
Asymmetric Hydroarylation Reactions Catalyzed by Transition Metals: Last 10 Years in a Mini Review




{kind=link}
{kind=link}
{kind=link}
{kind=link}
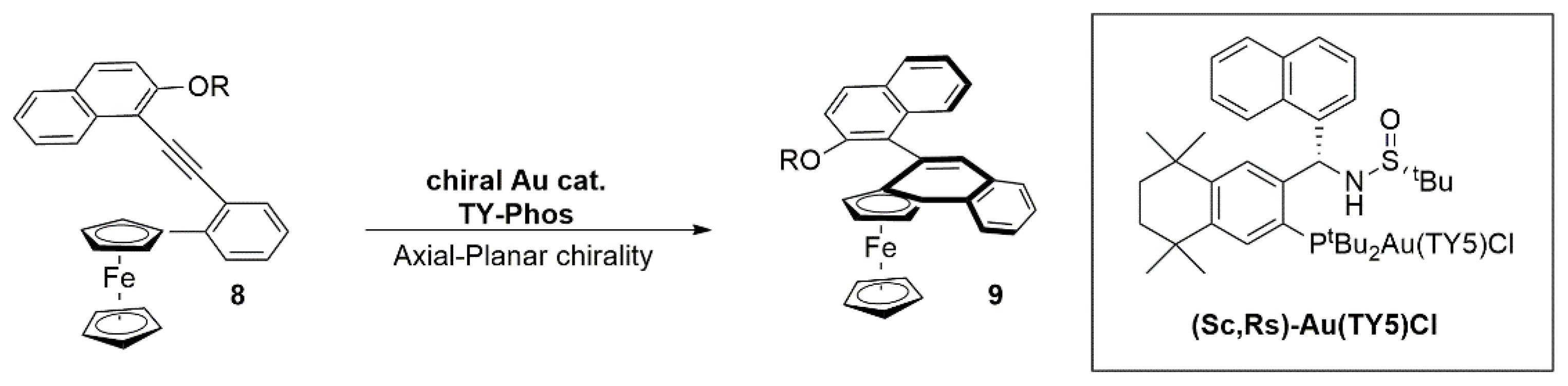{kind=link}
{kind=link}
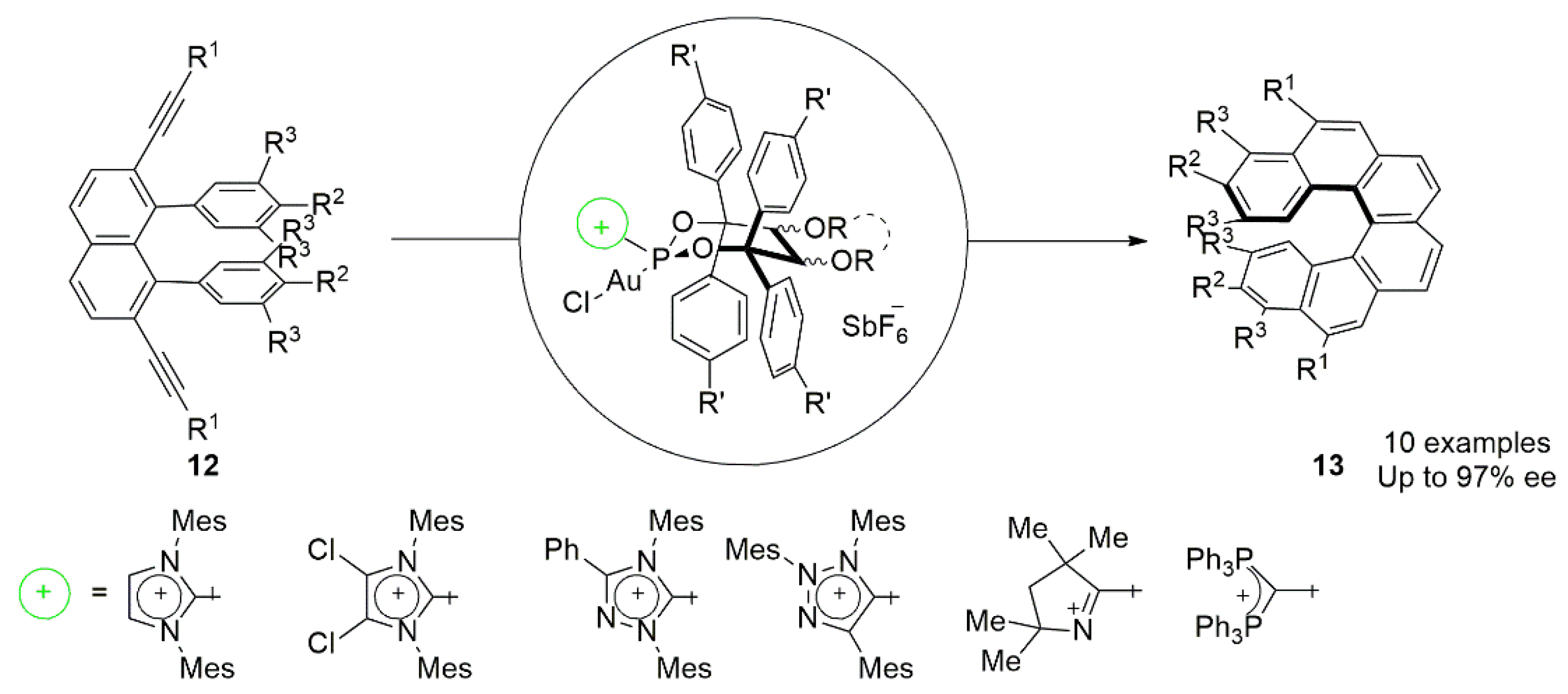{kind=link}
{kind=link}
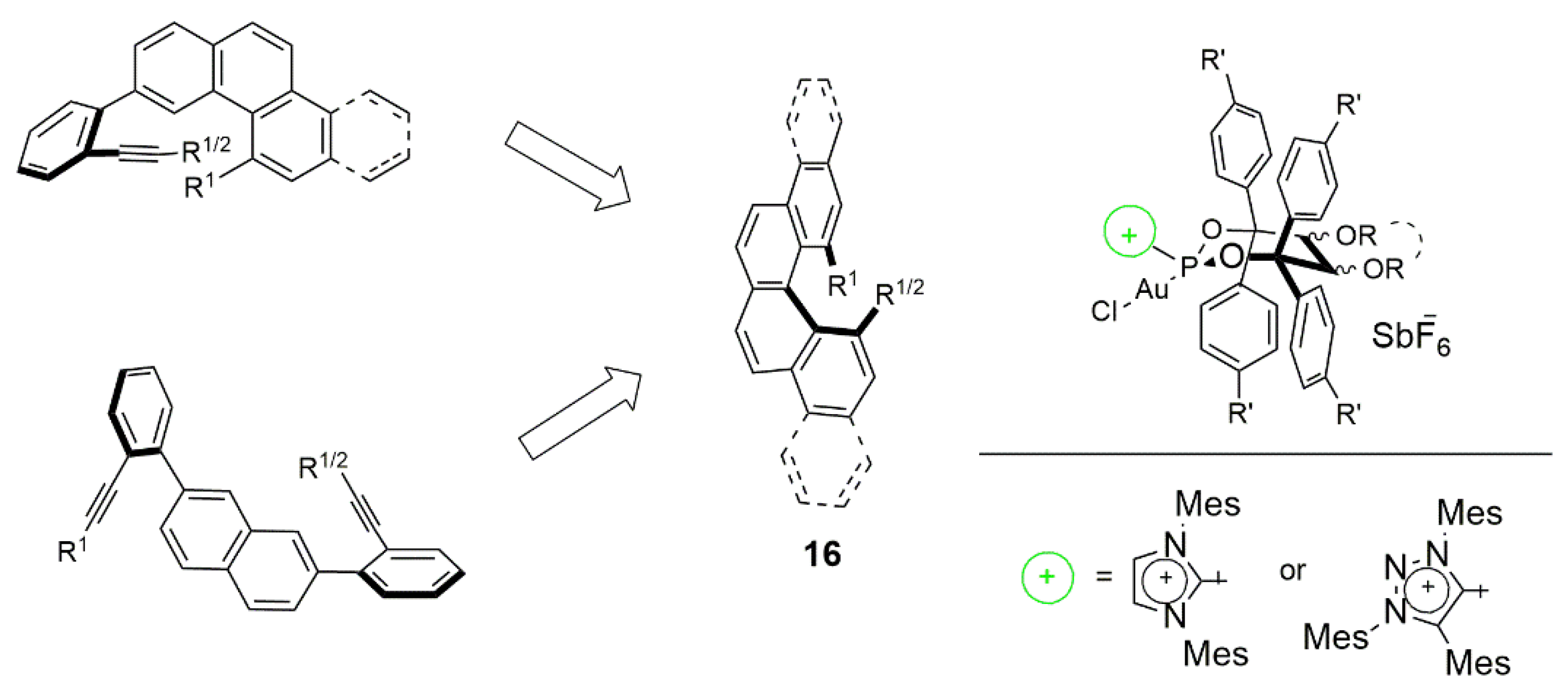{kind=link}
{kind=link}
{kind=link}
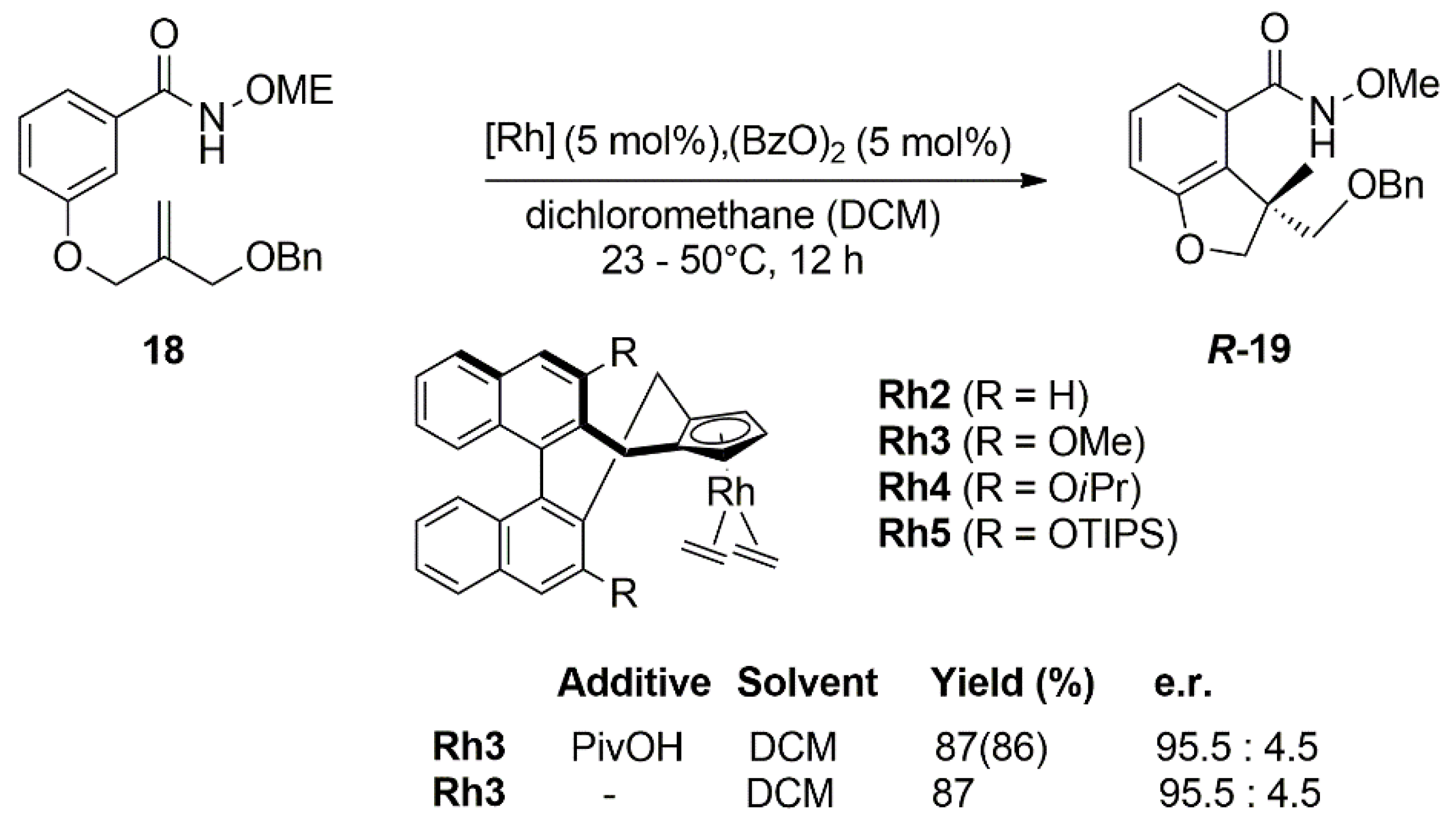{kind=link}
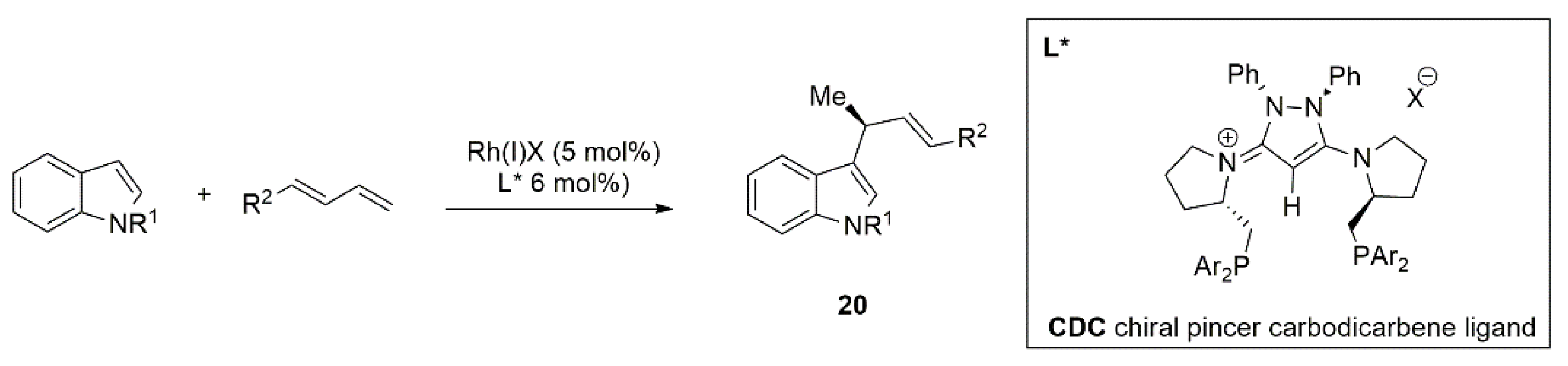{kind=link}
{kind=link}
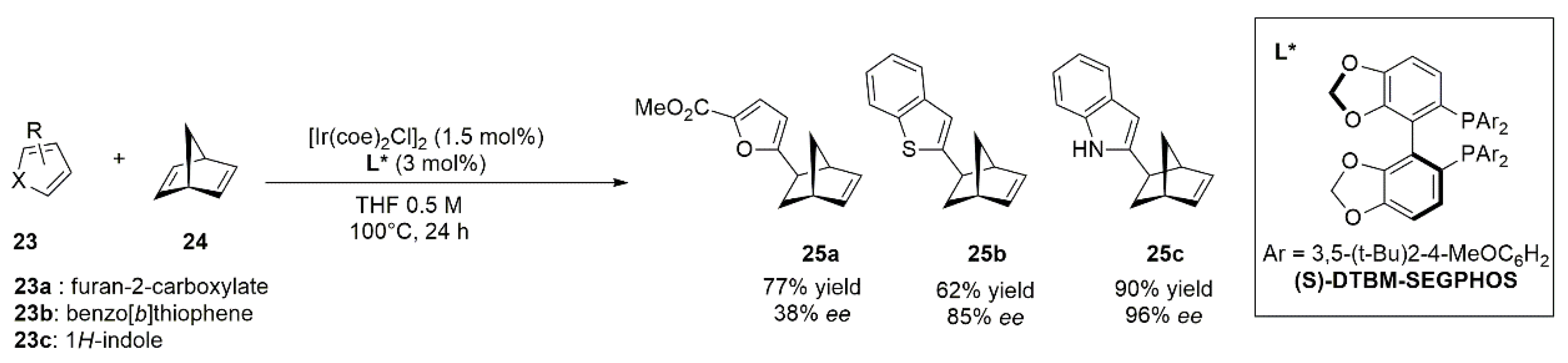{kind=link}
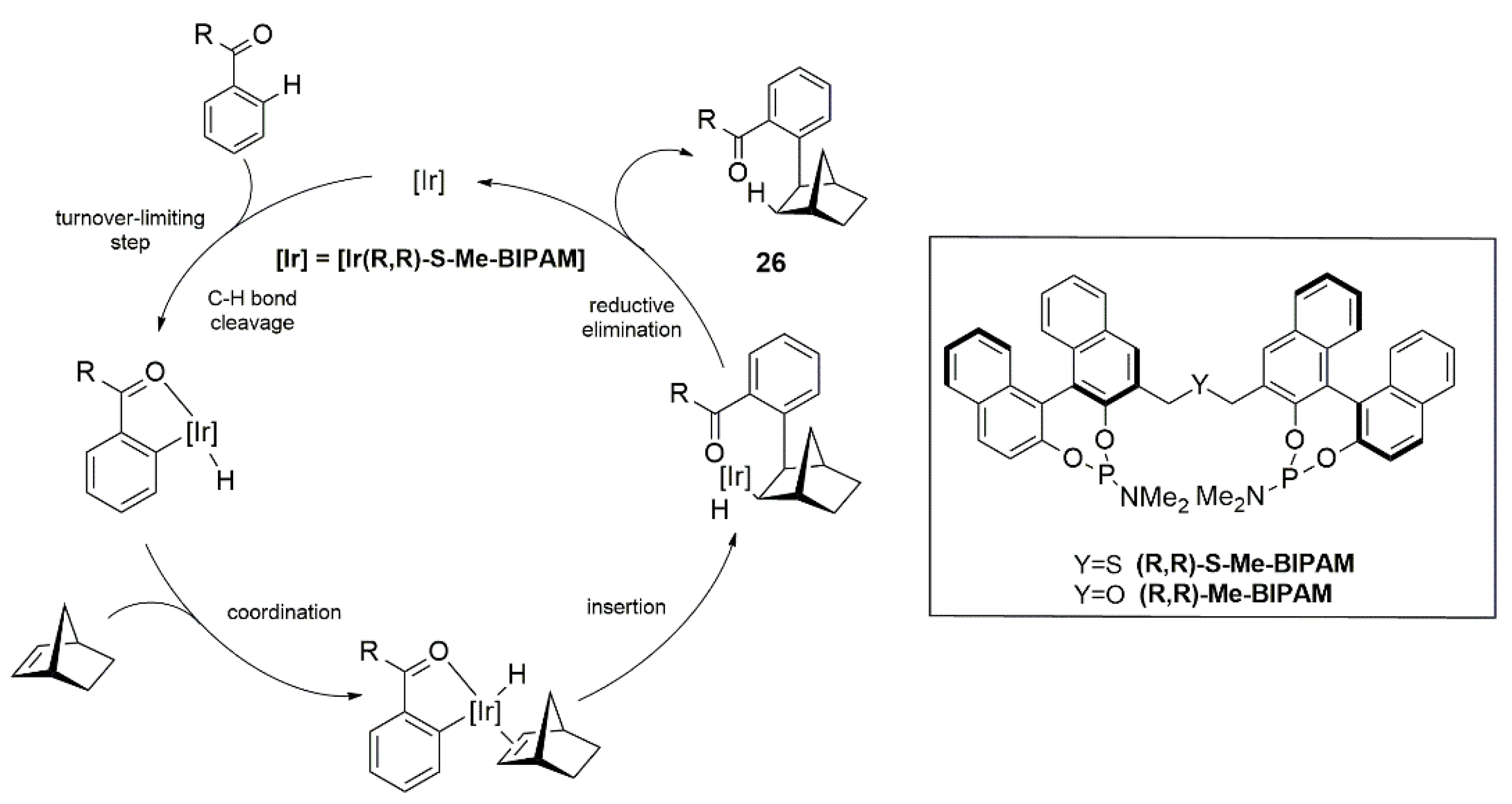{kind=link}
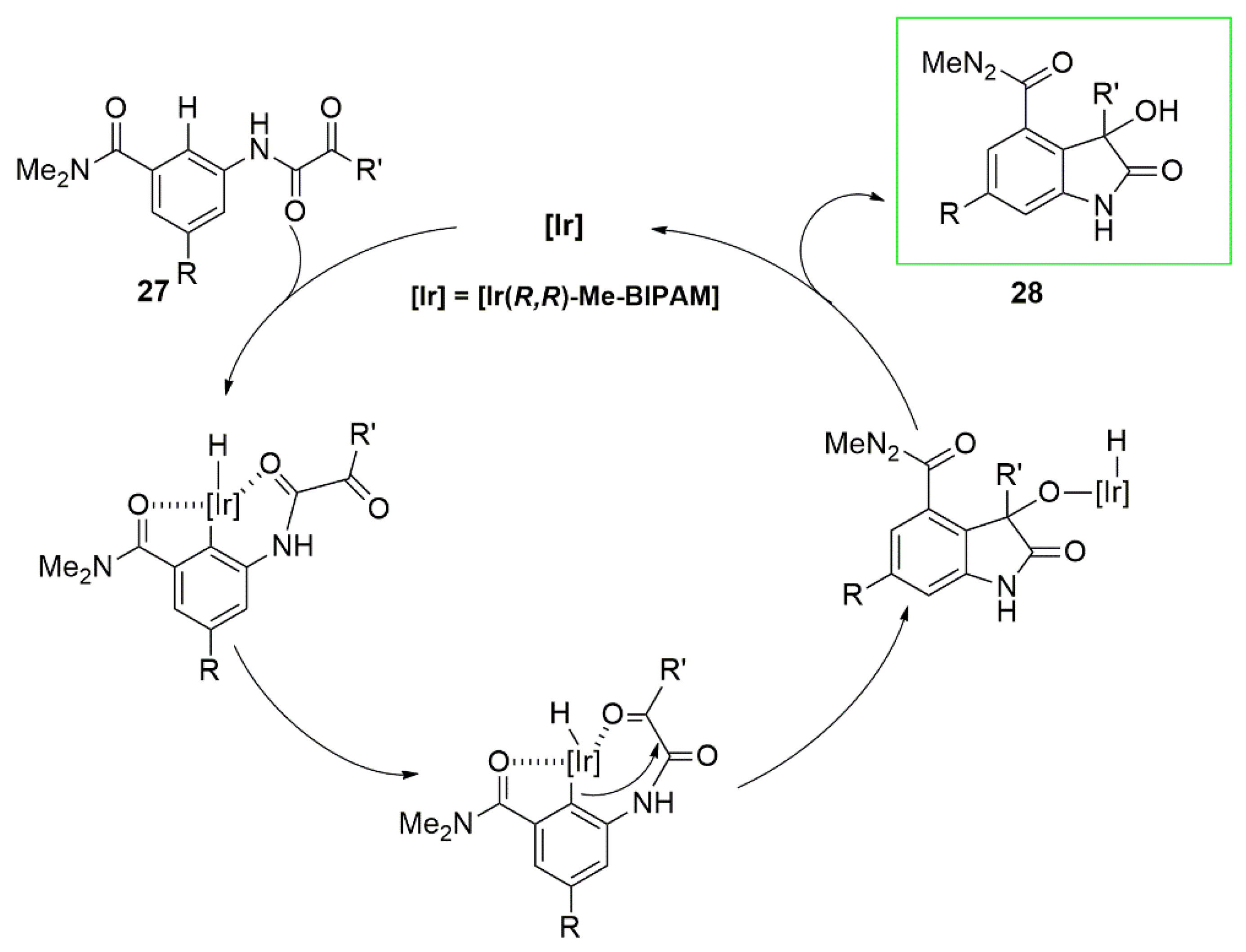{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Asymmetric Construction of Chiral Compounds Sorted by Metal: The Direct Asymmetric Hydroarylation
2.1. Gold
2.2. Rhodium
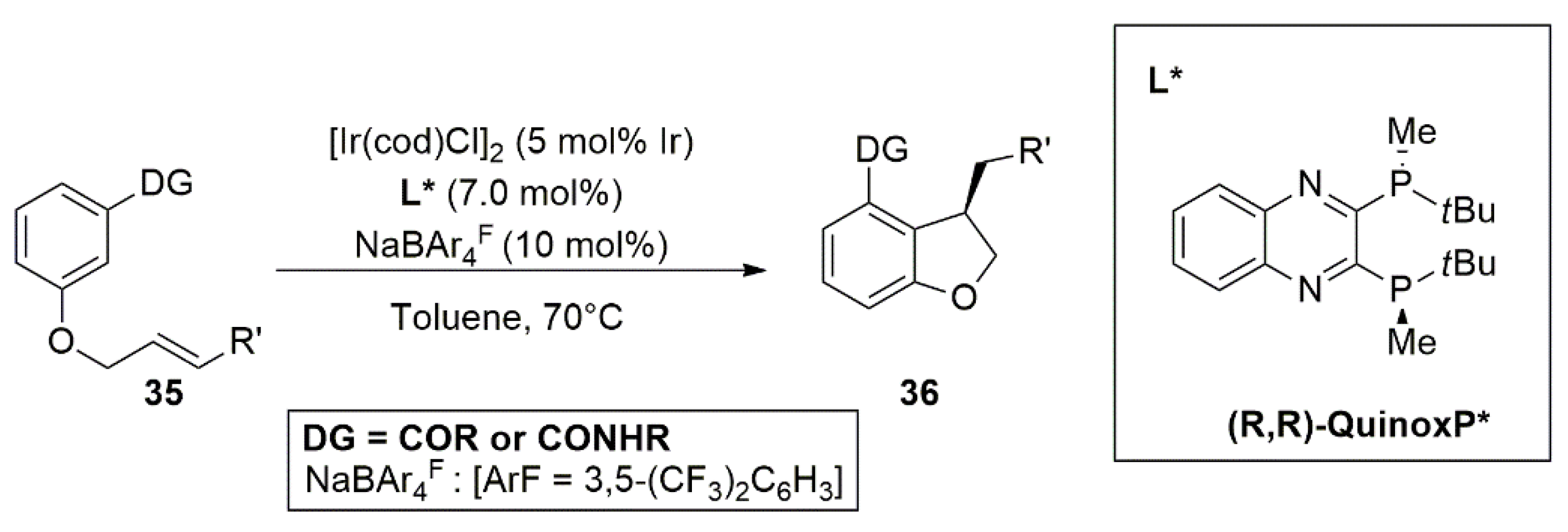
2.3. Iridium
2.4. Nickel
2.5. Cobalt, Platinum, and Ruthenium
3. Asymmetric Construction of Chiral Compounds Sorted by Metal: The Asymmetric Hydroarylation of Activated Aryl Portions
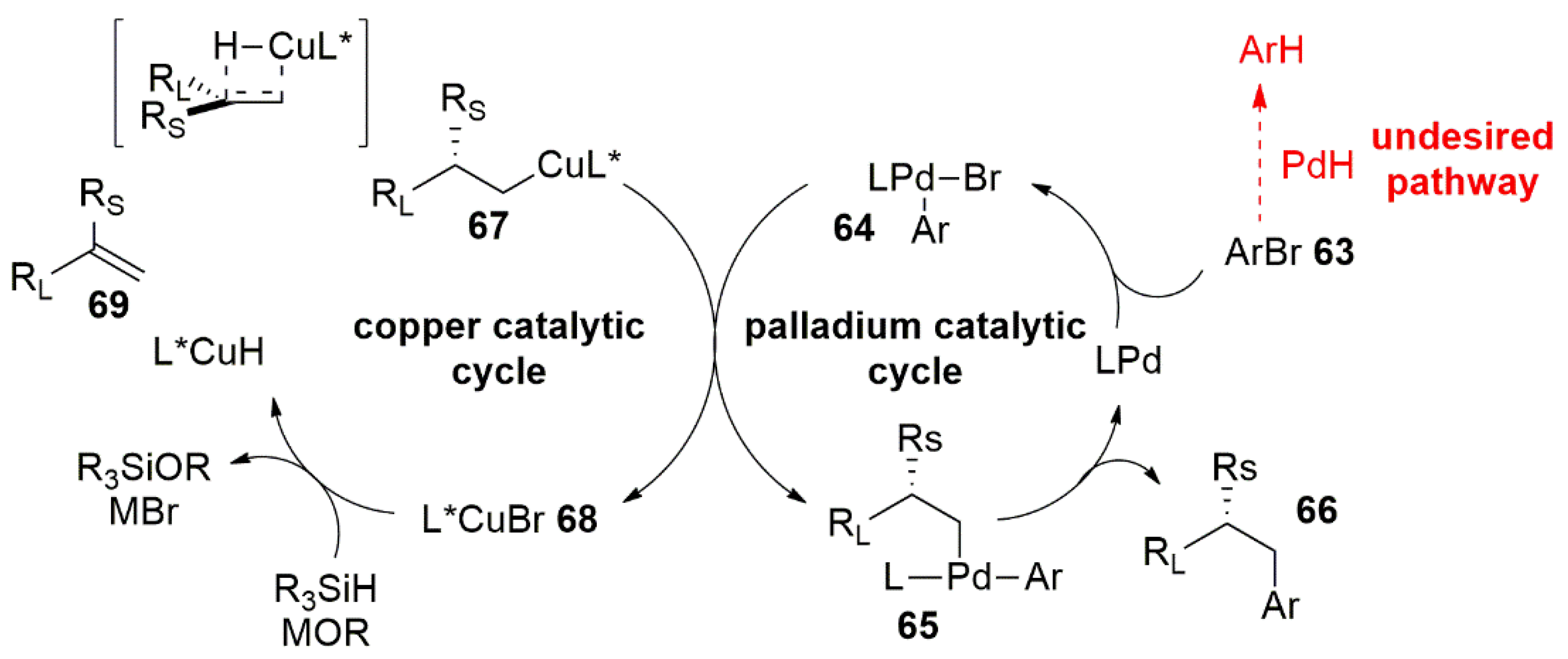
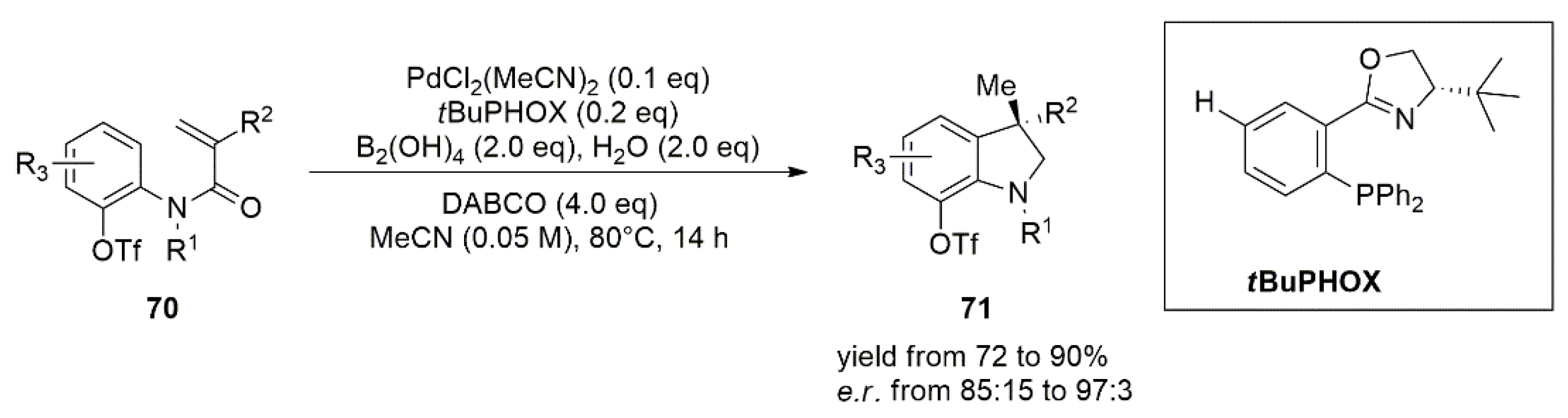
3.1. Palladium
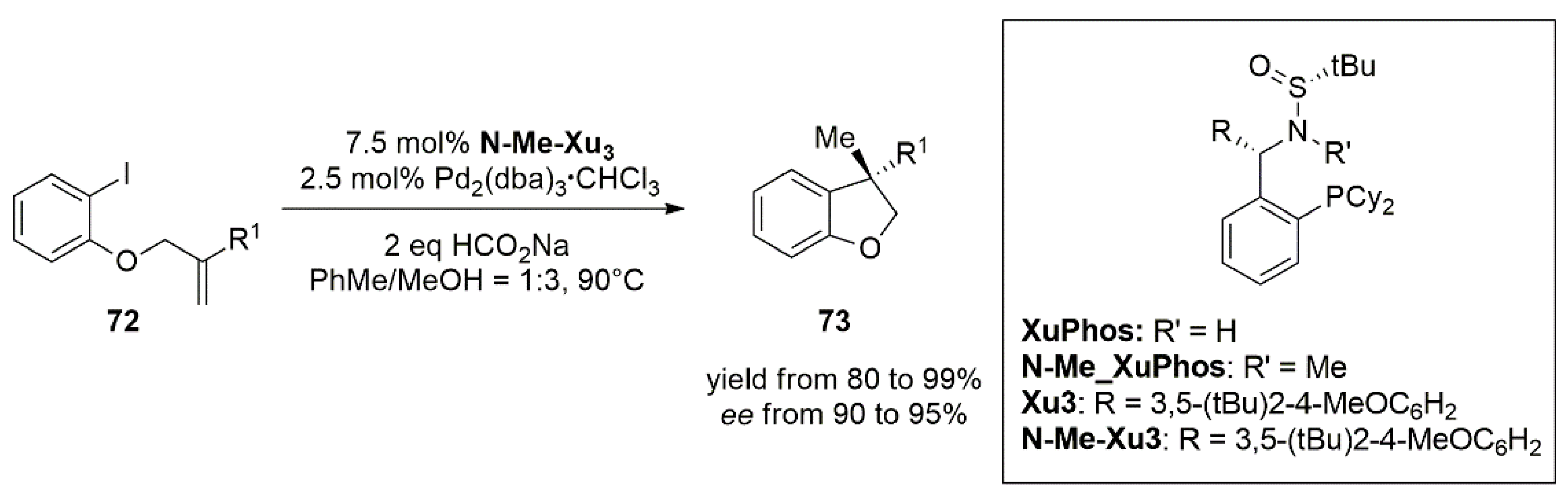
3.2. Nickel
3.3. Rhodium
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ackermann, L.; Gunnoe, T.B.; Habgood, L.G. Catalytic Hydroarylation of Carbon-Carbon Multiple Bonds; Wiley-VCH: Weinheim Germany, 2018. [Google Scholar]
- Pramanik, A.; Ghatak, A. Current Trends on C–C Bond Formation Through Regioselective Hydroarylation of Alkynes and Alkenes Using Metal Free Catalysts. Tetrahedron 2022, 112, 132757. [Google Scholar] [CrossRef]
- Zhang, P.; Tsuji, N.; Ouyang, J.; List, B. Strong and Confined Acids Catalyze Asymmetric Intramolecular Hydroarylations of Unactivated Olefins with Indoles. J. Am. Chem. Soc. 2021, 143, 675–680. [Google Scholar] [CrossRef]
- Arae, S.; Beppu, S.; Kawatsu, T.; Igawa, K.; Tomooka, K.; Irie, R. Asymmetric Synthesis of Axially Chiral Benzocarbazole Derivatives Based on Catalytic Enantioselective Hydroarylation of Alkynes. Org. Lett. 2018, 20, 4796–4800. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Lu, H.; Jiang, X.-L.; Gao, T.-T.; Shi, F. Organocatalytic asymmetric hydroarylation of o-hydroxyl styrenes via remote activation of phenylhydrazones. Tetrahedron Asymmetry 2015, 26, 109–117. [Google Scholar] [CrossRef]
- Snieckus, V.; Zhang, Y. Synthesis of α- and β-C-Glycosyl Arenes by Direct Iridium-Catalyzed Hydroarylation. Synfacts 2019, 15, 0599. [Google Scholar]
- Zhang, Y. Syntheses of vinylindoles via a Brønsted acid catalyzed highly regio- and stereoselective cis-hydroarylation of ynamides. Tetrahedron Lett. 2005, 46, 6483–6486. [Google Scholar] [CrossRef]
