
Catalytic Hydrofunctionalization Reactions of 1,3-Diynes


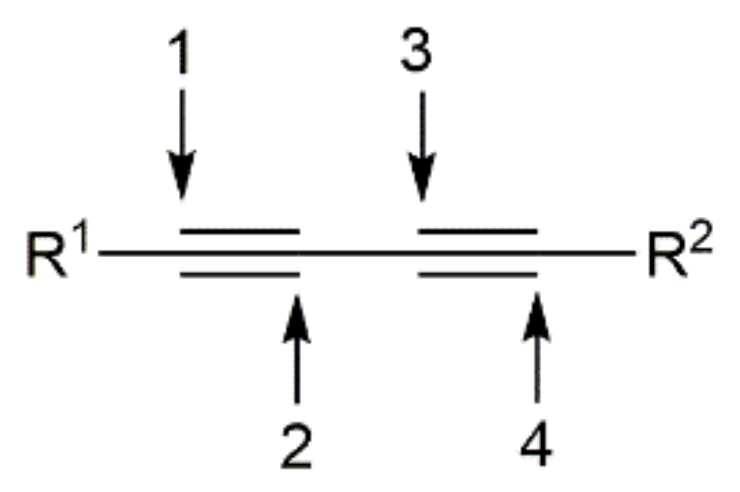{kind=link}
{kind=link}
{kind=link}
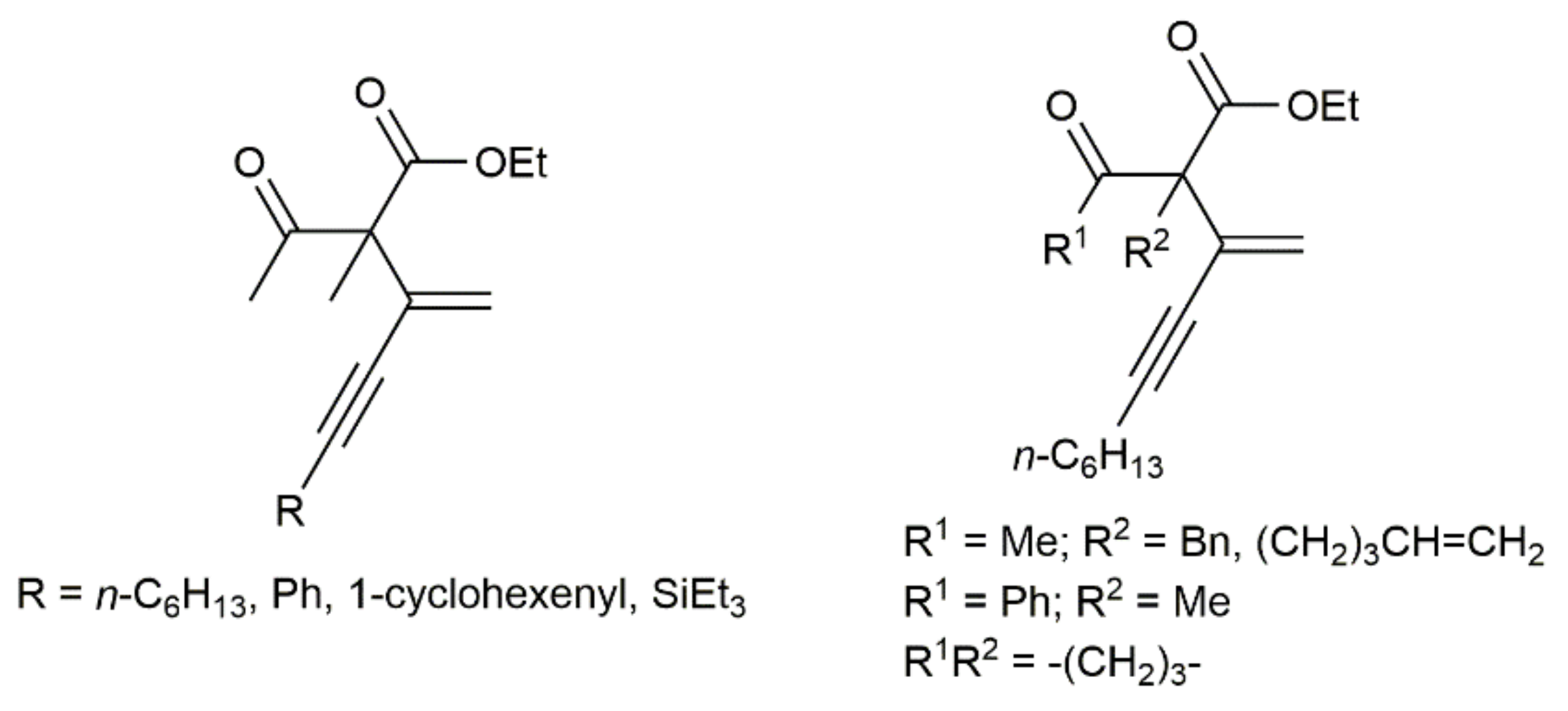{kind=link}
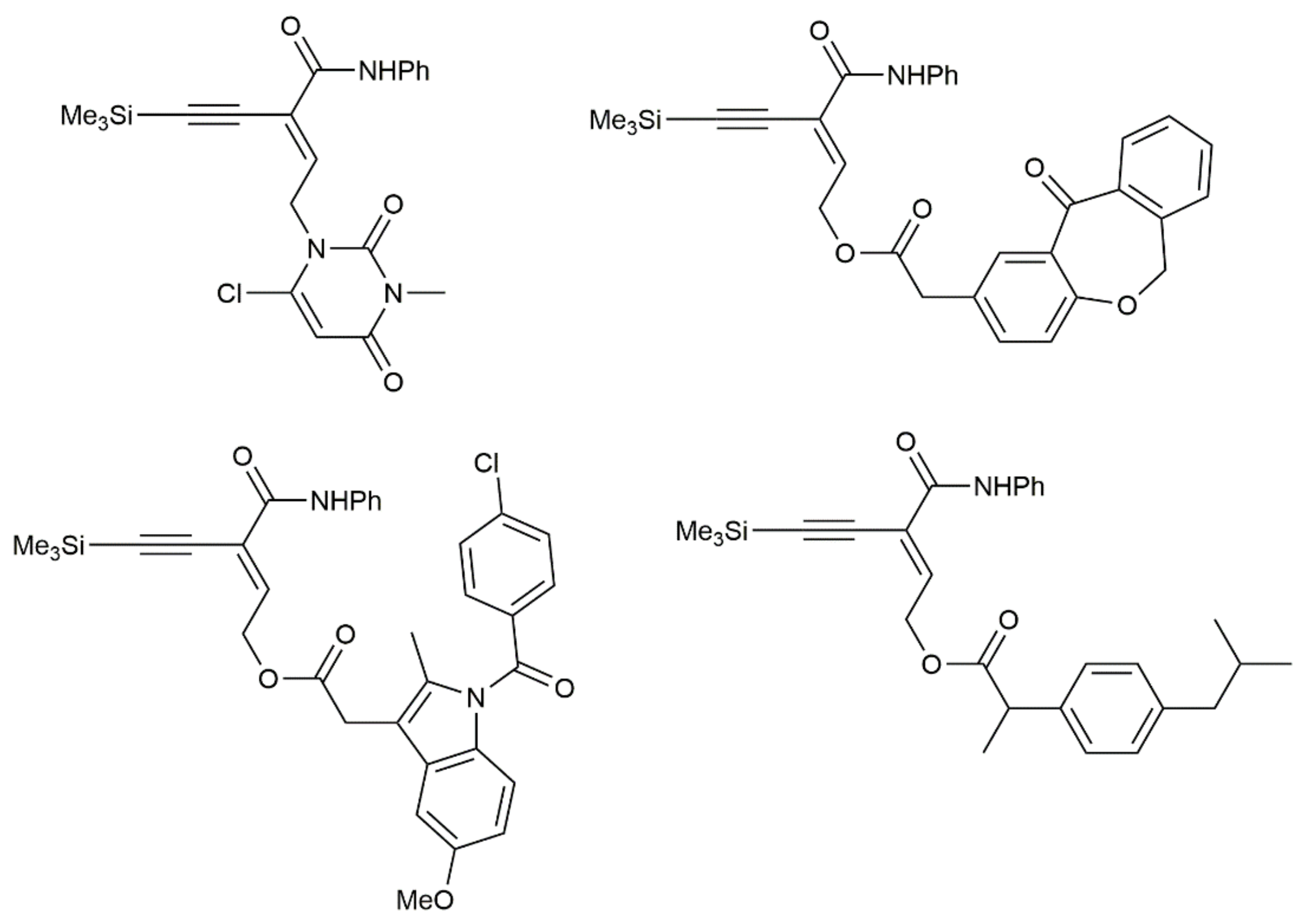{kind=link}
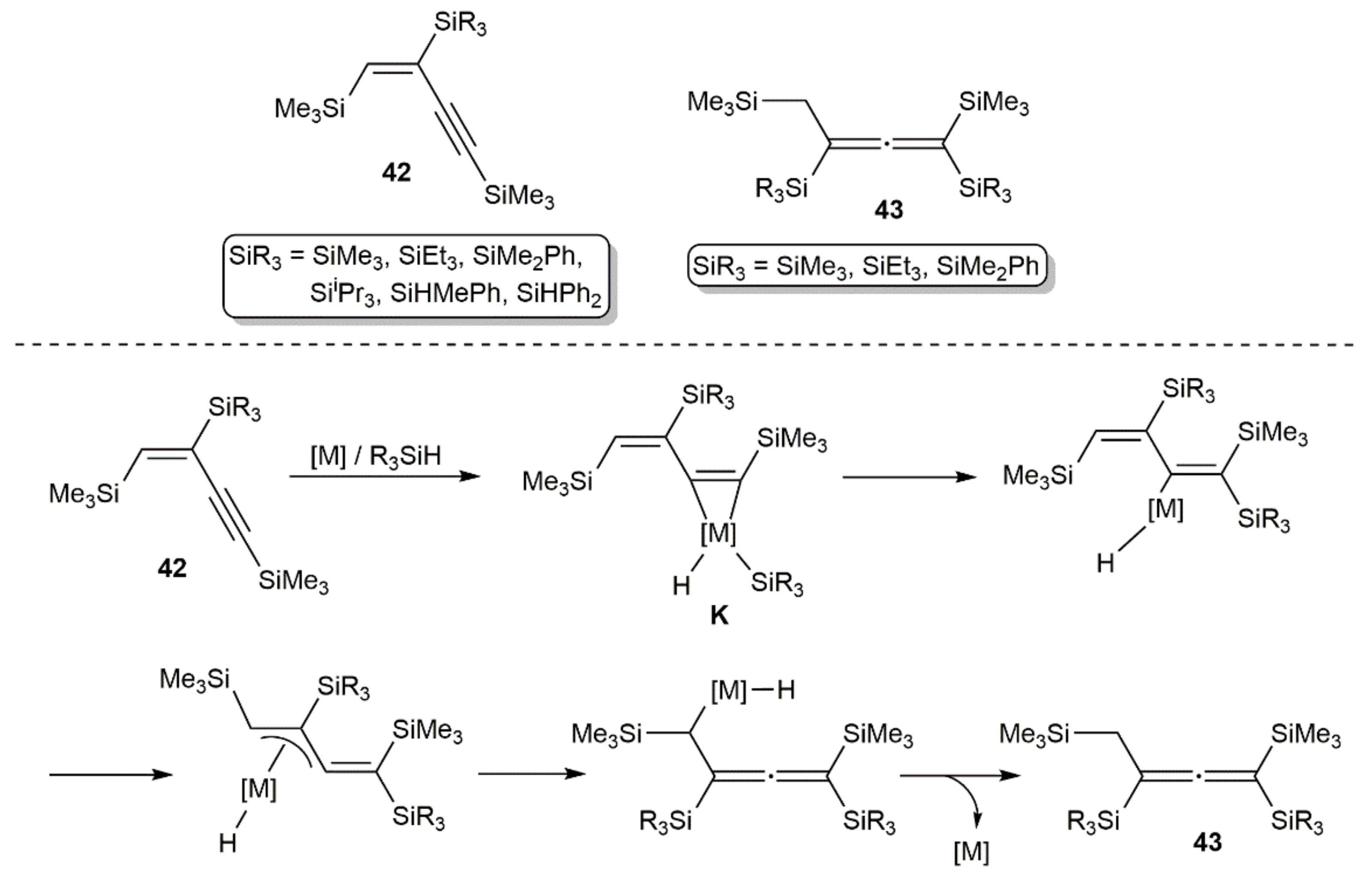{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
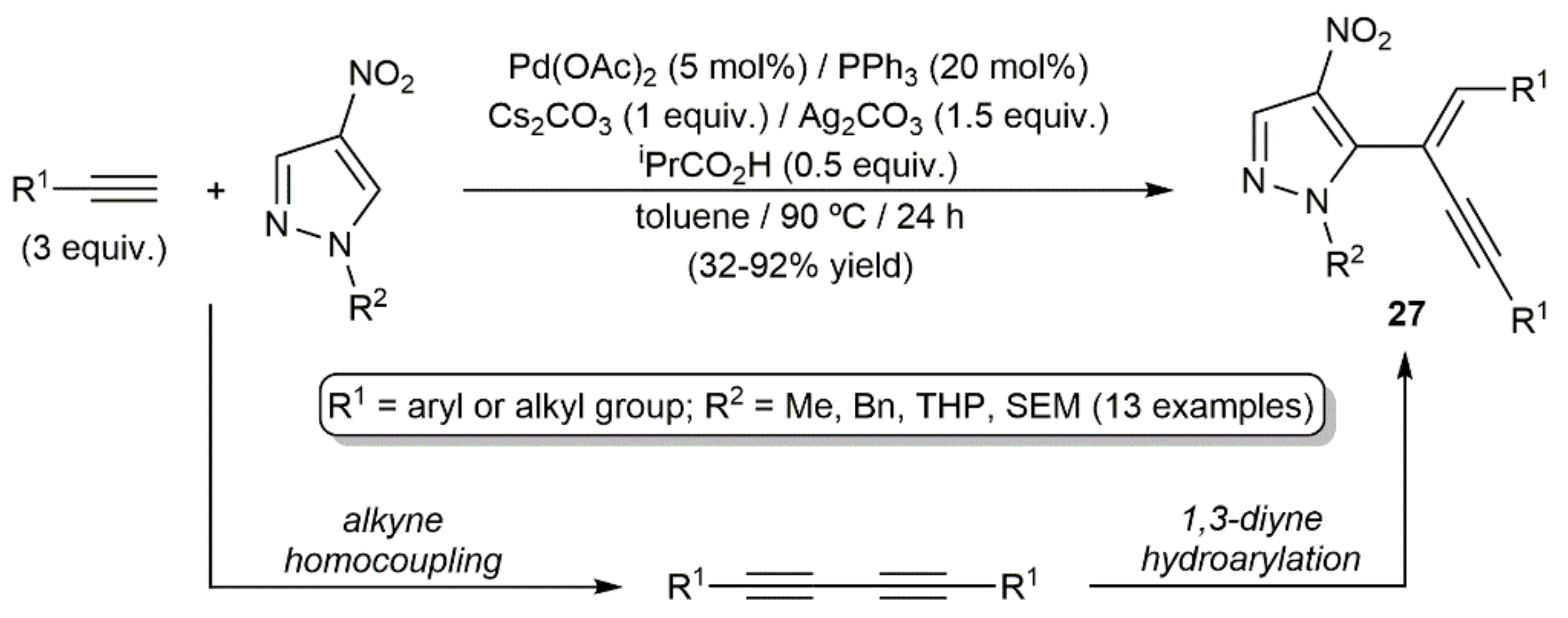{kind=link}
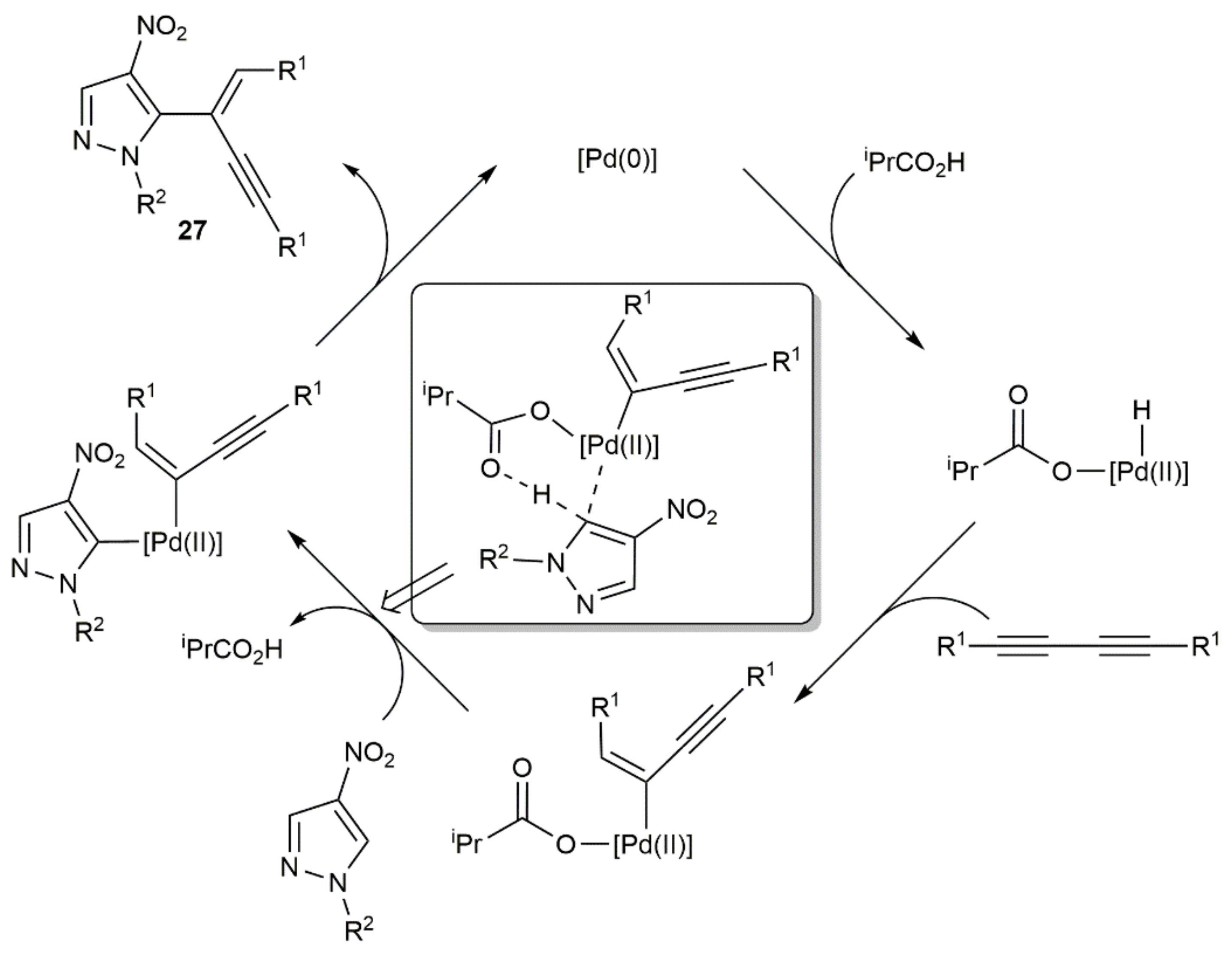{kind=link}
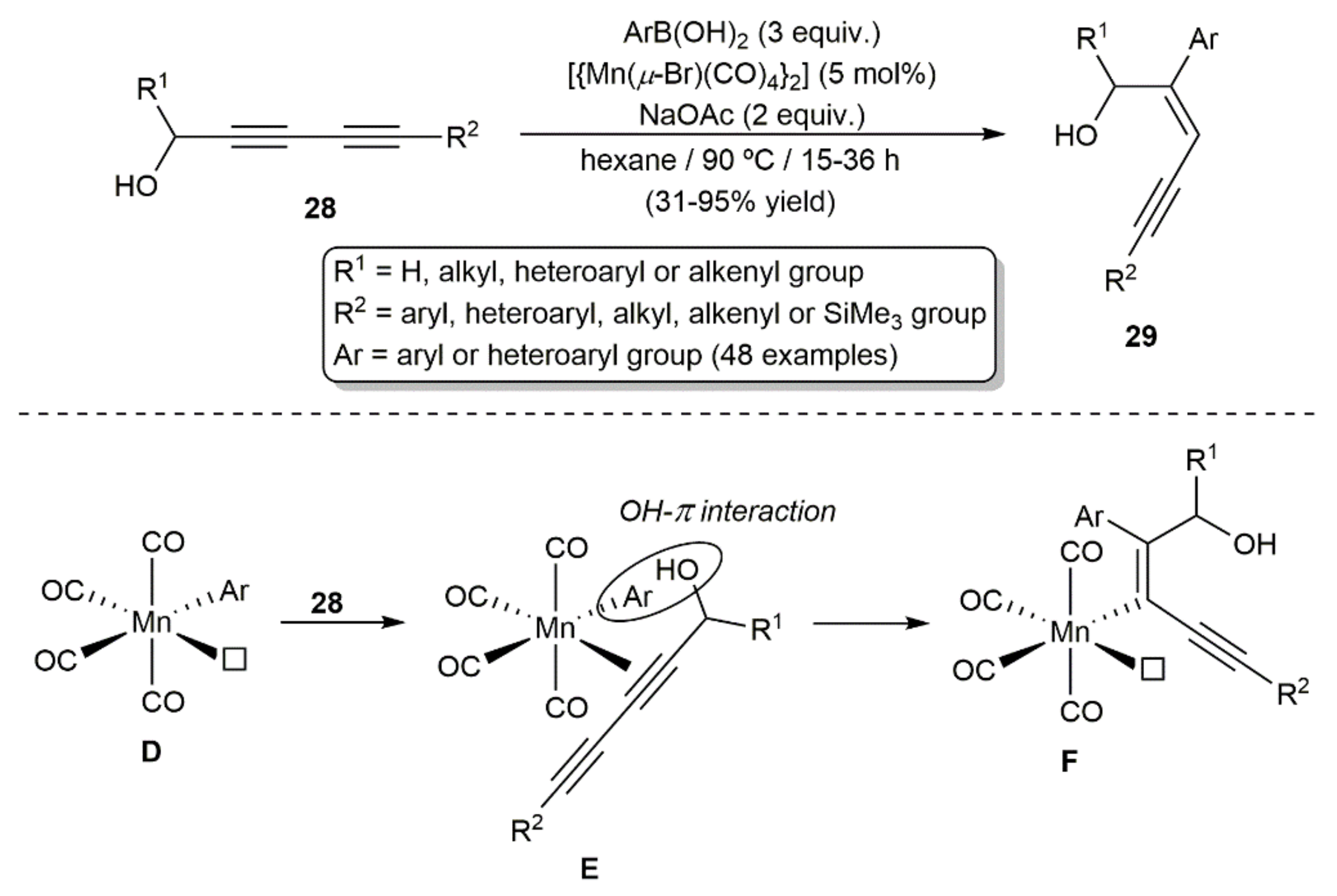{kind=link}
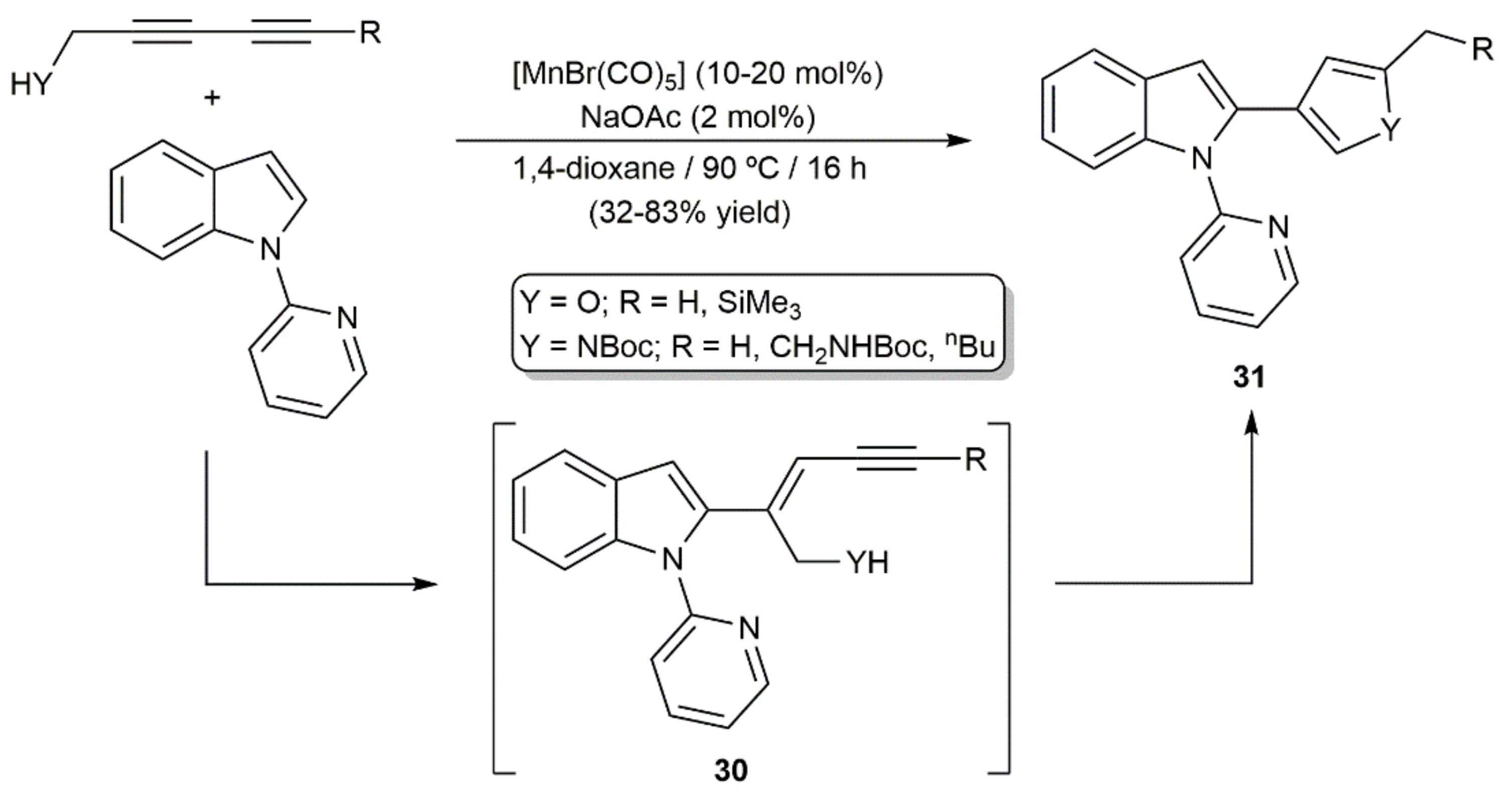{kind=link}
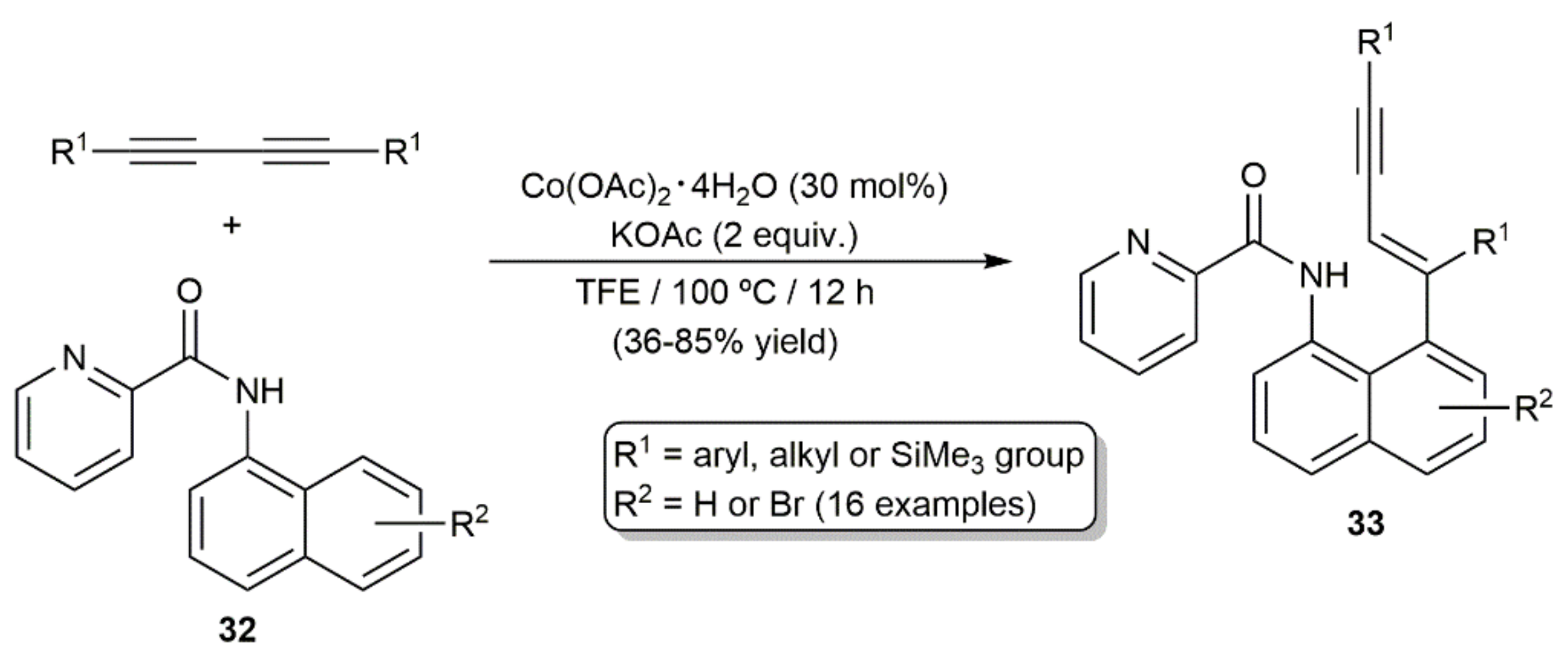
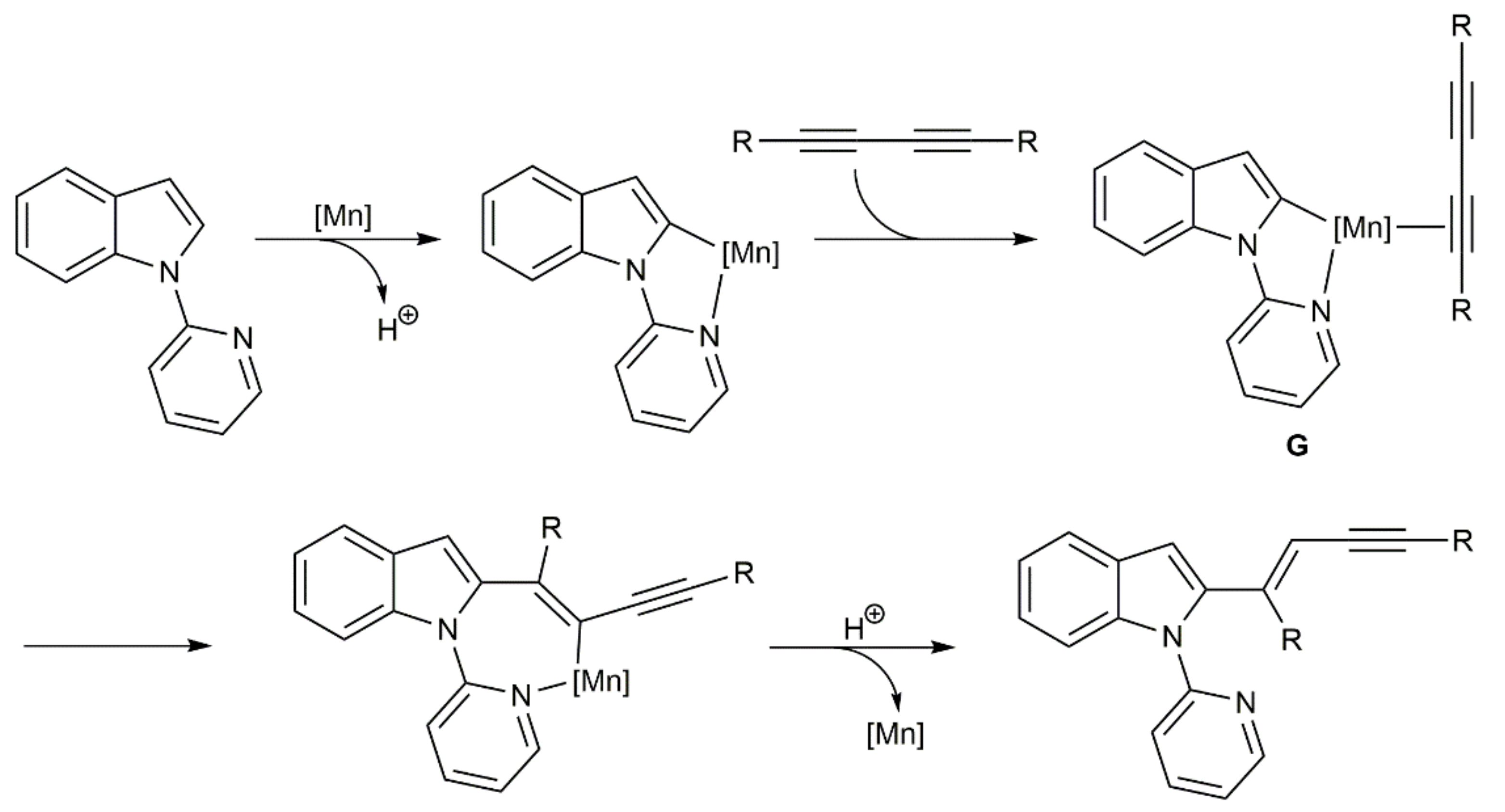{kind=link}
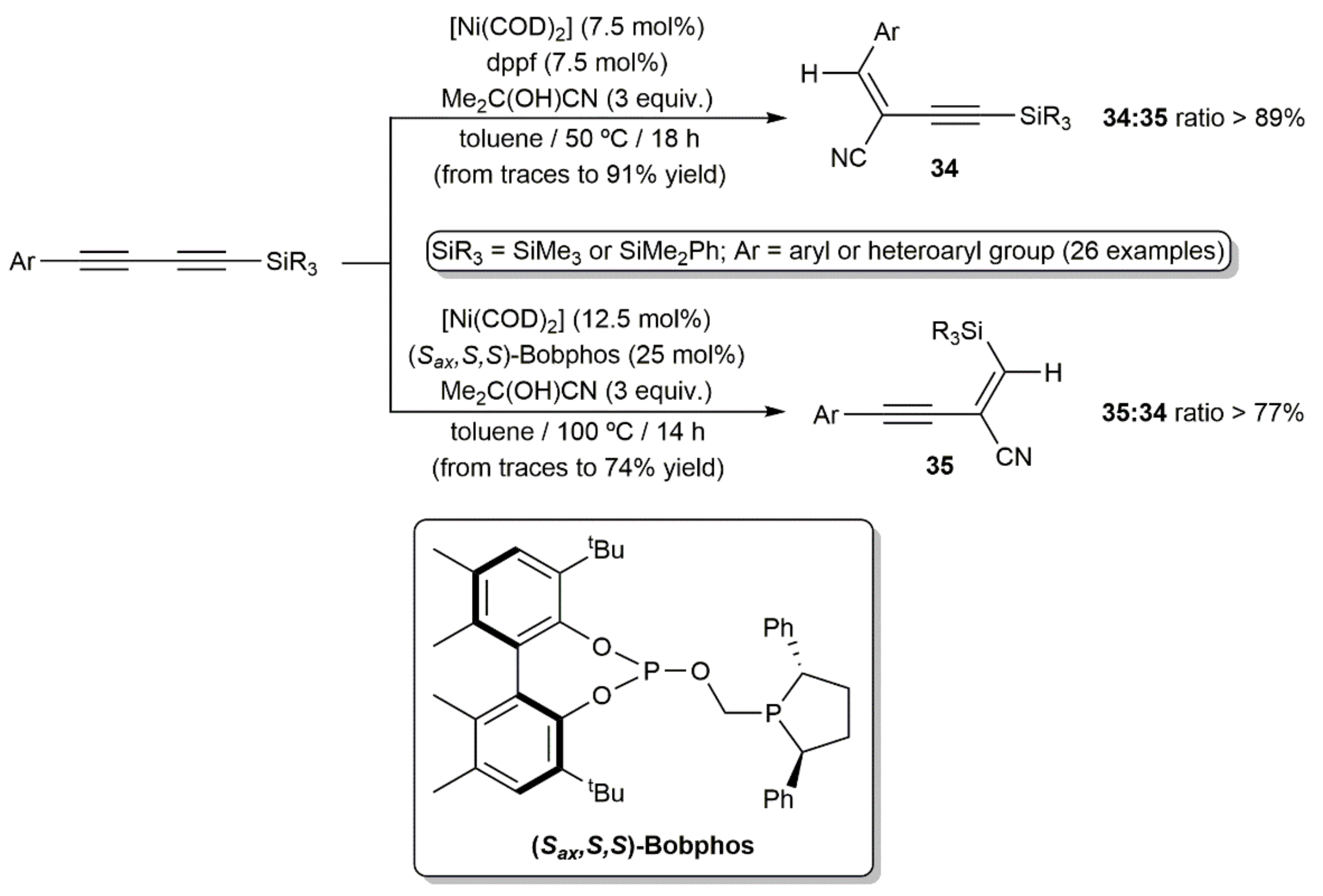
{kind=link}
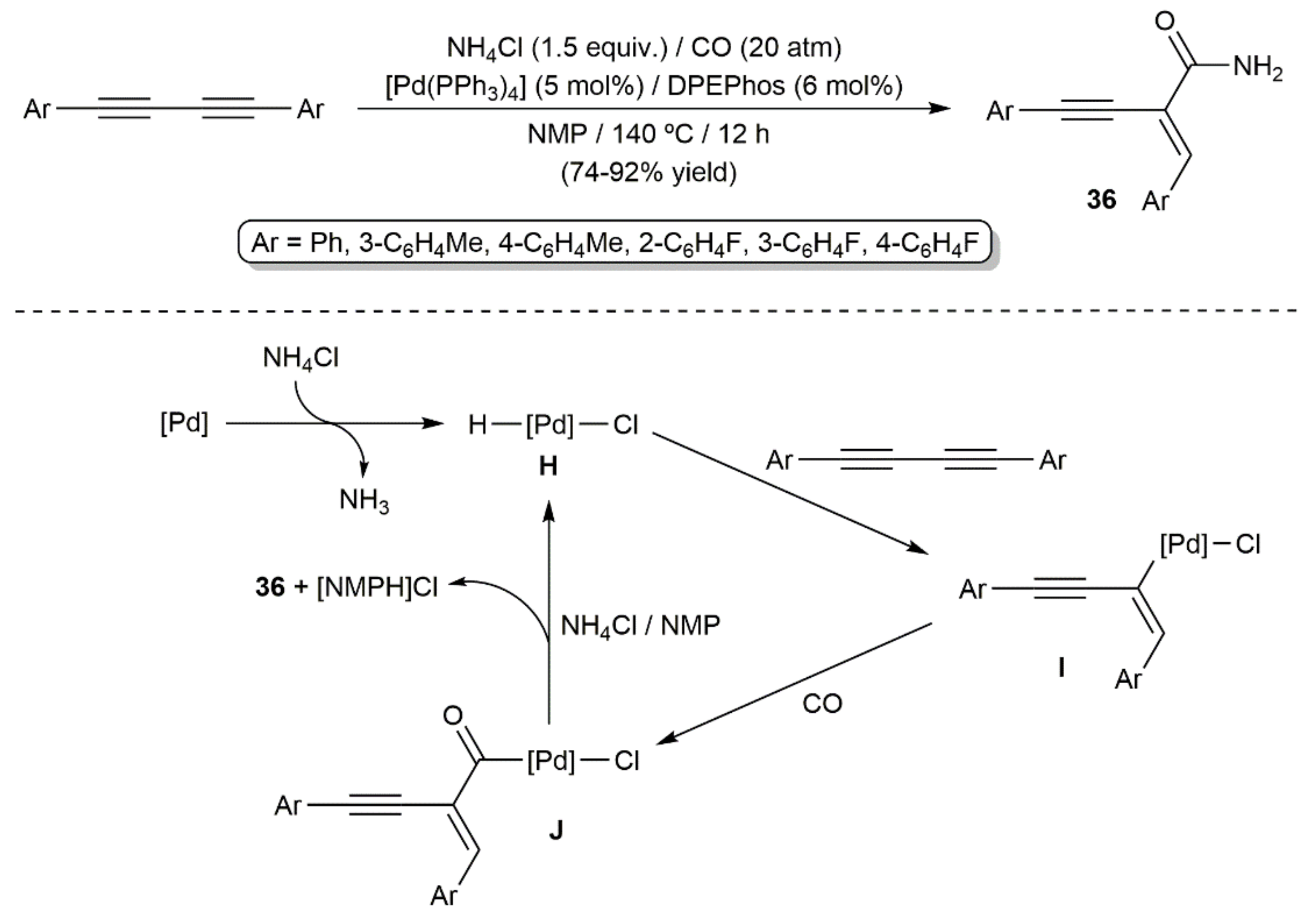
{kind=link}
{kind=link}
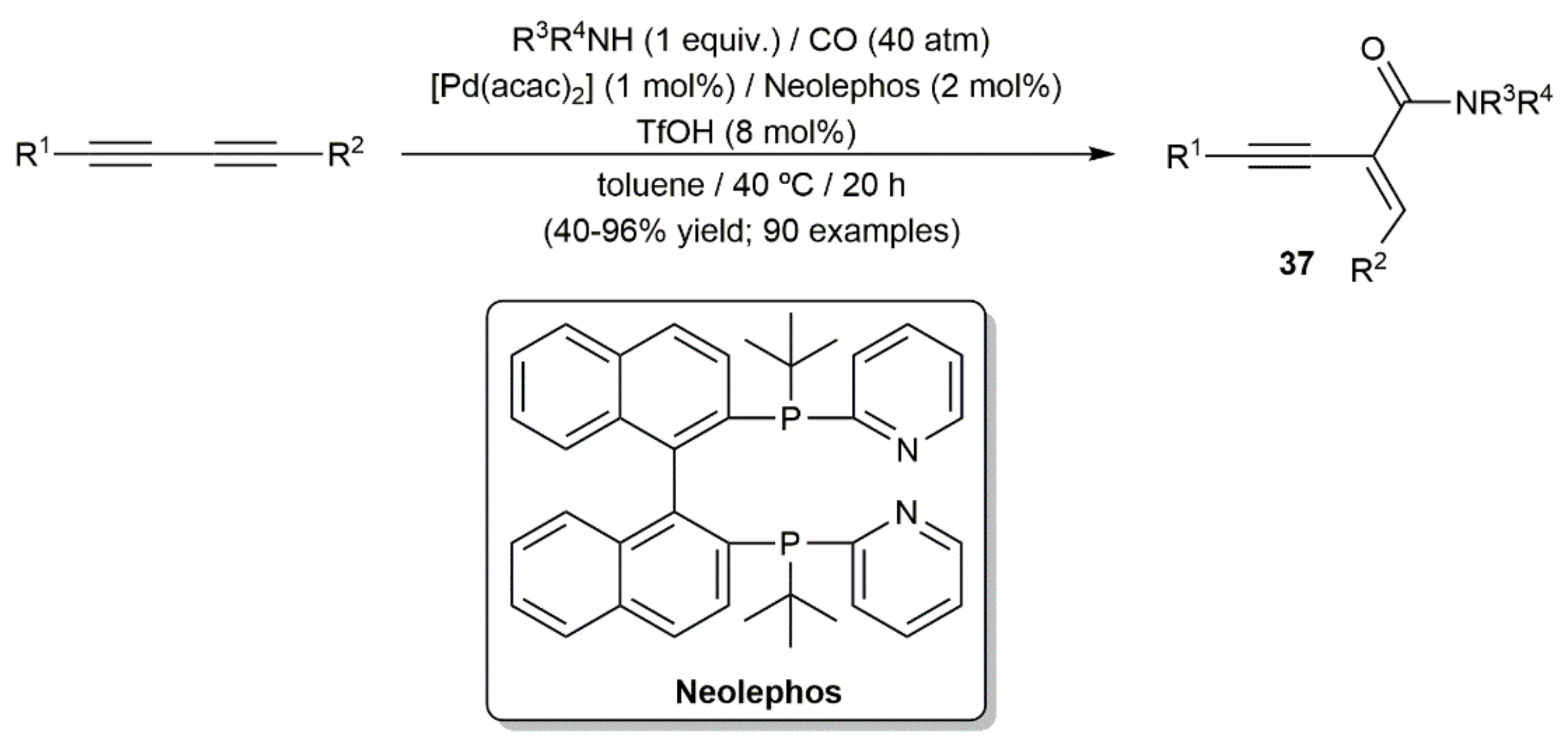{kind=link}
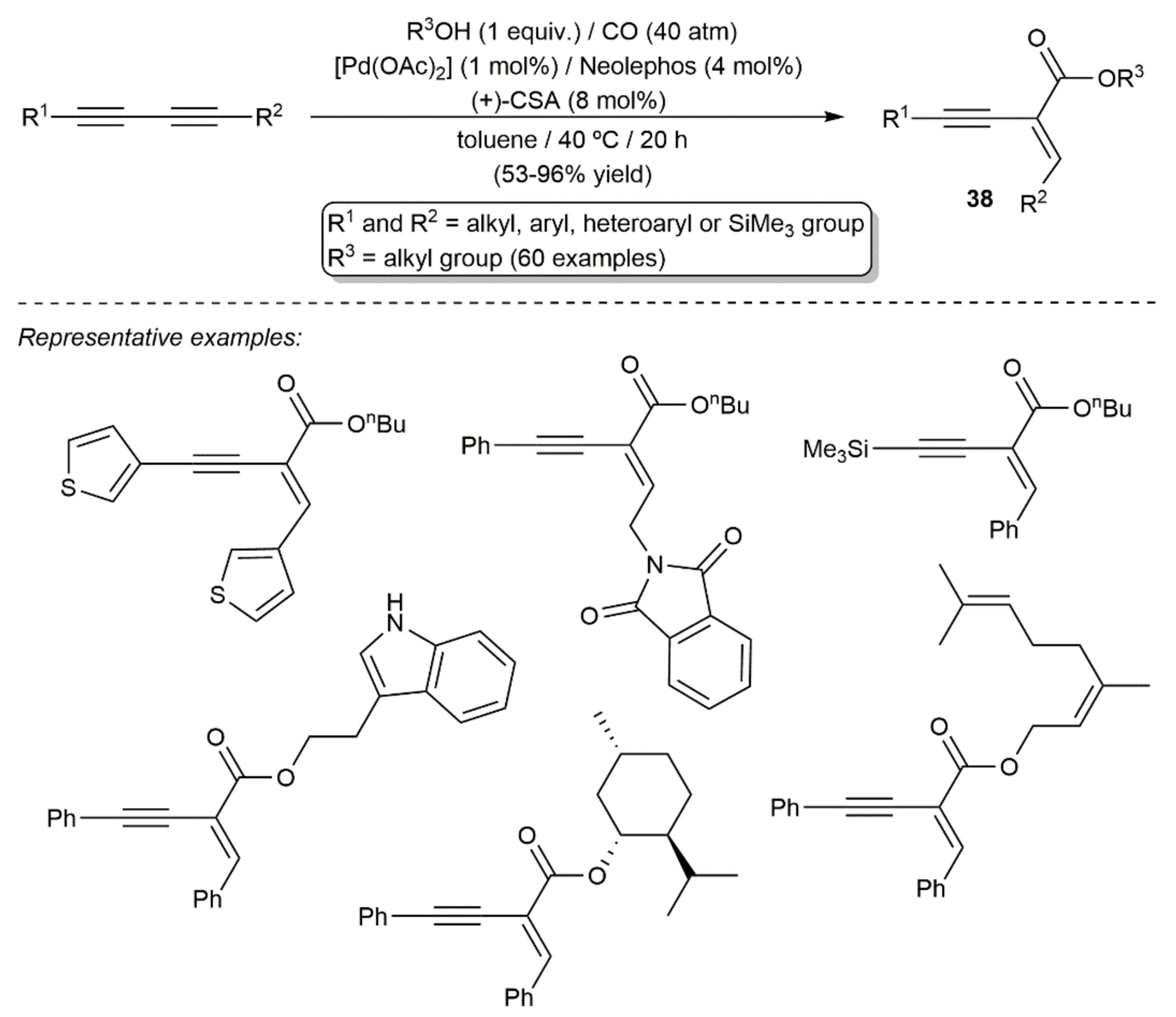{kind=link}
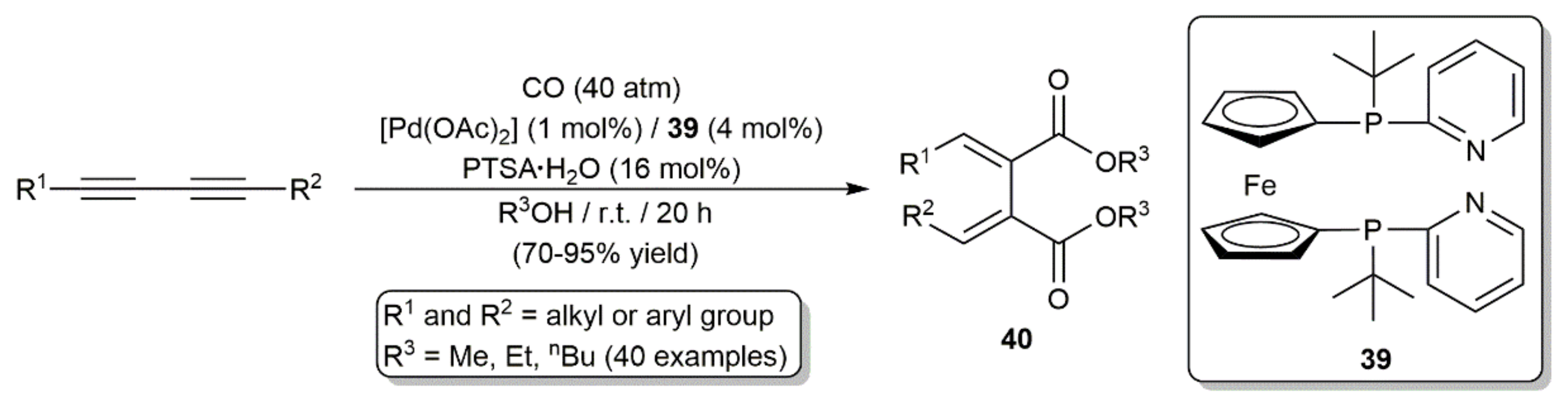{kind=link}
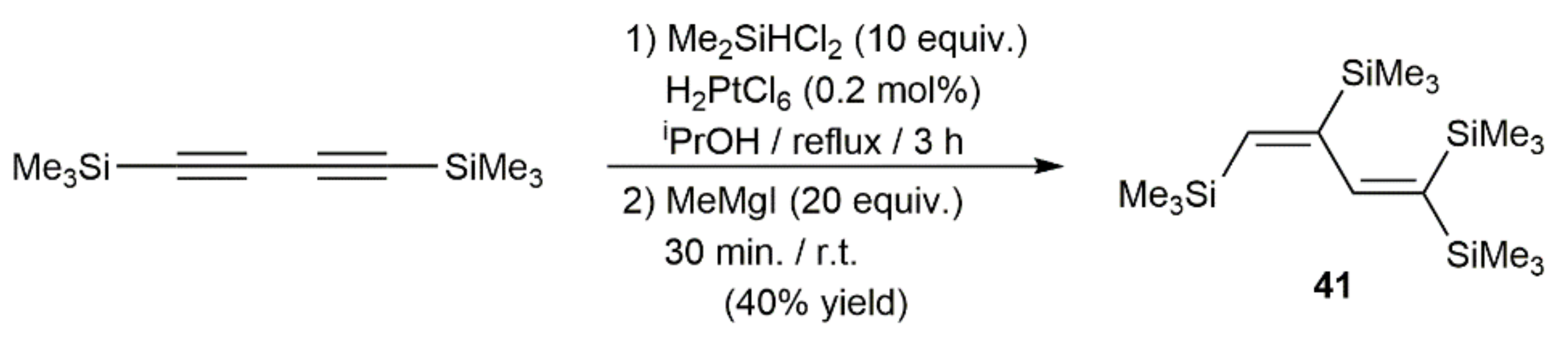{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hydroboration Processes
3. Hydroarylation and Hydroalkylation Processes
4. Hydrocyanation Processes
5. Other Formal C–H Bond Addition Processes
6. Hydrosilylation Processes
7. Hydroamination and Hydroamidation Processes
8. Hydrophosphination and Hydrophosphorylation Processes
9. Hydration Processes
10. Hydroalkoxylation Processes
11. Hydro-Oxycarbonylation Processes
12. Hydrothiolation, Hydroselenation, and Hydrotelluration Processes
13. Hydrohalogenation Processes
14. Summary
Funding
Conflicts of Interest
References
- Houk, K.; Hopf, H.; Stang, P.; Nicholas, K.M.; Schore, N.; Regitz, M.; Nicolaou, K.C.; Gleiter, R.; Scott, L.R.; Grubbs, H.; et al. Modern Acetylene Chemistry; Stang, P.J., Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Diederich, F.; Stang, P.J.; Tykwinski, R.R. (Eds.) Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Trost, B.M.; Li, C.-J. (Eds.) Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-metal-catalyzed addition of heteroatom-hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Beller, M.; Seayad, J.; Tillack, A.; Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004, 43, 3368–3398. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.T.; Kavthe, R.D.; Shinde, V.S. Transition metal-catalyzed addition of C-, N- and O-nucleophiles to unactivated C-C multiple bonds. Tetrahedron 2012, 68, 8079–8146. [Google Scholar] [CrossRef]
- Zeng, X. Recent advances in catalytic sequential reactions involving heteroelement addition to carbon-carbon multiple bonds. Chem. Rev. 2013, 113, 6864–6900. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zi, W. Transition-metal catalyzed enantioselective functionalization of alkynes. Tetrahedron Lett. 2018, 59, 2205–2213. [Google Scholar] [CrossRef]
- Cadierno, V. Metal-catalyzed hydrofunctionalization reactions of haloalkynes. Eur. J. Inorg. Chem. 2020, 2020, 886–898. [Google Scholar] [CrossRef]
- Siemsen, P.; Livingston, R.C.; Diederich, F. Acetylenic coupling: A powerful tool in molecular construction. Angew. Chem. Int. Ed. 2000, 39, 2632–2657. [Google Scholar] [CrossRef]
- Maretina, I.A.; Trofimov, B.A. Diacetylene and its derivatives in heterocyclization reactions. Adv. Heterocycl. Chem. 2002, 82, 157–259. [Google Scholar]
- Shi, W.; Lei, A. 1,3-Diyne Chemistry: Synthesis and derivatizations. Tetrahedron Lett. 2014, 55, 2763–2772. [Google Scholar] [CrossRef] [Green Version]
- Nizami, T.A.; Hua, R. Cycloaddition of 1,3-butadiynes: Efficient synthesis of carbo- and heterocycles. Molecules 2014, 19, 13788–13802. [Google Scholar] [CrossRef] [PubMed]
- Shi, W. Conjugated diyne chemistry: Synthesis, natural existence and applications. Curr. Organocatalysis 2015, 2, 2–13. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, Y.; Zhang, W.; Cheng, H.; Kuang, C.; Chen, Z. Synthesis of 1,3-diynes. Prog. Chem. 2016, 28, 507–527. [Google Scholar]
- Rej, S.; Das, A.; Panda, T.K. Overview of regioselective and steroselective catalytic hydroboration of alkynes. Adv. Synth. Catal. 2021, 363, 4818–4840. [Google Scholar] [CrossRef]
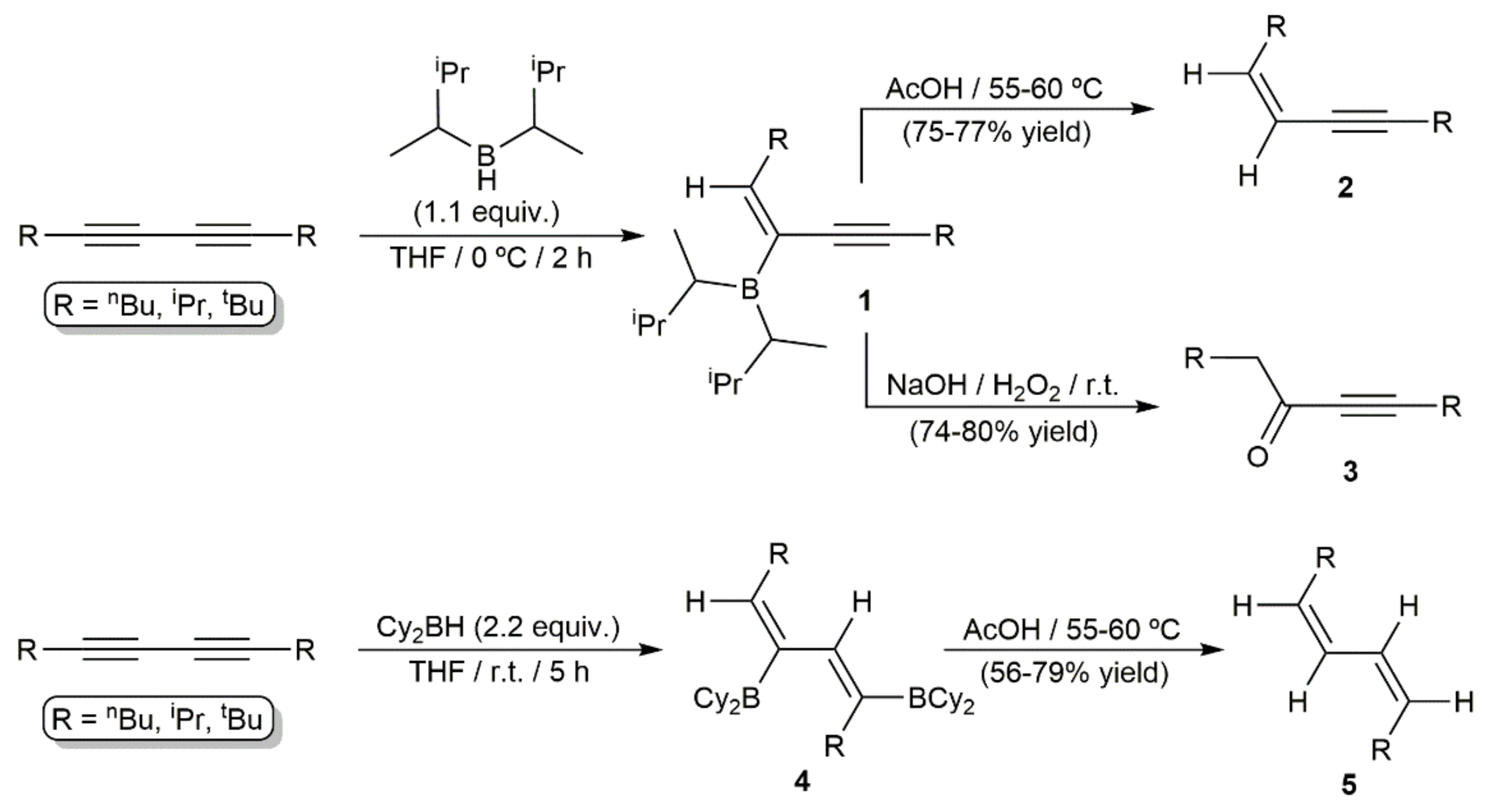
- Zweifel, G.; Polston, N.L. Selective hydroboration of conjugated diynes with dialkylboranes. A convenient route to conjugated cis-enynes, α,β-acetylenic ketones, and cis,cis-dienes. J. Am. Chem. Soc. 1970, 92, 4068–4071. [Google Scholar] [CrossRef]
- Stracker, E.C.; Leong, W.; Miller, J.A.; Shoup, T.M.; Zweifel, G. Directive effects in hydroborations of 1-(trimethylsilyl)-1,3-diynes. Syntheses of (Z)-enynes and α-ketocetylenes. Tetrahedron Lett. 1989, 30, 6487–6490. [Google Scholar] [CrossRef]
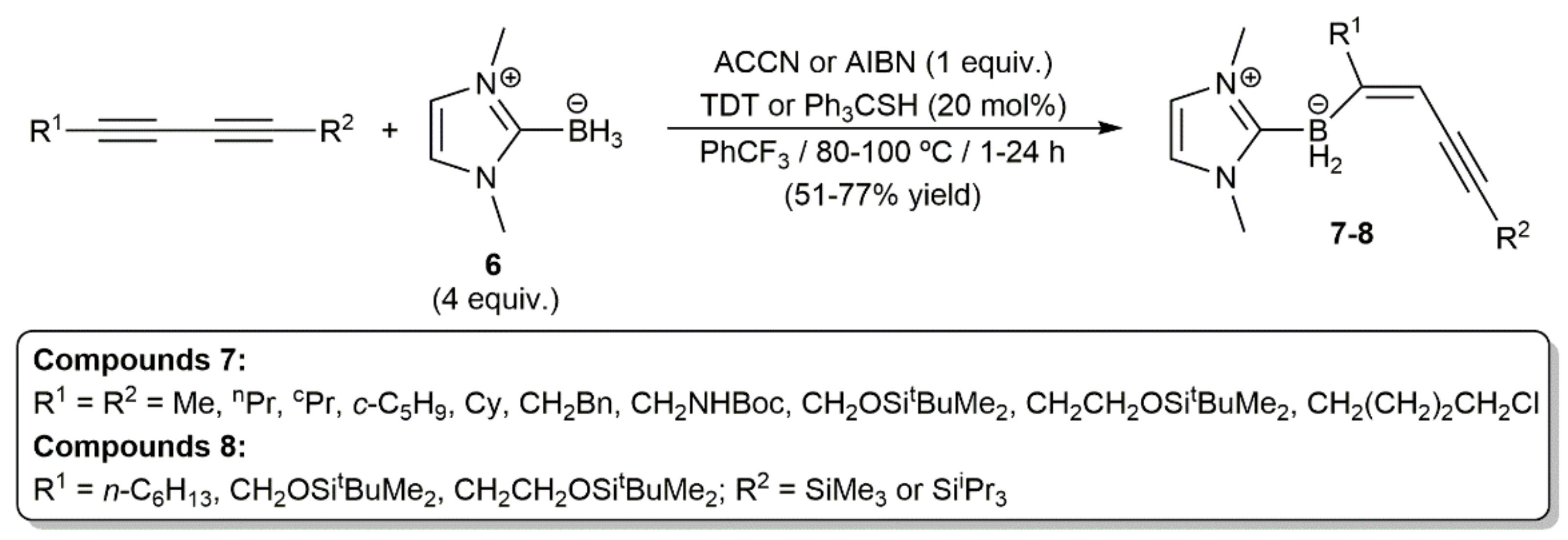
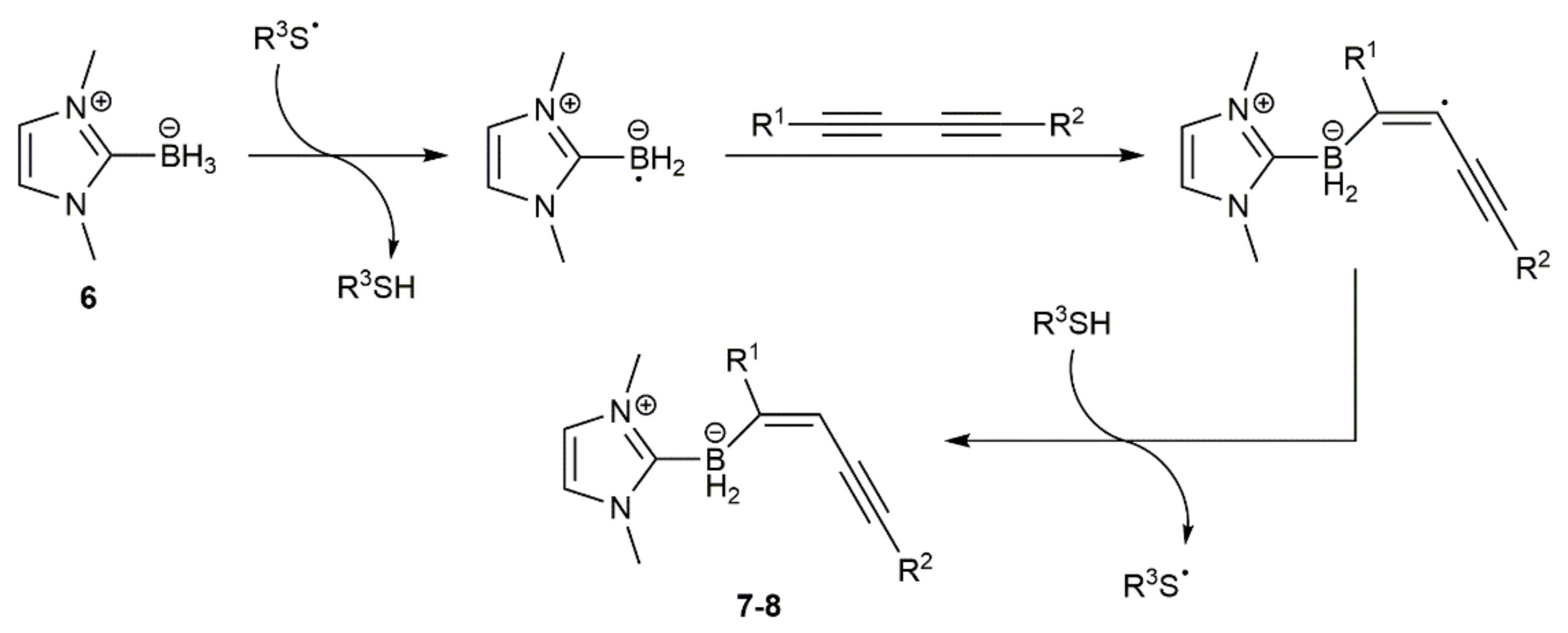
- Shimoi, M.; Watanabe, T.; Maeda, K.; Curran, D.P.; Taniguchi, T. Radical trans-hydroboration of alkynes with N-heterocyclic carbene boranes. Angew. Chem. Int. Ed. 2018, 57, 9485–9490. [Google Scholar] [CrossRef]
