


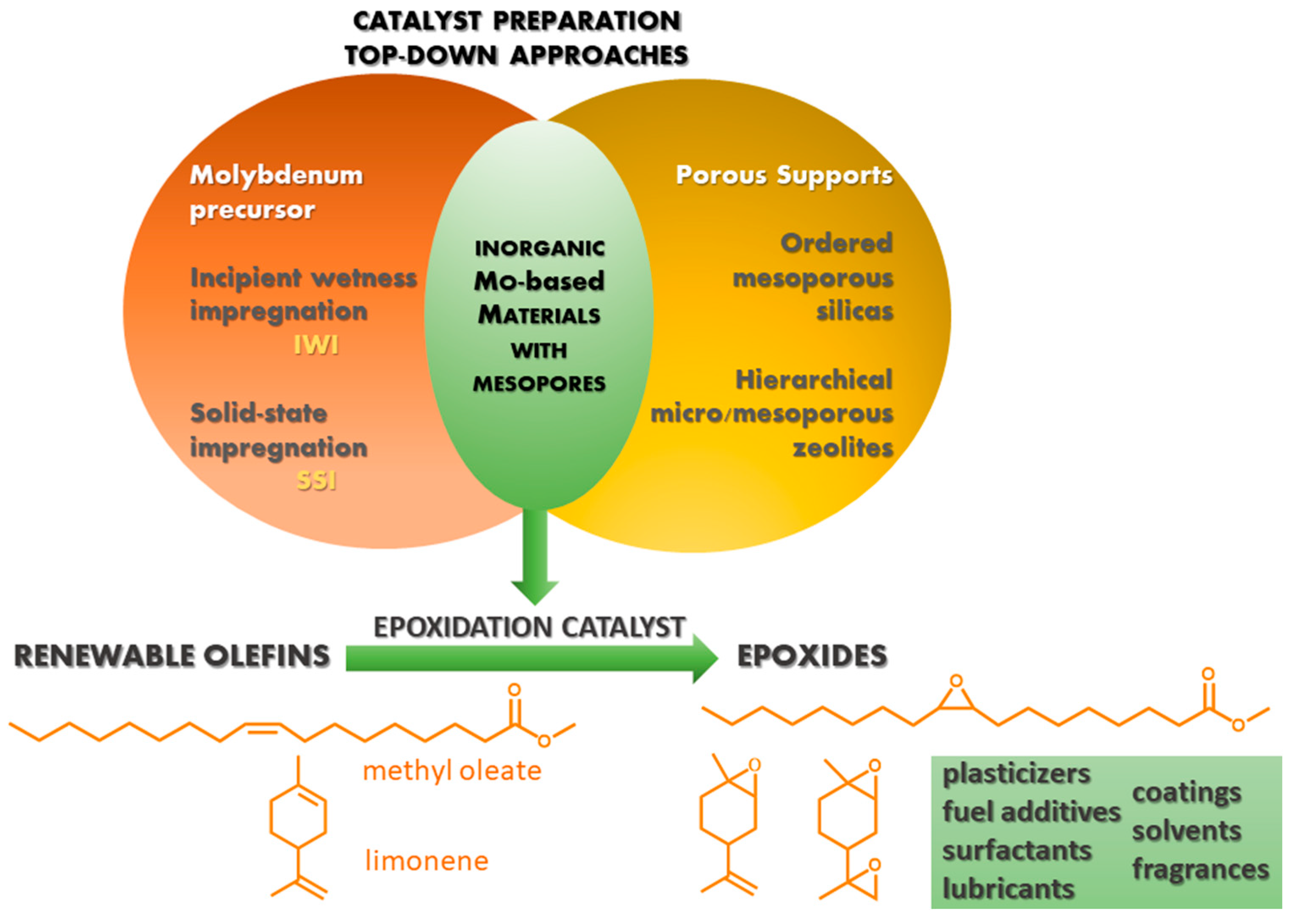
Post-Synthesis Strategies to Prepare Mesostructured and Hierarchical Silicates for Liquid Phase Catalytic Epoxidation

 , , , , and
, , , , and 
Abstract
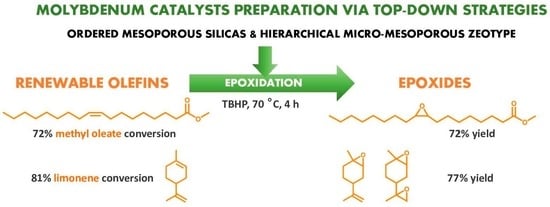
:
1. Introduction
2. Results and Discussion
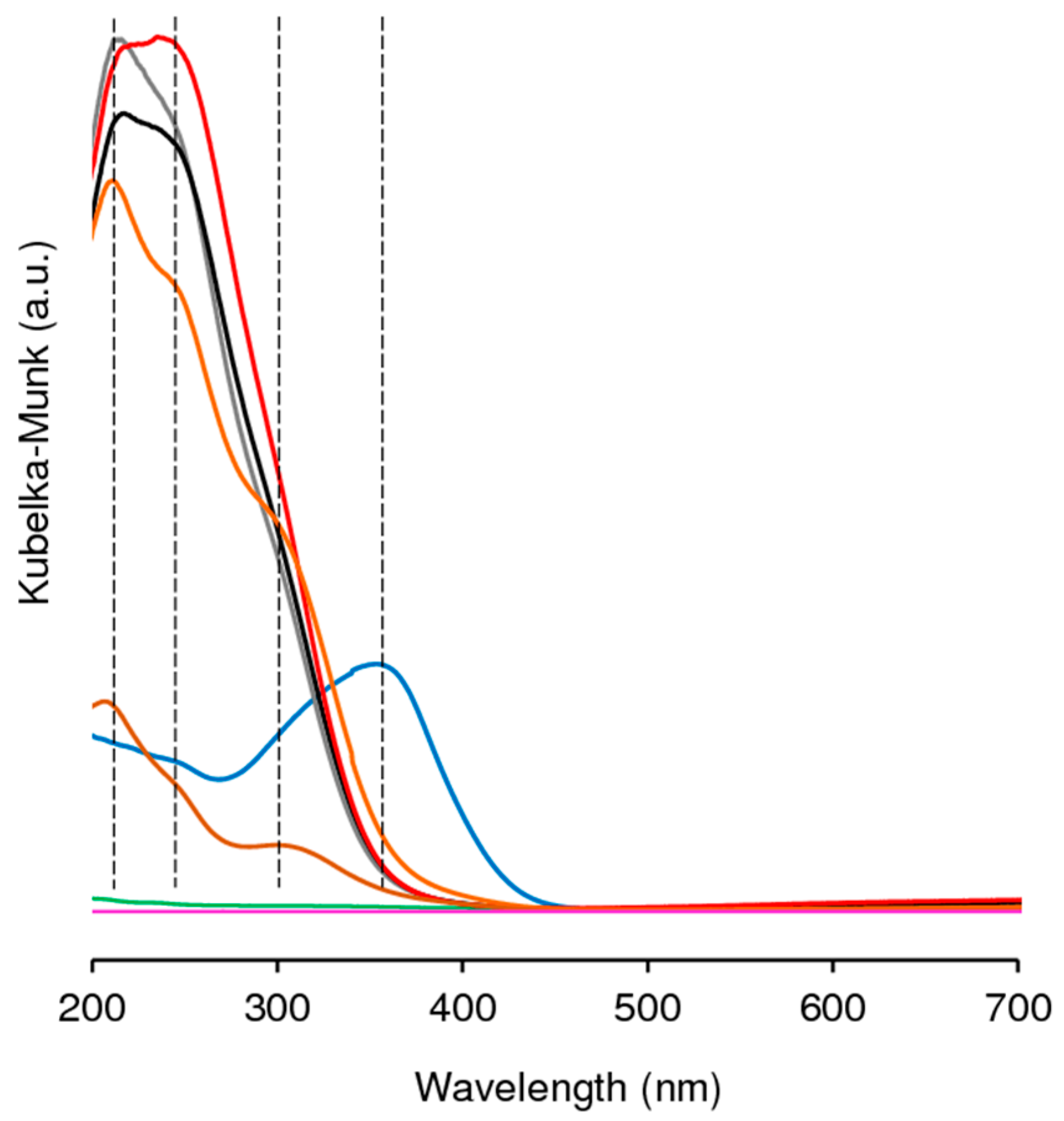
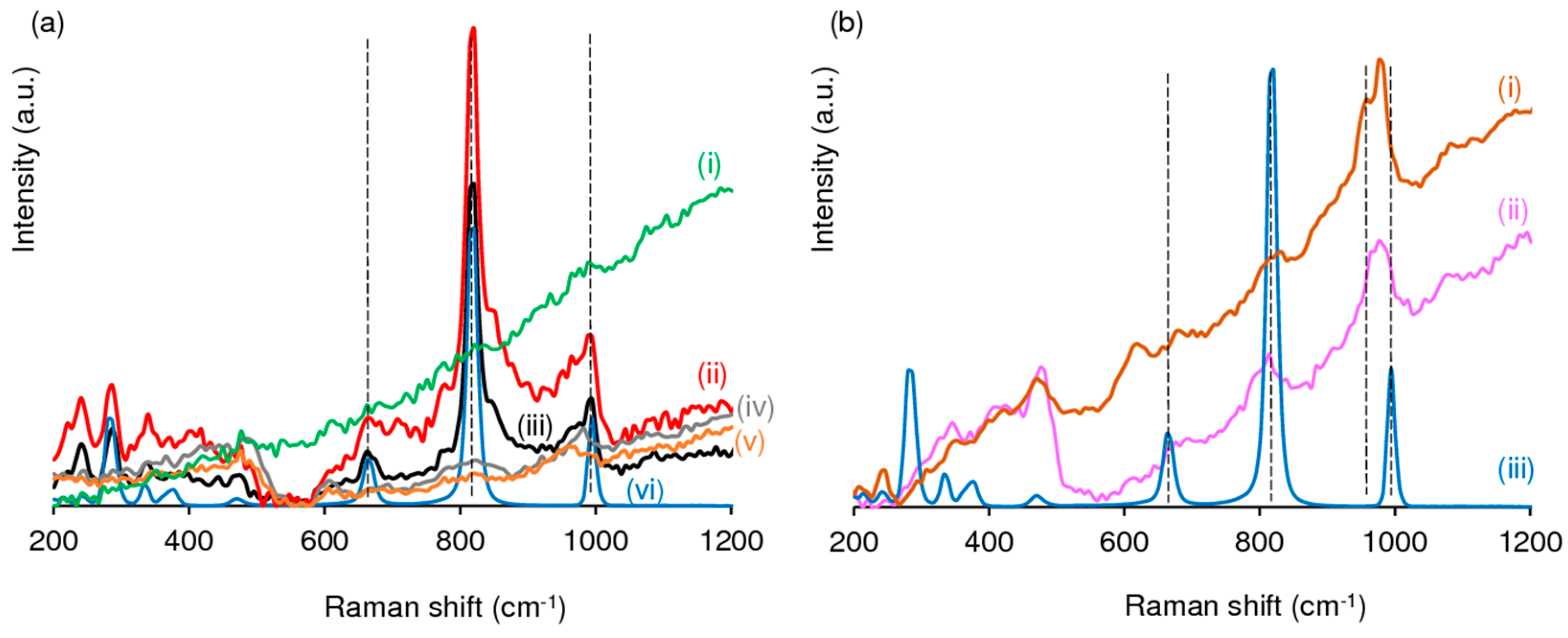
