzymatic Synthesis of Optically Active Alcohols Possessing 1,2,3,4-Tetrahydroquinoline Moiety Employing Lipases or Variants of the Acyltransferase from Mycobacterium smegmatis . Catalysts, 12(12), 1610. https://doi.org/10.3390/catal12121610