
Solid Acid Catalysts for the Hock Cleavage of Hydroperoxides


Abstract
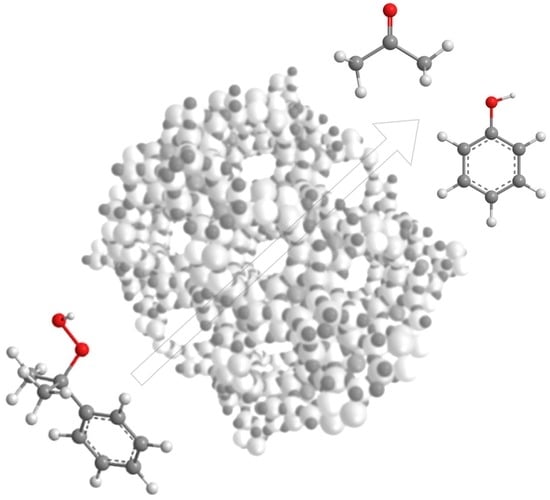
:
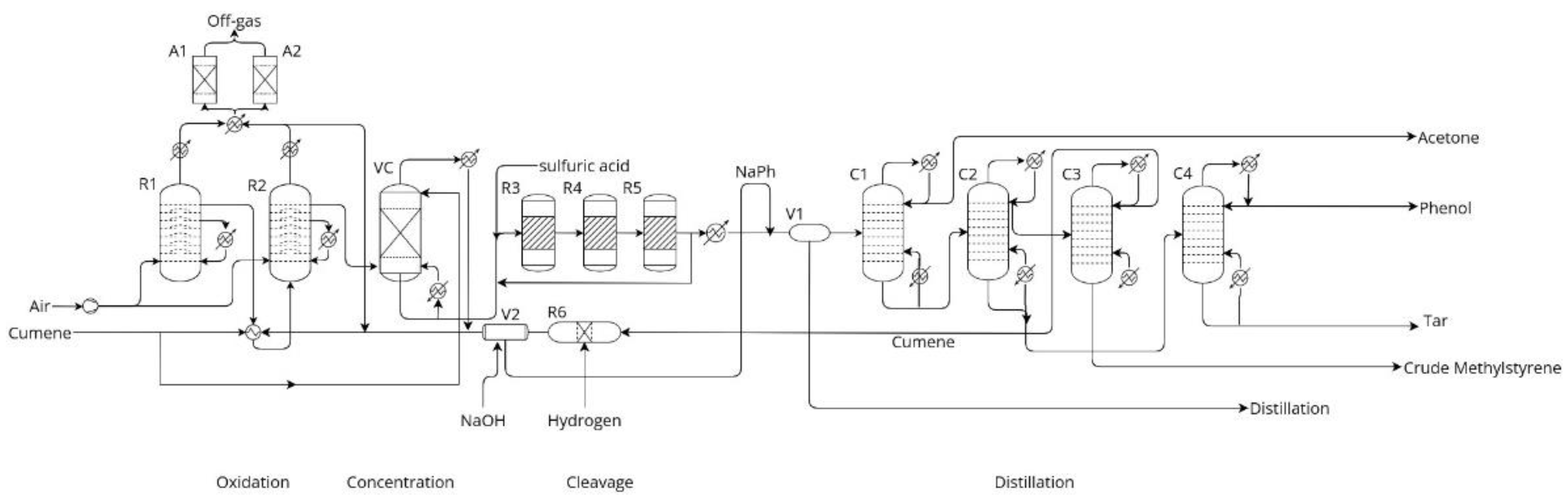
1. Introduction
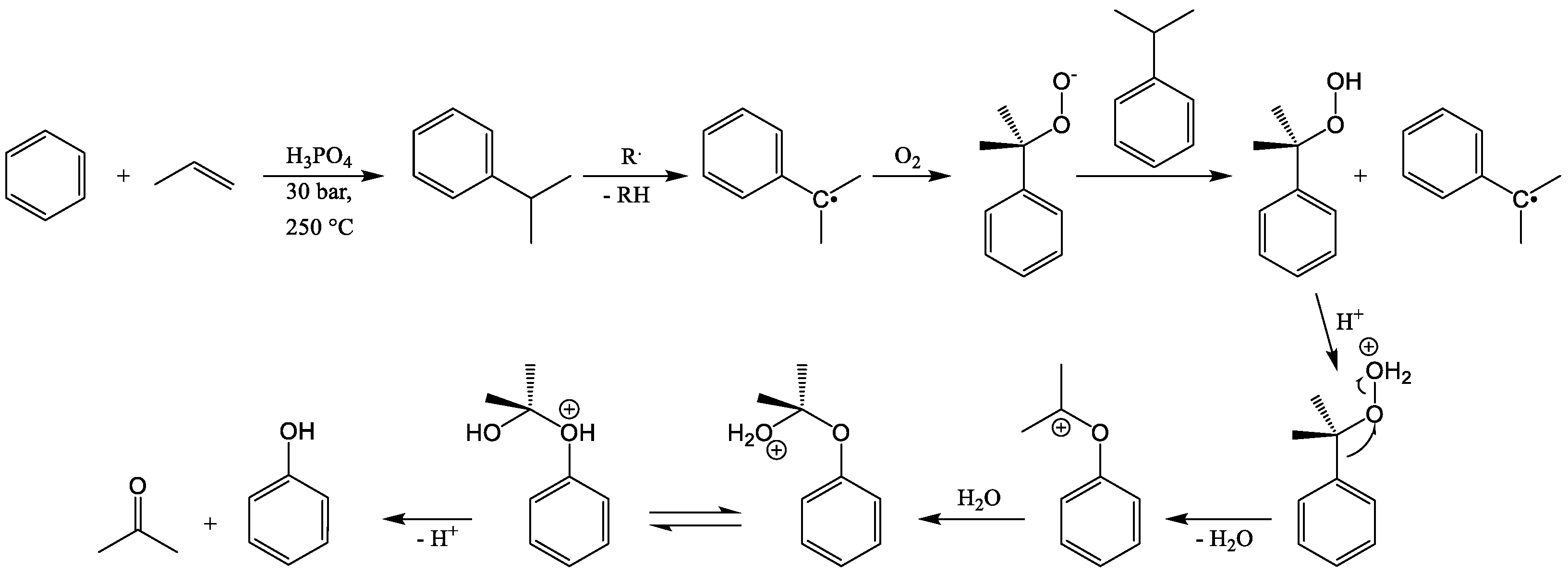
2. Mechanism, Kinetics, and Safety Aspects of the CHP Cleavage
3. Solid Acid Catalysts
3.1. Mineral Acid-Treated Clays
3.2. Heteropoly Acids (HPA) on Supports
3.3. Zeolites
3.4. Ion Exchange Resins
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Weber, M.M.; Weber, M.M.; Weber, V. Phenol; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2020; pp. 4–7. [Google Scholar] [CrossRef]
- Sheldon, R.A. Homogeneous and Heterogeneous Catalytic Oxidations with Peroxide Reagents. Org. Peroxygen Chem. 1993, 164, 21–43. [Google Scholar] [CrossRef]
- Alekar, N.A.; Indira, V.; Halligudi, S.B.; Srinivas, D.; Gopinathan, S.; Gopinathan, C. Kinetics and Mechanism of Selective Hydroxylation of Benzene Catalysed by Vanadium Substituted Heteropolymolybdates. J. Mol. Catal. A Chem. 2000, 164, 181–189. [Google Scholar] [CrossRef]
- Lee, C.W.; Lee, W.J.; Park, Y.K.; Park, S.E. Catalytic Hydroxylation of Benzene over Vanadium-Containing Molecular Sieves. Catal. Today 2000, 61, 137–141. [Google Scholar] [CrossRef]
- Olah, G.A.; Ohnishi, R. Oxyfunctionalization of Hydrocarbons. 8. Electrophilic Hydroxylation of Benzene, Alkylbenzenes, and Halobenzenes with Hydrogen Peroxide in Superacids. J. Org. Chem. 1978, 43, 865–867. [Google Scholar] [CrossRef]
- Kuznetsova, N.I.; Kuznetsova, L.I.; Kirillova, N.V.; Pokrovskii, L.M.; Detusheva, L.G. Oxidation of Cyclohexene and α Pinene with O2—H2 Mixture. Russ. Chem. Bull. 2003, 52, 1544–1551. [Google Scholar] [CrossRef]
- Niwa, S.I.; Eswaramoorthy, M.; Nair, J.; Raj, A.; Itoh, N.; Shoji, H.; Namba, T.; Mizukami, F. A One-Step Conversion of Benzene to Phenol with a Palladium Membrane. Science 2002, 295, 105–107. [Google Scholar] [CrossRef]
- Sobolev, V.I.; Dubkov, K.A.; Paukshtis, E.A.; Piratko, L.V.; Rodkin, M.A.; Kharitonov, A.S.; Panov, G.I. On the Role of Brønsted Acidity in the Oxidation of Benzene to Phenol by Nitrous Oxide. Appl. Catal. A Gen. 1996, 141, 185–192. [Google Scholar] [CrossRef]