- Takahashi, K.; Geib, S.J.; Maeda, K.; Curran, D.P.; Taniguchi, T. Radical trans-hydroboration of substituted 1,3-diynes with an N-heterocyclic carbene borane. Org. Lett. 2021, 23, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
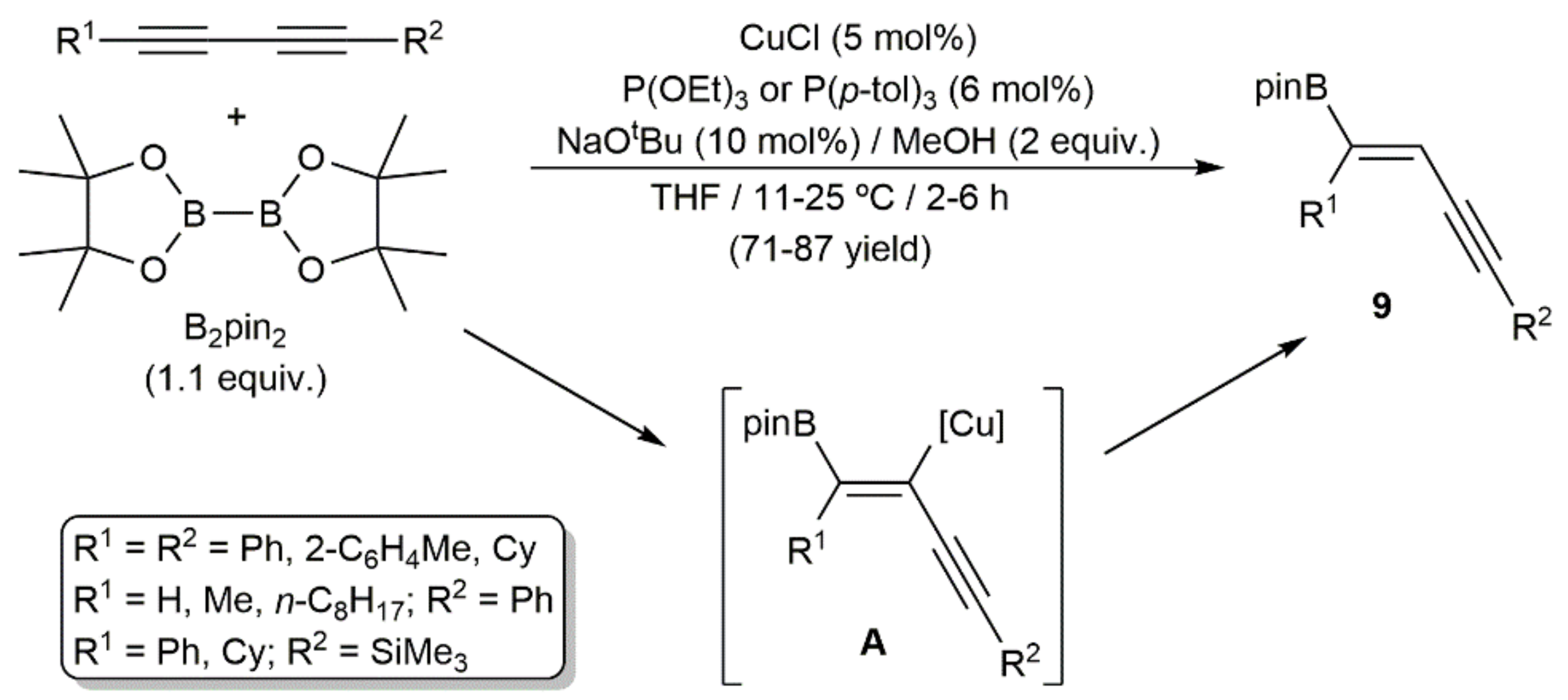
- Li, D.; Kim, Y.E.; Yun, J. Highly regio- and stereoselective synthesis of boron-substituted enynes via copper-catalyzed borylation of conjugated diynes. Org. Lett. 2015, 17, 860–863. [Google Scholar] [CrossRef]
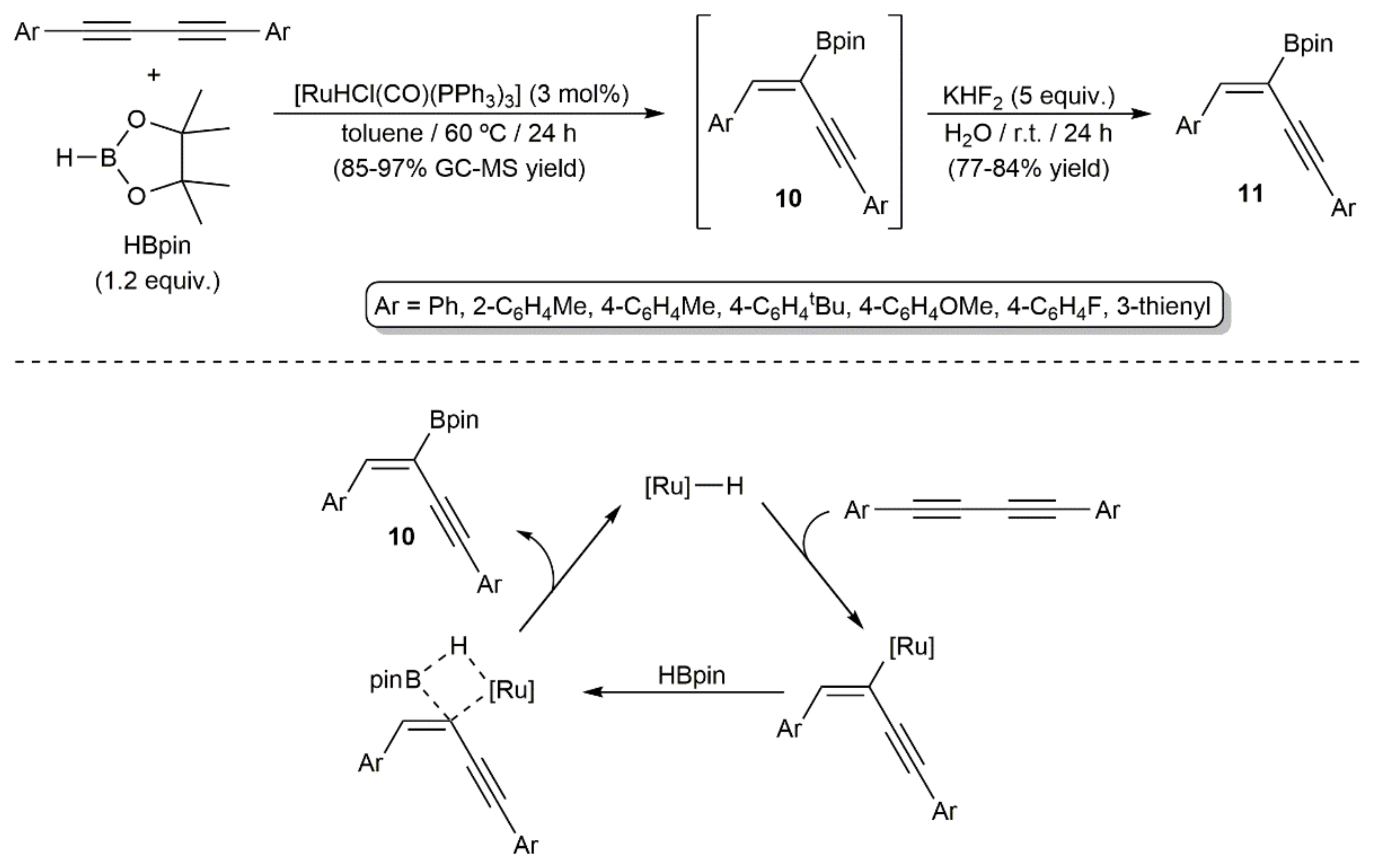
- Sokolnicki, T.; Szyling, J.; Franczyk, A.; Walkowiak, J. Regio- and stereoselective synthesis of enynyl boronates via ruthenium-catalyzed hydroboration of 1,4-diaryl-substituted 1,3-diynes. Adv. Synth. Catal. 2020, 362, 177–183. [Google Scholar] [CrossRef] [Green Version]
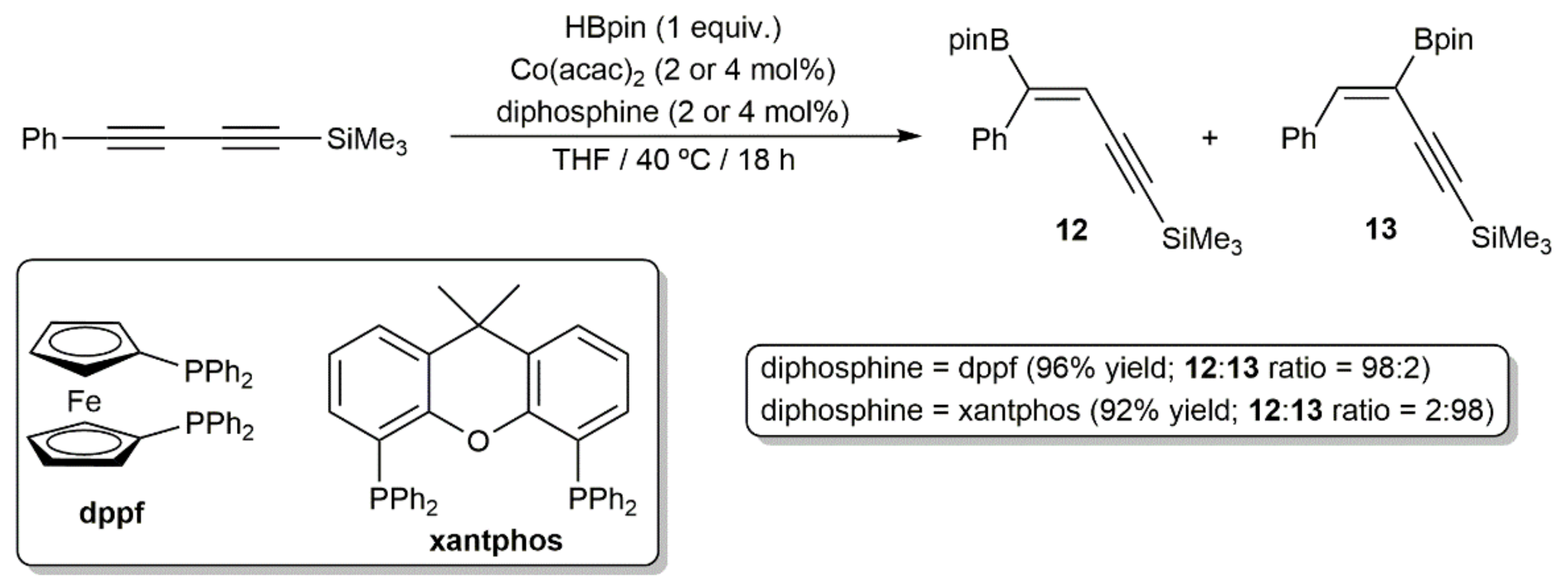
- Sang, H.L.; Wu, C.; Phua, G.G.D.; Ge, S. Cobalt-catalyzed regiodivergent setereoselective hydroboration of 1,3-diynes to access boryl-functionalized enynes. ACS Catal. 2019, 9, 10109–10114. [Google Scholar] [CrossRef]
- Nevado, C.; Echavarren, A.M. Transition metal-catalyzed hydroarylation of alkynes. Synthesis 2005, 2005, 167–182. [Google Scholar] [CrossRef]
- Kitamura, T. Transition-metal-catalyzed hydroarylation reactions of alkynes through direct functionalization of C-H bonds: A convenient tool for organic synthesis. Eur. J. Org. Chem. 2009, 2009, 1111–1125. [Google Scholar] [CrossRef]
- Yamamoto, Y. Synthesis of heterocycles via transition-metal-catalyzed hydroarylation of alkynes. Chem. Soc. Rev. 2014, 43, 1575–1600. [Google Scholar] [CrossRef]
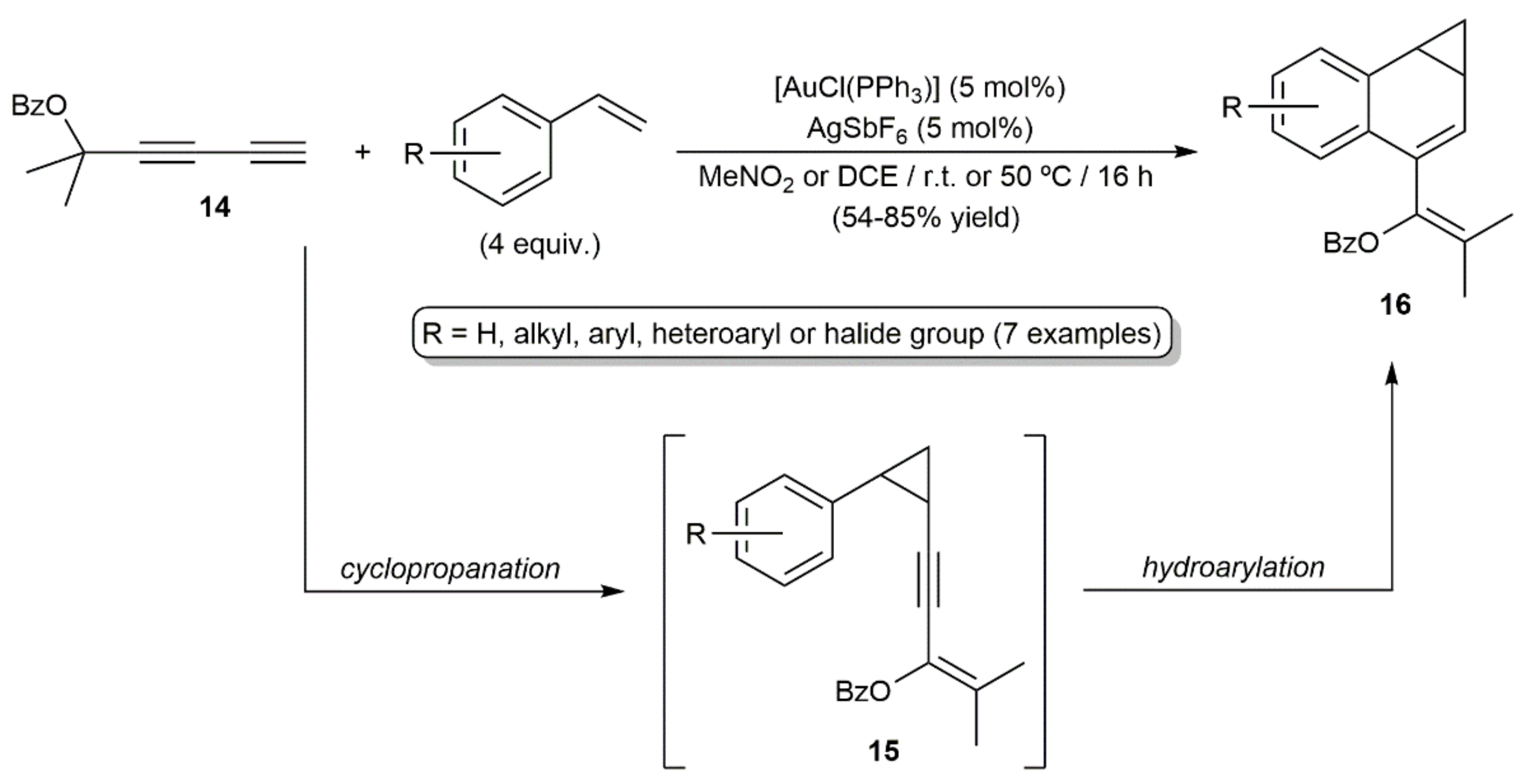
- Gorin, D.J.; Dubé, P.; Toste, F.D. Synthesis of benzonorcaradienes by gold(I)-catalyzed [4+3] annulation. J. Am. Chem. Soc. 2006, 128, 14480–14481. [Google Scholar] [CrossRef]
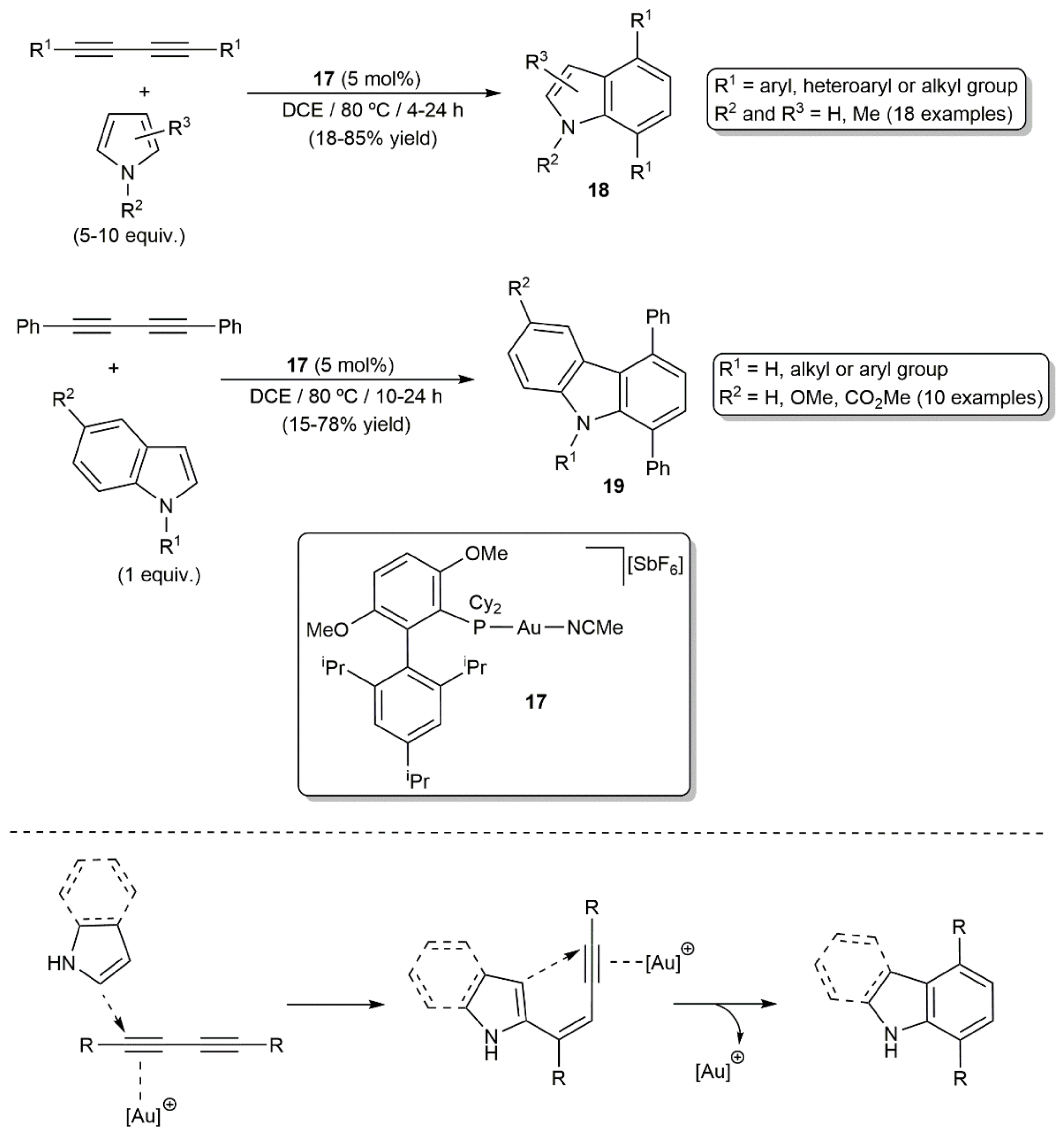
- Matsuda, Y.; Naoe, S.; Oishi, S.; Fujii, N.; Ohno, H. Formal [4+2] reaction of 1,3-diynes and pyrroles: Gold(I)-catalyzed indole synthesis by double hydroarylation. Chem. Eur. J. 2015, 21, 1463–1467. [Google Scholar] [CrossRef] [Green Version]
- Stylianakis, I.; Faza, O.N.; López, C.S.; Kolocouris, A. The key role of protodeauration in the gold-catalyzed reaction of 1,3-diynes with pyrrole and indole to form complex heterocycles. Org. Chem. Front. 2020, 7, 997–1005. [Google Scholar] [CrossRef]
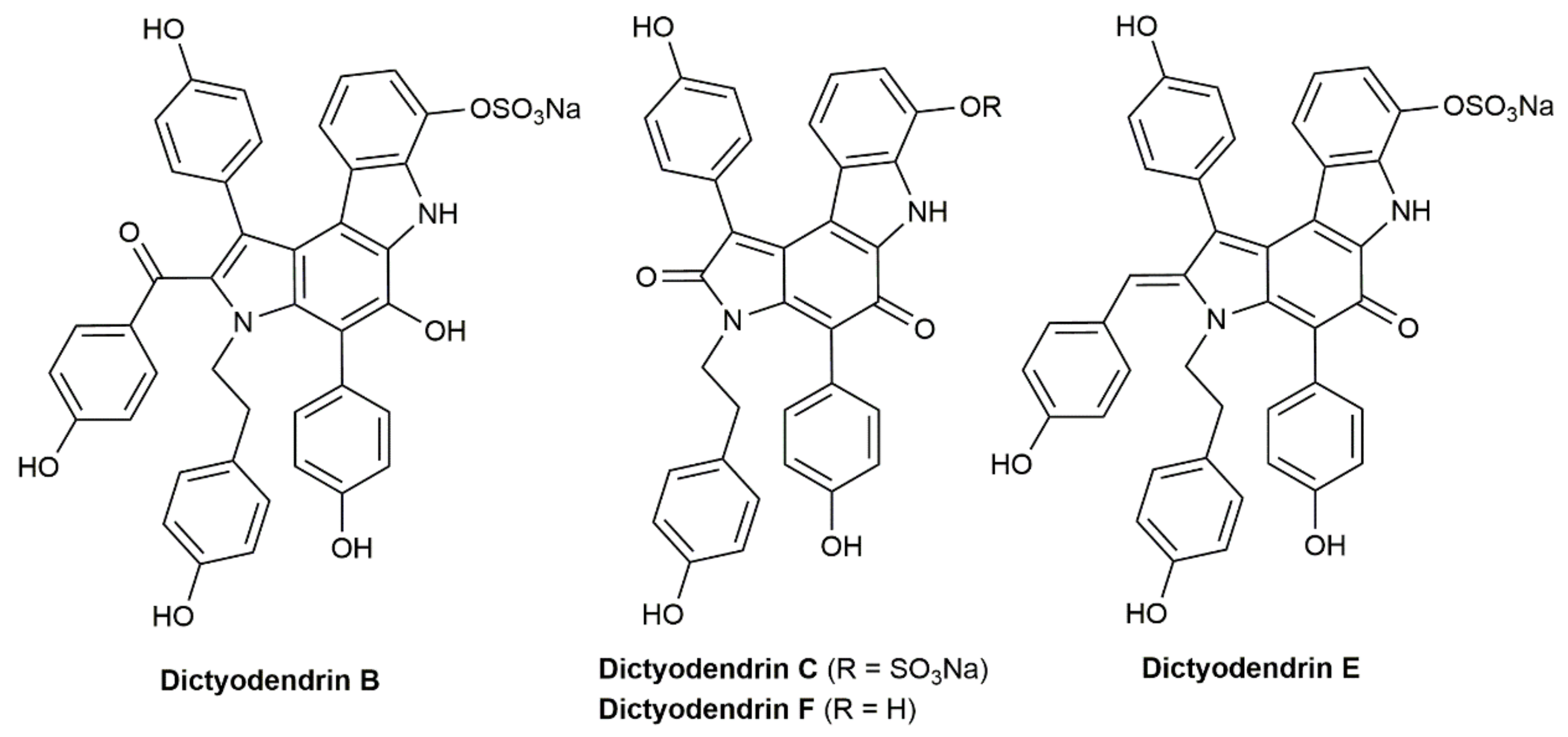
- Matsuoka, J.; Matsuda, Y.; Kawada, Y.; Oishi, S.; Ohno, H. Total synthesis of dictyodendrins by the gold-catalyzed cascade cyclization of conjugated diynes and pyrroles. Angew. Chem. Int. Ed. 2017, 56, 7444–7448. [Google Scholar] [CrossRef] [PubMed]
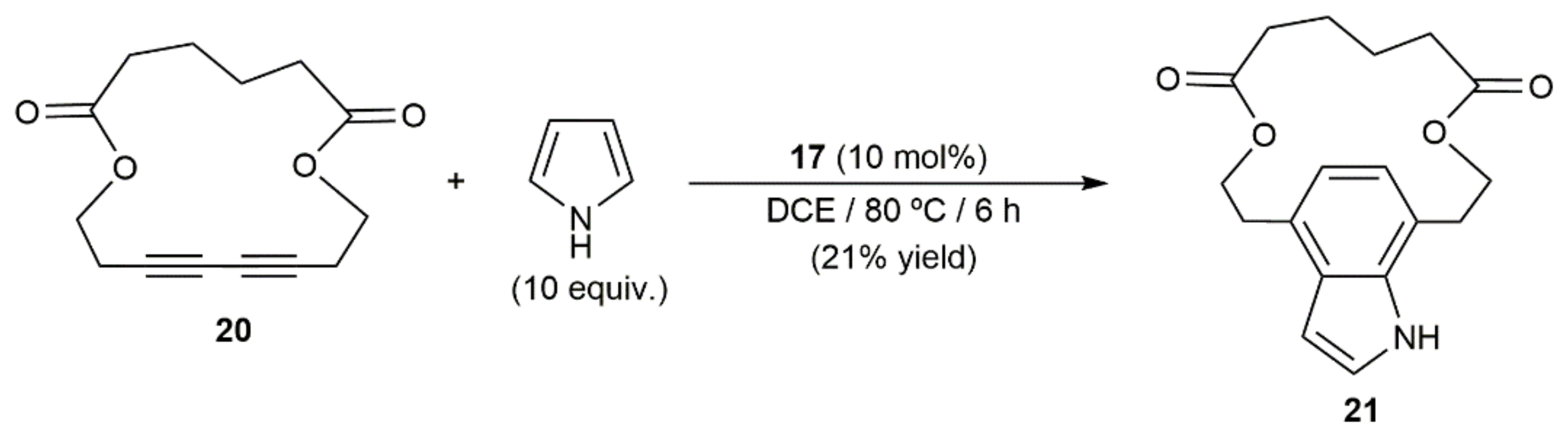
- Ungeheuer, F.; Fürstner, A. Concise total synthesis of Ivorenolide B. Chem. Eur. J. 2015, 21, 11387–11392. [Google Scholar] [CrossRef] [Green Version]
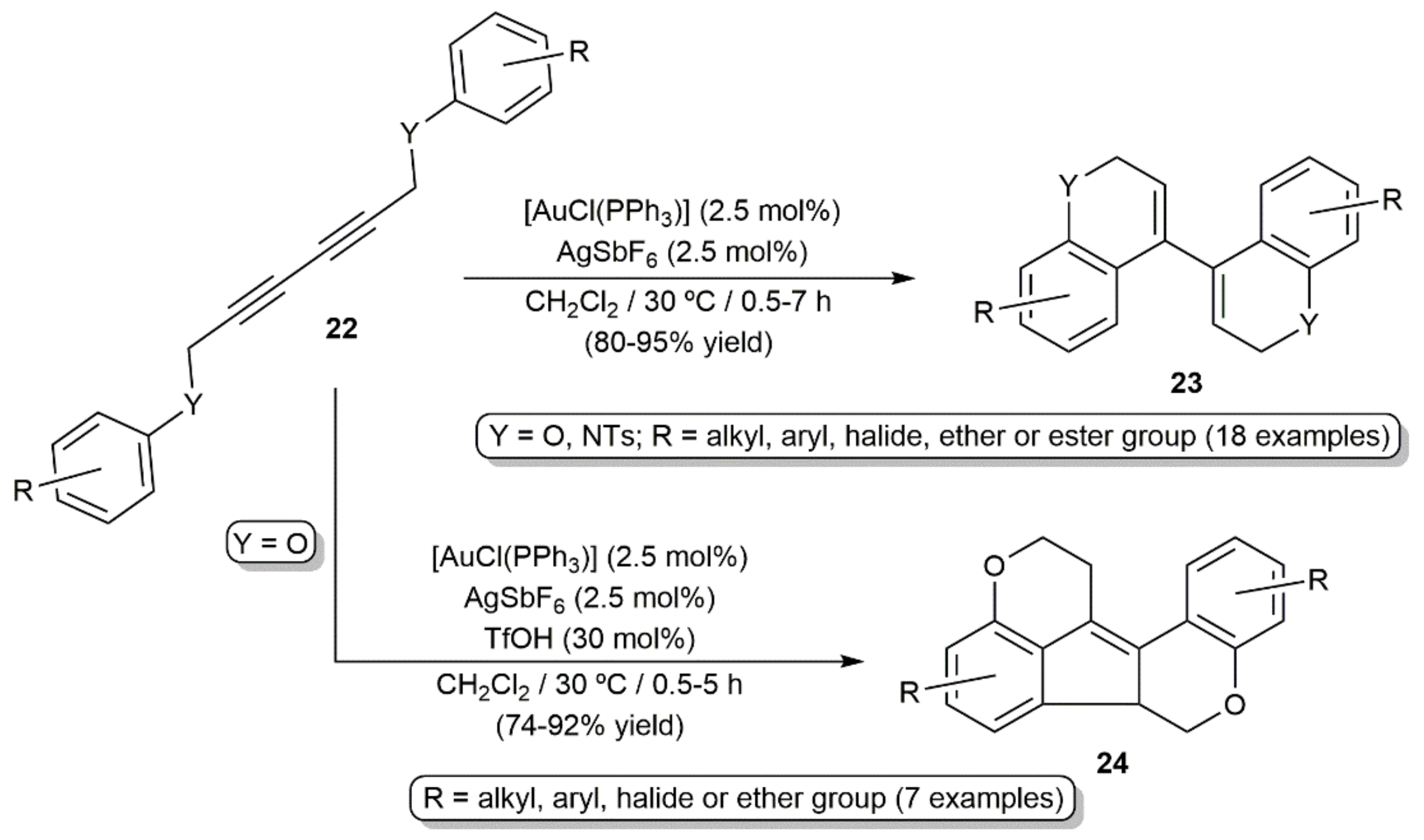
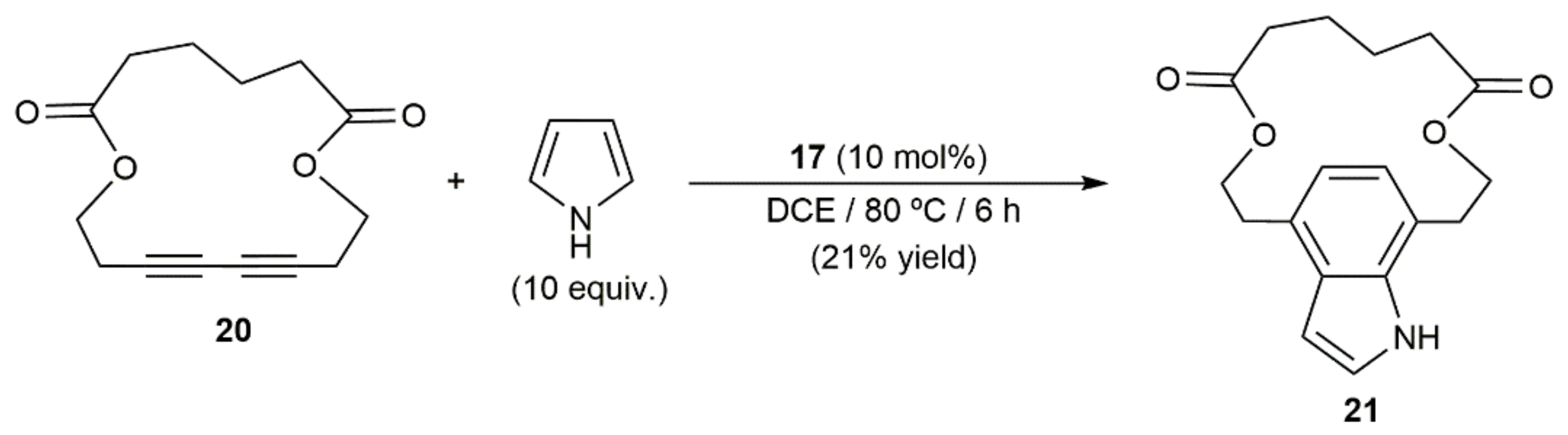
- Mo, J.; Eom, D.; Lee, E.; Lee, P.H. Hybrid system of metal/Brønsted acid relay catalysis for the intramolecular double hydroarylation and cationic cyclization of diyne diethers and diamines. Org. Lett. 2012, 14, 3684–3687. [Google Scholar] [CrossRef]
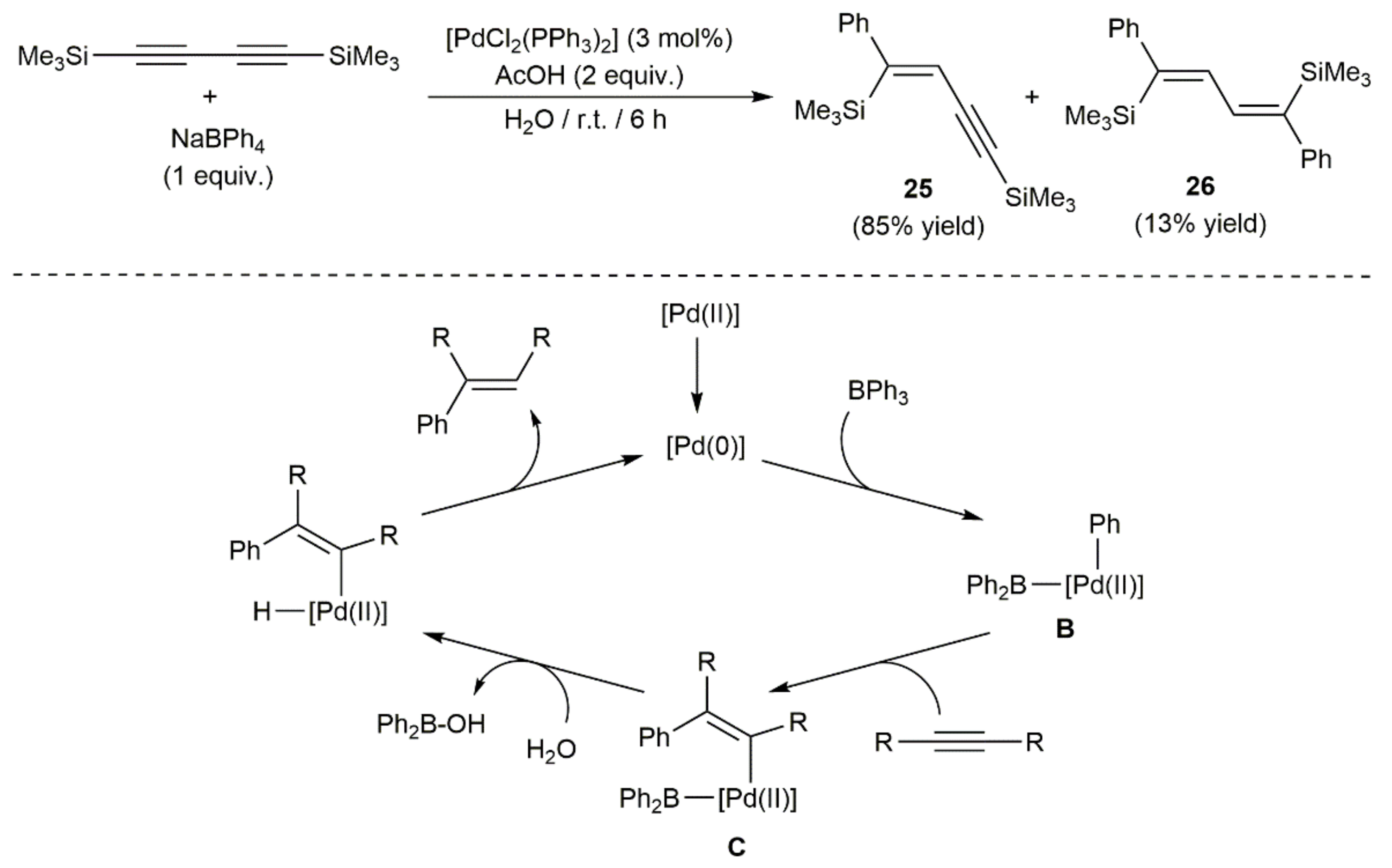
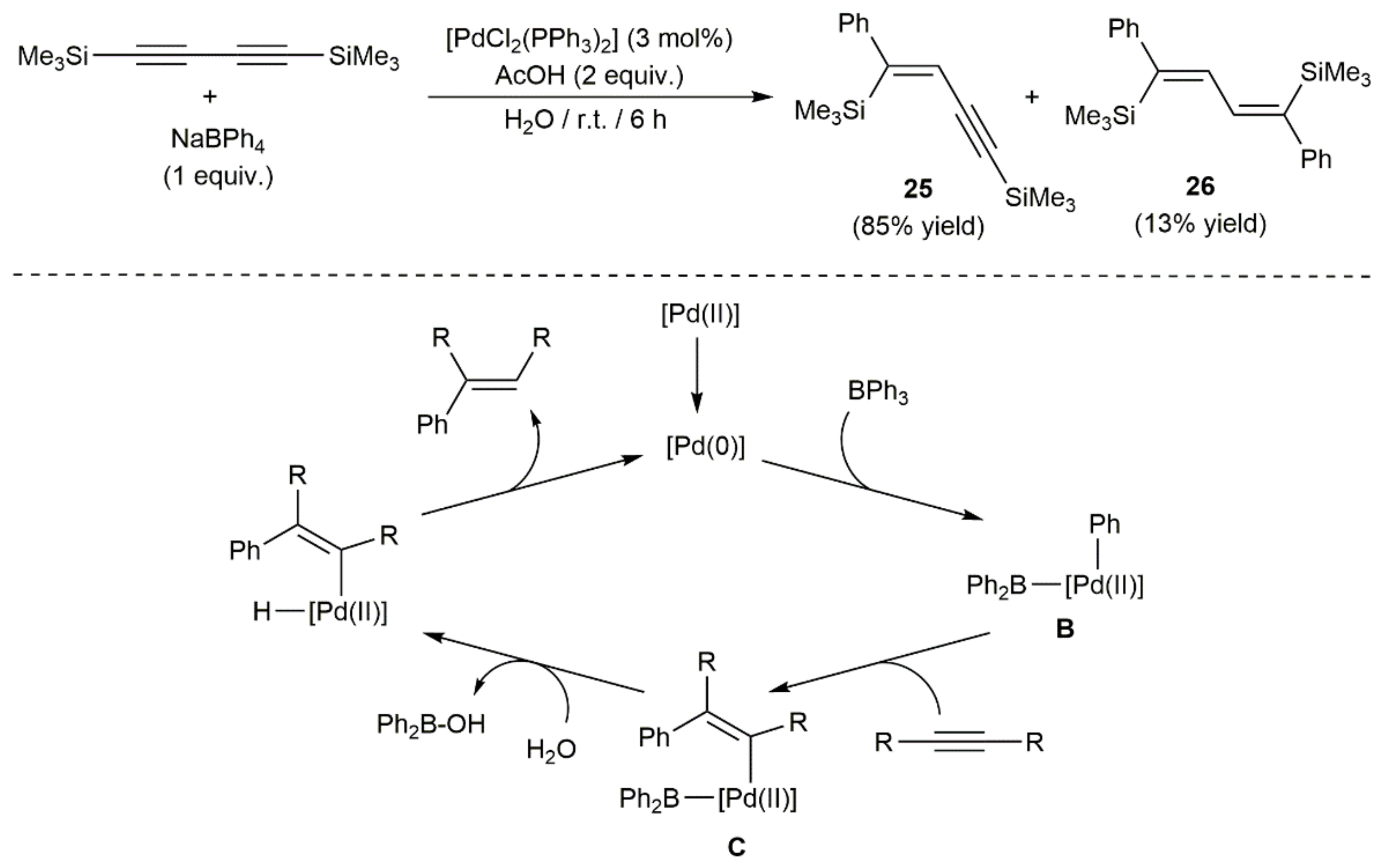
- Zeng, H.; Hua, R. Palladium-catalyzed hydrophenylation of alkynes with sodium tetraphenylborate under mild conditions. J. Org. Chem. 2008, 73, 558–562. [Google Scholar] [CrossRef] [PubMed]
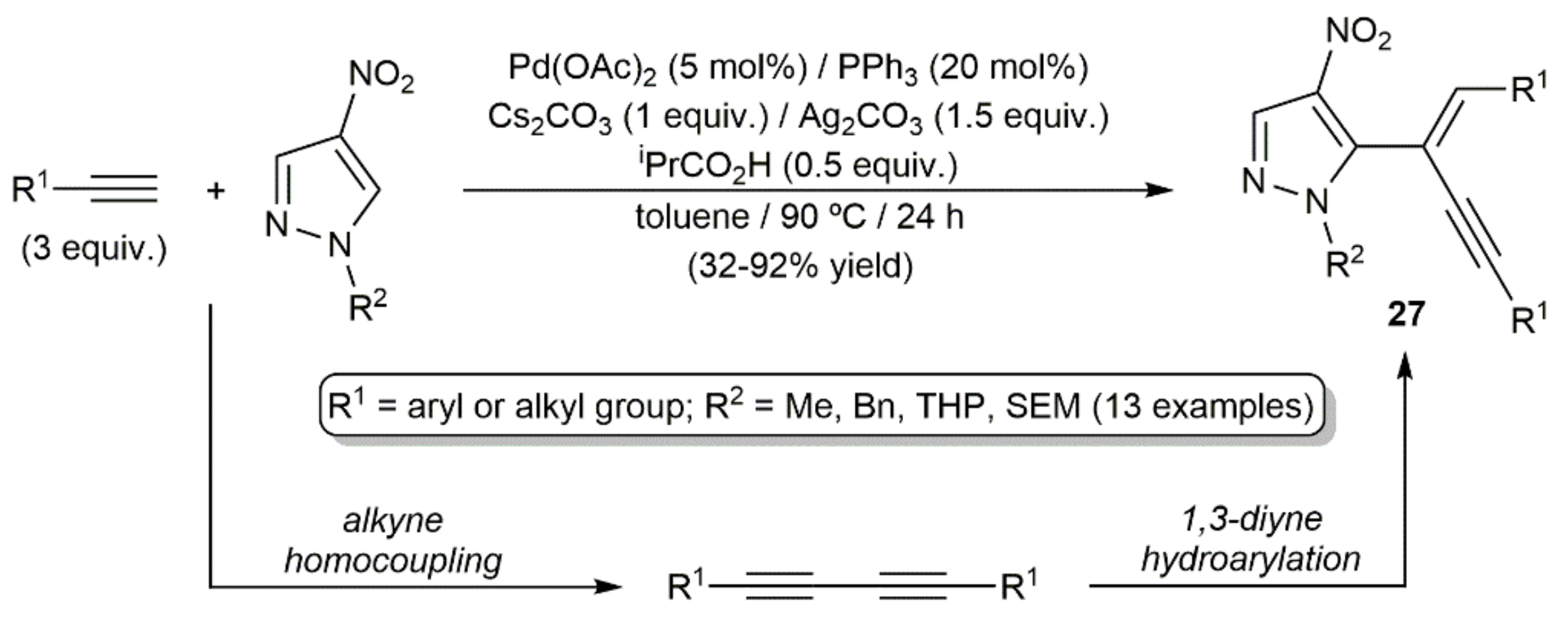
- Ha, H.; Shin, C.; Bae, S.; Joo, J.M. Divergent palladium-catalyzed cross-coupling of nitropyrazoles with terminal alkynes. Eur. J. Org. Chem. 2018, 2018, 2645–2650. [Google Scholar] [CrossRef]
- Yan, Z.; Yuan, X.-A.; Zhao, Y.; Zhu, C.; Xie, J. Selective hydroarylation of 1,3-diynes using dimeric manganese catalyst: Modular synthesis of Z-enynes. Angew. Chem. Int. Ed. 2018, 57, 12906–12910. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhu, C.; Xie, J. Manganese(I)-catalyzed selective functionalization of alkynes. Synlett 2018, 29, 124–128. [Google Scholar]
- Cembellin, S.; Dalton, T.; Pinkert, T.; Schäfers, F.; Glorius, F. Highly selective synthesis of 1,3-enynes, pyrroles, and furans by manganese(I)-catalyzed C-H activation. ACS Catal. 2020, 10, 197–202. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, M.; Wang, C.; Yang, Z.; Huang, X.; Feng, Q.C. Cobalt(II)-catalyzed hydroarylation of 1,3-diynes and internal alkynes with picolinamides promoted by alcohol. Chem. Commun. 2020, 56, 14231–14234. [Google Scholar] [CrossRef]
- Nakamura, M.; Endo, K.; Nakamura, E. Indium-catalyzed addition of active methylene compounds to 1-alkynes. J. Am. Chem. Soc. 2003, 125, 13002–13003. [Google Scholar] [CrossRef]