- Kumar, G.; Qu, Z.-W.; Grimme, S.; Chatterjee, I. Boron-Catalyzed Hydroarylation of 1,3-Dienes with Arylamines. Org. Lett. 2021, 23, 8952–8957. [Google Scholar] [CrossRef] [PubMed]
- Badir, S.-O.; Lipp, A.; Krumb, M.; Cabrera-Afonso, M.-J.; Kammer, L.-M.; Wu, V.-E.; Huang, M.; Csakai, A.; Marcaurelle, L.-A.; Molander, G.-A. Photoredox-mediated hydroalkylation and hydroarylation of functionalized olefins for DNA-encoded library synthesis. Chem. Sci. 2021, 12, 12036–12045. [Google Scholar] [CrossRef]
- Cai, B.; Yang, Q.; Meng, L.; Wang, J.-J. Kinetic Resolution of 2-Substituted 1,2-Dihydroquinolines by Rhodium-Catalyzed Asymmetric Hydroarylation. Chin. J. Chem. 2021, 39, 1606–1610. [Google Scholar] [CrossRef]
- Ito, Y.; Sawamura, M.; Hayashi, T.J. Catalytic asymmetric aldol reaction: Reaction of aldehydes with isocyanoacetate catalyzed by a chiral ferrocenylphosphine-gold(I) complex. J. Am. Chem. Soc. 1986, 108, 6405–6406. [Google Scholar] [CrossRef]
- Hashmi AS, K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, S.-A.; Sajid, M.-A.; Khan, Z.-A.; Canseco-Gonzalez, D. Gold catalysis in organic transformations: A review. Synth. Commun. 2017, 47, 735–755. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhou, J.; Li, J.; Fu, C.; Guo, Y.; Wang, H.; Ma, S. Asymmetric Construction of Six-Membered Rings by Cyclization of Allenes with Dinuclear Gold Catalysis. Chem. Eur. J. 2015, 21, 15939–15943. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Shibata, Y.; Kimura, Y.; Tanaka, K. Atroposelective Synthesis of Axially Chiral All-Benzenoid Biaryls by the Gold-Catalyzed Intramolecular Hydroarylation of Alkynones. Eur. J. Org. Chem. 2016, 2016, 4465–4469. [Google Scholar] [CrossRef]
- Sutherland, D.-R.; Kinsman, L.; Angiolini, S.-M.; Rosair, G.-M.; Lee, A.-L. Gold(I)-Catalysed Hydroarylation of 1,3-Disubstituted Allenes with Efficient Axial-to-Point Chirality Transfer. Chem. Eur. J. 2018, 24, 7002–7009. [Google Scholar] [CrossRef]
- Toups, K.-L.; Liu, G.-T.; Widenhoefer, R.-A. Gold(I)-catalyzed hydroarylation of allenes with indoles. J. Organomet. Chem. 2009, 694, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-Z.; Zhou, C.-Y.; Guo, Z.; Wong, E.-L.-M.; Wong, M.-K.; Che, C.-M. Gold(I)-Catalyzed Enantioselective Intermolecular Hydroarylation of Allenes with Indoles and Reaction Mechanism by Density Functional Theory Calculations. Chem. Asian J. 2011, 6, 812–824. [Google Scholar] [CrossRef]