- Dubkov, K.A.; Sobolev, V.I.; Talsi, E.P.; Rodkin, M.A.; Watkins, N.H.; Shteinman, A.A.; Panov, G.I. Kinetic Isotope Effects and Mechanism of Biomimetic Oxidation of Methane and Benzene on FeZSM-5 Zeolite. J. Mol. Catal. A Chem. 1997, 123, 155–161. [Google Scholar] [CrossRef]
- Zakoshansky, V. Phenol Process Celebrates Its 60th Anniversary: The Role of Chemical Principles in Technological Breakthroughs. Russ. J. Gen. Chem. 2009, 79, 2267–2271. [Google Scholar] [CrossRef]
- Morachevskii, A.G.; Nemtsov, M.S. Vospominaniya i Razmyshleniya (Zapiski Khimika) (Recollections and Reflection (Chemist’s Memoirs)). Russ. J. Appl. Chem. 2007, 80, 511–512. [Google Scholar] [CrossRef]
- Hock, H.; Lang, S. Autoxydation von Koblenwasserstoffen: Über Peroxyde von Benzol-Derivaten. Eur. J. Inorg. Chem. 1944, 77, 257–263. [Google Scholar]
- Udris, R.J.; Sergeyev, P.G.; Kruzhalov, B.D. Sposob Polucheniya Gidroperekisejj Alkilirovannykh-Proizvodnykh Benzola Ili Alicikloaromaticheskikh Uglevodorodov. USSR Patent 106,666, 7 January 1947. [Google Scholar]
- Yaremenko, I.A.; Vil’, V.A.; Demchuk, D.V.; Terent’ev, A.O. Rearrangements of Organic Peroxides and Related Processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748. [Google Scholar] [CrossRef] [Green Version]
- Duh, Y.S.; Kao, C.S.; Hwang, H.H.; Lee, W.W.L. Thermal Decomposition Kinetics of Cumene Hydroperoxide. Process Saf. Environ. Prot. 1998, 76, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.F.; Guo, S.J.; Wu, S.H. Fire and Thermal Hazard Analyses of Industrial Zeolite Catalysis for Phenol Production. Adv. Mater. Res. 2012, 560–561, 161–166. [Google Scholar] [CrossRef]
- The 100 Largest Losses 1972–2011; Marsh & McLennan: New York, NY, USA, 2011; pp. 1–42.
- Yadav, G.D.; Asthana, N.S. Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone by a Novel Cesium Substituted Heteropolyacid on Clay. Appl. Catal. A Gen. 2003, 244, 341–357. [Google Scholar] [CrossRef]
- Schmidt, R.J. Industrial Catalytic Processes—Phenol Production. Appl. Catal. A Gen. 2005, 280, 89–103. [Google Scholar] [CrossRef]
- Twigg, G.H. Liquid-Phase Oxidation (of Organic Compounds) by Molecular Oxygen. Chem. Ind. 1962, 1, 4–11. [Google Scholar]