- Nakamura, M.; Endo, K.; Nakamura, E. A modular approach to α-arylated carbonyl compounds via indium tris(bistriflylamide)-catalyzed regioselective addition of β-keto esters to 1,3-diynes. Adv. Synth. Catal. 2005, 347, 1681–1686. [Google Scholar] [CrossRef]
- Rajanbabu, T.V. Hydrocyanation of alkenes and alkynes. Org. React. 2011, 75, 1–73. [Google Scholar]
- Peng, L.; Hu, Z.; Wang, H.; Wu, L.; Jiao, Y.; Tang, Z.; Xu, X. Direct cyanation, hydrocyanation, dicyanation and cyanofunctionalization of alkynes. RSC Adv. 2020, 10, 10232–10244. [Google Scholar] [CrossRef]
- Zhang, H.; Su, X.; Dong, K. Recent progress in transition-metal-catalyzed hydrocyanation of nonpolar alkenes and alkynes. Org. Biomol. Chem. 2020, 18, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Yang, C.; Ni, J.; Cheng, G.-J.; Fang, X. Ligand-controlled regiodivergent nickel-catalyzed hydrocyanation of silyl-substituted 1,3-diynes. Org. Lett. 2021, 23, 4045–4050. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, H.; Huang, H. Transition-metal-catalyzed hydroaminocarbonylations of alkenes and alkynes. Trends Chem. 2021, 3, 218–230. [Google Scholar] [CrossRef]
- Ji, X.; Gao, B.; Zhou, X.; Liu, Z.; Huang, H. Palladium-catalyzed regioselective hydroaminocarbonylation of alkynes to α,β-unsaturated primary amides with ammonium chloride. J. Org. Chem. 2018, 83, 10134–10141. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Schneider, C.; Yang, J.; Wei, Z.; Jiao, H.; Franke, R.; Jackstell, R.; Beller, M. A general and highly selective palladium-catalyzed hydroamidation of 1,3-diynes. Angew. Chem. Int. Ed. 2021, 60, 371–379. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Schneider, C.; Franke, R.; Jackstell, R.; Beller, M. Tailored palladium catalyst for the selective synthesis of conjugated enynes by monocarbonylation of 1,3-diynes. Angew. Chem. Int. Ed. 2020, 59, 9032–9040. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Baumann, W.; Jackstell, R.; Beller, M. Stereoselective synthesis of highly substituted conjugated dienes via Pd-catalyzed carbonylation of 1,3-diynes. Angew. Chem. Int. Ed. 2019, 58, 10683–10687. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.C.; Trogler, W.C. Hydrosilylation of diynes as a route to functional polymers delocalized though silicon. Macromol. Chem. Phys. 2008, 209, 1527–1540. [Google Scholar] [CrossRef]
- Trost, B.M.; Ball, Z.T. Addition of metalloid hydrides to alkynes: Hydrometallation with boron, silicon, and tin. Synthesis 2005, 2005, 853–887. [Google Scholar] [CrossRef]
- He, P.; Hu, M.-Y.; Zhang, X.-Y.; Zhu, S.-F. Transition-metal-catalyzed stereo- and regioselective hydrosilylation of unsymmetrical alkynes. Synthesis 2022, 54, 49–66. [Google Scholar]
- Bock, H.; Seidl, H. “d-Orbital effects” in silicon-substituted π-electron systems. XII. Synthesis and properties of the isomeric bis(trimethylsilyl)-1,3-butadienes. J. Am. Chem. Soc. 1968, 90, 5694–5700. [Google Scholar] [CrossRef]
- Kusumoto, T.; Hiyama, T. Hydrosilylation of 1,4-bis(trimethylsilyl)-1,3-butadiyne. Chem. Lett. 1985, 14, 1405–1408. [Google Scholar] [CrossRef]
- Kusumoto, T.; Ando, K.; Hiyama, T. Hydrosilylation of 1,4-bis(trimethylsilyl)butadiyne and silyl-substituted butenynes. Bull. Chem. Soc. Jpn. 1992, 65, 1280–1290. [Google Scholar] [CrossRef]
- Tillack, A.; Pulst, S.; Baumann, W.; Baudisch, H.; Kortus, K.; Rosenthal, U. Hydrosilylierung von symmetrisch disubstituierten alkinen und butadiinen mit L2Ni(0)-butadiin-komplexen [Ph3P, (o-Tol-O)3P] als katalysatoren. J. Organomet. Chem. 1997, 532, 117–123. [Google Scholar] [CrossRef]
- Tillack, A.; Michalik, D.; Koy, C.; Michalik, M. Catalytic asymmetric hydrosilylation of butadiynes: A new synthesis of optically active allenes. Tetrahedron Lett. 1999, 40, 6567–6568. [Google Scholar] [CrossRef]
- Tillack, A.; Koy, C.; Michalik, D.; Fischer, C. A new asymmetric synthesis of optically active allenes via metal catalyzed hydrosilylation. J. Organomet. Chem. 2000, 603, 116–121. [Google Scholar] [CrossRef]
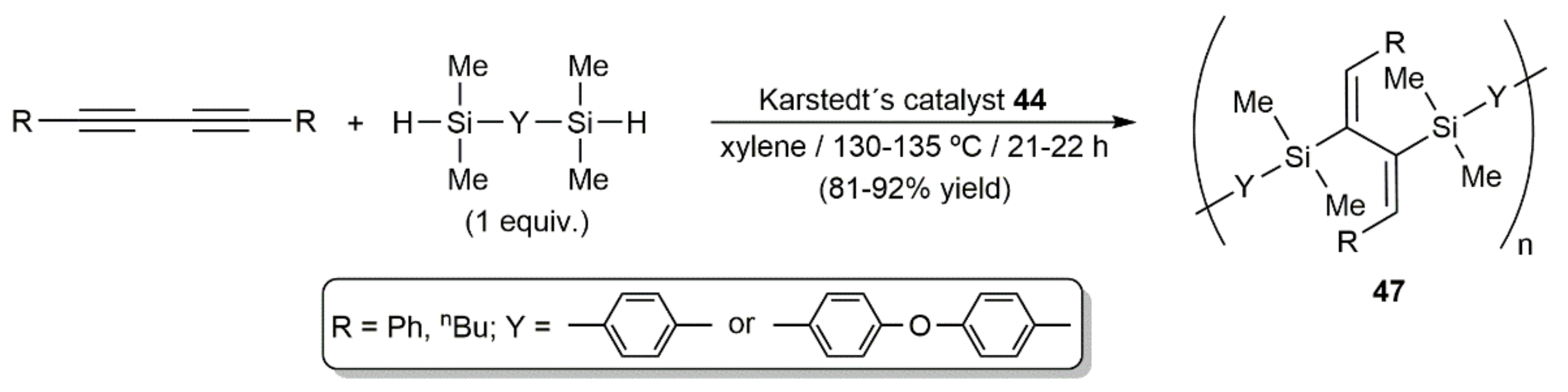
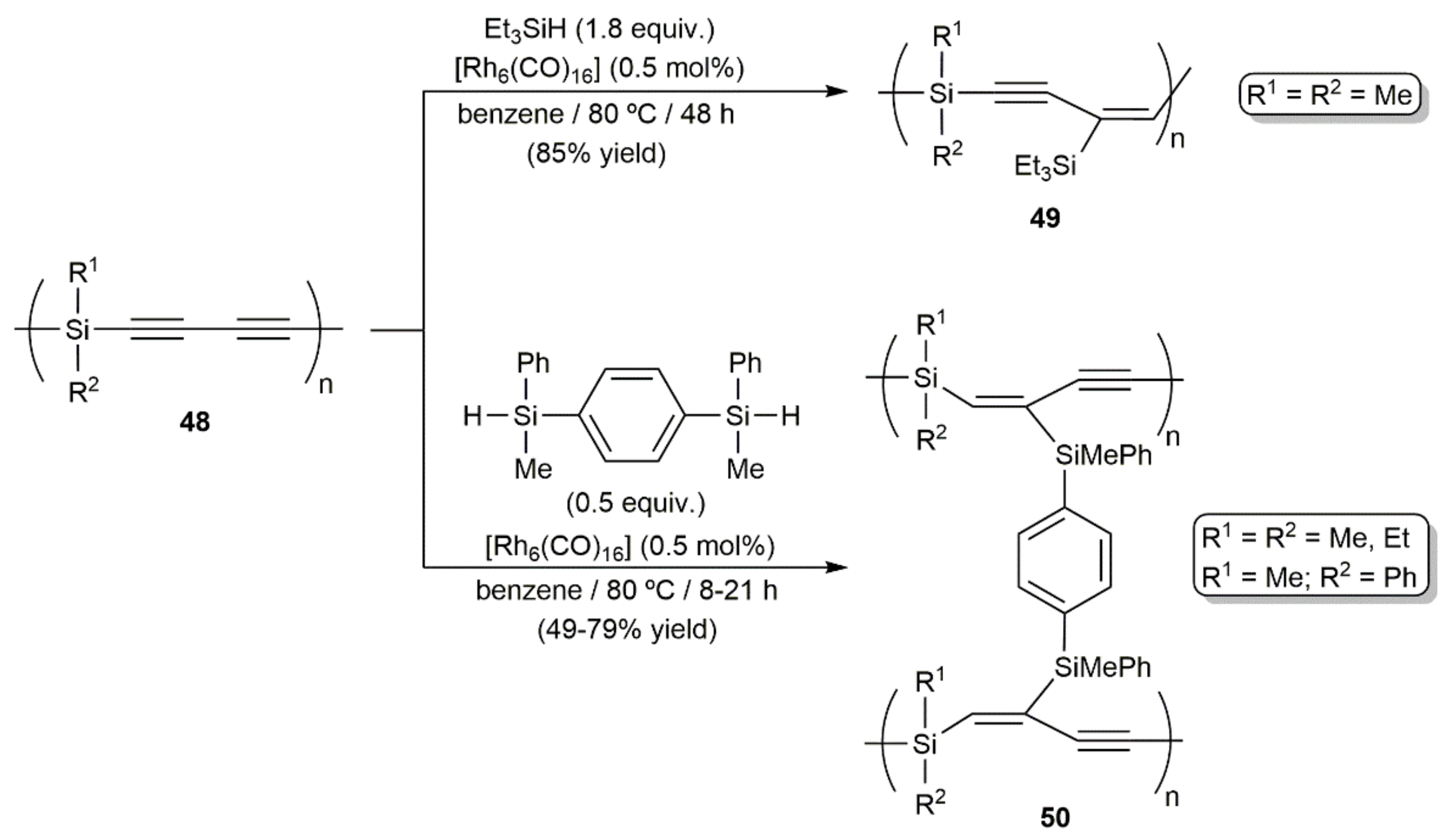
- Perry, R.J.; Karageorgis, M.; Hensler, J. Hydrosilylation reactions of 1,3-diynes and bis(silyl hydrides): Model studies and polymerizations. Macromolecules 2007, 40, 3929–3938. [Google Scholar] [CrossRef]
- Ishikawa, M.; Toyoda, E.; Horio, T.; Kunai, A. Polymeric organosilicon systems. 19. Preparation of branched polymers by selective hydrosilylation of poly[(silylene)but-1,3-diynes]. Organometallics 1994, 13, 26–27. [Google Scholar] [CrossRef]
- Kunai, A.; Toyoda, E.; Nagamoto, I.; Horio, T.; Ishikawa, M. Polymeric organosilicon systems. 25. Preparation of branched polymers by regiospecific hydrosilylation of poly[(silylene)diethynylenes] and their properties. Organometallics 1996, 15, 75–83. [Google Scholar] [CrossRef]
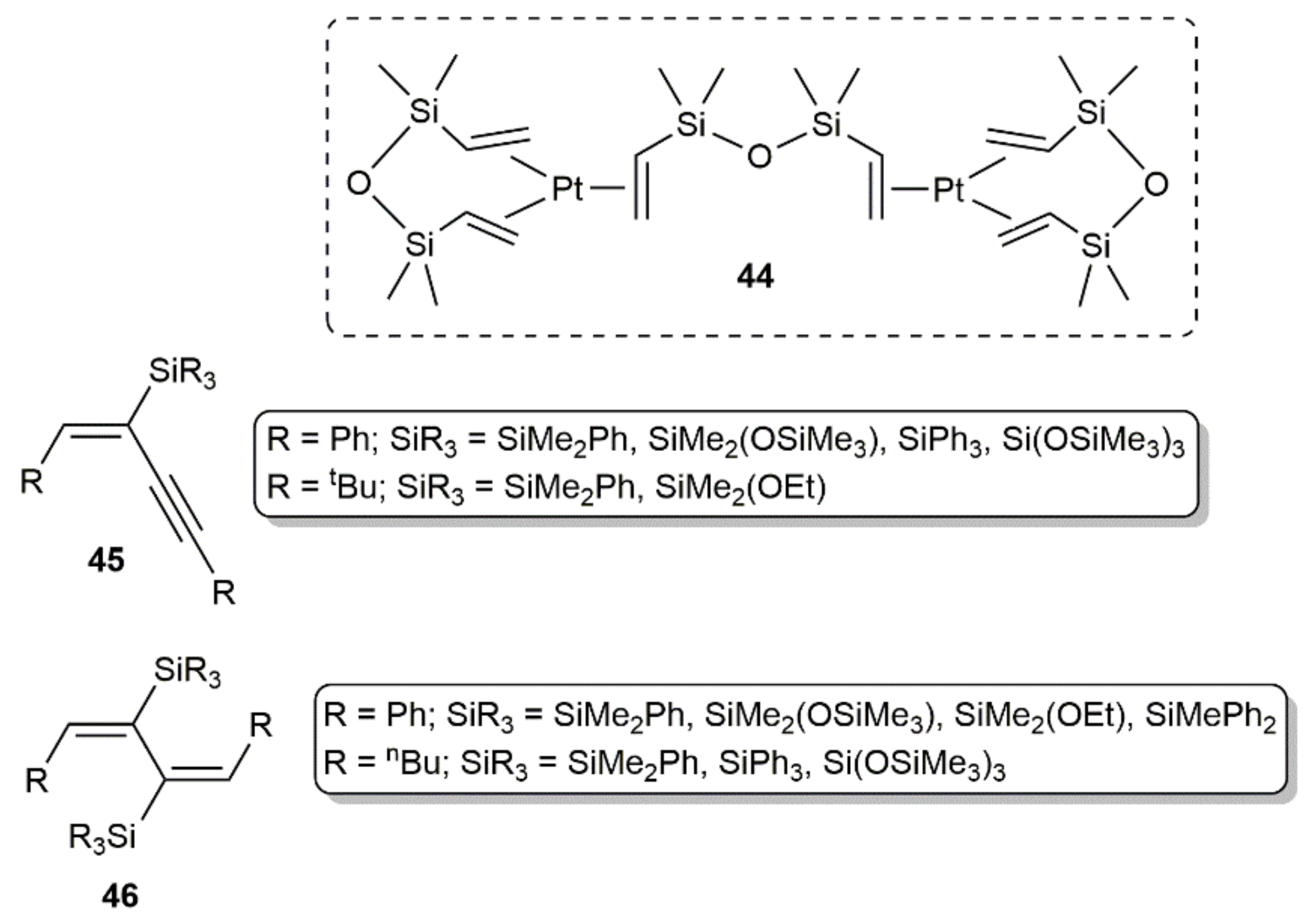