- Zhang, P.-C.; Li, Y.-L.-; He, J.; Wu, H.-H.; Li, Z.; Zhang, J. Simultaneous construction of axial and planar chirality by gold/TY-Phos-catalyzed asymmetric hydroarylation. Nat. Commun. 2021, 12, 4609. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Shibata, Y.; Nakamura, K.; Teraoka, K.; Uekusa, H.; Nakazono, K.; Takata, T.; Tanaka, K. Gold-Catalyzed Enantioselective Synthesis, Crystal Structure, and Photophysical/Chiroptical Properties of Aza [10]helicenes. Chem. Eur. J. 2016, 22, 9537–9541. [Google Scholar] [CrossRef]
- Satoh, M.; Shibata, Y.; Tanaka, K. Enantioselective Synthesis of Fully Benzenoid Single and Double Carbohelicenes via Gold-Catalyzed Intramolecular Hydroarylation. Chem. Eur. J. 2018, 24, 5434–5438. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, L.-D.-M.; Marx, M.; Hartung, T.; González-Fernández, E.; Golz, C.; Alcarazo, M. TADDOL-Derived Cationic Phosphonites: Toward an Effective Enantioselective Synthesis of [6]Helicenes via Au-Catalyzed Alkyne Hydroarylation. ACS Catal. 2018, 8, 6079–6085. [Google Scholar] [CrossRef]
- Usui, K.; Yamamoto, K.; Ueno, Y.; Igawa, K.; Hagihara, R.; Masuda, T.; Ojida, A.; Karasawa, S.; Tomooka, R.-K.; Hirai, G.; et al. Internal-Edge-Substituted Coumarin-Fused [6]Helicenes: Asymmetric Synthesis, Structural Features, and Control of Self-Assembly. Chem. Eur. J. 2018, 24, 14617–14621. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; Machleid, R.; Simon, M.; Golz, C.; Alcarazo, M. Enantioselective Synthesis of 1,12-Disubstituted [4]Helicenes. Angew. Chem. 2020, 132, 5709–5713. [Google Scholar] [CrossRef]
- Pelliccioli, V.; Hartung, T.; Simon, M.; Golz, C.; Licandro, E.; Cauteruccio, S.; Alcarazo, M. Enantioselective Synthesis of Dithia [5]helicenes and their Postsynthetic Functionalization to Access Dithia [9]helicenes. Angew. Chem. Int. Ed. 2022, 61, e202114577. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Dong, H.-Q.; Lin, G.-Q. Rhodium-Catalyzed Asymmetric Arylation. ACS Catal. 2012, 2, 95–119. [Google Scholar] [CrossRef]
- So, C.M.; Kume, S.; Hayashi, T. Rhodium-Catalyzed Asymmetric Hydroarylation of 3-Pyrrolines Giving 3-Arylpyrrolidines: Protonation as a Key Step. J. Am. Chem. Soc. 2013, 135, 10990–10993. [Google Scholar] [CrossRef]
- Ye, B.; Donets, P.-A.; Cramer, N. Chiral Cp-Rhodium(III)-Catalyzed Asymmetric Hydroarylations of 1,1-Disubstituted Alkenes. Angew. Chem. Int. Ed. 2014, 53, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Cramer, N. Chiral Cyclopentadienyls: Enabling Ligands for Asymmetric Rh(III)-Catalyzed C−H Functionalizations. Acc. Chem. Res. 2015, 48, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; You, S.-L. Construction of Axial Chirality by Rhodium-Catalyzed Asymmetric Dehydrogenative Heck Coupling of Biaryl Compounds with Alkenes. Angew. Chem. Int. Ed. 2014, 53, 13244–13247. [Google Scholar] [CrossRef]