- Weber, M.; Daldrup, J.B.G.; Weber, M. Noncatalyzed Radical Chain Oxidation: Cumene Hydroperoxide. In Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 15–31. [Google Scholar] [CrossRef]
- Hattori, H. Heterogeneous Basic Catalysis. Chem. Rev. 1995, 95, 537–558. [Google Scholar] [CrossRef]
- Cooke, M.D. The Distillers Company Limited Improvements in or relating to the Manufacture of Alkyl Benzene Peroxides. Br. Patent No. 610293A, 13 October 1948. [Google Scholar]
- Cooke, M.D. The Distillers Company Limited Verfahren zur Anreicherung Cumolhydroperoxid. DE1114817B, 12 October 1961. [Google Scholar]
- Hall, R.H.; Quin, D.C. Hercules Powder Company Manufacture of Alkyl Benzene Peroxides. U.S. Patent 2547938A, 10 April 1951. [Google Scholar]
- Lorand, E.J.; Reese, J.E. Hercules Powder Company Oxidation of Aromatic Hydrocarbons. U.S. Patent 2548435A, 10 April 1951. [Google Scholar]
- Sodomann, H.; Hauschulz, B. Phenolchemie Verfahren zur Herstellung von Aralkylmonohydroperoxyden. DE1131674B, 20 June 1960. [Google Scholar]
- Sifniades, S.; Tunick, A.A.; Koff, F.W. Decomposition of Cumene Oxidation Product. U.S. Patent 4358618A, 9 November 1982. [Google Scholar]
- Mategazza, A.; Reni, C.; Arsizio, B.; Societa Italiana Resine. Verfahren und Vorrichtung zum kontinuierlichen katalytischen Spalten von Alkylarylhydroperoxyden, besonders Cumolhydroperoxid, mit Schwefelsäure. DE1443329B1, 2 January 1970. [Google Scholar]
- Zakoshansky, V.M. Method for the Dewcompostion of Cujmene Hydroperoxide by Acidic Catalyst to Phenol and Acetone. U.S. Patent 5254751A, 19 October 1993. [Google Scholar]
- Zakoshansky, V.M.; Griaznov, A.K.; Vasilieva, I.I. Method for Preparing Phenol and Carbonyl Compounds. Russian Patent RU2142932C1, 20 December 1999. [Google Scholar]
- Zakoshansky, V.M.; Vasilieva, I.I.; Griaznov, A.K.; Youriev, Y.N.; Van Barnefeld, H.; Gerlich, O. Improved Method for Producing Phenol and Acetone from Cumol. International Patent WO98/27039, 25 June 1998. [Google Scholar]
- Zakoshansky, V.M.; Griaznov, A.K.; Vasilieva, I.I. High Selective Method of Phenol and Acetone Production. U.S. Patent 6057483A, 2 May 2000. [Google Scholar]