- Walkowiak, J.; Salamon, K.; Franczyk, A.; Stefanowska, K.; Szyling, J.; Kownacki, I. Pt-catalyzed hydrosilylation of 1,3-diynes with triorganosilanes: Regio- and stereoselective synthesis of mono- or bis-silylated adducts. J. Org. Chem. 2019, 84, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Stefanowska, K.; Franczyk, A.; Szyling, J.; Walkowiak, J. Synthesis of functional 3-buten-1-ynes and 1,3-butadienes with silsesquioxane moiety via hydrosilylation of 1,3-diynes. ChemCatChem 2019, 11, 4848–4853. [Google Scholar] [CrossRef]
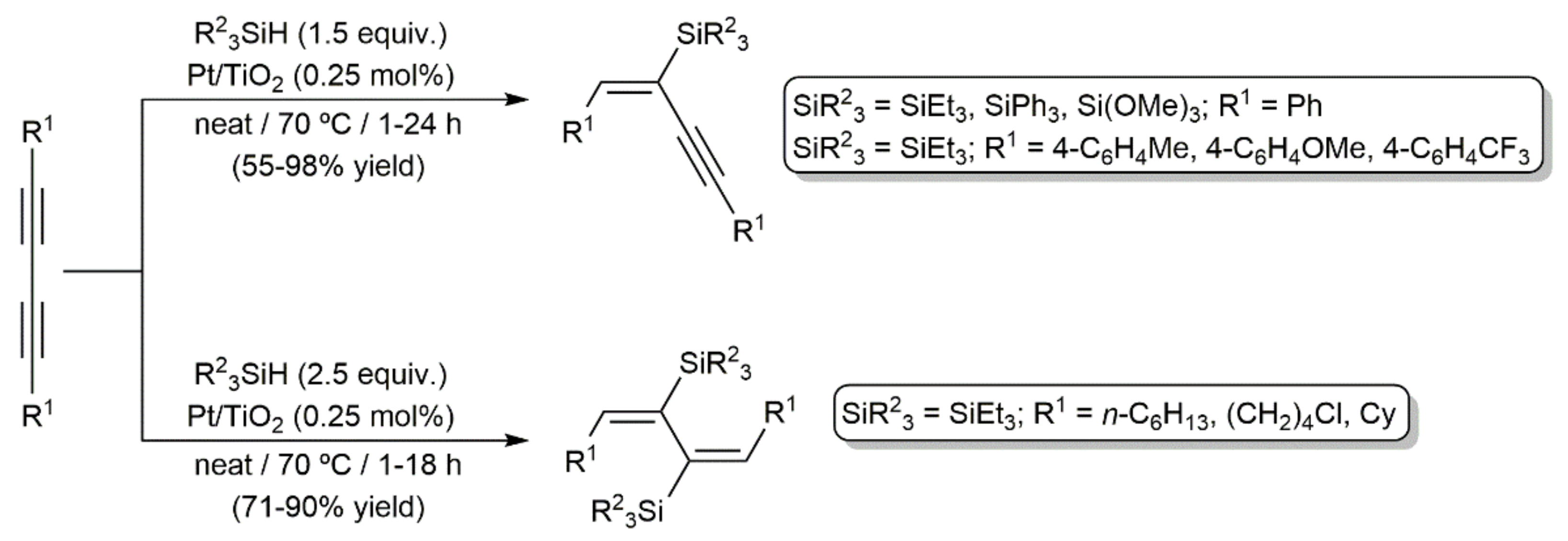
- Alonso, F.; Buitrago, R.; Moglie, Y.; Sepúlveda-Escribano, A.; Yus, M. Selective hydrosilylation of 1,3-diynes catalyzed by titania-supported platinum. Organometallics 2012, 31, 2336–2342. [Google Scholar] [CrossRef]
- Cano, R.; Yus, M.; Ramón, D.J. Impregnated platinum on magnetite as an efficient, fast, and reciclable catalyst for the hydrosilylation of alkynes. ACS Catal. 2012, 2, 1070–1078. [Google Scholar] [CrossRef]
- Reddy, C.B.; Shil, A.K.; Guha, N.R.; Sharma, D.; Das, P. Solid supported palladium(0) nanoparticles: An efficient heterogeneous catalyst for regioselective hydrosilylation of alkynes and Suzuki coupling of β-arylvinyl iodides. Catal. Lett. 2014, 144, 1530–1536. [Google Scholar] [CrossRef]
- Planellas, M.; Guo, W.; Alonso, F.; Yus, M.; Shafir, A.; Pleixats, R.; Parella, T. Hydrosilylation of internal alkynes catalyzed by tris-imidazolium salt-stabilized palladium nanoparticles. Adv. Synth. Catal. 2014, 356, 179–188. [Google Scholar] [CrossRef]
- Guo, W.; Pleixats, R.; Shafir, A.; Parella, T. Rhodium nanoflowers stabilized by a nitrogen-rich PEG-tagged substrate as recyclable catalyst for the stereoselective hydrosilylation of internal alkynes. Adv. Synth. Catal. 2015, 357, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jiang, Y.-N.; Lin, Z.-Y.; Zeng, J.-H.; Liu, Z.-K.; Zhan, Z.-P. Highly regio- and stereo-selective heterogeneous 1,3-diyne hydrosilylation controlled by a nickel-metalated porous organic polymer. Org. Chem. Front. 2021, 8, 4826–4832. [Google Scholar] [CrossRef]
- Sun, J.; Deng, L. Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 2016, 6, 290–300. [Google Scholar] [CrossRef]
- Tamang, S.R.; Findlater, M. Emergence and applications of base metals (Fe, Co and Ni) in hydroboration and hydrosilylation. Molecules 2019, 24, 3194. [Google Scholar] [CrossRef] [Green Version]
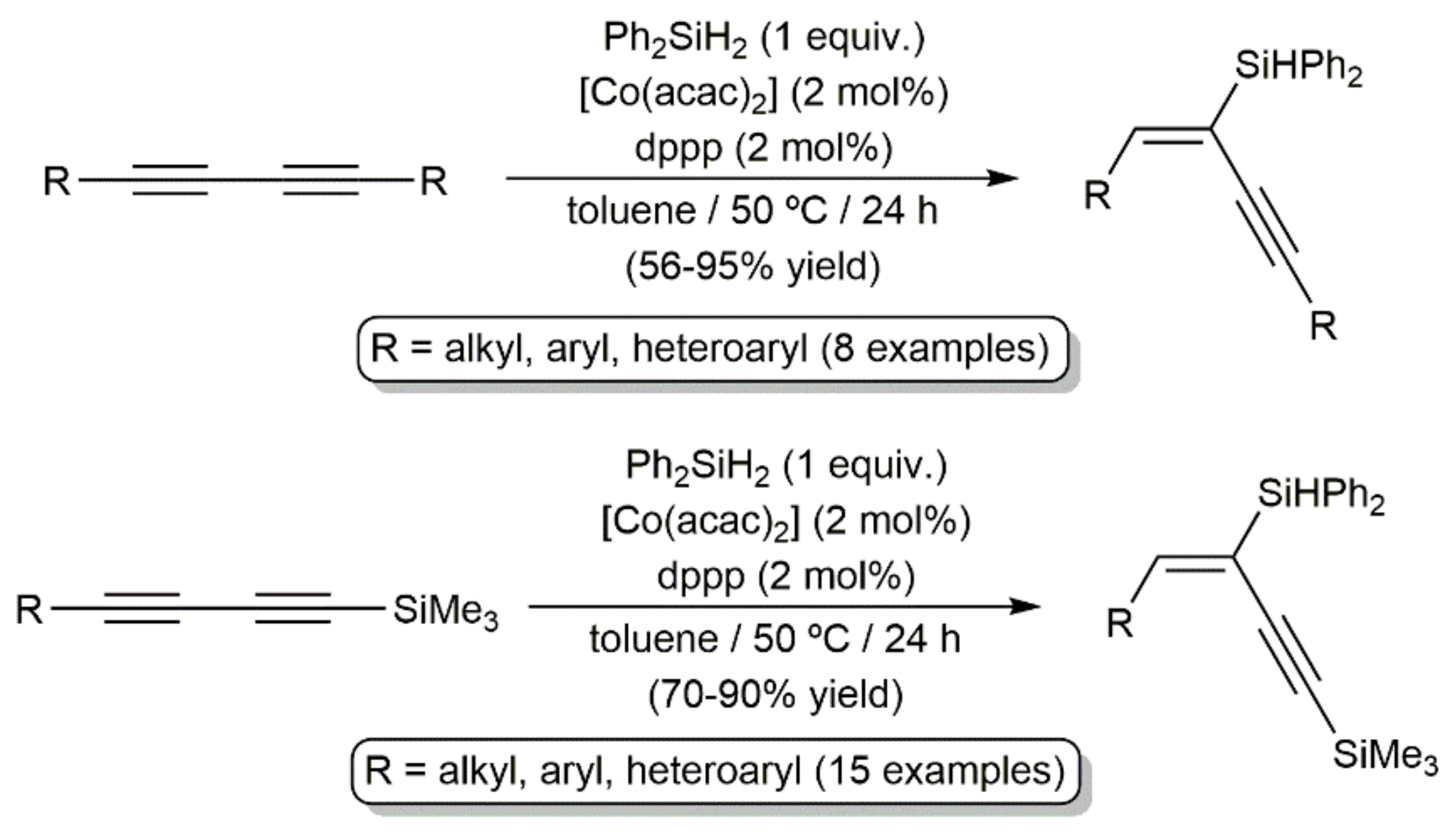
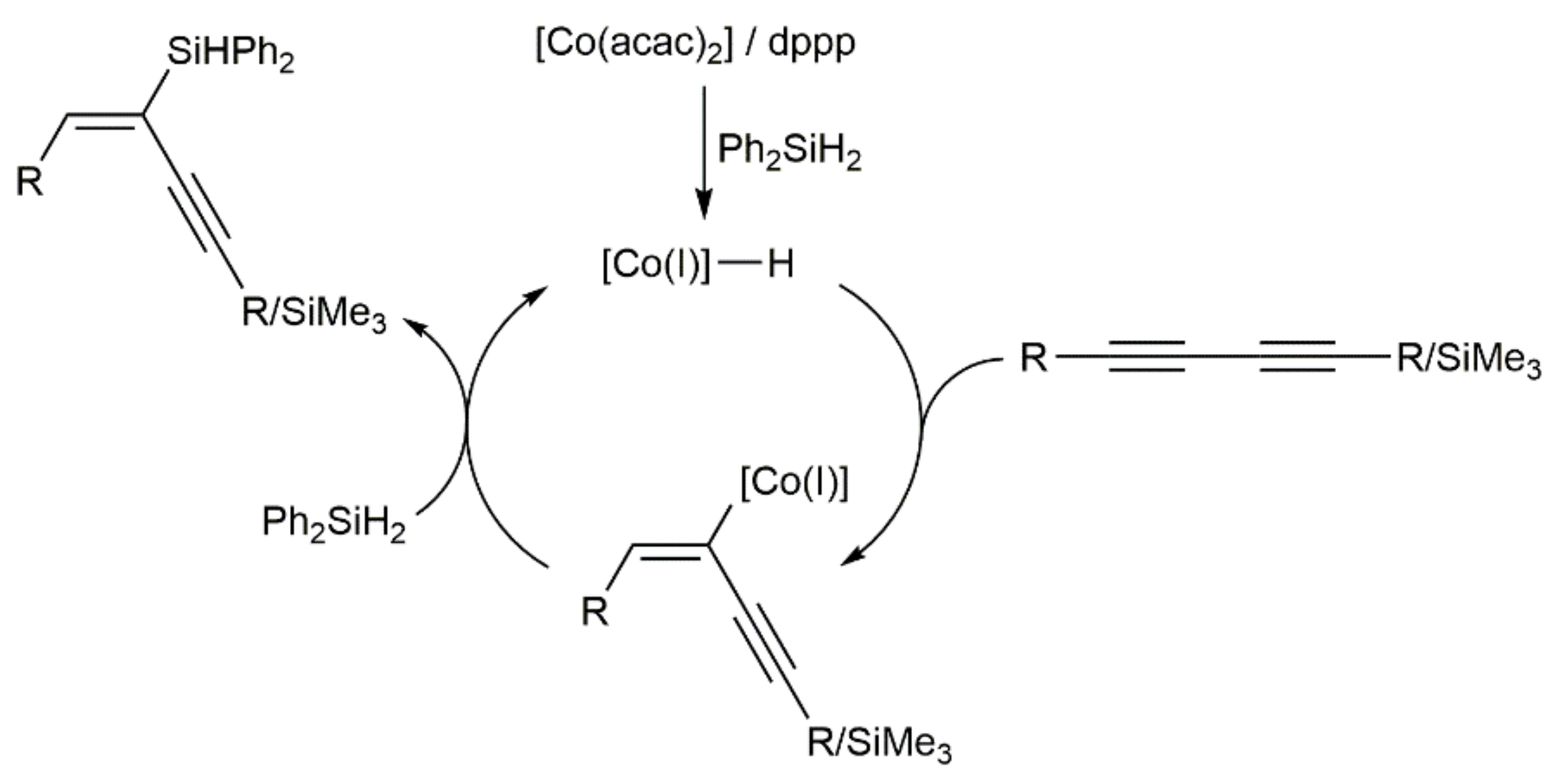
- Sang, H.L.; Hu, Y.; Ge, S. Cobalt-catalyzed regio- and stereoselective hydrosilylation of 1,3-diynes to access silyl-functionalized 1,3-enynes. Org. Lett. 2019, 21, 5234–5237. [Google Scholar] [CrossRef] [PubMed]
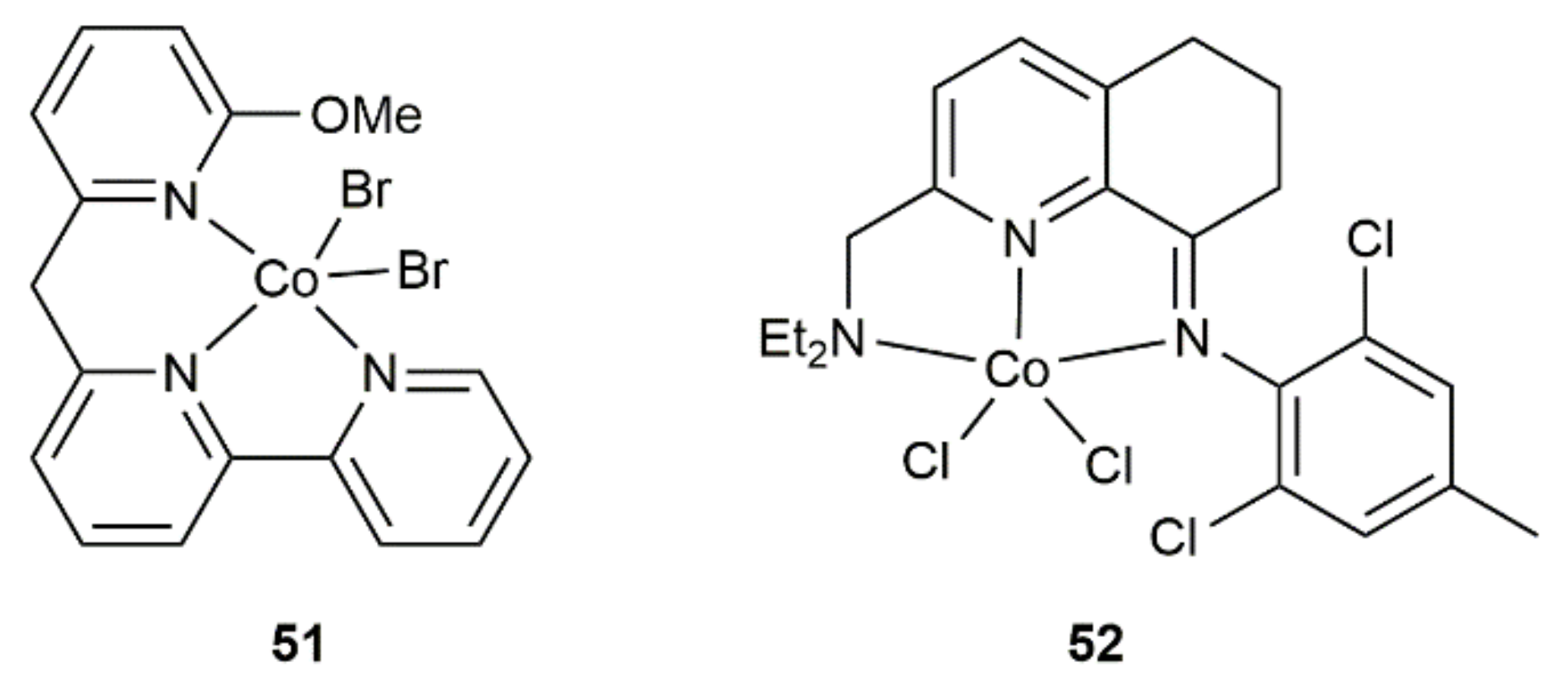
- Kong, D.; Hu, B.; Yang, M.; Chen, D.; Xia, H. Highly regio- and stereoselective tridentate NCNN cobalt-catalyzed 1,3-diyne hydrosilylation. Organometallics 2019, 38, 4341–4350. [Google Scholar] [CrossRef]
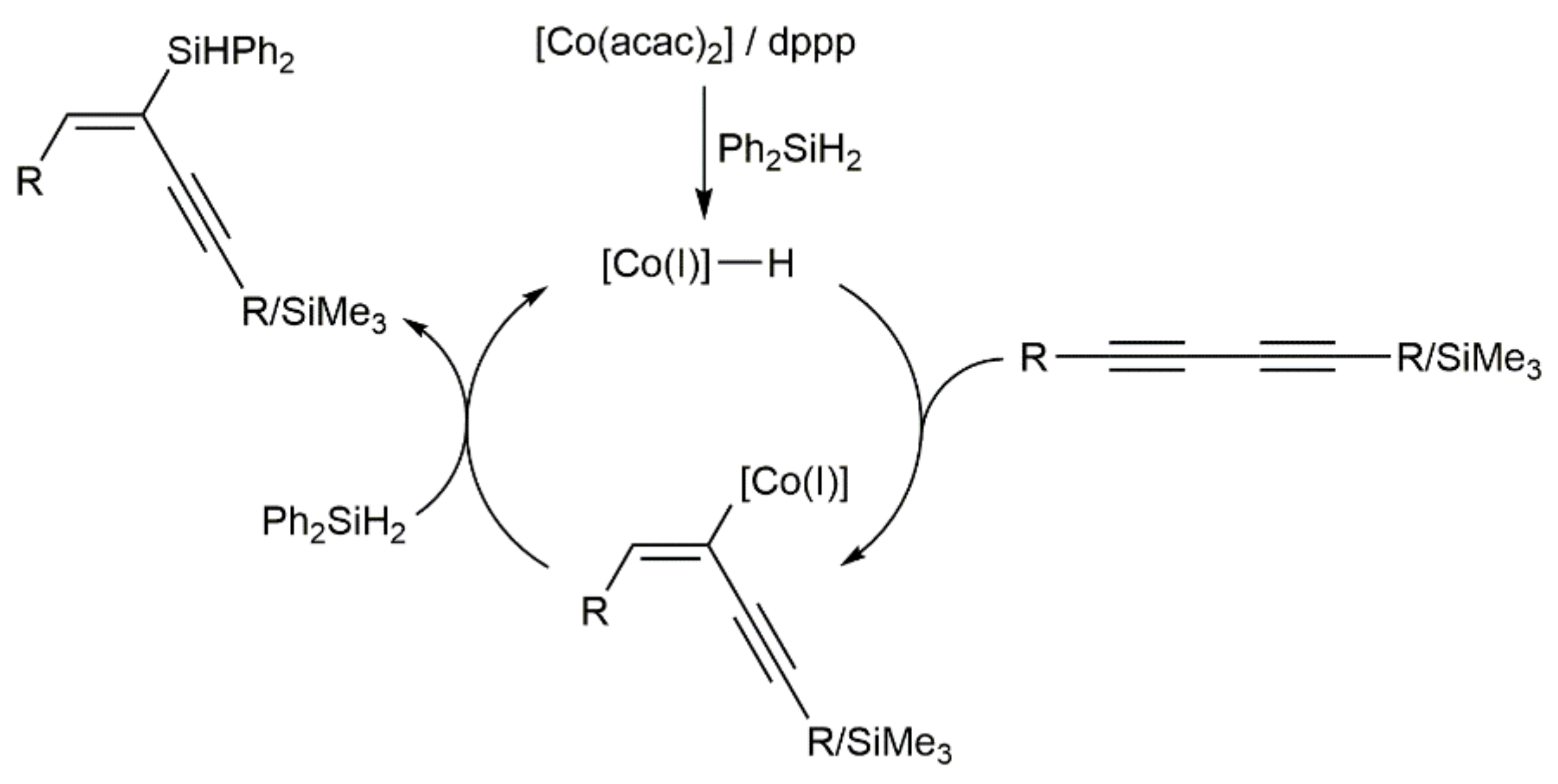
- Shen, S.; Zong, Z.; Sun, N.; Hu, B.; Shen, Z.; Hu, X.; Jin, L. Regio- and stereoselective cobalt-catalyzed hydroilylation of 1,3-diynes with primary and secondary silanes. Org. Chem. Front. 2021, 8, 6317–6322. [Google Scholar] [CrossRef]
- Kong, D.; Hu, B.; Yang, M.; Gong, D.; Xia, H.; Chen, D. Bis(phosphine)cobalt-catalyzed highly regio- and stereoselective hydrosilylation of 1,3-diynes. Organometallics 2020, 39, 4437–4443. [Google Scholar] [CrossRef]
- Matsuda, T. Synthesis of heterocycles via X-H bond addition to diynes. In Transition-Metal-Mediated Aromatic Ring Construction, 1st ed.; Tanaka, K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 537–547. [Google Scholar]
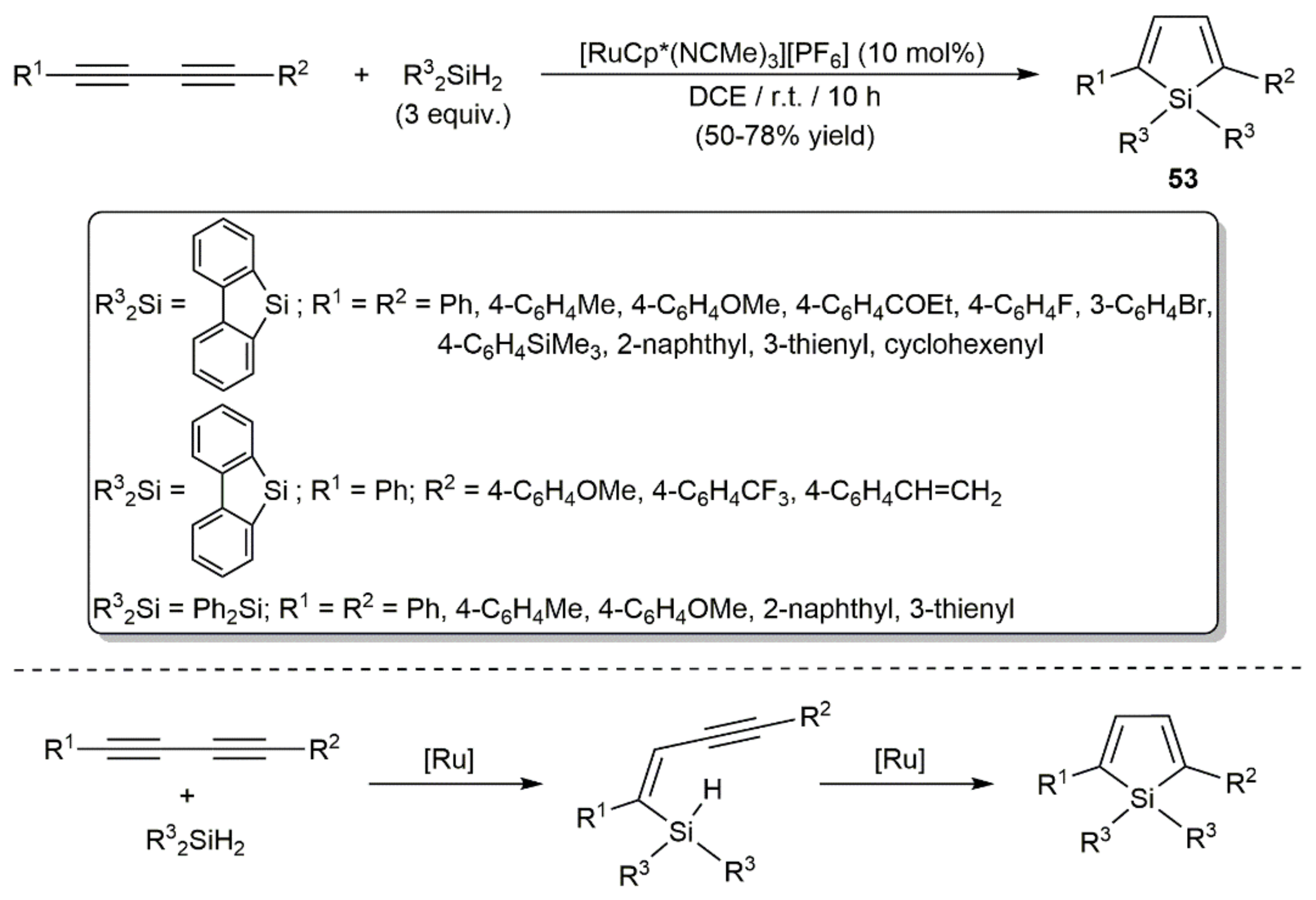
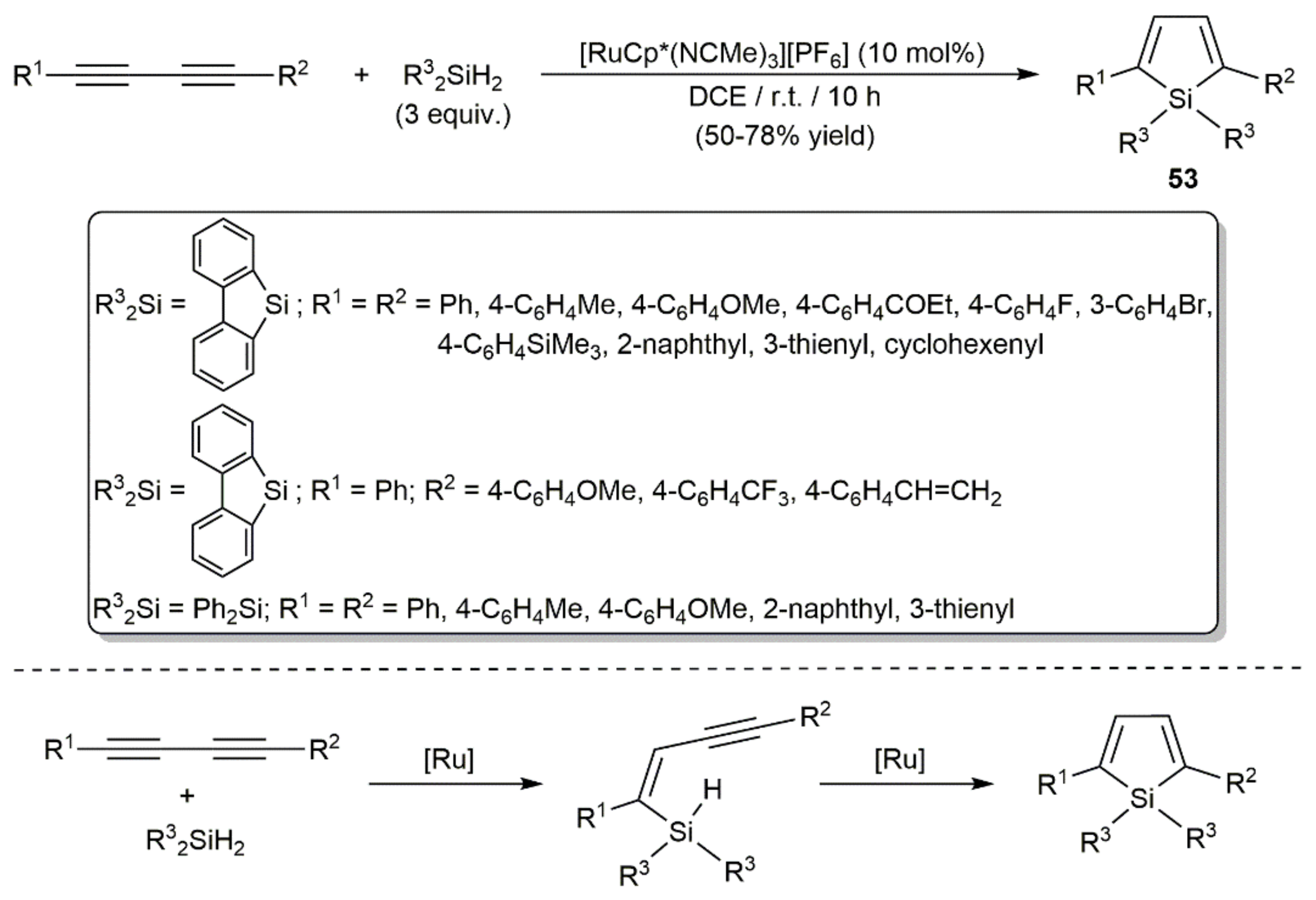
- Matsuda, T.; Kadowaki, S.; Murakami, M. Ruthenium-catalyzed trans-hydrosilylation of 1,4-diaryl-1,3-diynes leading to 2,5-diarylsiloles. Chem. Commun. 2007, 2627–2629. [Google Scholar] [CrossRef]
- Matsuda, T.; Kadowaki, S.; Yamaguchi, Y.; Murakami, M. Ruthenium-catalyzed trans-hydrogermylation of alkynes: Formation of 2,5-disubstituted germoles through double trans-hydrogermylation of 1,3-diynes. Org. Lett. 2010, 12, 1056–1058. [Google Scholar] [CrossRef]
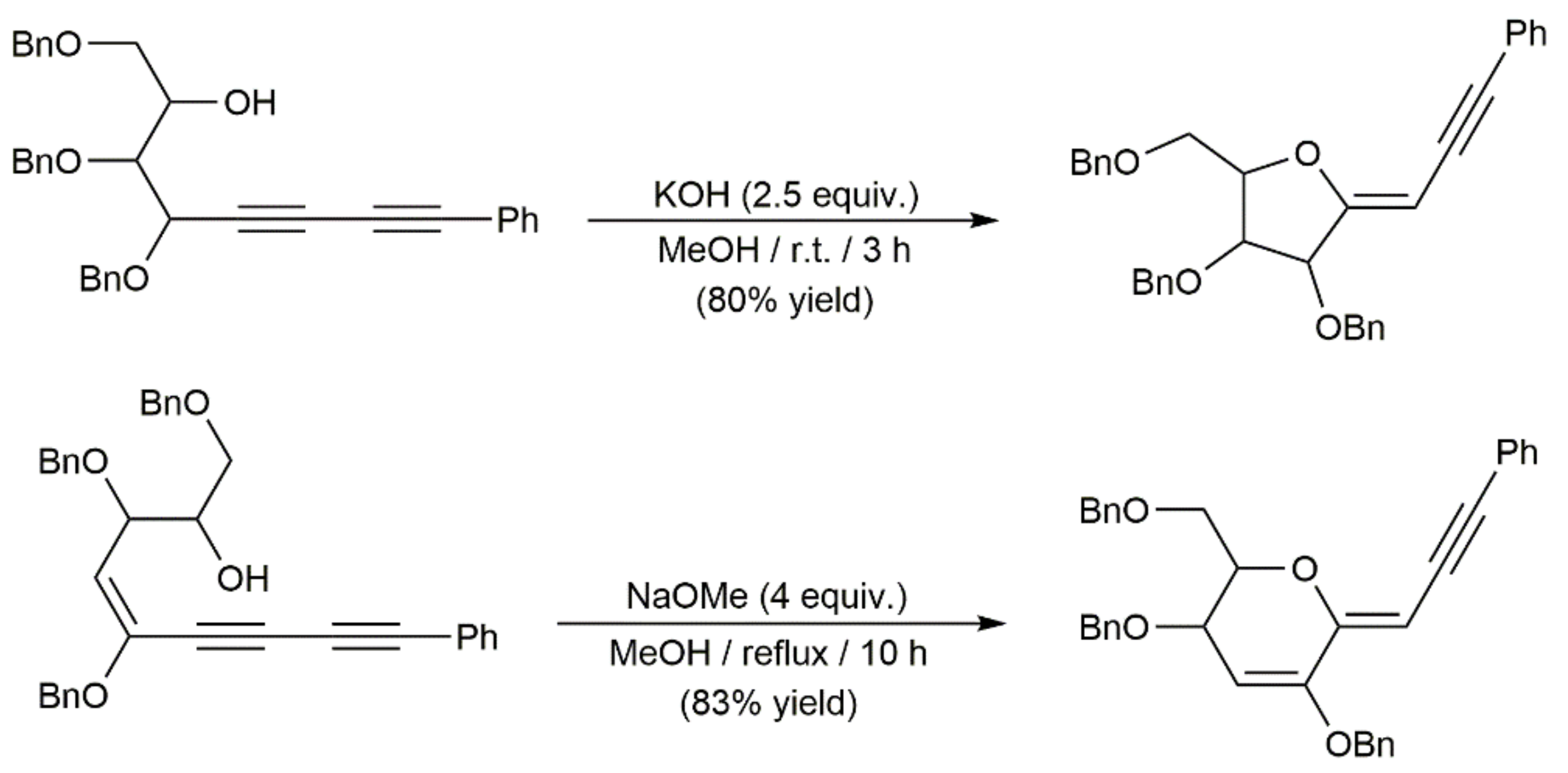
- Trost, B.M.; Chan, V.S.; Yamamoto, D. Enantioselective ProPhenol-catalyzed addition of 1,3-diynes to aldehydes to generate synthetically versatile building blocks and diyne natural products. J. Am. Chem. Soc. 2010, 132, 5186–5192. [Google Scholar] [CrossRef] [Green Version]
- Reisch, J.; Schulte, K.E. Pyrrol-derivate aus diacetylenen. Angew. Chem. 1961, 73, 241. [Google Scholar] [CrossRef]
- Schulte, K.E.; Reisch, J.; Walker, H. Eine neue pyrrolsynthese aus butadiin-derivaten. Chem. Ber. 1965, 98, 98–103. [Google Scholar] [CrossRef]
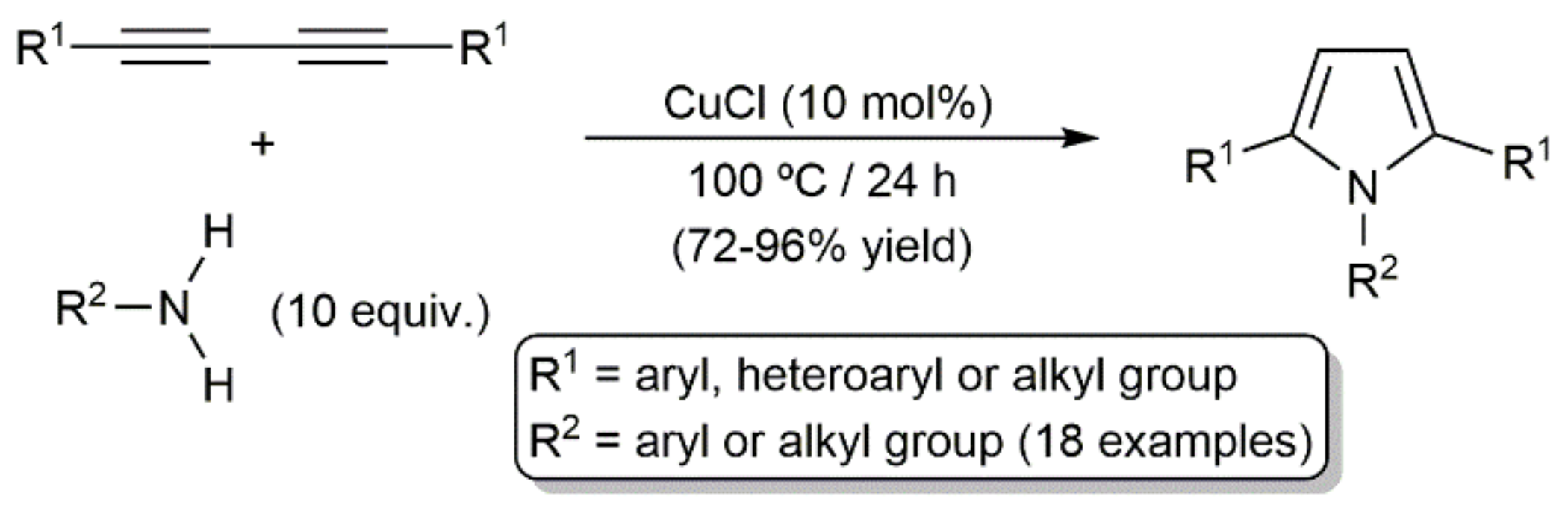
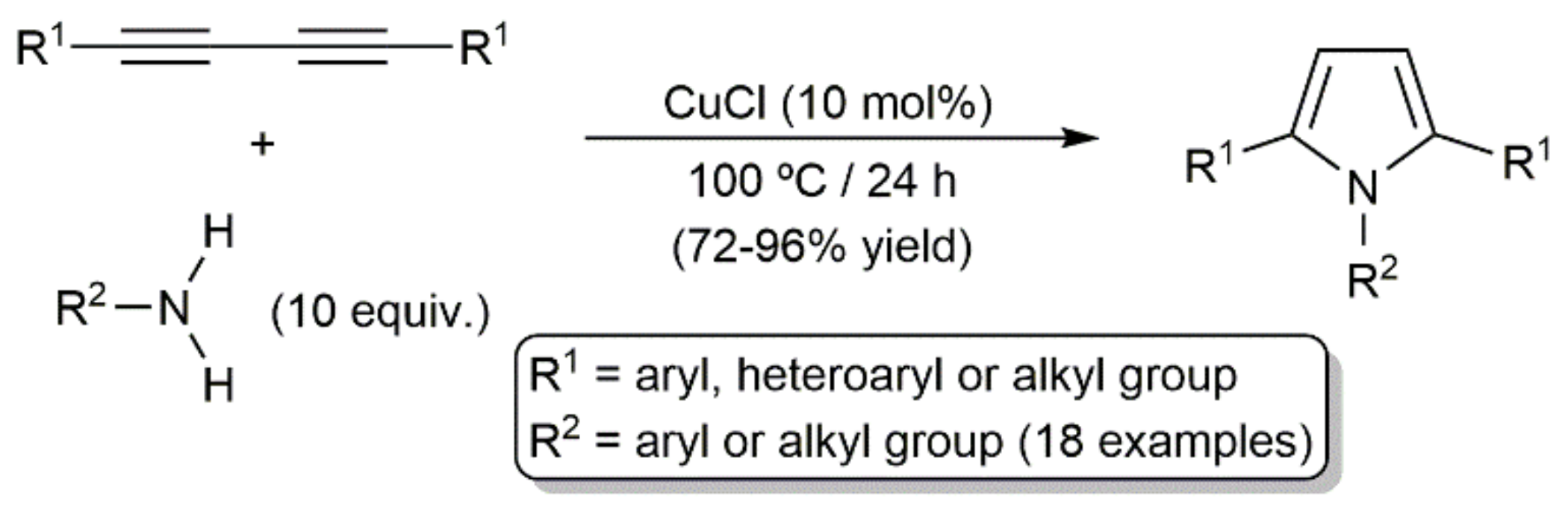
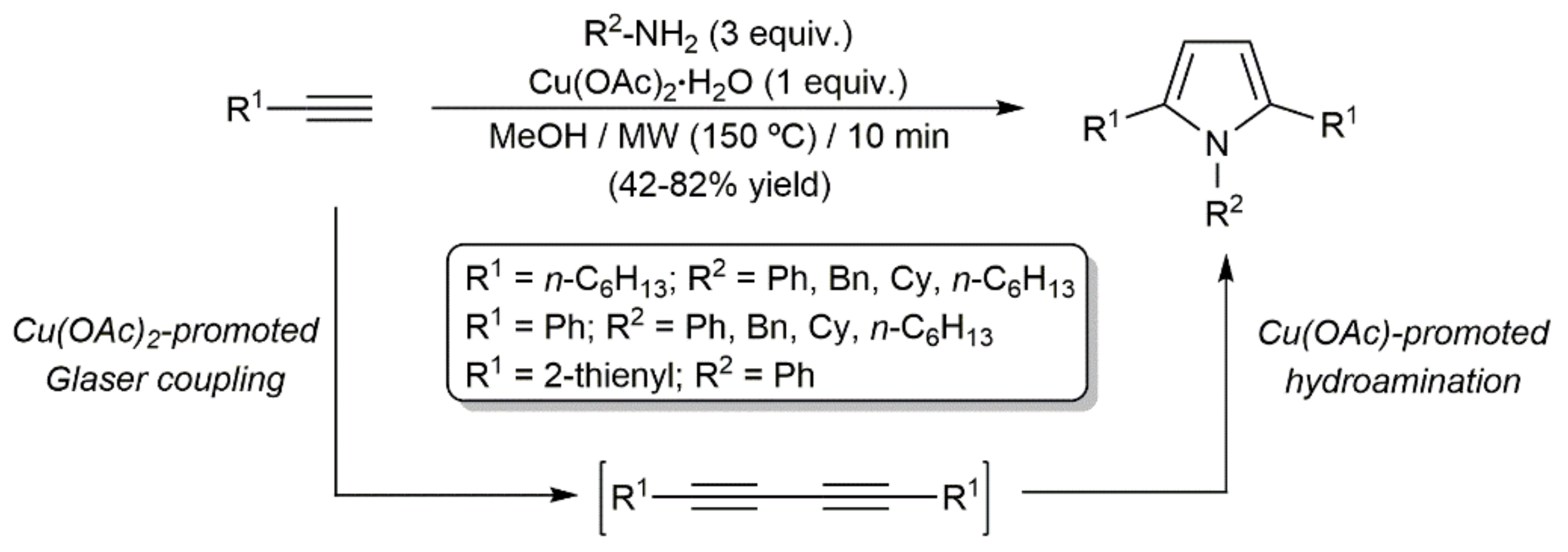