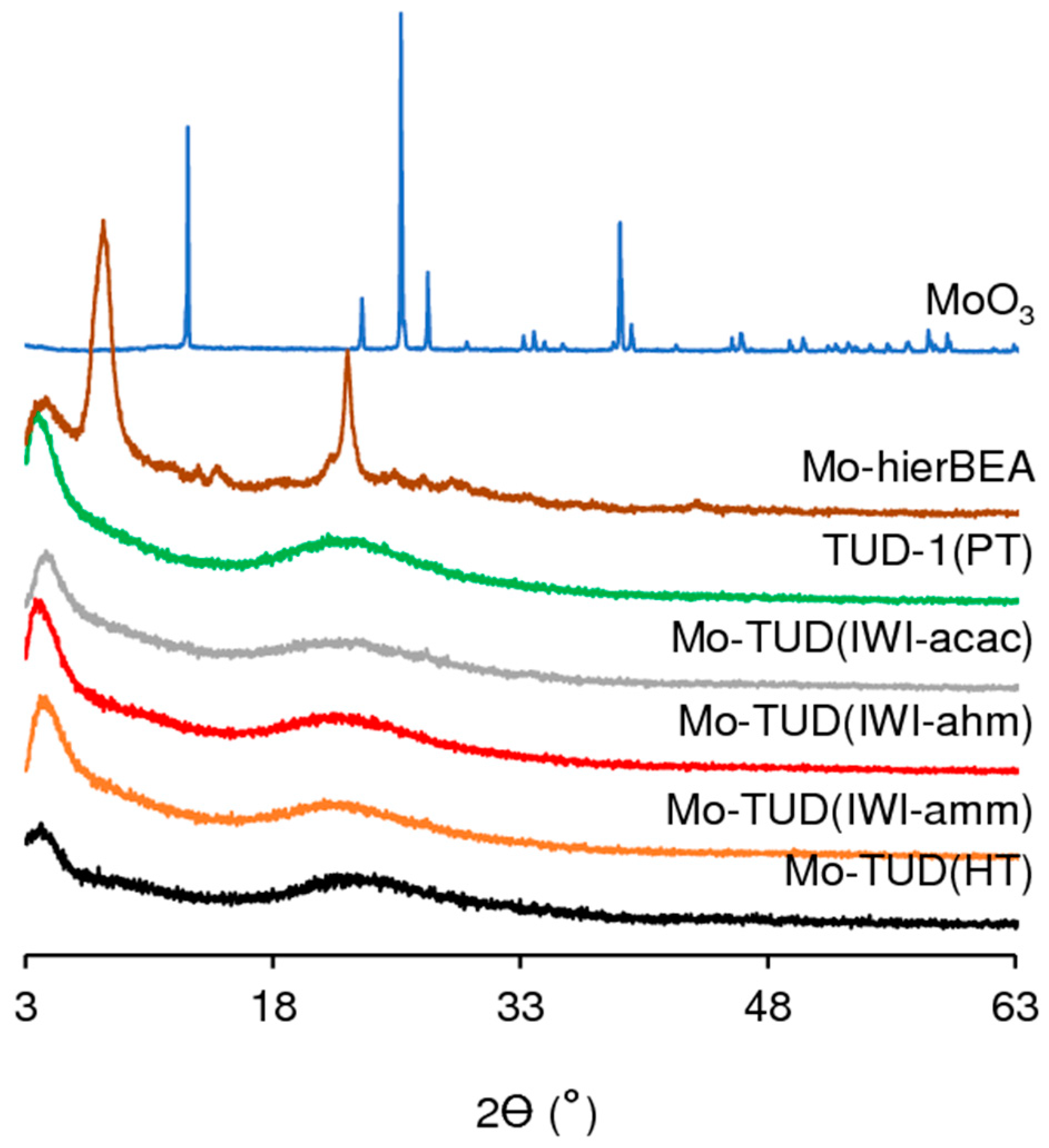
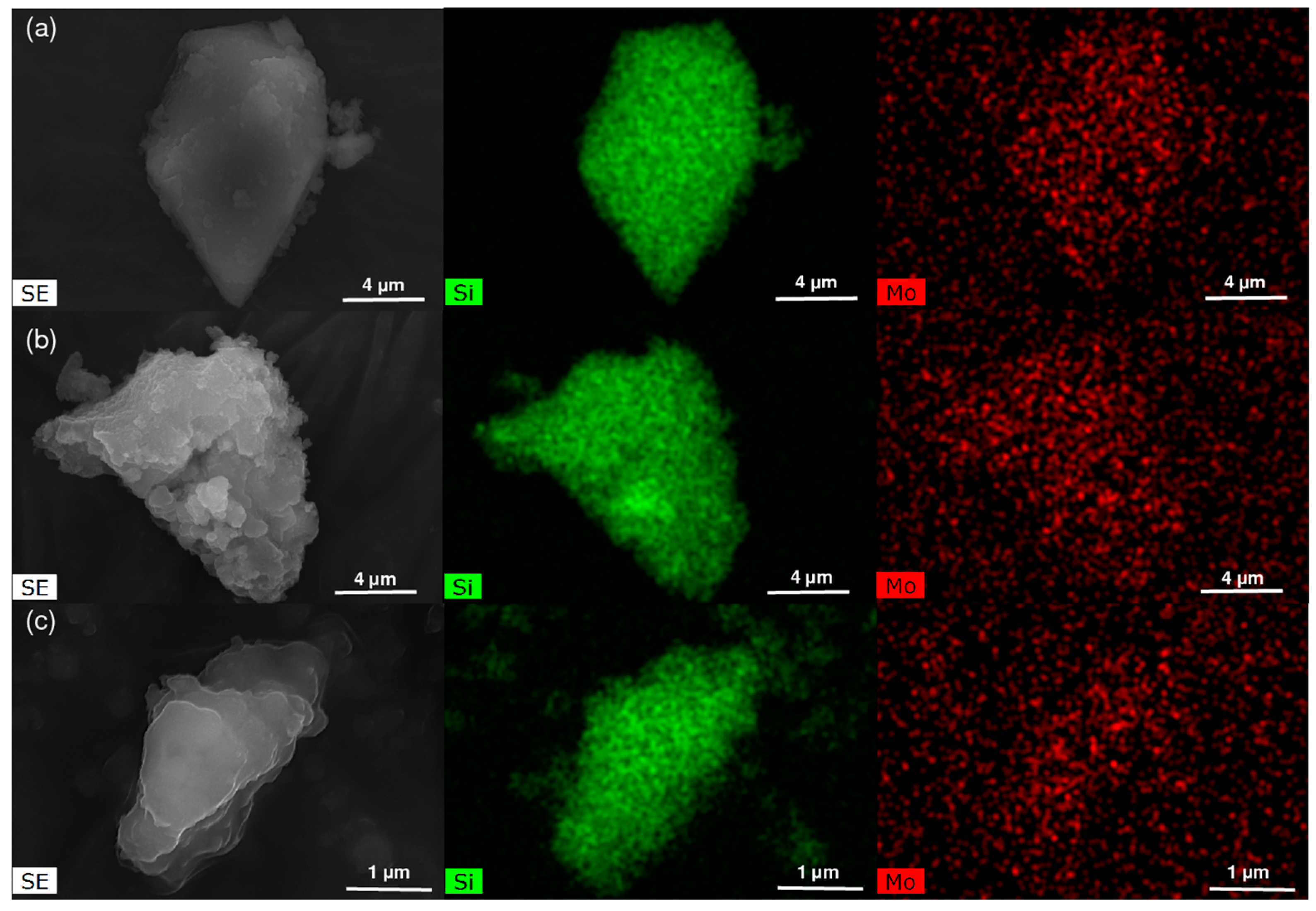
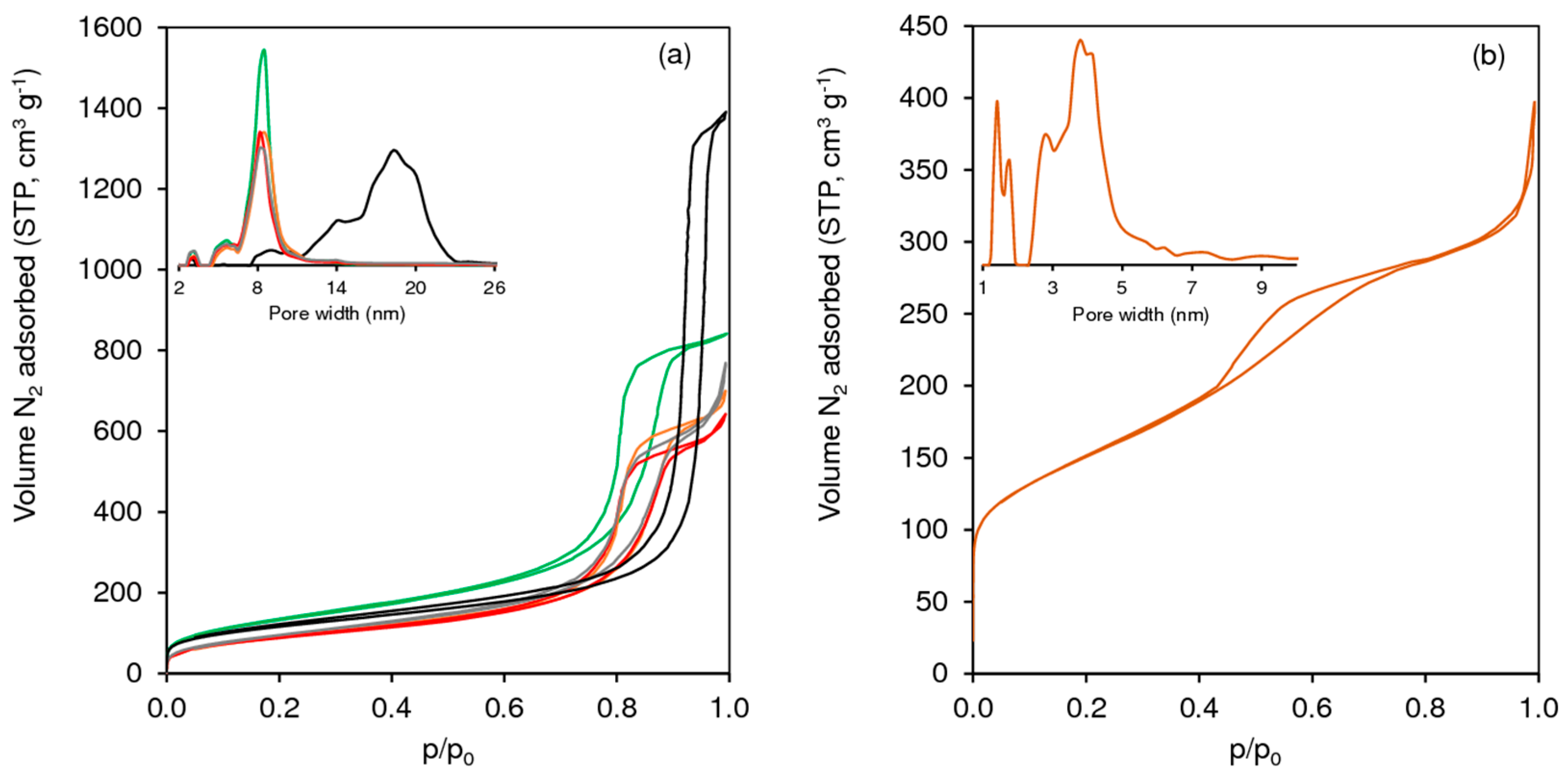
2.1. Characterization of the Materials
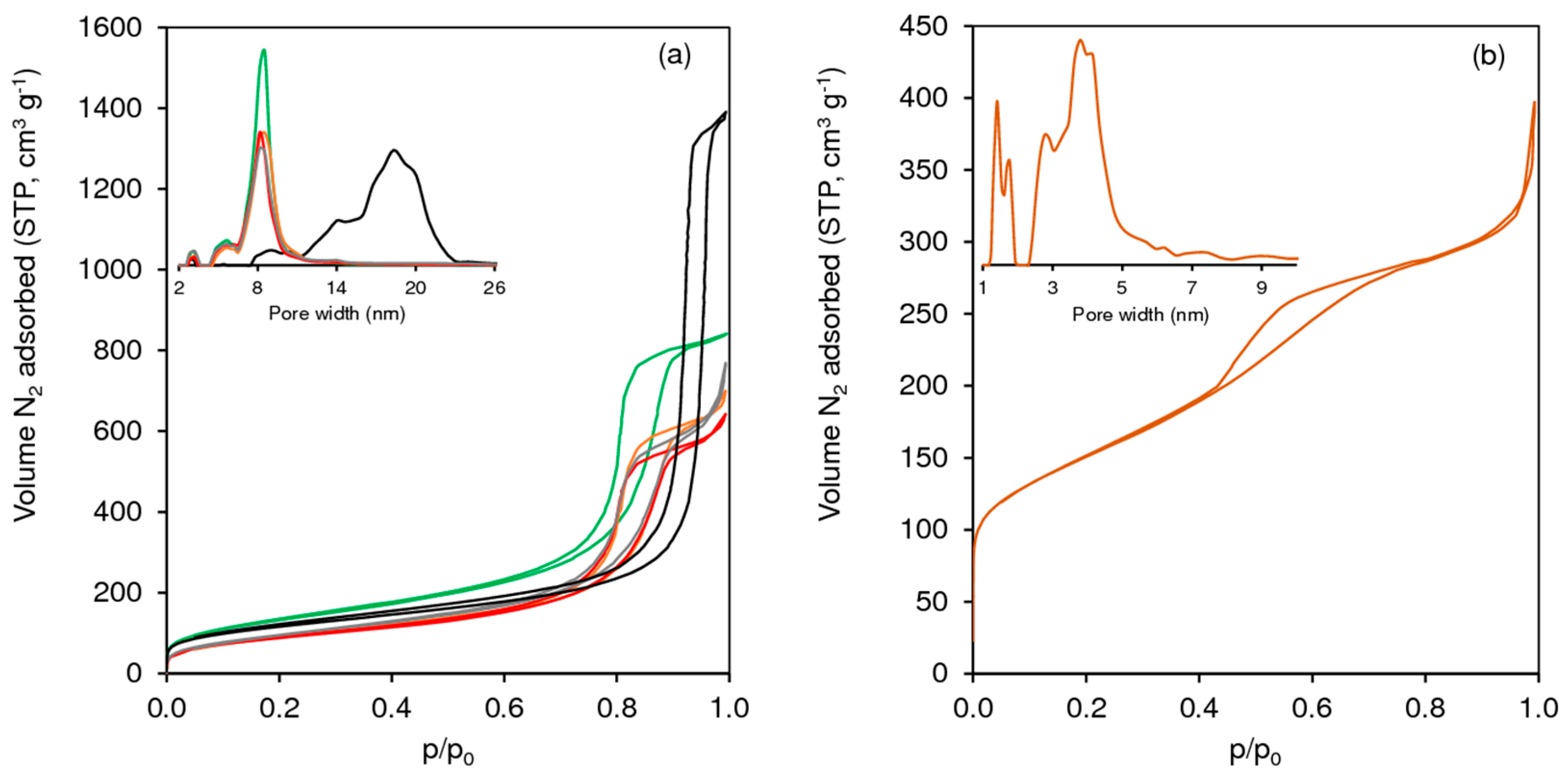
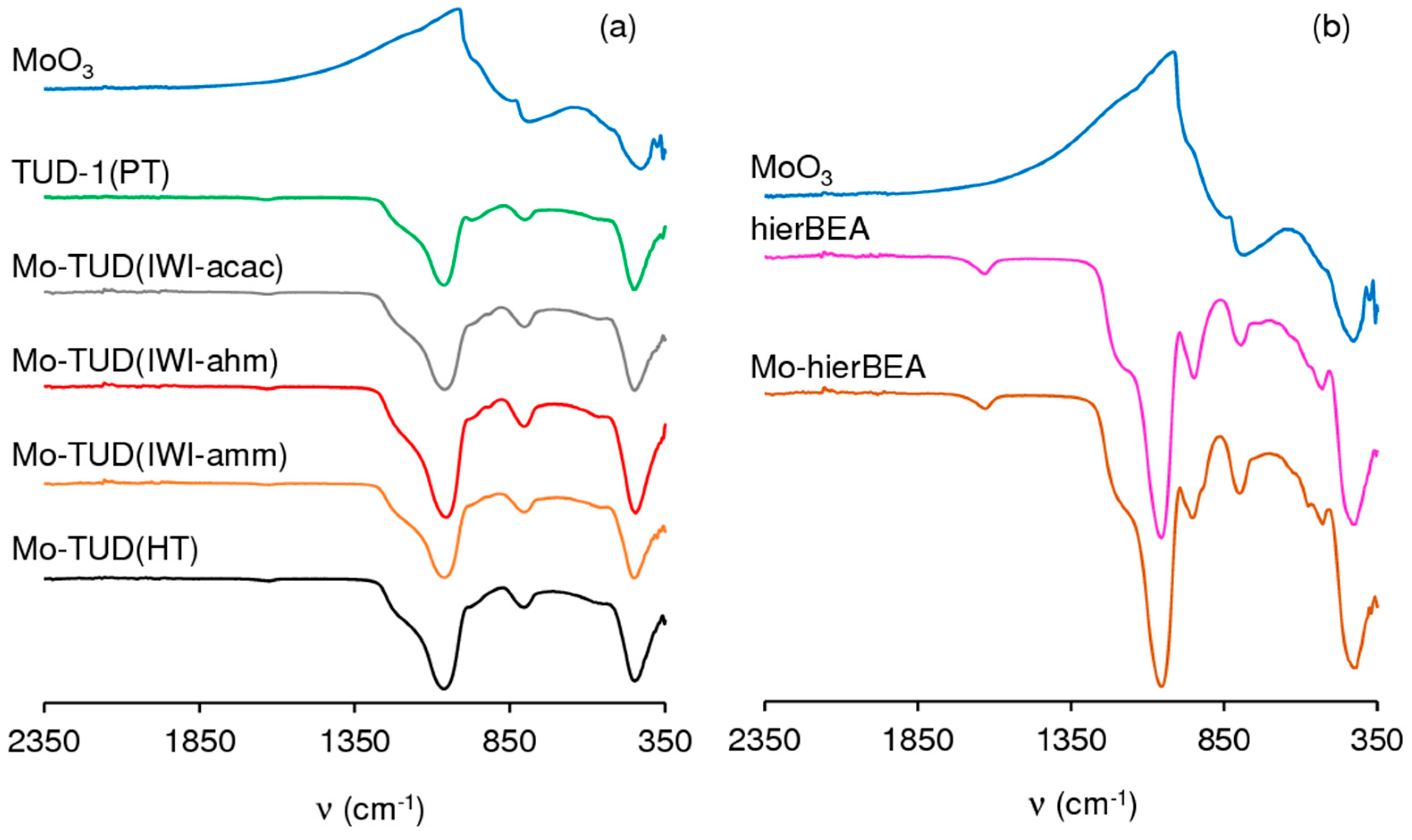
2.1.1. Ordered Mesoporous TUD-1 Type Materials
2.1.2. Hierarchical Micro-Mesoporous BEA Zeotype