- Marcum, J.-S.; Roberts, C.-C.; Manan, R.-S.; Cervarich, T.-N.; Meek, S.-J. Chiral Pincer Carbodicarbene Ligands for Enantioselective Rhodium-Catalyzed Hydroarylation of Terminal and Internal 1,3-Dienes with Indoles. J. Am. Chem. Soc. 2017, 139, 15580–15583. [Google Scholar] [CrossRef] [PubMed]
- Berthold, D.; Klett, J.; Breit, B. Rhodium-catalyzed asymmetric intramolecular hydroarylation of allenes: Access to functionalized benzocycles. Chem. Sci. 2019, 10, 10048. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Taube, D.-J.; Periana, R.-A.; Taube, H.; Yoshida, H. Anti-Markovnikov Olefin Arylation Catalyzed by an Iridium Complex. J. Am. Chem. Soc. 2000, 122, 7414–7415. [Google Scholar] [CrossRef]
- Aufdenblatten, R.; Diezi, S.; Togni, A. Iridium(I)-Catalyzed Asymmetric Intermolecular Hydroarylation of Norbornene with Benzamide. Mon. Für Chem. 2000, 131, 1345–1350. [Google Scholar] [CrossRef]
- Bhalla, G.; Oxgaard, J.; Goddard, W.-A., III; Periana, R.-A. Anti-Markovnikov Hydroarylation of Unactivated Olefins Catalyzed by a Bis-tropolonato Iridium(III) Organometallic Complex. Organometallics 2005, 24, 3229–3232. [Google Scholar] [CrossRef]
- Bischof, S.-M.; Ess, D.-H.; Meier, S.-K.; Oxgaard, J.; Nielsen, R.-J.; Bhalla, G.; Goddard, W.-A., III; Periana, R.-A. Benzene C-H Bond Activation in Carboxylic Acids Catalyzed by O-Donor Iridium(III) Complexes: An Experimental and Density Functional Study. Organometallics 2010, 29, 742–756. [Google Scholar] [CrossRef]
- Sevov, C.-S.; Hartwig, J.-F. Iridium-Catalyzed Intermolecular Asymmetric Hydroheteroarylation of Bicycloalkenes. J. Am. Chem. Soc. 2013, 135, 2116–2119. [Google Scholar] [CrossRef]
- Yamamoto, Y. Cationic Iridium/S-Me-BIPAM-Catalyzed Direct Asymmetric Intermolecular Hydroarylation of Bicycloalkenes. Angew. Chem. Int. Ed. 2015, 54, 9894–9897. [Google Scholar]
- Shirai, T.; Ito, H.; Yamamoto, Y. Cationic Ir/Me-BIPAM-Catalyzed Asymmetric Intramolecular Direct Hydroarylation of a-Ketoamides. Angew. Chem. 2014, 126, 2696–2699. [Google Scholar] [CrossRef]
- Shirai, T.; Yamamoto, Y. Scope and Mechanistic Studies of the Cationic Ir/Me-BIPAMCatalyzed Asymmetric Intramolecular Direct Hydroarylation Reaction. Organometallics 2015, 34, 3459–3463. [Google Scholar] [CrossRef]
- Yamauchi, D.; Nishimura, T.; Yorimitsu, H. Asymmetric hydroarylation of vinyl ethers catalyzed by a hydroxoiridium complex: Azoles as effective directing groups. Chem. Commun. 2017, 53, 2760–2763. [Google Scholar] [CrossRef]