- Zheng, Q.; Hua, R. CuCl-catalyzed cycloaddition of 1,3-butadiynes with primary amines: An atom-economic process for the synthesis of 1,2,5-trisubstituted pyrroles. Tetrahedron Lett. 2010, 51, 4512–4514. [Google Scholar] [CrossRef]
- Zheng, Q.; Hua, R.; Jiang, J.; Zhang, L. A general approach to arylated furans, pyrroles, and thiophenes. Tetrahedron 2014, 70, 8252–8256. [Google Scholar] [CrossRef]
- Feng, X.; Tong, B.; Shen, J.; Shi, J.; Han, T.; Chen, L.; Zhi, J.; Lu, P.; Ma, Y.; Dong, Y. Aggregation-induced emission enhancement of aryl-substituted pyrrole derivatives. J. Phys. Chem. B 2010, 114, 16731–16736. [Google Scholar] [CrossRef]
- Krompiec, S.; Filapek, M.; Grudzka-Flak, I.; Slodek, A.; Kula, S.; Malecki, J.G.; Malarz, J.; Szafraniec-Gorol, G.; Penkala, M.; Schab-Balcerzak, E.; et al. Multifaceted strategy for the synthesis of diverse 2,2´-bithiophene derivatives. Molecules 2015, 20, 4565–4593. [Google Scholar] [CrossRef] [Green Version]
- Yang, C. Pyrrole-cored push-pull single chromophore. Tetrahedron Lett. 2010, 51, 2007–2009. [Google Scholar] [CrossRef]
- Kowada, T.; Kuwabara, T.; Ohe, K. Synthesis, structure, and optical properties of heteroarene-fused dispiro compounds. J. Org. Chem. 2010, 75, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Kobayashi, T.; Ogura, K. Novel 1,1′,5,5′-tetraaryl-2,2′-bipyrroles: Their synthesis and physical properties. Heterocycles 2005, 66, 319–332. [Google Scholar] [CrossRef]
- Maeda, C.; Shinokubo, H.; Osuka, A. Synthesis of meso,meso’-pyrrole-bridged diporphyrins by Cu(I)-mediated annulation. Org. Lett. 2010, 12, 1820–1823. [Google Scholar] [CrossRef] [PubMed]
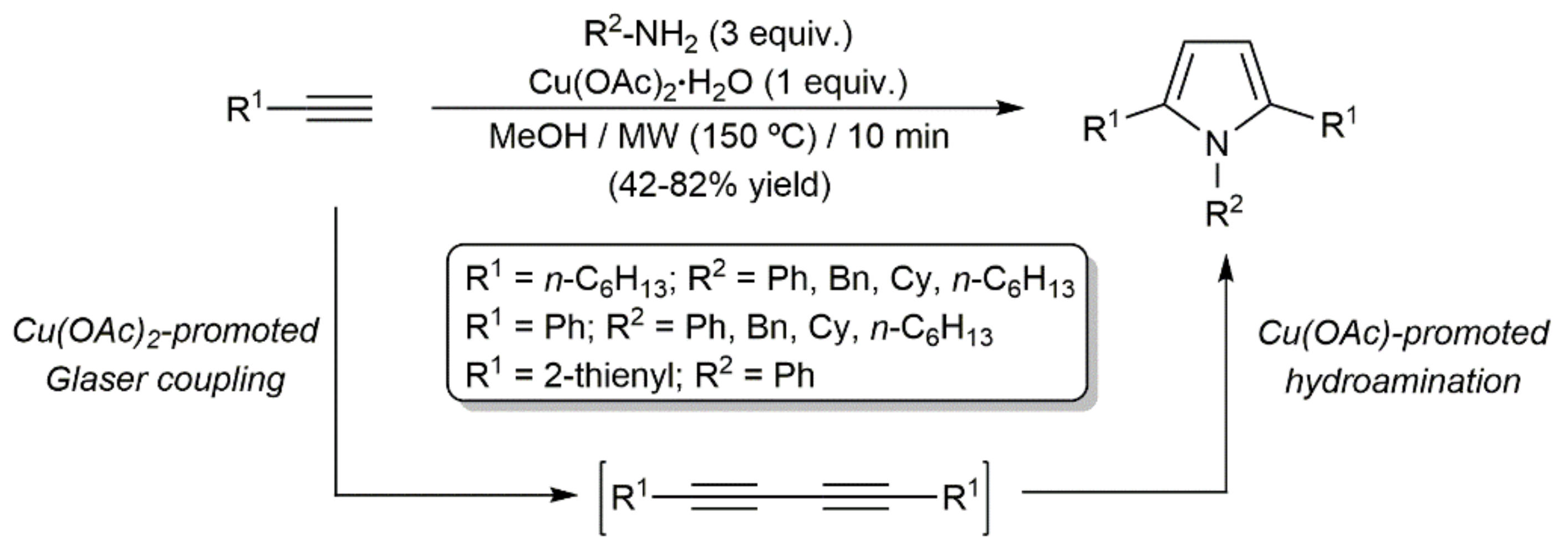
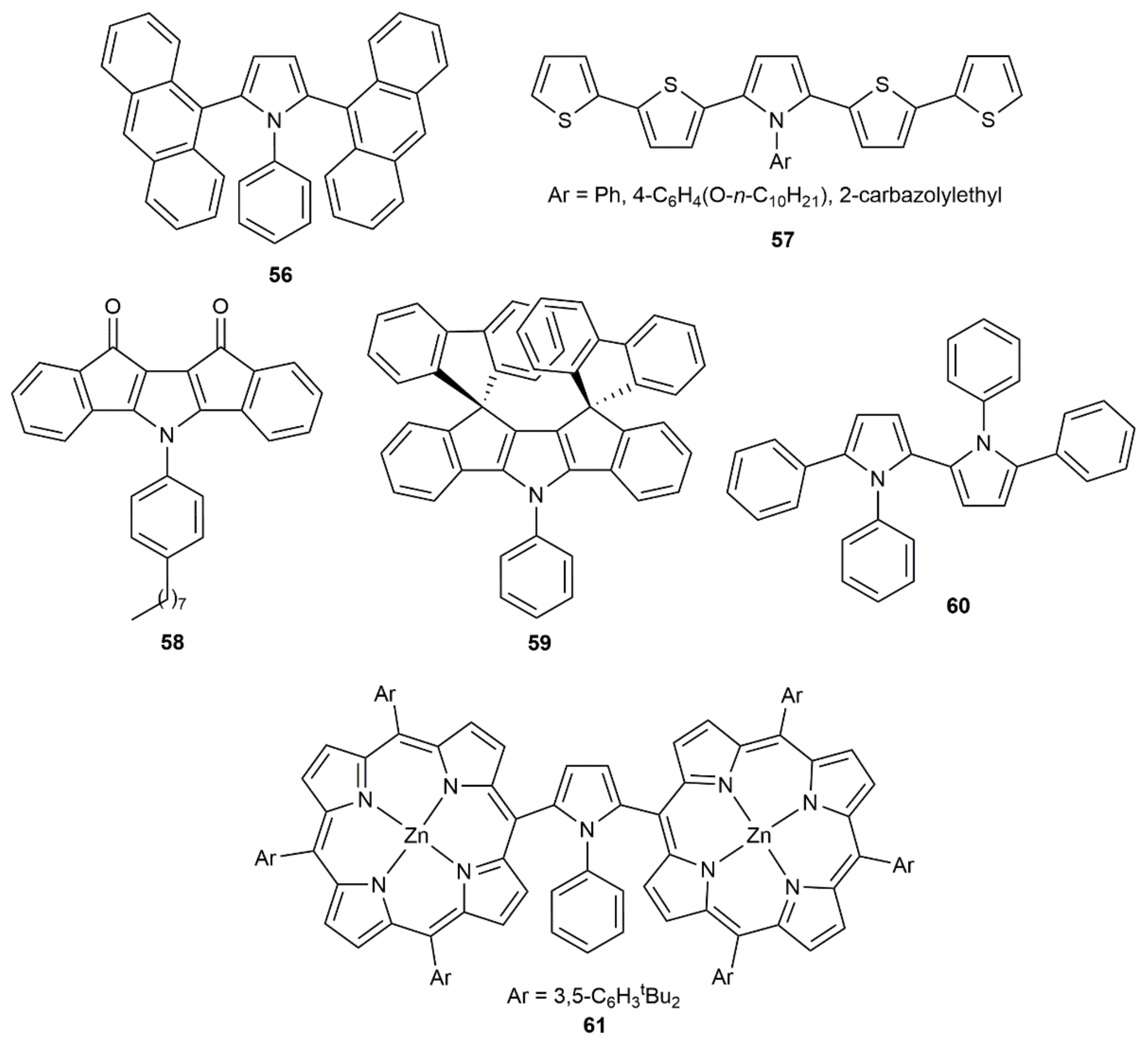
- Lee, H.; Yi, L.; Jun, C.-H. Copper(II)-promoted, one-pot conversion of 1-alkynes with anhydrides or primary amines to the respective 2,5-disubstituted furans or pyrroles under microwave irradiation conditions. Adv. Synth. Catal. 2015, 357, 3485–3490. [Google Scholar] [CrossRef]
- Widenhoefer, R.A.; Han, X. Gold-catalyzed hydroamination of C-C multiple bonds. Eur. J. Org. Chem. 2006, 2006, 4555–4563. [Google Scholar] [CrossRef]
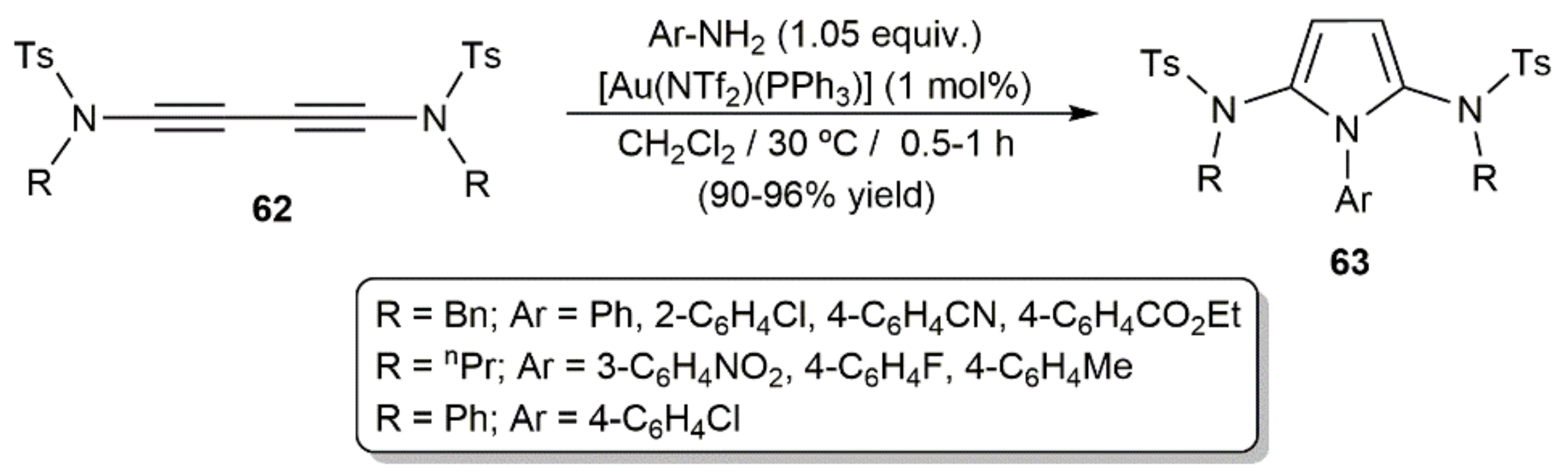
- Kramer, S.; Madsen, J.L.H.; Rottländer, M.; Skrydstrup, T. Access to 2,5-diamidopyrroles and 2,5-diamidofurans by Au(I)-catalyzed double hydroamination or hydration of 1,3-diynes. Org. Lett. 2010, 12, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Num, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S.P. Gold(I)-catalyzed synthesis of furans and pyrroles via alkyne hydration. Catal. Sci. Technol. 2011, 1, 58–61. [Google Scholar]
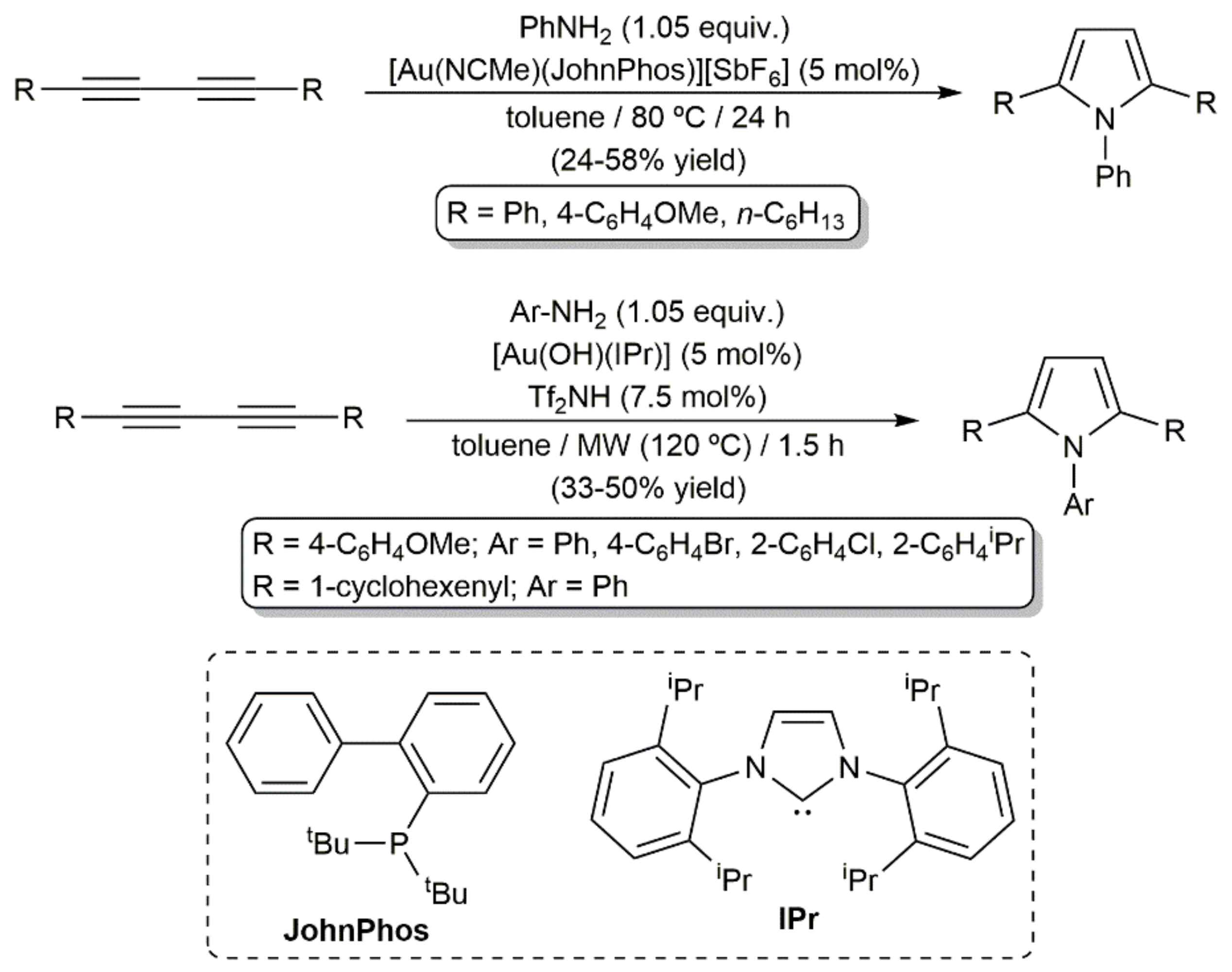
- Lavallo, V.; Frey, G.D.; Donnadieu, B.; Soleilhavoup, M.; Bertrand, G. Homogeneous catalytic hydroamination of alkynes and allenes with ammonia. Angew. Chem. Int. Ed. 2008, 47, 5224–5228. [Google Scholar] [CrossRef] [Green Version]
- Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-catalyzed hydroamination of alkynes and allenes with parent hydrazine. Angew. Chem. Int. Ed. 2011, 50, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Paudler, W.W.; Zeiler, A.G. 1,3-butadiynes in the synthesis of heterocyclic compounds. I. 2,3-Dihydro-1,4-diazepine, pyrazole, and isoxazole derivatives. J. Org. Chem. 1969, 34, 999–1001. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Feng, X.; Bao, M. Synthesis of 3,5-disubstituted pyrazoles via Cope-type hydroamination of 1,3-dialkynes. J. Org. Chem. 2013, 78, 1693–1698. [Google Scholar] [CrossRef]
- Yu, X.; Huang, N.; Feng, X.; Yamamoto, Y.; Bao, M. Synthesis of 1,3,5-trisubstituted pyrazoles by the Cope-type hydroamination of 1,3-dialkynes with alkylhydrazines. Synthesis 2014, 46, 2422–2429. [Google Scholar] [CrossRef]
- Bassaco, M.M.; Fortes, M.P.; Kaufman, T.S.; Silveira, C.C. Metal-free synthesis of 3,5-disubstituted 1H- and 1-aryl-1H-pyrazoles from 1,3-diyne-indole derivatives employing two successive hydroaminations. RSC Adv. 2015, 5, 21112–21124. [Google Scholar] [CrossRef]
- Shen, R.; Yang, J.; Luo, B.; Zhang, L.; Han, L.-B. Copper-catalyzed allenylation-isomerization sequence of 1,4-diyn-3-yl acetates with P(O)H compounds: Facile synthesis of 1-phosphonyl 2,4-diynes. Adv. Synth. Catal. 2016, 358, 3897–3906. [Google Scholar] [CrossRef]
- Verlinden, S.; Ballet, S.; Verniest, G. Synthesis of heterocycle-bridged peptidic macrocycles through 1,3-diyne transformations. Eur. J. Org. Chem. 2016, 2016, 5807–5812. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Georgiádes, Á.; Ozsvár, D.; Fülöp, F. Continuous-flow synthesis of 3,5-disubstituted pyrazoles via sequential alkyne homocoupling and Cope-type hydroamination. RSC Adv. 2019, 9, 8197–8203. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yu, X.; Feng, X.; Bao, M. Synthesis of 3,5-disubstituted isoxazoles via Cope-type hydroamination of 1,3-dialkynes. Org. Lett. 2012, 14, 2418–2421. [Google Scholar] [CrossRef]