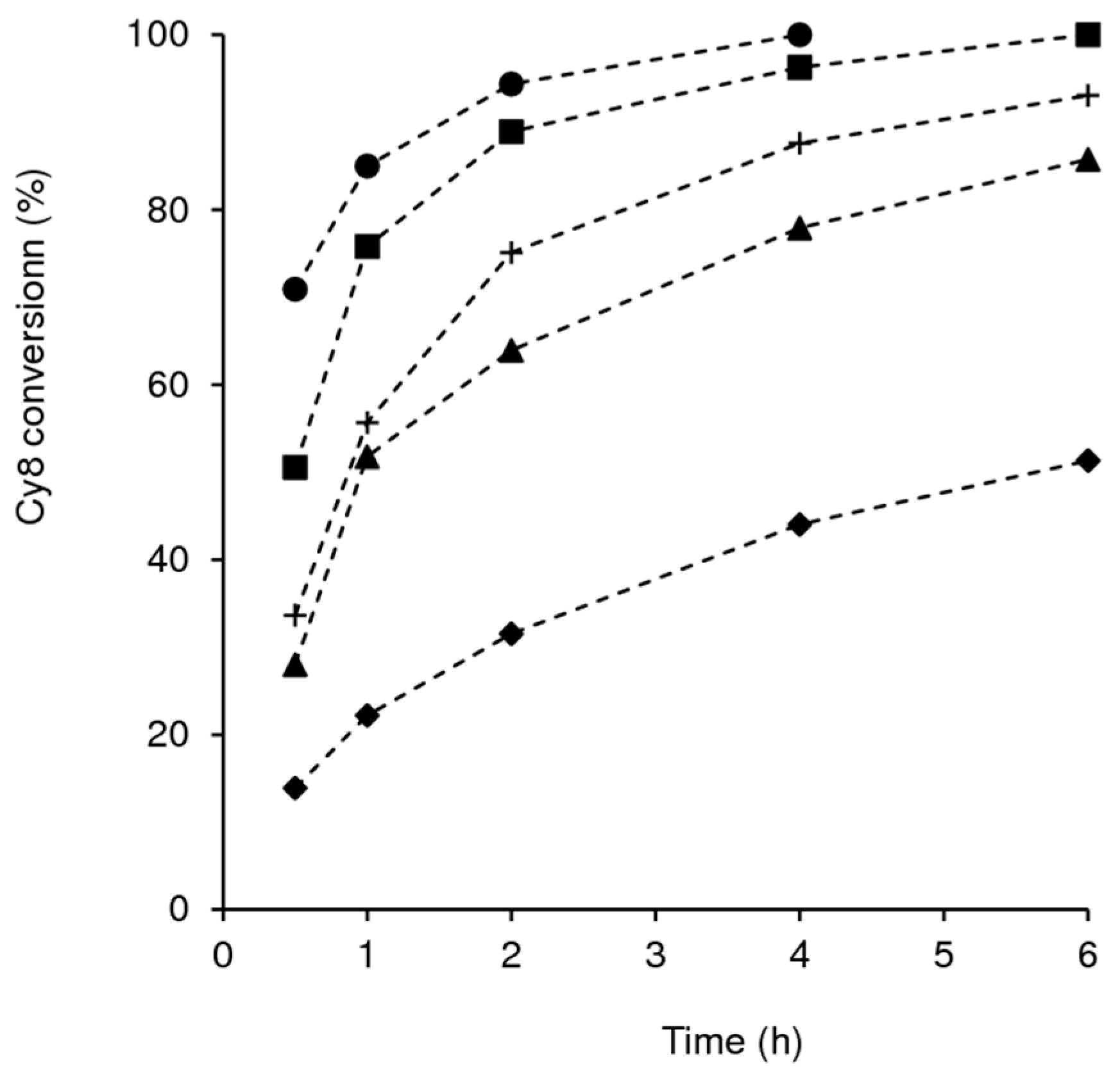
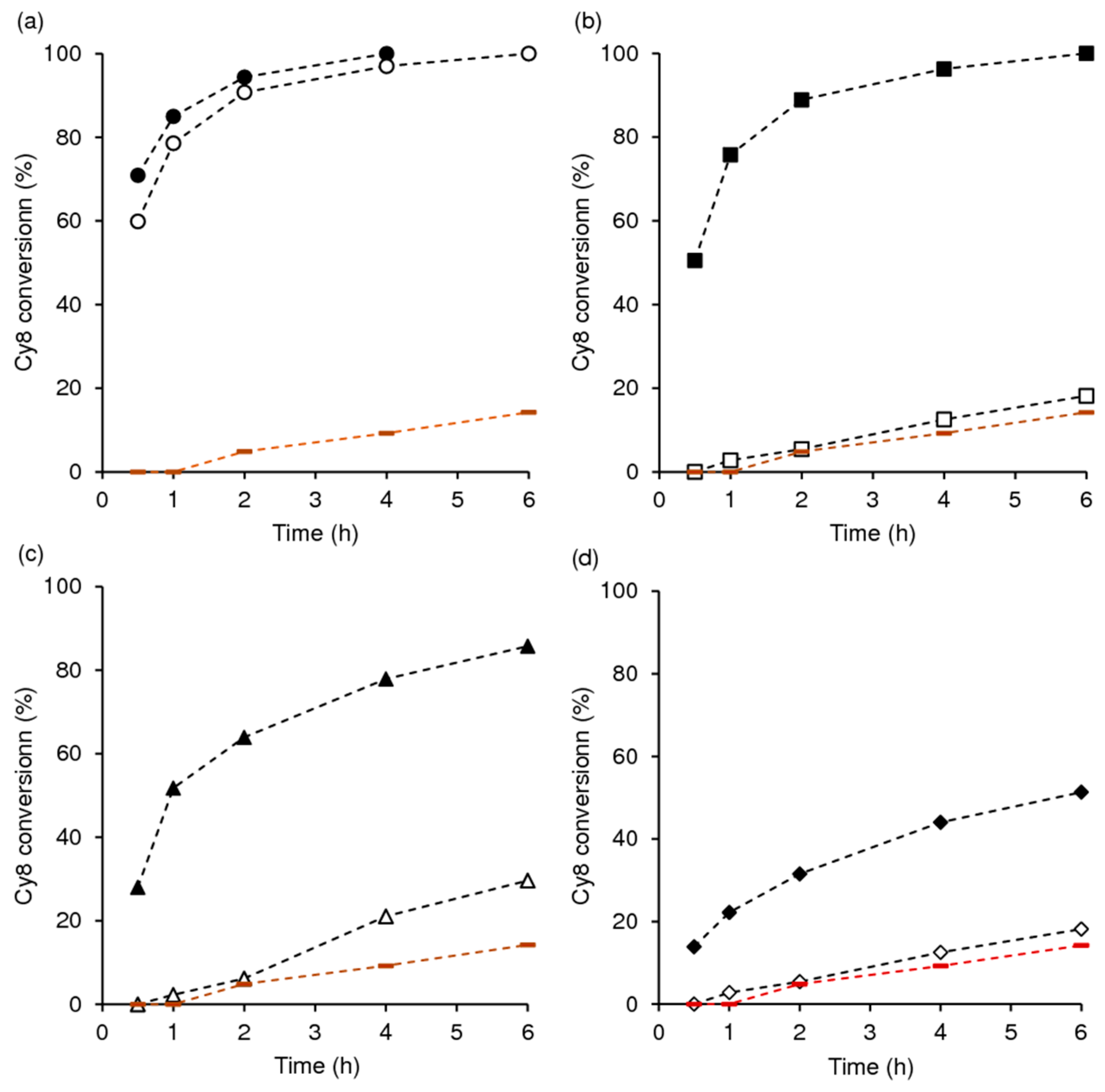
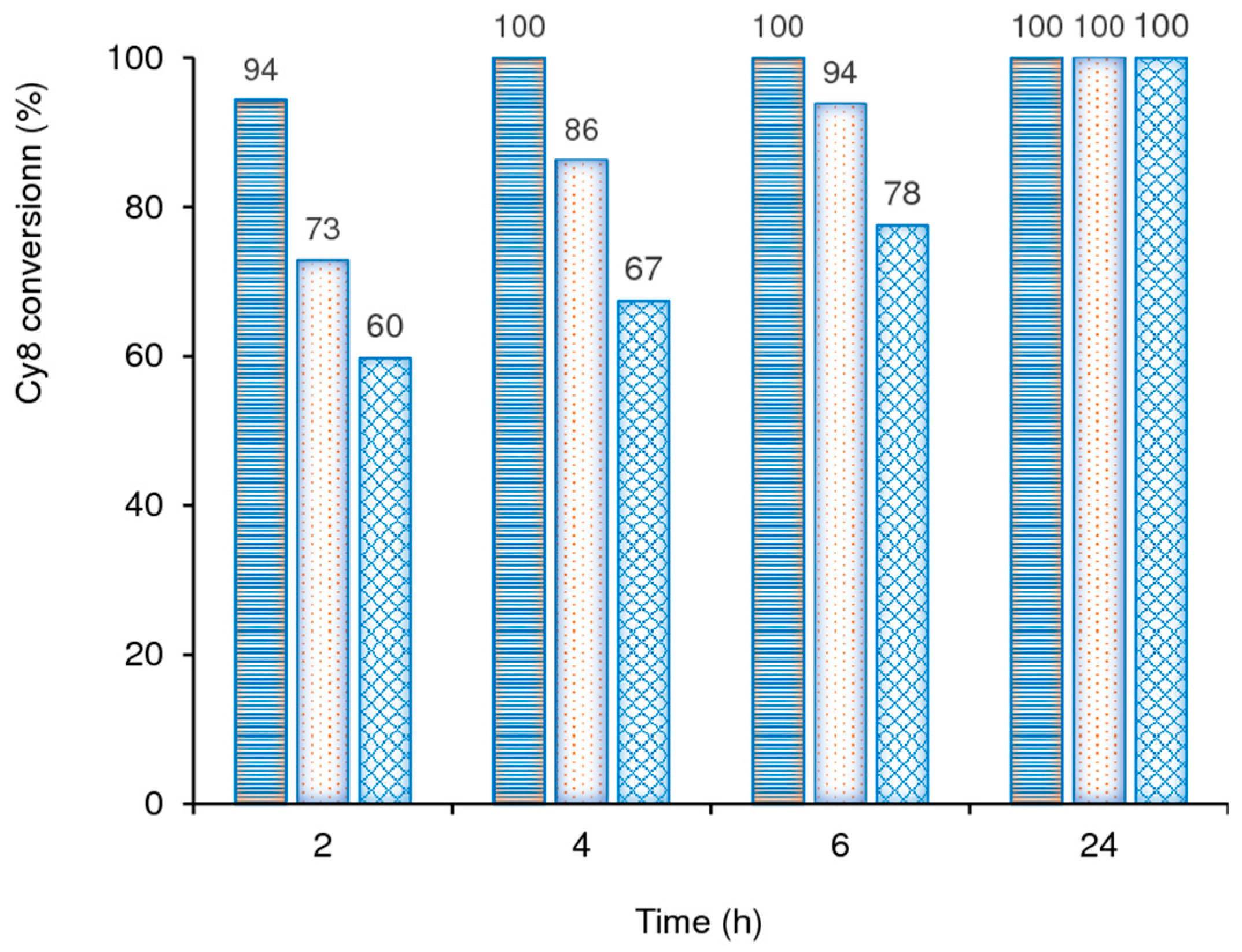
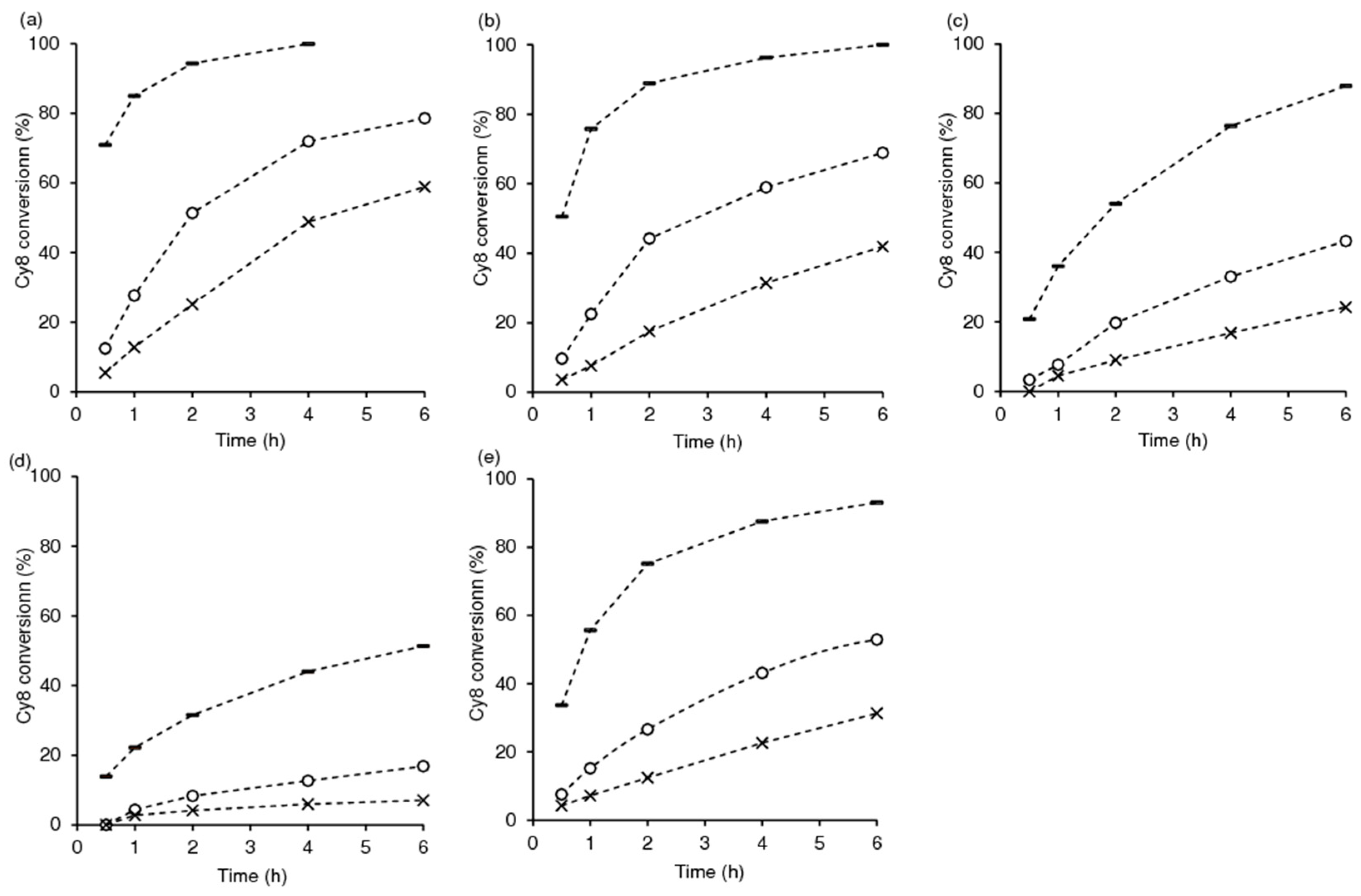
2.2. Catalytic Studies
2.2.1. General Considerations
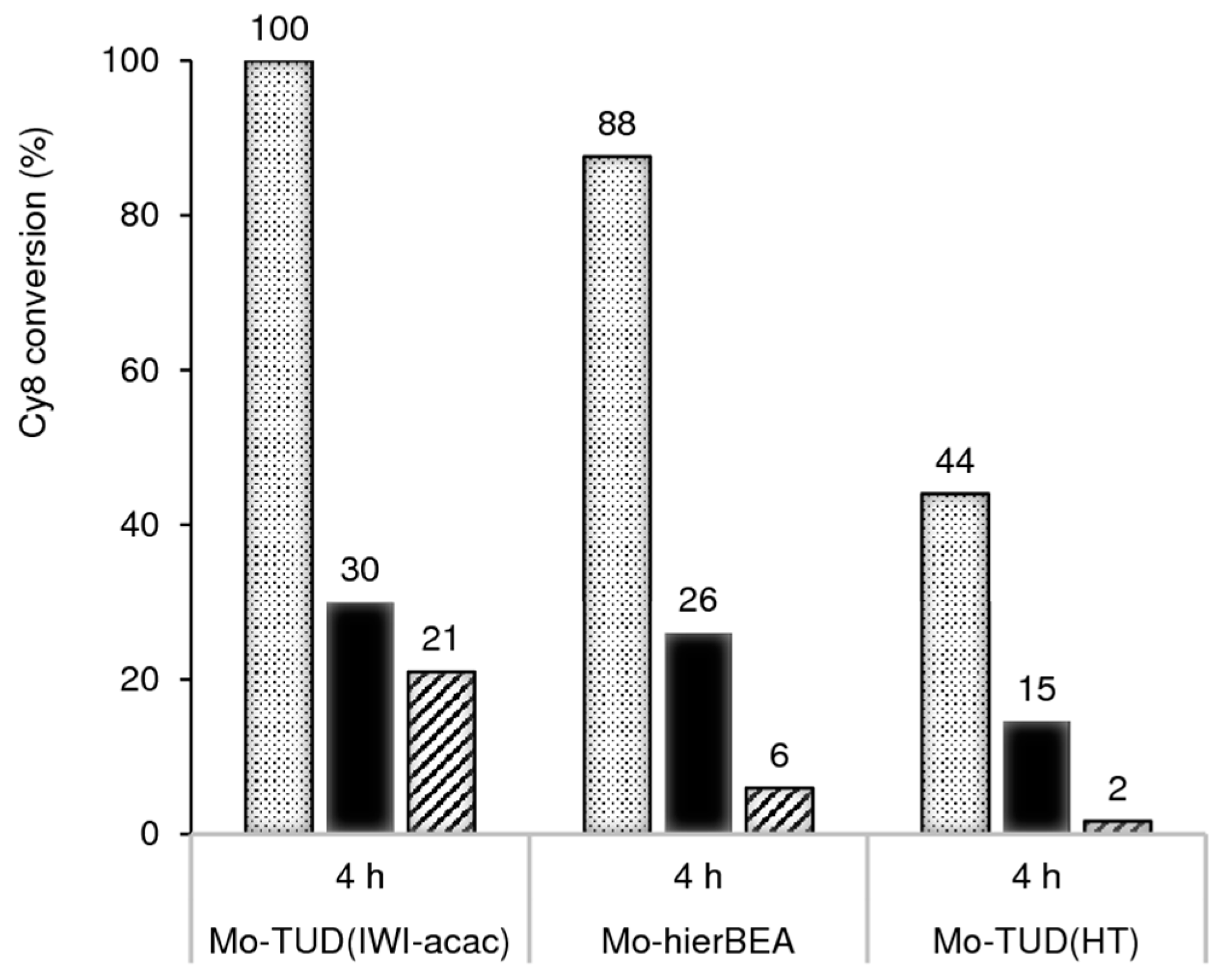
2.2.2. Mo-TUD(IWI) Type Catalysts
2.2.3. Hydrothermal Synthesis Versus Post-Synthesis Strategies (TUD-1)