- Sakamoto, K.; Nishimura, T. Iridium-Catalyzed Asymmetric Hydroarylation of Chromene Derivatives with Aromatic Ketones: Enantioselective Synthesis of 2-Arylchromanes. Adv. Synth. Catal. 2019, 361, 2124–2128. [Google Scholar] [CrossRef]
- Shinde, V.-S.; Mane, M.-V.; Cavallo, L.; Rueping, M. Iridium-Catalyzed Enantioselective Hydroarylation of Alkenes through C-H bond Activation: Experiment and Computation. Chem. Eur. J. 2020, 26, 8308–8313. [Google Scholar] [CrossRef] [PubMed]
- Diesel, J.; Finogenova, A.-M.; Cramer, N. Nickel-Catalyzed Enantioselective Pyridone C–H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand. J. Am. Chem. Soc. 2018, 140, 4489–4493. [Google Scholar] [CrossRef]
- Cai, Y.; Ye, X.; Liu, S.; Shi, S.-L. Nickel/NHC-Catalyzed Asymmetric C-H Alkylation of Fluoroarenes with Alkenes: Synthesis of Enantioenriched Fluorotetralins. Angew. Chem. 2019, 131, 13567–13571. [Google Scholar] [CrossRef]
- Qin, X.; Lee, M.-W.-Y.-; Zhou, J.-S. Asymmetric Hydroarylation of Enones via Nickel-Catalyzed 5-Endo-Trig Cyclization. Org. Lett. 2019, 21, 5990–5994. [Google Scholar] [CrossRef]
- Pradal, A.; Gladiali, S.; Michelet, V.; Toullec, P.-Y. Combinatorial Approach to Chiral Tris-ligated Carbophilic Platinum Complexes: Application to Asymmetric Catalysis. Chem. Eur. J. 2014, 20, 7128–7135. [Google Scholar] [CrossRef]
- Li, G.; Liu, Q.; Vasamsetty, L.; Guo, W.; Wang, J. Ruthenium(II)-Catalyzed Asymmetric Inert C-H Bond Activation Assisted by a Chiral Transient Directing Group. Angew. Chem. Int. Ed. 2020, 59, 3475–3479. [Google Scholar] [CrossRef]
- Whyte, A.; Torelli, A.; Mirabi, B.; Prieto, L.; Rodríguez, J.-F.; Lautens, M. Cobalt-Catalyzed Enantioselective Hydroarylation of 1,6-Enynes. J. Am. Chem. Soc. 2020, 142, 9510–9517. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Xie, P.-P.; Liu, L.; Fan, J.; Zhang, Z.-Z.; Hong, X.; Shi, B.-F. Cp*Co(III)-Catalyzed Enantioselective Hydroarylation of Unactivated Terminal Alkenes via C−H Activation. J. Am. Chem. Soc. 2021, 143, 19112–19120. [Google Scholar] [CrossRef]
- Cacchi, S. The palladium-catalyzed hydroarylation and hydrovinylation of carbon-carbon multiple bonds: New perspectives in organic synthesis. Pure Appl. Chem. 1990, 62, 713–722. [Google Scholar] [CrossRef]
- Cacchi, S.; Arcadi, A. Palladium-Catalyzed Conjugate Addition Type Reaction of Aryl Iodides with α,β-Unsaturated Ketones. J. Org. Chem. 1983, 48, 4236–4240. [Google Scholar] [CrossRef]
- Fernández-Alvarez, F.-J.; Iglesias, M.; Oro, L.-A.; Passarelli, V. Bond Activation and Catalysis. In Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevie: Amsterdam, The Netherlands, 2013; pp. 399–432. [Google Scholar]