- Kozhevnikov, I.V.; Mastikhin, V.M.; Matveev, K.I.; Kirichenko, G.N.; Churkin, Y.V.; Glazunova, V.I. Kinetics of Decomposition of Isopropylbenzene Hydroperoxide Catalyzed By Dodecamolybdophosphoric Acid. React. Kinet. Catal. Lett. 1977, 7, 291–296. [Google Scholar] [CrossRef]
- Levin, M.E.; Gonzales, N.O.; Zimmerman, L.W.; Yang, J. Kinetics of Acid-Catalyzed Cleavage of Cumene Hydroperoxide. J. Hazard. Mater. 2006, 130, 88–106. [Google Scholar] [CrossRef]
- Jung, G.; Just, G. Thermokinetische Untersuchung Der Cumolhydroperoxidzersetzung. J. Prakt. Chem. 1971, 313, 377–394. [Google Scholar] [CrossRef]
- Weber, M.; Hoffmann, R.; Weber, M. Some Safety Aspects on the Cleavage of Cumene Hydroperoxide in the Phenol Process. Process Saf. Prog. 2019, 38, 1–4. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Doorn, J.A. Observation by PMR Spectroscopy of the Intermediate Alkoxycarbonium Ions in the Acid-Catalysed Decomposition of Organic Hydroperoxides. Tetrahedron Lett. 1973, 14, 1021–1022. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Delanghe, N.C. New Catalysts for the Conversion of Cumene Hydroperoxide into Phenol. Tetrahedron Lett. 1997, 38, 73–78. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Fono, A.; Nudenberg, W. The Chemistry Of Hydroperoxides I. The Acid-Catalyzed Decomposition of α,α-Dimethylbenzyl (α-Cumyl) Hydroperxoide. J. Org. Chem. 1950, 15, 748–752. [Google Scholar] [CrossRef]
- Seubold, F.H.; Vaughan, W.E. Acid-Catalyzed Decomposition of Cumene Hydroperoxide. J. Am. Chem. Soc. 1953, 75, 3790–3792. [Google Scholar] [CrossRef]
- Turner, J.O. The Acid-Catalyzed Decomposition of Aliphatic Hydroperoxides: Reactions in the Presence of Alcohols. Tetrahedron Lett. 1971, 12, 887–890. [Google Scholar] [CrossRef]
- Deno, N.C.; Billups, W.E.; Kramer, K.E.; Lastomirsky, R.R. The Rearrangement of Aliphatic Primary, Secondary, and Tertiary Alkyl Hydroperoxides in Strong Acid. J. Org. Chem. 1970, 35, 3080–3082. [Google Scholar] [CrossRef]
- Hock, H.; Kropf, H. Autoxydation von Kohlenwasserstoffen Und Die Cumol-Phenol-Synthese. Angew. Chem. 1957, 69, 313–321. [Google Scholar] [CrossRef]