- Bassaco, M.M.; Fortes, M.P.; Back, D.F.; Kaufman, T.S.; Silveira, C.C. An eco-friendly synthesis of novel 3,5-disubstituted-1,2-isoxazoles in PEG-400, employing the Et3N-promoted hydroamination of symmetric and unsymmetric 1,3-diyne-indole derivatives. RSC Adv. 2014, 4, 60785–60797. [Google Scholar] [CrossRef]
- Kirillova, M.A.; Maretina, I.A.; Petrov, A.A. Reactions of alkadiynes with guanidine and its derivatives. Russ. J. Org. Chem. 1971, 7, 14–16. [Google Scholar]
- Singha, R.; Ray, J.K. Transition metal free synthesis of 2,4,6-trisubstitutd pyrimidines via Cope-type hydroamination of 1,4-diarylbuta-1,3-diynes. RSC Adv. 2014, 4, 44052–44056. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, M.; Zhao, X. The synthesis of carbonyl 2-amino-pyrimidines via tandem regioselective heterocyclization of 1,3-diynes with guanidine and selective oxidation. Chem. Commun. 2015, 51, 9370–9373. [Google Scholar] [CrossRef]
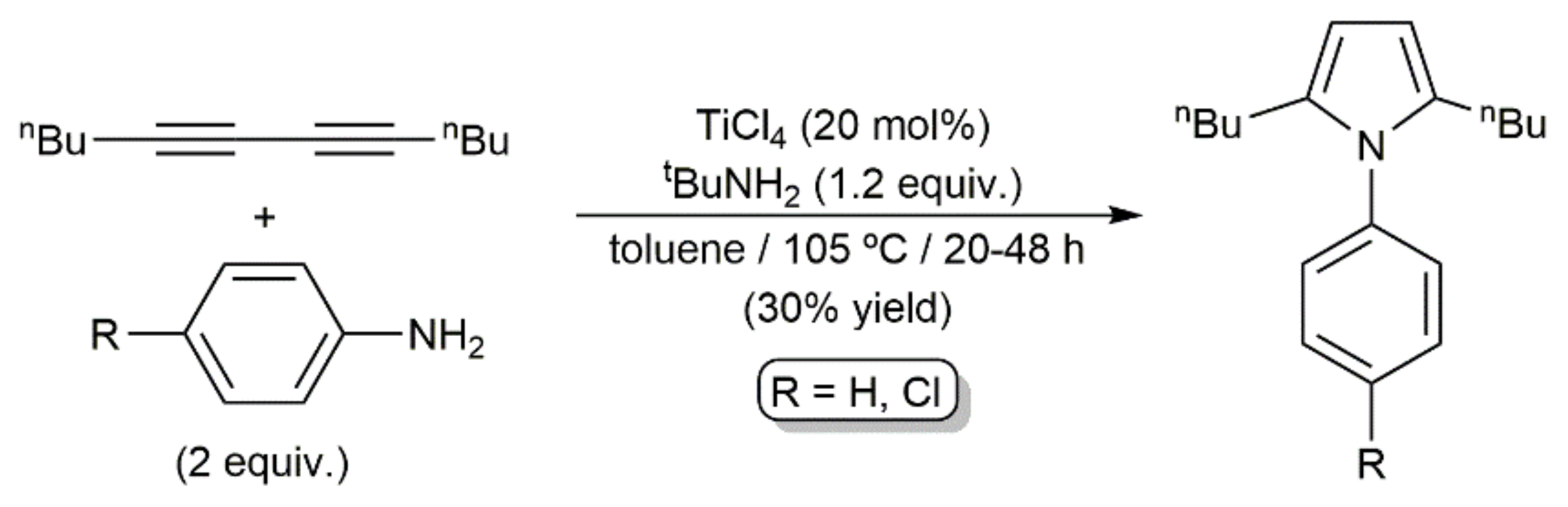
- Ackermann, L.; Born, R. TiCl4/t-BuNH2 as the sole catalyst for a hydroamination-based Fischer indole synthesis. Tetrahedron Lett. 2004, 45, 9541–9544. [Google Scholar] [CrossRef]
- Gupta, S.; Agarwal, P.K.; Saifuddin, M.; Kundu, B. Hydro-amination/-amidation of 1,3-diynes with indoles/azoles/amides under modified Ullmann conditions: Stereo- and regio-selective synthesis of N-alkenynes via N-H bond activation. Tetrahedron Lett. 2011, 52, 5752–5757. [Google Scholar] [CrossRef]
- Sun, H.; Wu, X.; Hua, R. Copper(I)-Catalyzed reaction of diaryl buta-1,3-diynes with cyclic amines: An atom-economic approach to amino-substituted naphthalene derivatives. Tetrahedron Lett. 2011, 52, 4408–4411. [Google Scholar] [CrossRef]
- Choi, J.; Park, K.; Lim, J.; Jung, H.M.; Lee, S. Copper-catalyzed synthesis of amino-substituted polycyclic aromatic hydrocarbons by the sequential reaction between aryl alkynyl carboxylic acids and amines. Asian J. Org. Chem. 2015, 4, 969–974. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, S.; Fan, L.; Yin, G. Cooperative palladium and copper catalysis: One-pot synthesis of diamino-substituted naphthalenes from aryl halides, 1,4-bis(trimethylsilyl)butadiene and amines. ChemCatChem 2020, 12, 1230–1235. [Google Scholar] [CrossRef]
- Glock, C.; Görls, H.; Westerhausen, M. Calcite-mediated intermolecular hydroamination of diphenylbutadiyne with secondary anilines. Chem. Commun. 2012, 48, 7094–7096. [Google Scholar] [CrossRef] [Green Version]
- Younis, F.M.; Krieck, S.; Görls, H.; Westerhausen, M. Hydroamination of diphenylbutadiyne with secondary N-methyl-anilines using the dipotassium tetrakis(2,6-diisopropylanilino)calciate precatalyst. Dalton Trans. 2016, 45, 6241–6250. [Google Scholar] [CrossRef]
- Younis, F.M.; Krieck, S.; Görls, H.; Westerhausen, M. s-Block-metal-mediated hydroamination of diphenylbutadiyne with primary arylamines using dipotassium tetrakis(amino)calciate precatalyst. Organometallics 2015, 34, 3577–3585. [Google Scholar] [CrossRef]
- Glock, C.; Younis, F.M.; Ziemann, S.; Görls, H.; Imhof, W.; Kriek, S.; Westerhausen, M. 2,6-Diisopropylphenylamides of potassium and calcium: A primary amido ligand in s-block metal chemistry with an unprecedented catalytic reactivity. Organometallics 2013, 32, 2649–2660. [Google Scholar] [CrossRef]
- Ziemann, S.; Krieck, S.; Görls, H.; Westerhausen, M. 1,2-Bis(anilido)ethane complexes of calcium and potassium: Synthesis, structures, and catalytic activity. Organometallics 2018, 37, 924–933. [Google Scholar] [CrossRef]
- Koradin, C.; Dohle, W.; Rodriguez, A.L.; Schmid, B.; Knochel, P. Synthesis of polyfunctional indoles and related heterocycles mediated by cesium and potassium bases. Tetrahedron 2003, 59, 1571–1587. [Google Scholar] [CrossRef]
- Fiandanese, V.; Bottalico, D.; Marchese, G.; Punzi, A. A rapid synthesis of 2-alkynylindoles and 2-alkynylbenzofurans. Tetrahedron 2008, 64, 7301–7306. [Google Scholar] [CrossRef]
- Inamdar, S.M.; Gonnade, R.G.; Patil, N.T. Synthesis of annulated bis-indoles through Au(I)/Brønsted acid-catalyzed reactions of (1H-indol-3-yl)(aryl)methanols with 2-(arylethynyl)-1H-indoles. Org. Biomol. Chem. 2017, 15, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wei, Y.; Shi, M. Rhodium(III)-catalyzed cross coupling of sulfoxonium ylides and 1,3-diynes to produce naphthol-indole derivatives: An arene ortho C-H activation/annulation cascade. ChemCatChem 2020, 12, 5903–5906. [Google Scholar] [CrossRef]
- Perea-Buceta, J.E.; Wirtanen, T.; Laukkanen, O.-V.; Mäkelä, M.K.; Nieger, M.; Melchionna, M.; Huittinen, N.; Lopez-Sanchez, J.A.; Heleja, J. Cycloisomerization of 2-alkynylanilines to indoles by carbon-supported gold nanoparticles and subsequent homocoupling to 3,3′-biindoles. Angew. Chem. Int. Ed. 2013, 52, 11835–11839. [Google Scholar] [CrossRef]