- Cacchi, S.; Fabrizi, G. Carbopalladation of Alkynes Followed by Trapping with Nucleophilic Reagents. In Handbook of Organopalladium Chemistry for Organic Synthesis; Wiley Online: Hoboken, NJ, USA, 2002; pp. 1335–1359. [Google Scholar]
- Liao, L.; Sigman, M.-S. Palladium-Catalyzed Hydroarylation of 1,3-Dienes with Boronic Esters via Reductive Formation of π-Allyl Palladium Intermediates under Oxidative Conditions. J. Am. Chem. Soc. 2010, 132, 10209–10211. [Google Scholar] [CrossRef] [PubMed]
- Podhajsky, S.-M.; Iwai, Y.; Cook-Sneathen, A.; Sigman, M.-S. Asymmetric palladium-catalyzed hydroarylation of styrenes and dienes. Tetrahedron 2011, 67, 4435–4441. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, J.-S. Desymmetrization of cyclic olefins via asymmetric Heck reaction and hydroarylation. Chem. Commun. 2013, 49, 11758–11760. [Google Scholar] [CrossRef]
- Friis, S.-D.; Pirnot, M.-T.; Buchwald, S.-L. Asymmetric Hydroarylation of Vinylarenes Using a Synergistic Combination of CuH and Pd Catalysis. J. Am. Chem. Soc. 2016, 138, 8372–8375. [Google Scholar] [CrossRef]
- Lu, Z.; Buchwald, S.-L. Enantioselective Preparation of Arenes with b-Stereogenic Centers: Confronting the 1,1-Disubstituted Olefin Problem Using CuH/Pd Cooperative Catalysis. Angew. Chem. Int. Ed. 2020, 59, 16128–16132. [Google Scholar] [CrossRef]
- Schuppe, A.-W.; Borrajo-Calleja, G.-M.; Buchwald, S.-L. Enantioselective Olefin Hydrocyanation without Cyanide. J. Am. Chem. Soc. 2019, 141, 18668–18672. [Google Scholar] [CrossRef]
- Chang, J.; Reiner, J.; Xie, J. Progress on the Chemistry of Dibenzocyclooctadiene Lignans. J. Chem. Rev. 2005, 105, 4581–4609. [Google Scholar] [CrossRef]
- Lednicer, D. The Organic Chemistry of Drug Synthesis. In Organic Chem. Synthesis; Wiley: Hoboken, NJ, USA, 2007; Volume 7. [Google Scholar]
- Wang, R.; Liu, D.; Li, X.; Zhang, J.; Cui, D.; Wan, X. Synthesis and Stereospecific Polymerization of a Novel Bulky Styrene Derivative. Macromolecules 2016, 49, 2502–2510. [Google Scholar] [CrossRef]
- Kong, W.; Wang, Q.; Zhu, J. Water as a Hydride Source in Palladium-Catalyzed Enantioselective Reductive Heck Reactions. Angew. Chem. Int. Ed. 2017, 56, 3987–3991. [Google Scholar] [CrossRef]
- Zhang, Z.-M.; Xu, B.; Qian, Y.; Wu, L.; Wu, Y.; Zhou, L.; Liu, Y.; Zhang, J. Palladium-Catalyzed Enantioselective Reductive Heck Reactions:Convenient Access to 3,3-Disubstituted 2,3-Dihydrobenzofuran. Angew. Chem. Int. Ed. 2018, 130, 10530–10534. [Google Scholar] [CrossRef]