- Pinnavaia, T.J. Intercalated Clay Catalysts. Science 1983, 220, 365–371. [Google Scholar] [CrossRef]
- Varma, R.S. Clay and Clay-Supported Reagents in Organic Synthesis. Tetrahedron 2002, 58, 1235–1255. [Google Scholar] [CrossRef]
- Theocharis, C.R.; S’Jacob, K.J.; Gray, A.C. Enhancement of Lewis Acidity in Layer Aluminosilicates. Treatment with Acetic Acid. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 1509–1515. [Google Scholar] [CrossRef]
- Cativiela, C.; Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Figueras, F.; De Menorval, L.C.; Alonso, P.J. Factors Influencing the K10 Montmorillonite-Catalyzed Diels-Alder Reaction between Methyl Acrylate and Cyclopentadiene. J. Catal. 1992, 137, 394–407. [Google Scholar] [CrossRef]
- Rhodes, C.N.; Brown, D.R. Surface Properties and Porosities of Silica and Acid-Treated Montmorillonite Catalyst Supports: Influence on Activities of Supported ZnCl2 Alkylation Catalysts. J. Chem. Soc. Faraday Trans. 1993, 89, 1387–1391. [Google Scholar] [CrossRef]
- Selvin, R.; Hsu, H.L.; Aneesh, P.; Chen, S.H.; Hung, L.H. Preparation of Acid-Modified Bentonite for Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone. React. Kinet. Mech. Catal. 2010, 100, 197–204. [Google Scholar] [CrossRef]
- Sasidharan, M.; Kumar, R. Zeolite-Catalysed Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone. J. Chem. Res.-Part S 1997, 2, 52–53. [Google Scholar] [CrossRef]
- Knifton, J.F.; Sanderson, J.R. Phenol/Acetone Cogeneration via Solid Acid Catalysis. Appl. Catal. A Gen. 1997, 161, 199–211. [Google Scholar] [CrossRef]
- Han, L.; Wang, Y.; Zhang, J.; Lei, Z.; Huang, C.; Chen, B. Acidic Montmorillonite/Cordierite Monolithic Catalysts for Cleavage of Cumene Hydroperoxide. Chin. J. Chem. Eng. 2014, 22, 854–860. [Google Scholar] [CrossRef]
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Izumi, Y.; Urabe, K.; Onaka, M. Zeolites, Calys and Heteropoly Acids; VSH Publishers Inc.: London, UK, 1992. [Google Scholar]
- Okuhara, T.; Mizuno, N.; Misono, M. Catalytic Chemistry of Heteropoly Compounds. Adv. Catal. 1996, 41, 113–252. [Google Scholar] [CrossRef]