- Sharp, P.P.; Banwell, M.G.; Renner, J.; Lohmann, K.; Willis, A.C. Consecutive gold(I)-catalyzed cyclization reactions of o-(buta-1,3-diyn-1-yl-)-substituted N-aryl ureas: A one-pot synthesis of pyrimido[1,6-a]indol-1(2H)-ones and related systems. Org. Lett. 2013, 15, 2616–2619. [Google Scholar] [CrossRef]
- Naoe, S.; Saito, T.; Uchiyama, M.; Oishi, S.; Fujii, N.; Ohno, H. Direct construction of fused indoles by gold-catalyzed cascade cyclization of conjugated diynes. Org. Lett. 2015, 17, 1774–1777. [Google Scholar] [CrossRef]
- Märkl, G.; Potthast, R. A simple synthesis of phospholes. Angew. Chem. Int. Ed. 1967, 6, 86. [Google Scholar] [CrossRef]
- Arkhypchuk, A.I.; Orthaber, A.; Ott, S. Tuning the optical properties of 1,1´-biphospholes by chemical alterations of the P-P bridge. Eur. J. Inorg. Chem. 2014, 2014, 1760–1766. [Google Scholar] [CrossRef]
- Ogasawara, M.; Yoshida, K.; Hayashi, T. Synthesis and characterization of a novel chiral phosphole and its derivatives. Organometallics 2001, 20, 1014–1019. [Google Scholar] [CrossRef]
- Ogasawara, M.; Ito, A.; Yoshida, K.; Hayashi, T. Synthesis of 2,5-bis(binaphthyl)phospholes and phosphametallocene derivatives and their application in palladium-catalyzed asymmetric hydrosilylation. Organometallics 2006, 25, 2715–2718. [Google Scholar] [CrossRef]
- Miesel, D.; Hildebrandt, A.; Korb, M.; Low, P.J.; Lang, H. Synthesis and (spectro)electrochemical properties of 2,5-diferrocenyl-1-phenyl-1H-phosphole. Organometallics 2013, 32, 2993–3002. [Google Scholar] [CrossRef] [Green Version]
- Miesel, D.; Hildebrandt, A.; Korb, M.; Wild, D.A.; Low, P.J.; Lang, H. Influence of P-bonded bulky substituents on electronic interactions in ferrocenyl-substituted phospholes. Chem. Eur. J. 2015, 21, 11545–11559. [Google Scholar] [CrossRef]
- Miesel, D.; Korb, M.; Hildebrandt, A.; Lang, H. Synthesis and crystal structure of an acetylenic ferrocenyl substituted phosphaalkene. Inorg. Chim. Acta 2018, 471, 741–745. [Google Scholar] [CrossRef]
- Klintuch, D.; Krekić, K.; Bruhn, C.; Benkő, Z.; Pietschnig, R. A rational synthetic approach to 2,5-diphenyl-β-silyl phospholes. Eur. J. Inorg. Chem. 2016, 2016, 718–725. [Google Scholar] [CrossRef]
- Roesler, F.; Kaban, B.; Klintuch, D.; Ha, U.-M.; Bruhn, C.; Hillmer, H.; Pietschnig, R. Tailoring phospholes for imprint of fluorescent 3D structures. Eur. J. Inorg. Chem. 2019, 2019, 4820–4825. [Google Scholar] [CrossRef] [Green Version]
- Klintuch, D.; Kirchmeier, A.; Bruhn, C.; Pietschnig, R. Synthetic access and luminescence tuning in a seies of β-H and β-silyl substituted phospholes. Dye. Pigm. 2020, 180, 108443. [Google Scholar] [CrossRef]
- Märkl, G.; Hauptmann, H. 1.2.5-Trisubstituierte arsole. Tetrahedron Lett. 1968, 9, 3257–3260. [Google Scholar] [CrossRef]
- Märkl, G.; Hauptmann, H.; Merz, A. Synthese von 1-phenyl-2,5-diaryl(dialkyl)-arsolen; umsetzung der arsole mit alkalimetallen und lithiumorganylen. J. Organomet. Chem. 1983, 249, 335–363. [Google Scholar] [CrossRef]
- Manhart, S.; Schier, A.; Paul, M.; Riede, J.; Schmidbaur, H. New organophosphorus ligands: Cyclopropanation of cumulenes bearing diphenylphosphanyl substituents. Chem. Ber. 1995, 128, 365–371. [Google Scholar] [CrossRef]
- Trifonov, A.A.; Basalov, I.V.; Kisel, A.A. Organolanthanides in catalytic intermolecular hydrophosphination and hydroamination of multiple C-C bonds. Dalton Trans. 2016, 45, 19172–19193. [Google Scholar] [CrossRef]
- Takaki, K.; Takeda, M.; Koshoji, G.; Shishido, T.; Takehira, K. Intermolecular hydrophosphination of alkynes and related carbon-carbon multiple bonds catalyzed by ytterbium-imine complexes. Tetrahedron Lett. 2001, 42, 6357–6360. [Google Scholar] [CrossRef] [Green Version]
- Takaki, K.; Koshoji, G.; Komeyama, K.; Takeda, M.; Shishido, T.; Kitani, A.; Takehira, K. Intermolecular hydrophosphination of alkynes and related carbon-carbon multiple bonds catalyzed by organoytterbiums. J. Org. Chem. 2003, 68, 6554–6565. [Google Scholar] [CrossRef]
- Komeyama, K.; Kobayashi, D.; Yamamoto, Y.; Takehira, K.; Takaki, K. Ytterbium-catalyzed dual intermolecular hydrophosphination: Synthesis of bis(phosphinyl)dienes and bis(alkenyl)phosphine oxides. Tetrahedron 2006, 62, 2511–2519. [Google Scholar] [CrossRef] [Green Version]
- Al-Shboul, T.M.A.; Görls, H.; Westerhausen, M. Calcium-mediated hydrophosphination of diphenylethyne and diphenylbutadiyne as well as crystal structure of 1,4-diphenyl-1,4-bis(diphenylphosphanyl)buta-1,3-diene. Inorg. Chem. Commun. 2008, 11, 1419–1421. [Google Scholar] [CrossRef]
- Al-Shboul, T.M.A.; Pálfi, V.K.; Yu, L.; Kretschmer, R.; Wimmer, K.; Fischer, R.; Görls, H.; Reiher, M.; Westerhausen, M. Catalytic synthesis of vinylphosphanes via calcium-mediated intermolecular hydrophosphanylation of alkynes and butadiynes. J. Organomet. Chem. 2011, 696, 216–227. [Google Scholar] [CrossRef]