2.2.4. Hierarchical Mo-hierBEA
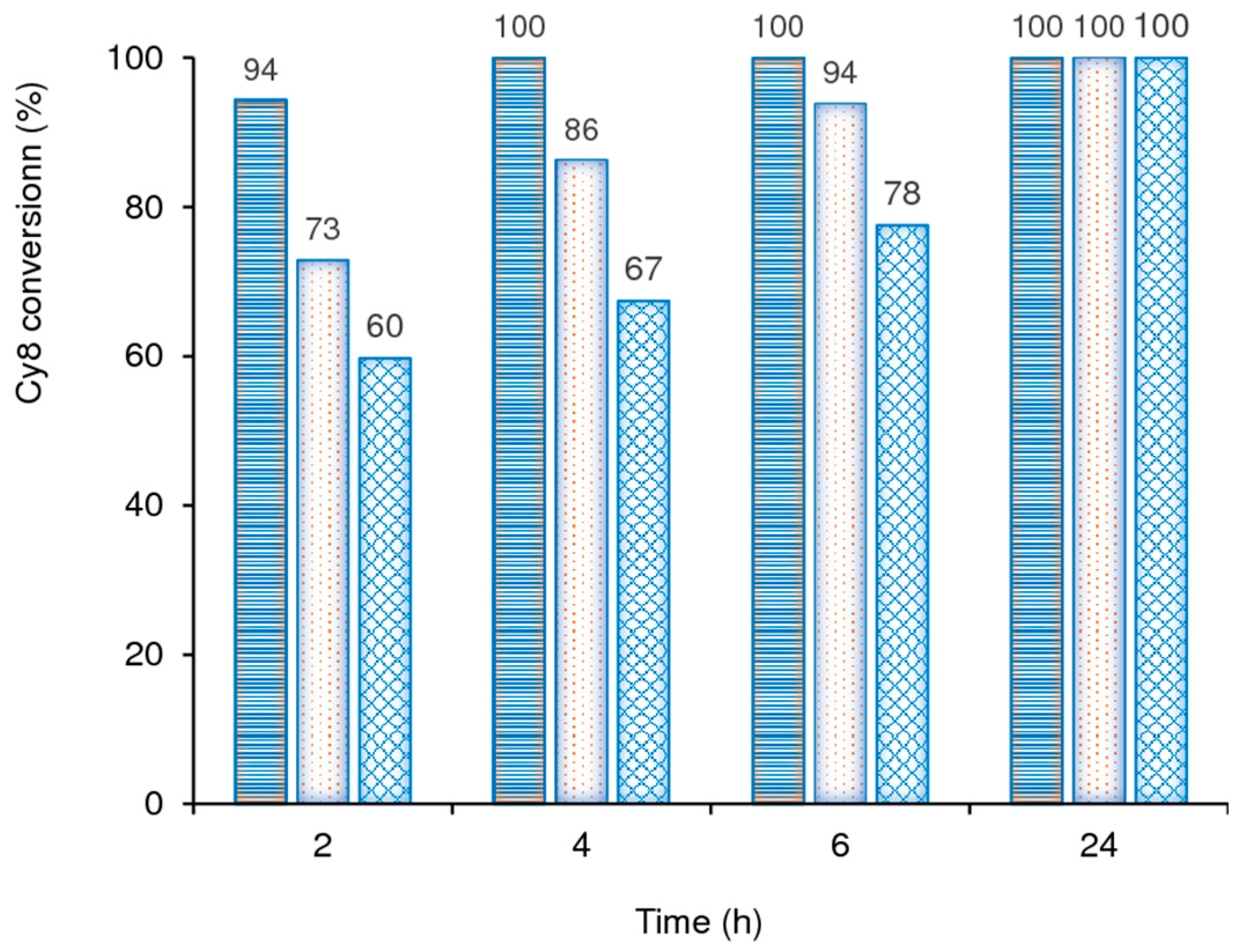
2.2.5. Substrate Scope
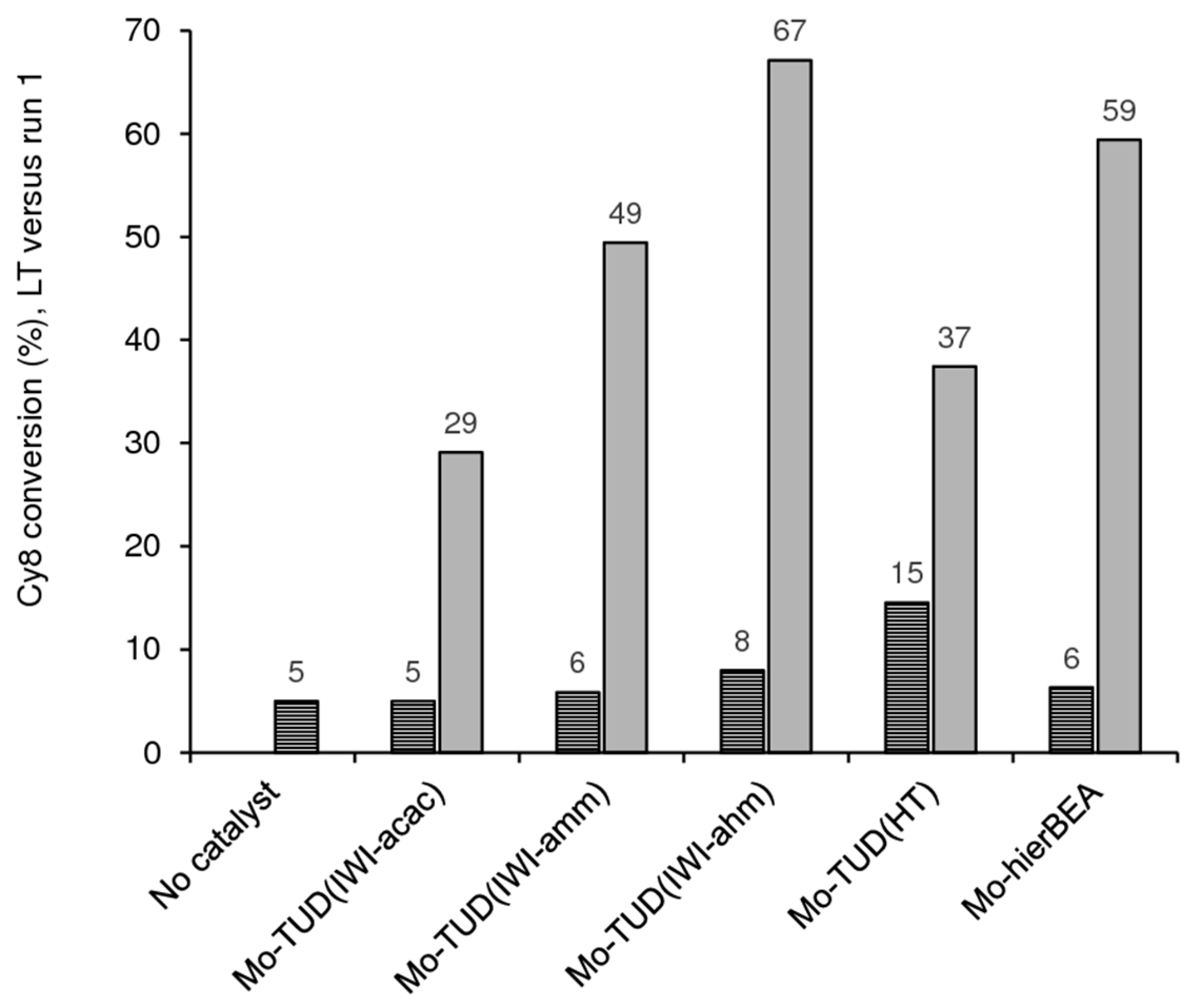
2.2.6. Catalyst Stability
3. Materials and Methods
3.1. Materials
3.2. Synthesis of the Catalysts
3.2.1. Mo-TUD(IWI-x) Materials
3.2.2. Mo-TUD(HT)
3.2.3. Mo-hierBEA
3.3. Characterization of the Catalysts
3.4. Catalytic Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakhtyari, A.; Makarem, M.A.; Rahimpour, M.R. Light Olefins/Bio-Gasoline Production from Biomass. In Bioenergy Systems for the Future; Elsevier: Amsterdam, The Netherlands, 2017; pp. 87–148. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; Makkee, M.; Moulijn, J.A.; Weckhuysen, B.M. The Production of Propene Oxide: Catalytic Processes and Recent Developments. Ind. Eng. Chem. Res. 2006, 45, 3447–3459. [Google Scholar] [CrossRef] [Green Version]
- Tert-Butyl Hydroperoxide|Ataman Kimya A.Ş. Available online: https://www.ataman-Chemicals.com/en/products/tert-butyl-hydroperoxide-1817.html (accessed on 22 October 2022).