- Oxtoby, L.-J.; Li, Z.-Q.; Tran, V.-T.; Erbay, T.-G.; Deng, R.; Liu, P.; Engle, K.-M. A Transient-Directing-Group Strategy Enables Enantioselective Reductive Heck Hydroarylation of Alkenes. Angew. Chem. Int. Ed. 2020, 132, 8970–8975. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, B.; Zhu, S. Rapid Access to Highly Functionalized Alkyl Boronates by NiH-Catalyzed Remote Hydroarylation of Boron-Containing Alkenes. Angew. Chem. Int. Ed. 2019, 131, 13998–14002. [Google Scholar] [CrossRef]
- He, Y.; Liu, C.; Yu, L.; Zhu, S. Enantio- and Regioselective NiH-Catalyzed Reductive Hydroarylation of Vinylarenes with Aryl Iodides. Angew. Chem. Int. Ed. 2020, 59, 21530–21534. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Chen, J.; Song, P.; He, Y.; Zhu, S. Nickel-Catalyzed Regiodivergent Reductive Hydroarylation of Styrenes. Synlett 2021, 32, 1647–1651. [Google Scholar]
- Zhang, Y.; Ma, J.; Chen, J.; Meng, L.; Liang, Y.; Zhu, S. A relay catalysis strategy for enantioselective nickel-catalyzed migratory hydroarylation forming chiral a-aryl alkylboronates. Chem 2021, 7, 3171–3188. [Google Scholar] [CrossRef]
- He, Y.; Song, H.; Chen, J.; Zhu, S. NiH-catalyzed asymmetric hydroarylation of N-acyl enamines to chiral benzylamines. Nat. Commun. 2021, 12, 638. [Google Scholar] [CrossRef]
- Cuesta-Galisteo, S.; Schçrgenhumer, J.; Wei, X.; Merino, E.; Nevado, C. Nickel-Catalyzed Asymmetric Synthesis of a-Arylbenzamides. Angew. Chem. Int. Ed. 2021, 133, 1629–1633. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Shuai, B.; Xu, X.-T.; Li, Y.-Q.; Yang, Q.-L.; Qiu, H.; Zhang, K.; Fang, P.; Mei, T.-S. Nickel-catalyzed Enantioselective Hydroarylation and Hydroalkenylation of Styrenes. J. Am. Chem. Soc. 2019, 141, 3395–3399. [Google Scholar] [CrossRef]
- Lv, X.-Y.; Fan, C.; Xiao, L.-J.; Xie, J.-H.; Zhou, Q.-L. Ligand-Enabled Ni-Catalyzed Enantioselective Hydroarylation of Styrenes and 1,3-Dienes with Arylboronic Acids. CCS Chem. 2019, 1, 328–334. [Google Scholar] [CrossRef]
- Marcum, J.-S.; Taylor, T.-R.; Meek, S.-J. Enantioselective Synthesis of Functionalized Arenes by Nickel-Catalyzed Site-Selective Hydroarylation of 1,3-Dienes with Aryl Boronates. Angew. Chem. Int. Ed. 2020, 59, 14070–14075. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.-N.; Burgett, R.-W.; Stanley, L.-M. Nickel-Catalyzed Asymmetric Hydroarylation of Vinylarenes: Direct Enantioselective Synthesis of Chiral 1,1-Diarylethanes. J. Org. Chem. 2021, 86, 3836–3849. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Hayashi, H.; Miyaura, N. Rhodium-catalyzed conjugate addition of aryl-or 1-alkenylboronic acids to enones. Organometallics 1997, 16, 4229. [Google Scholar] [CrossRef]