- Misono, M.; Nojiri, N. Recent Progress in Catalytic Technology in Japan. Appl. Catal. 1990, 64, 1–30. [Google Scholar] [CrossRef]
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Heteropoly Acids as Catalysts for Fine Chemicals Synthesis; Woodhead Publishing Ltd.: Sawston, UK, 2005; Volume 6. [Google Scholar] [CrossRef]
- Chang, C.D.; Pelrine, P.P. Mobil Oil Corporation Production of Phenol. U.S. Patent 4490565A, 25 December 1984. [Google Scholar]
- Romano, U.; Clerici, M.G.; Bellussi, G.; Buonomo, F. Catalyst for the Selective Decomposition of Cumene Hydroperoxide. U.S. Patent 47543573A, 10 May 1988. [Google Scholar]
- Romano, U.; Clerici, M.G.; Bellussi, G.; Buonomo, F. Catalyst for the Selective Decompostion of Cumene Hydroperoxide and Process Using it. U.S. Patent 4849387A, 18 July 1989. [Google Scholar]
- Xu, R.; Pang, W.; Yu, J.; Huo, Q.; Chen, J. Chemistry of Zeolites and Related Porous Materials; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Cejka, J.; Corma, A.; Zones, S. Zeolites and Catalysis; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Koltunov, K.Y.; Sobolev, V.I. Efficient Cleavage of Cumene Hydroperoxide over HUSY Zeolites: The Role of Brønsted Acidity. Appl. Catal. A Gen. 2008, 336, 29–34. [Google Scholar] [CrossRef]
- Kumar, K.P.; Selvin, R.; Kumari, P.; Roselin, L.S.; Arul, N.S.; Bououdina, M. Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone over Nanocrystalline ZSM-5. Int. J. Mater. Eng. Innov. 2010, 1, 417–425. [Google Scholar] [CrossRef]
- Hedge, S.G.; Ratnasamy, P.; Kustov, L.M.; Kazansky, V.B. Acidity and Catalytic Activity of SAPO-5 and AlPO-5 Molecular Sieves. Zeolites 1988, 8, 137–141. [Google Scholar] [CrossRef]
- Chaudhary, V.R.; Singh, A.P.; Kumar, R. Acidity and Sorbate Shape Selectivity of H-ZSM-22, H-ZSM-48, and H-ZSM-50 Zeolites. J. Catal. 1991, 129, 293–296. [Google Scholar] [CrossRef]
- Guisnet, M.; Gilson, J.-P. Zeolites for Cleaner Technologies; Imperial College Press: London, UK, 2002. [Google Scholar] [CrossRef]