- Brégeault, J.M. Transition-Metal Complexes for Liquid-Phase Catalytic Oxidation: Some Aspects of Industrial Reactions and of Emerging Technologies. J. Chem. Soc. Dalton Trans. 2003, 3, 3289–3302. [Google Scholar] [CrossRef]
- Smith, M.B. Functional Group Exchange Reactions: Oxidations. In Organic Synthesis; Elsevier (Academic Press): London, UK, 2017. [Google Scholar] [CrossRef]
- Serge Hub, V.; Philippe Maj, B. Manufacture of Tertiobutyl Hydroperoxide from Renewable Materials, Tertiobutyl Hydroperoxide Thus Obtained, and Used Thereof. U.S. Patent US8536379B2, 17 September 2013. Available online: https://patents.google.com/patent/US8536379B2/en (accessed on 22 October 2022).
- Ayla, E.Z.; Potts, D.S.; Bregante, D.T.; Flaherty, D.W. Alkene Epoxidations with H2O2 over Groups 4–6, Metal-Substituted BEA Zeolites: Reactive Intermediates, Reaction Pathways, and Linear Free-Energy Relationships. ACS Catal. 2021, 11, 139–154. [Google Scholar] [CrossRef]
- Juwiler, D.; Blum, J.; Neumann, R. Metal Silicates by a Molecular Route as Catalysts for Epoxidation of Alkenes with tert-Butyl Hydroperoxide. Chem. Commun. 1998, 10, 1123–1124. [Google Scholar] [CrossRef]
- Dusi, M.; Mallat, T.; Baiker, A. Epoxidation of Functionalized Olefins over Solid Catalysts. Catal. Rev.-Sci. Eng. 2000, 42, 213–278. [Google Scholar] [CrossRef]
- Shen, Y.; Jiang, P.; Wai, P.T.; Gu, Q.; Zhang, W. Recent Progress in Application of Molybdenum-Based Catalysts for Epoxidation of Alkenes. Catalysts 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Guo, T.; Peng, X.; Chen, F.; Lin, M.; Xia, C.; Zhu, B.; Liao, W.; Luo, Y.; Shu, X. Molybdenum-Confined Hierarchical Titanium Silicalite-1: The Synthesis, Characterization, and Catalytic Activity in Alkene Oxidation. Ind. Eng. Chem. Res. 2020, 59, 1093–1100. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Furuichi, N.; Seki, H.; Yamashita, H. One-Pot Synthesis of Molybdenum Oxide Nanoparticles Encapsulated in Hollow Silica Spheres: An Efficient and Reusable Catalyst for Epoxidation of Olefins. J. Mater. Chem. A 2017, 5, 18518–18526. [Google Scholar] [CrossRef]
- Hamdy, M.S.; Mul, G. Synthesis, Characterization and Catalytic Performance of Mo-TUD-1 Catalysts in Epoxidation of Cyclohexene. Catal. Sci. Technol. 2012, 2, 1894–1900. [Google Scholar] [CrossRef]
- Adam, F.; Iqbal, A. Silica Supported Amorphous Molybdenum Catalysts Prepared via Sol-Gel Method and Its Catalytic Activity. Microporous Mesoporous Mater. 2011, 141, 119–127. [Google Scholar] [CrossRef]
- Bakala, P.C.; Briot, E.; Piquemal, J.Y.; Brégeault, J.M.; Beaunier, P. Comparison of the Conventional Impregnation Method Using Ammonium Heptamolybdate with a Simple Route to Silica-Supported Molybdenum(VI) Materials. Catal. Commun. 2007, 8, 1447–1451. [Google Scholar] [CrossRef]
- Bakala, P.C.; Briot, E.; Salles, L.; Brégeault, J.M. Comparison of Liquid-Phase Olefin Epoxidation over MoOx Inserted within Mesoporous Silica (MCM-41, SBA-15) and Grafted onto Silica. Appl. Catal. A Gen. 2006, 300, 91–99. [Google Scholar] [CrossRef]
- Niederer, J.P.M.; Hölderich, W.F. Oxidation Capabilities of BEA Isomorphously Substituted with Molybdenum, Vanadium and Titanium: An Explorative Study. Appl. Catal. A Gen. 2002, 229, 51–64. [Google Scholar] [CrossRef]
- Piquemal, J.Y.; Manoli, J.M.; Beaunier, P.; Ensuque, A.; Tougne, P.; Legrand, A.P.; Brégeault, J.M. Using Inorganic Silicate Precursor/Molybdenum Peroxo Complexes/Onium Salt Interfaces in Aqueous Acidic Media to Design Mesoporous Silica with High Molybdenum Content and High Dispersion. Microporous Mesoporous Mater. 1999, 29, 291–304. [Google Scholar] [CrossRef]
- Telalovi, S. TUD-1: Synthesis and Application of a Versatile Catalyst, Carrier, Material. J. Mater. Chem. 2010, 20, 642–658. [Google Scholar] [CrossRef] [Green Version]
- Angevine, P.J.; Gaffney, A.M.; Shan, Z.; Koegler, J.H.; Yeh, C.Y. TUD-1: A Generalised Mesoporous Catalyst Family for Industrial Applications. In Proceedings of the 235th ACS National Meeting, New Orleans, LA, USA, 6–10 April 2008. [Google Scholar]
- Ooi, Y.K.; Hussin, F.; Yuliati, L.; Lee, S.L. Comparison Study on Molybdena-Titania Supported on TUD-1 and TUD-C Synthesized via Sol-Gel Templating Method: Properties and Catalytic Performance in Olefins Epoxidation. Mater. Res. Express 2019, 6, 074001. [Google Scholar] [CrossRef]
- Ooi, Y.K.; Yuliati, L.; Hartanto, D.; Nur, H.; Lee, S.L. Mesostructured TUD-C Supported Molybdena Doped Titania as High Selective Oxidative Catalyst for Olefins Epoxidation at Ambient Condition. Microporous Mesoporous Mater. 2016, 225, 411–420. [Google Scholar] [CrossRef]
- Sato, T.; Dakka, J.; Sheldon, R.A. Titanium-Substituted Zeolite Beta(Ti-Al-β)-Catalysed Epoxidation of Oct-1-ene with tert-Butyl Hydroperoxide (TBHP). J. Chem. Soc. Chem. Commun. 1994, 16, 1887–1888. [Google Scholar] [CrossRef]
- Corma, A.; Esteve, P.; Martinez, A.; Valencia, S. Oxidation of Olefins with Hydrogen Peroxide and Tert-Butyl Hydroperoxide on Ti-Beta Catalyst. J. Catal. 1995, 152, 18–24. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.W.C.E.; Lempers, H.E.B. Activities and Stabilities of Redox Molecular Sieve Catalysts in Liquid Phase Oxidations. A Review. Collect. Czech. Chem. Commun. 1998, 63, 1724–1742. [Google Scholar] [CrossRef]
- Sotomayor, F.J.; Cychosz, K.A.; Thommes, M.; Sotomayor, F.; Cychosz, K.A.; Thommes, M. Characterization of Micro/Mesoporous Materials by Physisorption: Concepts and Case Studies. Acc. Mater. Surf. Res. 2018, 3, 34–50. [Google Scholar]
- Schlumberger, C.; Thommes, M. Characterization of Hierarchically Ordered Porous Materials by Physisorption and Mercury Porosimetry—A Tutorial Review. Adv. Mater. Interfaces 2021, 8, 2002181. [Google Scholar] [CrossRef]
- Antunes, M.M.; Silva, A.F.; Fernandes, A.; Pillinger, M.; Ribeiro, F.; Valente, A.A. Renewable Bio-Based Routes to γ-Valerolactone in the Presence of Hafnium Nanocrystalline or Hierarchical Microcrystalline Zeotype Catalysts. J. Catal. 2022, 406, 56–71. [Google Scholar] [CrossRef]
- Mehmood, Y.; Khan, I.U.; Shahzad, Y.; Khalid, S.H.; Yousaf, A.M.; Hussain, T. Facile Synthesis of Mesoporous Silica Nanoparticles Using Modified Sol- Gel Method: Optimization and in Vitro Cytotoxicity Studies. Pak. J. Pharm. Sci. 2019, 32, 1805–1812. [Google Scholar] [PubMed]
- Shawky, S.M.; Abo-alhassan, A.A.; Lill, H.; Bald, D.; El-khamisy, S.F.; Ebeid, E.M. Efficient Loading and Encapsulation of Anti-Tuberculosis Drugs Using Multifunctional Mesoporous Silicate Nanoparticles Running Title: Mesoporous Silicate Nanoparticles as Smart Drug Delivery System. J. Nanosci. Curr. Res. 2016, 1, 1–8. [Google Scholar] [CrossRef]
- Ayu, G.; Kusumah, P.; Azizah, M. Silica Content and Structure from Corncob Ash with Various Acid Treatment (HCl, HBr, and Citric Acid). Molekul 2017, 12, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Jiang, P.P.; Shen, Y.; Zhang, K.; Wai, P.T.; Haryono, A. High-Dispersed MoO3 Nanoparticles in 3D-Dendritic Mesoporous Silica Nanospheres: Heterogeneous Catalysts for the Epoxidation of Olefins. J. Porous Mater. 2021, 28, 779–789. [Google Scholar] [CrossRef]
- Uchagawkar, A.; Ramanathan, A.; Hu, Y.; Subramaniam, B. Highly Dispersed Molybdenum Containing Mesoporous Silicate (Mo-TUD-1) for Olefin Metathesis. Catal. Today 2020, 343, 215–225. [Google Scholar] [CrossRef]
- Hahn, T.; Bentrup, U.; Armbrüster, M.; Kondratenko, E.V.; Linke, D. The Enhancing Effect of Brønsted Acidity of Supported MoOx Species on Their Activity and Selectivity in Ethylene/Trans-2-Butene Metathesis. ChemCatChem 2014, 6, 1664–1672. [Google Scholar] [CrossRef]