- Takaya, Y.; Ogasawara, M.; Hayashi, T.; Sakai, M.; Miyaura, N. Rhodium-catalyzed asymmetric 1, 4-addition of aryl-and alkenylboronic acids to enones. J. Am. Chem. Soc. 1998, 120, 5579–5580. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamasaki, K. Rhodium-catalyzed asymmetric 1, 4-addition and its related asymmetric reactions. Chem. Rev. 2003, 103, 2829–2844. [Google Scholar] [CrossRef]
- Berthon, G.; Hayashi, T. Catalytic Asymmetric Conjugate Reactions; Córdova, A., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Chapter 1; p. 1. [Google Scholar]
- Wang, Z.; Hayashi, T. Rhodium-Catalyzed Enantioposition-Selective Hydroarylation of Divinylphosphine Oxides with Aryl Boroxines. Angew. Chem. Int. Ed. 2018, 130, 1718–1722. [Google Scholar] [CrossRef]
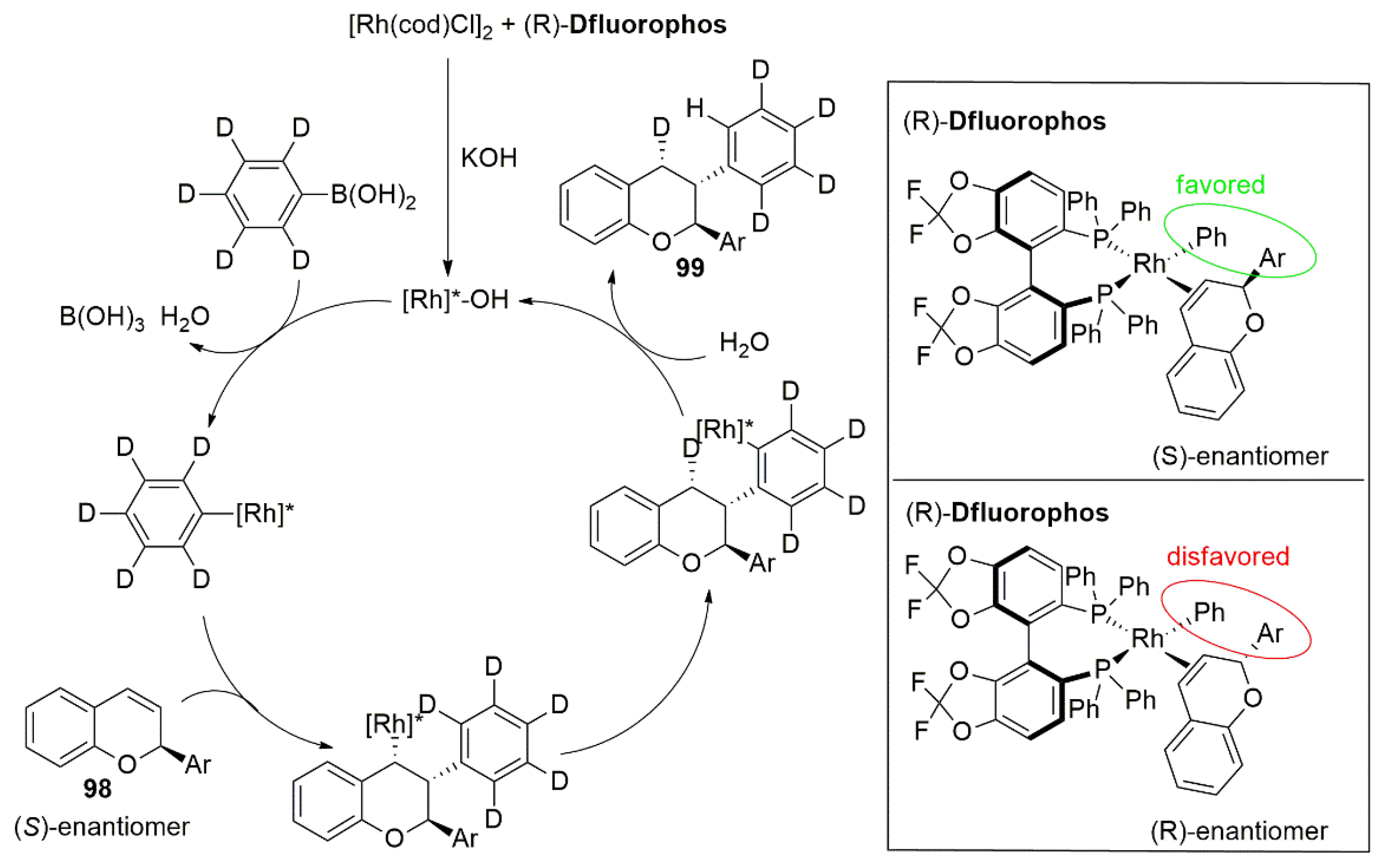
- Yang, Q.; Wang, Y.; Luo, S.; Jun Wang, J. Kinetic Resolution and Dynamic Kinetic Resolution of Chromene by Rhodium-Catalyzed Asymmetric Hydroarylation. Angew. Chem. Int. Ed. 2019, 131, 5397–5401. [Google Scholar] [CrossRef]
- Chen, J.P.; Li, Y.; Liu, C.; Wang, T.; Chung, L.W.; Xu, M.H. Water as a Direct Proton Source for Asymmetric Hydroarylation Catalyzed by a Rh(I)−Diene: Access to Nonproteinogenic β2/γ2/δ2-Amino Acid Derivatives. Org. Lett. 2021, 23, 571–577. [Google Scholar] [CrossRef]
- Hu, F.; Jia, J.; Li, X.; Xia, Y. Enantioselective Hydroarylation or Hydroalkenylation of Benzo[b]thiophene 1,1-Dioxides with Organoboranes. Org. Lett. 2021, 23, 896–901. [Google Scholar] [CrossRef]









































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Angelis, M.; Iazzetti, A.; Serraiocco, A.; Ciogli, A. Asymmetric Hydroarylation Reactions Catalyzed by Transition Metals: Last 10 Years in a Mini Review. Catalysts 2022, 12, 1289. https://doi.org/10.3390/catal12101289
De Angelis M, Iazzetti A, Serraiocco A, Ciogli A. Asymmetric Hydroarylation Reactions Catalyzed by Transition Metals: Last 10 Years in a Mini Review. Catalysts. 2022; 12(10):1289. https://doi.org/10.3390/catal12101289
Chicago/Turabian StyleDe Angelis, Martina, Antonia Iazzetti, Andrea Serraiocco, and Alessia Ciogli. 2022. "Asymmetric Hydroarylation Reactions Catalyzed by Transition Metals: Last 10 Years in a Mini Review" Catalysts 12, no. 10: 1289. https://doi.org/10.3390/catal12101289
APA StyleDe Angelis, M., Iazzetti, A., Serraiocco, A., & Ciogli, A. (2022). Asymmetric Hydroarylation Reactions Catalyzed by Transition Metals: Last 10 Years in a Mini Review. Catalysts, 12(10), 1289. https://doi.org/10.3390/catal12101289