- Louis, B.; Walspurger, S.; Sommer, J. Quantitative Determination of Brönsted Acid Sites on Zeolites: A New Approach towards the Chemical Composition of Zeolites. Catal. Lett. 2004, 93, 81–84. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Superelectrophilic Activation of Polyfunctional Organic Compounds Using Zeolites and Other Solid Acids. Chem. Commun. 2004, 15, 1754. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Cyclization of 1-Phenyl-2-Propen-1-Ones into 1-Indanones Using H-Zeolite and Other Solid Acids. The Role of Mono- and Dicationic Intermediates. Tetrahedron Lett. 2005, 46, 8391–8393. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Selective, C,C-Double Bond Reduction of α,β-Unsaturated Carbonyl Compounds with Cyclohexane Using Zeolites. J. Mol. Catal. A Chem. 2006, 245, 231–234. [Google Scholar] [CrossRef]
- Ye, J.; Li, J.; Sha, Y.; Xu, Y.; Zhou, D. Novel Reactive Distillation Process for Phenol Production with a Dry Cation Exchange Resin as the Catalyst. Ind. Eng. Chem. Res. 2014, 53, 12614–12621. [Google Scholar] [CrossRef]
- Huang, D.; Han, M.; Wang, J.; Jin, Y. Catalytic Decomposition Process of Cumene Hydroperoxide Using Sulfonic Resins as Catalyst. Chem. Eng. J. 2002, 88, 215–223. [Google Scholar] [CrossRef]
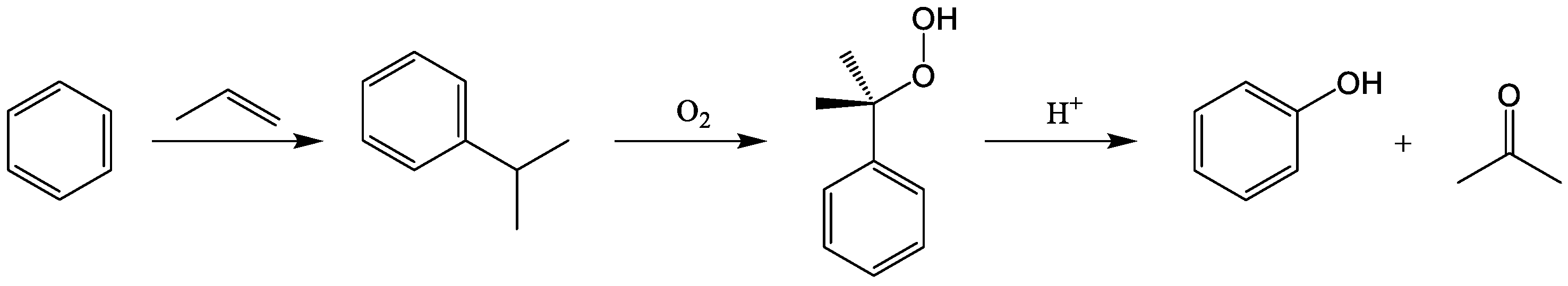



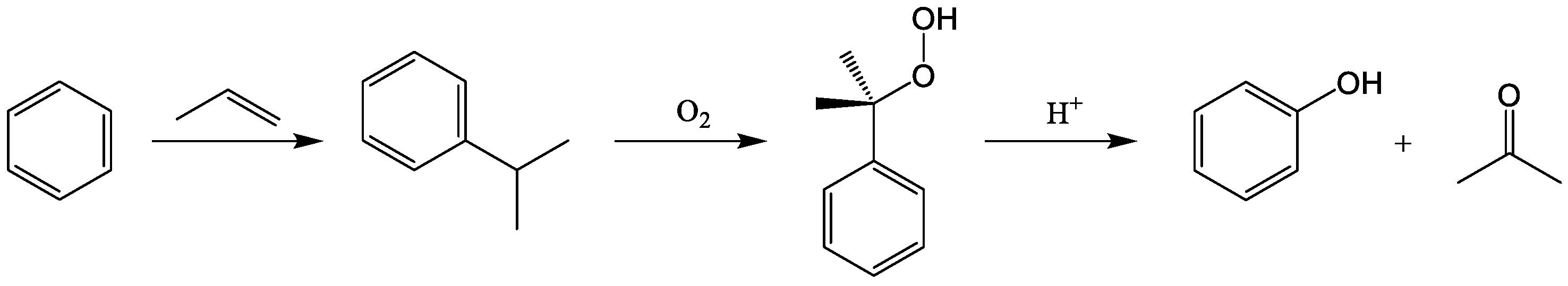
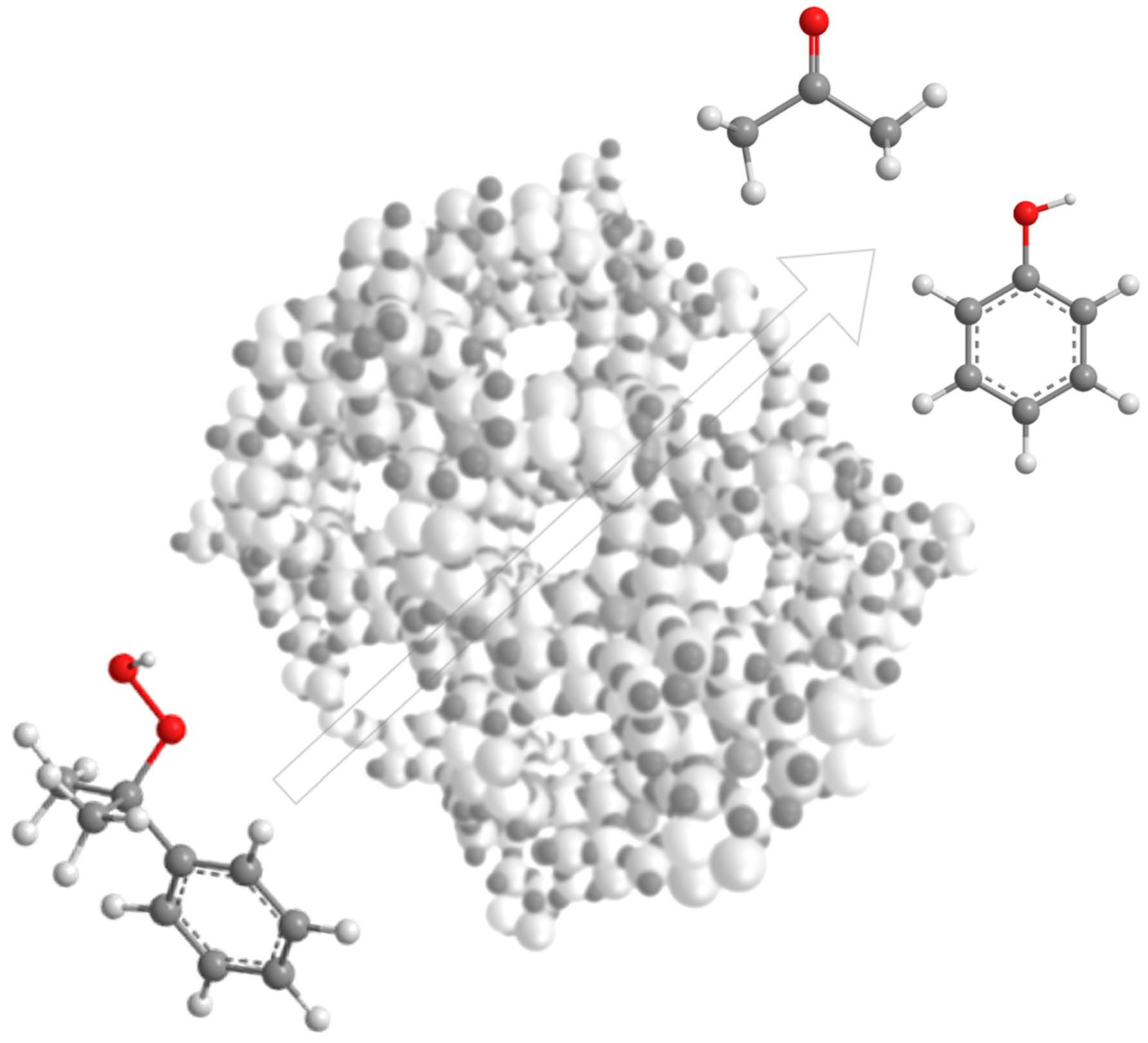{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Si/Al Ratio | Reaction Time/min | Reaction Temperature/°C | Conversion of CHP/% | Y (phenol)/% | S (phenol)/% |
|---|---|---|---|---|---|---|
| Clays | ||||||
| K1051 | n/a | 30 | 50 | 100 | n/a | n/a |
| K1051 with ZnCl2 | n/a | 30 | 50 | 80 | n/a | n/a |
| K1051 with FeCl3 | n/a | 30 | 50 | 92 | n/a | n/a |
| K1051 with CeCl3 | n/a | 30 | 50 | 100 | n/a | n/a |
| K1051 with LaCl3 | n/a | 30 | 50 | 95 | n/a | n/a |
| K1018 | n/a | 30 | 40 | 99 | n/a | n/a |
| Grade F2452 | n/a | 120 | 57–80 | 100 | 98 | 98 |
| Grade F11352 | n/a | 120 | 57–80 | 100 | 95 | 95 |
| Grade F1352 | n/a | 120 | 57–80 | 100 | 99 | 99 |
| Grade F6252 | n/a | 120 | 57–80 | 76 | 37 | 49 |
| Acidic montmorillonite53 | n/a | 90 | 90 | n/a | 95 | n/a |
| HPA on supports | ||||||
| 12-tungstophosphoric acid52 | n/a | 120 | 57–80 | 100 | 99 | n/a |
| 12-molybdophosphoric acid52 | n/a | 120 | 57–80 | 100 | 99 | n/a |
| 12-tungstosilicic acid52 | n/a | 120 | 57–80 | 100 | 99 | n/a |