- Thielemann, J.P.; Ressler, T.; Walter, A.; Tzolova-müller, G.; Hess, C. Applied Catalysis A: General Structure of Molybdenum Oxide Supported on Silica SBA-15 Studied by Raman, UV-Vis and X-Ray Absorption Spectroscopy. Appl. Catal. A Gen. 2011, 399, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Mimura, N.; Bravo-Suárez, J.J.; Akita, T.; Tsubota, S.; Oyama, S.T. Gas-Phase Epoxidation of Propylene through Radicals Generated by Silica-Supported Molybdenum Oxide. Appl. Catal. A Gen. 2007, 316, 142–151. [Google Scholar] [CrossRef]
- Ramanathan, A.; Wu, J.; Maheswari, R.; Hu, Y. Microporous and Mesoporous Materials Synthesis of Molybdenum-Incorporated Mesoporous Silicates by Evaporation-Induced Self-Assembly: Insights into Surface Oxide Species and Corresponding Olefin Metathesis Activity. Microporous Mesoporous Mater. 2017, 245, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Gulec, A.; Johnson, A.M.; Drake, T.L.; Wu, W.; Lin, Y.; Weitz, E.; Marks, L.D.; Stair, P.C. Highly Efficient Activation, Regeneration, and Active Site Identification of Oxide-Based Olefin Metathesis Catalysts. ACS Catal. 2016, 5, 5740–5746. [Google Scholar] [CrossRef]
- Chandra, P.; Doke, D.S.; Umbarkar, B.; Biradar, A.V. One-Pot Synthesis of Ultrasmall MoO3. J. Mater. Chem. A 2014, 2, 19060–19066. [Google Scholar] [CrossRef]
- Chen, C.; Cai, L.; Zhang, L.; Fu, W.; Hong, Y.; Gao, X.; Jiang, Y.; Li, L. Transesterification of Rice Bran Oil to Biodiesel Using Mesoporous NaBeta Zeolite-Supported Molybdenum Catalyst: Experimental and Kinetic Studies. Chem. Eng. J. 2020, 382, 122839. [Google Scholar] [CrossRef]
- Tian, H.; Roberts, C.A.; Wachs, I.E. Molecular Structural Determination of Molybdena in Different Environments: Aqueous Solutions, Bulk Mixed Oxides, and Supported MoO3 Catalysts. J. Phys. Chem. C 2010, 114, 14110–14120. [Google Scholar] [CrossRef]
- Lee, E.L.; Wachs, I.E. In Situ Spectroscopic Investigation of the Molecular and Electronic Structures of SiO2 Supported Surface Metal Oxides. J. Phys. Chem. C 2007, 111, 14410–14425. [Google Scholar] [CrossRef]
- Higgins, J.B.; La Pierre, R.B.; Schlenker, J.L.; Rohrman, A.C.; Wood, J.D.; Ker, G.T.; Rohrbaugh, W.J. The framework topology of zeolite beta. Zeolites 1988, 8, 446–452. [Google Scholar] [CrossRef]
- Hajjar, R.; Millot, Y.; Man, P.P.; Che, M.; Dzwigaj, S.J. Two Kinds of Framework Al Sites Studied in BEA Zeolite by X-ray Diffraction, Fourier Transform Infrared Spectroscopy, NMR Techniques, and V Probe. Phys. Chem. C 2008, 112, 20167–20175. [Google Scholar] [CrossRef]
- Costa, D.G.; Capaz, R.B. Structural analysis of zeolite beta through periodic ab initio simulations of XRD and 29Si and 17O NMR spectra. J. Mol. Struct. 2015, 1097, 112–116. [Google Scholar] [CrossRef]
- Li, C.; Xin, Q.; Wang, K.-L.; Gu, X. FT-IR Emission Spectroscopy Studies of Molybdenum Oxide and Supported Molybdena on Alumina, Silica, Zirconia, and Titania. Appl. Spectrosc. 1991, 45, 874–882. [Google Scholar] [CrossRef]
- Veiros, L.F.; Prazeres, A.; Costa, P.J.; Romão, C.C.; Kühn, F.E.; José Calhorda, M. Olefin Epoxidation with Tert-Butyl Hydroperoxide Catalyzed by MoO2X2L Complexes: A DFT Mechanistic Study. Dalton Trans. 2006, 11, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.; Montilla, F.; Álvarez, E.; Galindo, A. New Insights into the Mechanism of Oxodiperoxomolybdenum Catalysed Olefin Epoxidation and the Crystal Structures of Several Oxo-Peroxo Molybdenum Complexes. Dalton Trans. 2012, 41, 6942. [Google Scholar] [CrossRef] [PubMed]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert-Butyl hydroperoxide Activation. ChemCatChem 2013, 5, 601–611. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Costa, P.J. Unveiling the Mechanisms of Catalytic Oxidation Reactions Mediated by Oxo-Molybdenum Complexes: A Computational Overview. Curr. Org. Chem. 2012, 16, 65–72. [Google Scholar] [CrossRef]
- Lempers HE, B.; Van Crey, M.J.; Sheldon, R.A. Molybdenum-Catalyzed Epoxidations of Oct-1-ene and Cyclohexene with Organic Hydroperoxides: Steric Effects of the Alkyl Substituents of the Hydroperoxide on the Reaction Rate. Recl. Trav. Chim. Pays-Bas 1996, 115, 542–546. [Google Scholar] [CrossRef]
- Sharbatdaran, M.; Farzaneh, F.; Larijani, M.M. Epoxidation of Alkenes Using Inorganic Polymer of Silica Zirconia Molybdate as Catalyst. J. Mol. Catal. A Chem. 2014, 382, 79–85. [Google Scholar] [CrossRef]
- Shen, Y.; Jiang, P.; Zhang, J.; Bian, G.; Zhang, P.; Dong, Y. Highly Dispersed Molybdenum Incorporated Hollow Mesoporous Silica Spheres as an Efficient Catalyst on Epoxidation of Olefins. Mol. Catal. 2017, 433, 212–223. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Liu, L.; Chen, Z. The Interaction of Molybdenum and Titanium in Mesoporous Materials for Olefin Epoxidation. React. Kinet. Mech. Catal. 2022, 135, 317–331. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, J.; Li, X.; Yang, Y.; Yang, Q.; Li, C. Assembly of ZIF Nanostructures around Free Pt Nanoparticles: Efficient Size-Selective Catalysts for Hydrogenation of Alkenes under Mild Conditions. Chem Comm. 2013, 49, 3330–3332. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, T.; Zhang, X.; Liu, H.; Yeung, K.L.; Qiu, J. New Pd/SiO2@ZIF-8 Core–Shell Catalyst with Selective, Antipoisoning, and Antileaching Properties for the Hydrogenation of Alkenes. Ind. Eng. Chem. Res. 2014, 53, 10906–10913. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Li, A.; Ye, H.; Shang, Q.; Shi, X.; Shen, Y. Enhanced Catalytic Activity over Mo-Containing Hierarchical Macro-Mesoporous SBA-15 Catalysts for Epoxidation of Olefins. J. Porous Mater. 2019, 26, 869–880. [Google Scholar] [CrossRef]
- Lima, S.; Antunes, M.M.; Fernandes, A.; Pillinger, M.; Ribeiro, M.F.; Valente, A.A. Catalytic Cyclodehydration of Xylose to Furfural in the Presence of Zeolite H-Beta and a Micro/Mesoporous Beta/TUD-1 Composite Material. Appl. Catal. A Gen. 2010, 388, 141–148. [Google Scholar] [CrossRef]
- Mahtabani, A.; La Zara, D.; Anyszka, R.; He, X.; Paajanen, M.; van Ommen, J.R.; Dierkes, W.; Blume, A. Gas Phase Modification of Silica Nanoparticles in a Fluidized Bed: Tailored Deposition of Aminopropylsiloxane. Langmuir 2021, 37, 4481–4492. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mo 1 (mmolMo g−1) | SBET 2 (m2 g−1) | Vp 3 (cm3 g−1) | Dp 4 (nm) |
|---|---|---|---|---|
| TUD-1 | - | 471 | 1.23 | 8.5 |
| TUD-1(PT) | - | 485 | 1.24 | 8.5 |
| Mo-TUD(IWI-acac) | 0.21 | 355 | 1.19 | 8.2 |
| Mo-TUD(IWI-amm) | 0.21 | 398 | 1.08 | 8.5 |
| Mo-TUD(IWI-ahm) | 0.21 | 322 | 0.99 | 8.2 |
| Mo-TUD(SSI-acac) | 0.21 | 318 | 0.74 | 8.2 |
| Mo-TUD(HT) | 0.08 5 | 418 | 2.15 | 18.3 |
| hierBEA 6 | - | 759 (468) 6 | 0.73 (0.12) 6 | 2.5–5 6 |
| Mo-hierBEA | 0.21 | 532 (427) | 0.61 (0.05) | 2.5–5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, D.M.; Neves, P.; Antunes, M.M.; Fernandes, A.J.S.; Pillinger, M.; Valente, A.A. Post-Synthesis Strategies to Prepare Mesostructured and Hierarchical Silicates for Liquid Phase Catalytic Epoxidation. Catalysts 2022, 12, 1513. https://doi.org/10.3390/catal12121513
Gomes DM, Neves P, Antunes MM, Fernandes AJS, Pillinger M, Valente AA. Post-Synthesis Strategies to Prepare Mesostructured and Hierarchical Silicates for Liquid Phase Catalytic Epoxidation. Catalysts. 2022; 12(12):1513. https://doi.org/10.3390/catal12121513
Chicago/Turabian StyleGomes, Diana M., Patrícia Neves, Margarida M. Antunes, António J. S. Fernandes, Martyn Pillinger, and Anabela A. Valente. 2022. "Post-Synthesis Strategies to Prepare Mesostructured and Hierarchical Silicates for Liquid Phase Catalytic Epoxidation" Catalysts 12, no. 12: 1513. https://doi.org/10.3390/catal12121513
APA StyleGomes, D. M., Neves, P., Antunes, M. M., Fernandes, A. J. S., Pillinger, M., & Valente, A. A. (2022). Post-Synthesis Strategies to Prepare Mesostructured and Hierarchical Silicates for Liquid Phase Catalytic Epoxidation. Catalysts, 12(12), 1513. https://doi.org/10.3390/catal12121513