| 12-molybdosilicic acid52 | n/a | 120 | 57–80 | 100 | 99 | n/a |
| Cs2.5H0.5PW12O40 on K1018 | n/a | 30 | 40 | 99 | n/a | |
| Zeolites | ||||||
| H-[Al]-Beta65 | 14 | 5 | 25 | 100 | 88 | 88 |
| H-[Ga]-Beta65 | 20 | 5 | 25 | 100 | 92 | 92 |
| H-[Fe]-Beta65 | 22 | 5 | 25 | 100 | 91 | 91 |
| H-[B]-Beta65 | 30 | 5 | 25 | 100 | 92 | 92 |
| H-[Al]-ZSM-565 | 30 | 5 | 25 | 100 | 86 | 86 |
| H-[Ga]-ZSM-565 | 35 | 5 | 25 | 100 | 89 | 89 |
| H-[Fe]-ZSM-565 | 30 | 5 | 25 | 100 | 88 | 88 |
| H-Mordenite65 | 7 | 5 | 25 | 100 | 87 | 87 |
| H-Y65 | 2.5 | 10 | 40 | 96 | 82 | 85 |
| H-[Al]-ZSM-1265 | 40 | 30 | 40 | 95 | 78 | 82 |
| H-[Al]-NCL-165 | 40 | 15 | 40 | 85 | 71 | 84 |
| H-[Al]-ZSM-2265 | 60 | 15 | 40 | 65 | 57 | 88 |
| H-[Al]-MCM-2265 | 30 | 15 | 40 | 90 | 78 | 87 |
| H-[Al]-ZSM-4865 | 50 | 60 | 60 | 45 | 36 | 80 |
| H-[Al]-EU-165 | 50 | 30 | 60 | 80 | 71 | 89 |
| H-SAPO-565 | n/a | 60 | 60 | 10 | 9 | 88 |
| H-AlPO-565 | n/a | 60 | 60 | 25 | 22 | 86 |
| H-[Al]-Betac65 | 14 | 60 | 60 | 99 | 94 | 95 |
| ZSM-5-0NC70 | n/a | 25 | 50 | 60 | n/a | n/a |
| ZSM-5-30NC70 | n/a | 25 | 50 | 94 | n/a | n/a |
| HUSY66 | 2.5 | 10 | 60 | 32 | n/a | n/a |
| HUSY66 | 15 | 10 | 60 | 90 | n/a | n/a |
| HUSY66 | 40 | 10 | 60 | 85 | n/a | n/a |
| HY66 | n/a | 20 | 60 | 10 | n/a | n/a |
| Beta66 | n/a | 600 | 20 | 89 | n/a | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drönner, J.; Hausoul, P.; Palkovits, R.; Eisenacher, M. Solid Acid Catalysts for the Hock Cleavage of Hydroperoxides. Catalysts 2022, 12, 91. https://doi.org/10.3390/catal12010091
Drönner J, Hausoul P, Palkovits R, Eisenacher M. Solid Acid Catalysts for the Hock Cleavage of Hydroperoxides. Catalysts. 2022; 12(1):91. https://doi.org/10.3390/catal12010091
Chicago/Turabian StyleDrönner, Jan, Peter Hausoul, Regina Palkovits, and Matthias Eisenacher. 2022. "Solid Acid Catalysts for the Hock Cleavage of Hydroperoxides" Catalysts 12, no. 1: 91. https://doi.org/10.3390/catal12010091
APA StyleDrönner, J., Hausoul, P., Palkovits, R., & Eisenacher, M. (2022). Solid Acid Catalysts for the Hock Cleavage of Hydroperoxides. Catalysts, 12(1), 91. https://doi.org/10.3390/catal12010091