- Al-Shboul, T.M.A.; Görls, H.; Krieck, S.; Westerhausen, M. Regiospecific calcium-mediated intermolecular hydrophosphanylation of butadiynes with diphenylphosphane oxide. Eur. J. Inorg. Chem. 2012, 2012, 5451–5455. [Google Scholar] [CrossRef]
- Younis, F.M.; Krieck, S.; Al-Shboul, T.M.A.; Görls, H.; Westerhausen, M. Calcium-mediated synthesis of 1-(diorganylamino)-1,4-diphenyl-4-(diphenylphosphanyl)buta-1,3-dienes. Inorg. Chem. 2016, 55, 4676–4682. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, L.; Labonne, A. Catalytic hydration of alkynes and its application in synthesis. Synthesis 2007, 2007, 1121–1150. [Google Scholar] [CrossRef] [Green Version]
- Turbanova, E.S.; Porfir´eva, Y.I.; Petrov, A.A. Rules in the addition reactions of diacetylenes. VIII. Orientation of hydration of disubstituted diacetylenes. Russ. J. Gen. Chem. 1966, 2, 772–777. [Google Scholar]
- Sokolov, L.B.; Petrov, A.A. Rules in the addition reactions of diacetylenes. X. Orientation of hydration of conjugated monoalkyldiacetylenes. Russ. J. Gen. Chem. 1966, 2, 1003–1005. [Google Scholar]
- Constantino, M.G.; Donate, P.M.; Petragnani, N. Hydration of diacetylene compounds. Synthesis of a merine natural product: (±)-1-(2,6,6-trimethyl-4-hydroxycyclohexenyl)-1,3-butanedione. J. Org. Chem. 1986, 51, 387–390. [Google Scholar] [CrossRef]
- Shim, S.C.; Lee, T.S. Photohydration reaction of 1-(1-naphthyl)buta-1,3-diynes. J. Chem. Soc. Perkin Trans. 1990, 2, 1739–1743. [Google Scholar] [CrossRef]
- Baek, E.K.; Shim, S.C. Photohydration reaction of 1-(p-nitrophenyl)-5,5-dimethyl-1,3-hexadiyne. J. Phys. Org. Chem. 1995, 8, 699–705. [Google Scholar] [CrossRef]
- Baek, E.K.; Lee, S.T.; Chae, Y.S.; Shim, S.C. The photohydration of 1-(nitrophenyl)-5,5-dimethyl-1,3-hexadiynes: The nitro substituent effect on the excited states of diacetylenes. J. Photosci. 1995, 2, 73–76. [Google Scholar]
- Shim, S.C.; Chae, Y.S.; Baek, E.K.; Park, S.K. Substituent effects on the photohydration of 1-aryl-5,5-diemthyl-1,3-hexadiynes. J. Photochem. Photobiol. A 1997, 106, 155–160. [Google Scholar] [CrossRef]
- Jiang, H.; Zeng, W.; Li, Y.; Wu, W.; Huang, L.; Fu, W. Copper(I)-catalyzed synthesis of 2,5-disubstituted furans and thiophenes from haloalkynes or 1,3-diynes. J. Org. Chem. 2012, 77, 5179–5183. [Google Scholar] [CrossRef]
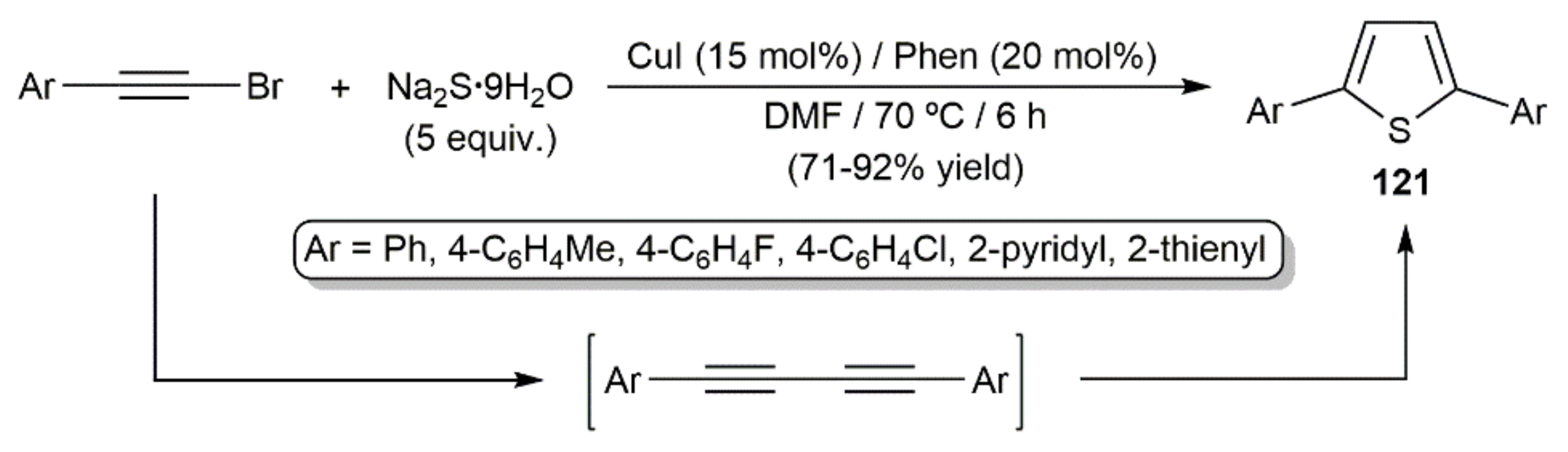
- Irudayanathan, F.M.; Raja, G.C.E.; Lee, S. Copper-catalyzed direct synthesis of furans and thiophenes via decarboxylative coupling of alkynyl carboxylic acids with H2O or Na2S. Tetrahedron 2015, 71, 4418–4425. [Google Scholar] [CrossRef]
- Zheng, Q.; Hua, R.; Yin, T. Palladium-catalyzed cycloaddition of 1,3-butadiynes with water: An alternative efficient catalytic system for atom-economic synthesis of 2,5-disubstituted furans. Curr. Org. Synth. 2013, 10, 161–164. [Google Scholar]
- Pérez, J.M.; Cano, R.; Yus, M.; Ramón, D.J. Copper-impregnated magnetite as a heterogeneous catalyst for the homocoupling of terminal alkynes. Synthesis 2013, 45, 1373–1379. [Google Scholar]
- Klukas, F.; Grunwald, A.; Menschel, F.; Müller, T.J.J. Rapid pseudo five-component synthesis of intensively blue luminescent 2,5-di(hetero)arylfurans via a Sonogashira-Glaser cyclization sequence. Beilstein J. Org. Chem. 2014, 10, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Guo, P. Gold-catalyzed formation of C-O and C-C bonds: An efficient domino reaction synthesis of functionalized furans. Catal. Commun. 2015, 68, 58–60. [Google Scholar] [CrossRef]
- Camacho, D.H.; Saito, S.; Yamamoto, Y. “Anti-Wacker”-type hydroalkoxylation of diynes catalyzed by palladium(0). Tetrahedron Lett. 2002, 43, 1085–1088. [Google Scholar] [CrossRef]
- Lee, T.S.; Shim, S.C.; Kim, S.S. Photoreaction of 1,4-disubstituted-1,3-butadiyne with alcohols. Bull. Korean Chem. Soc. 1986, 7, 116–120. [Google Scholar]
- Schermann, G.; Vostrowsky, O.; Hirsch, A. Addition chemistry of rod-shaped 1,6-dicyanohexatriyne: Regioselectivity control by the remote cyano function. Eur. J. Org. Chem. 1999, 1999, 2491–2500. [Google Scholar] [CrossRef]
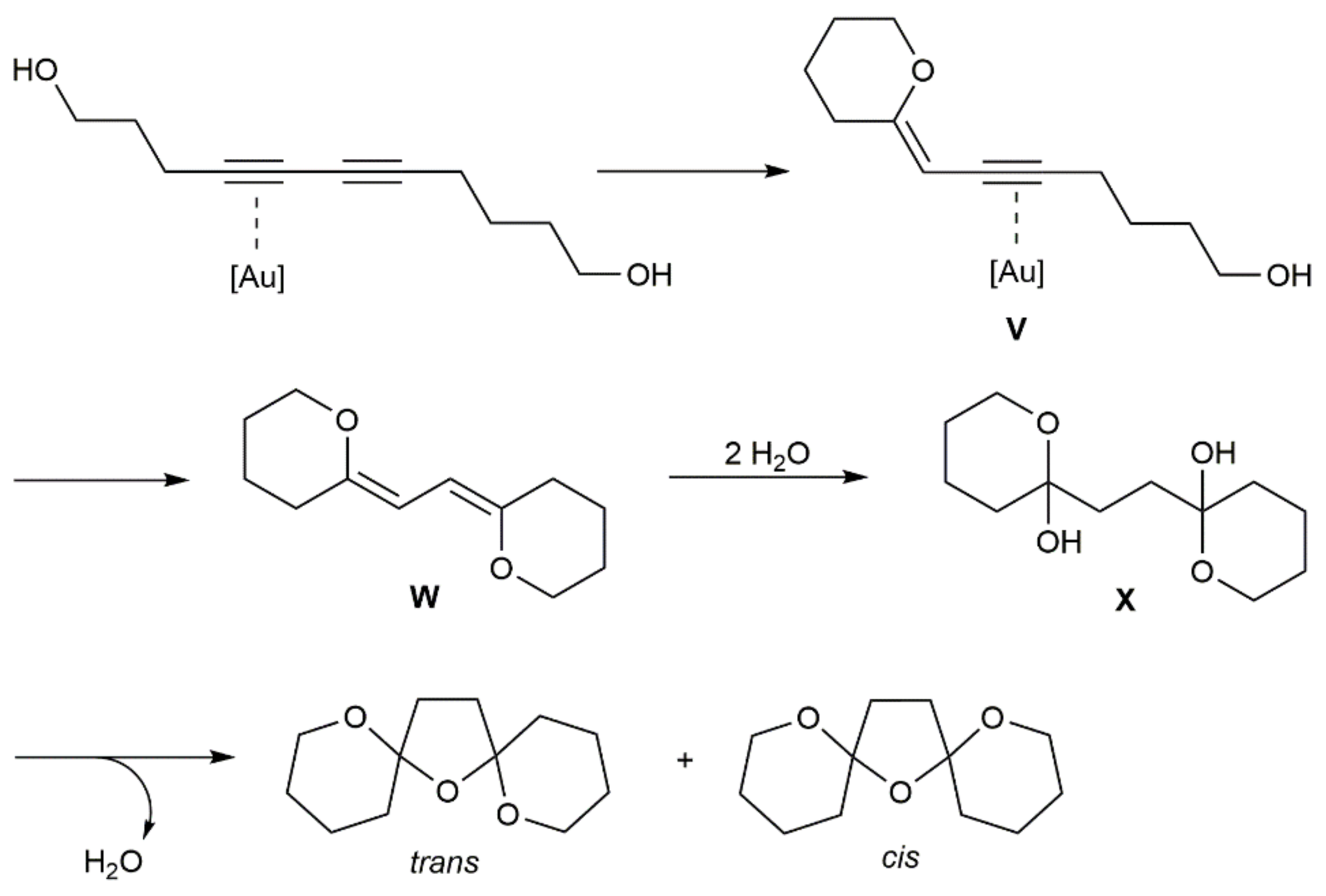
- Volchkov, I.; Sharma, K.; Cho, E.J.; Lee, D. Efficient one-step synthesis of bis-spiroketals from diynediols by π-Lewis acid-catalyzed hydroalkoxylation/hydration. Chem. Asian J. 2011, 6, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Unraveling the mechanism of tricyclic bis-spiroketal formation from diyne diol by DFT study. Lett. Org. Chem. 2019, 16, 392–395. [Google Scholar] [CrossRef]
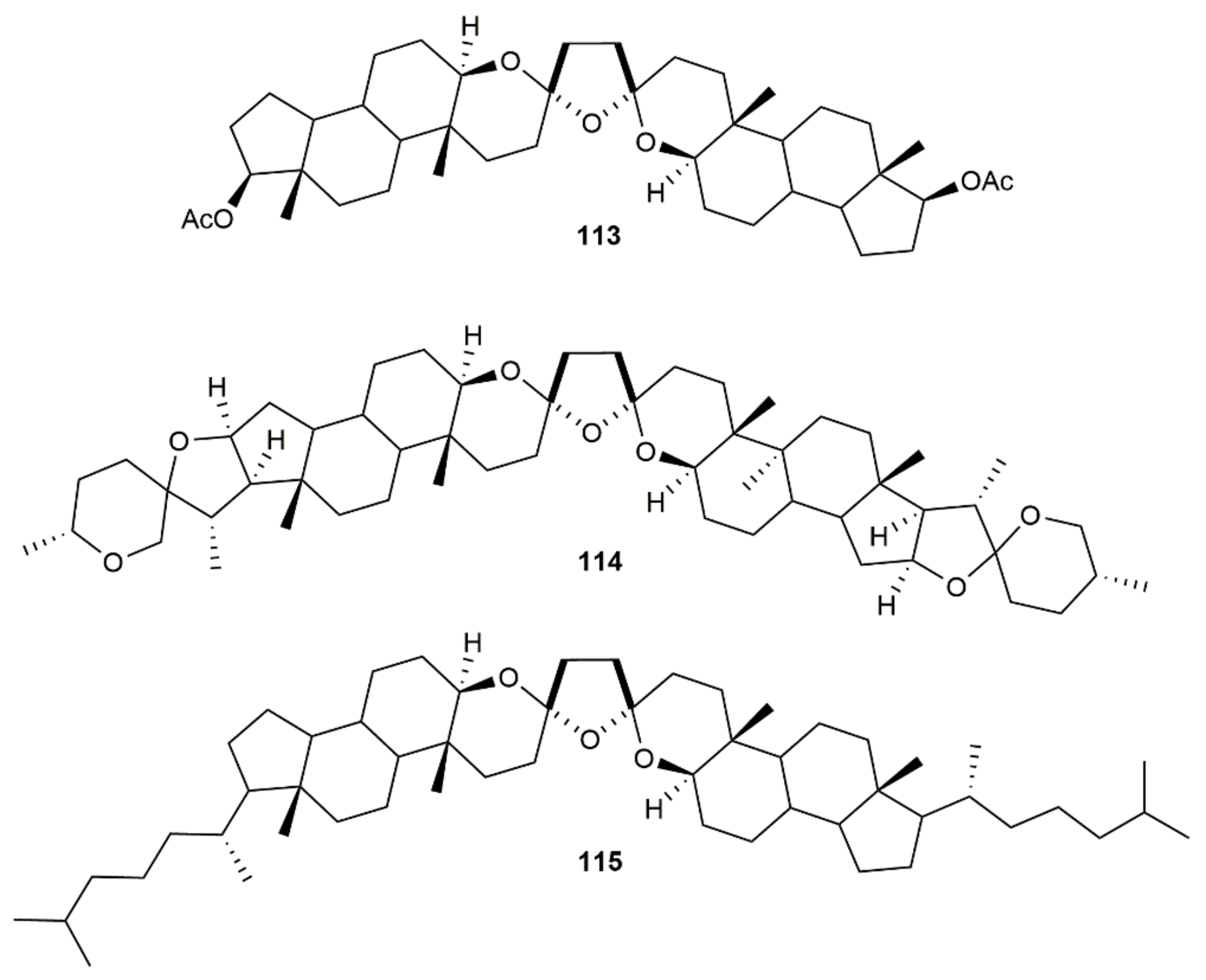
- Valdez-García, R.M.; Alarcón-Manjarrez, C.; Galano, A.; Rodríguez-Molina, B.; Flores-Álamo, M.; Iglesias-Arteaga, M.A. Synthesis of dimeric steroid trioxabispiroacetals scaffolds by gold(I)-catalyzed hydroalkoxylation-hydration of diynediols. Eur. J. Org. Chem. 2019, 2019, 4916–4927. [Google Scholar] [CrossRef]
- Jones, E.R.H.; Stephenson, J.S. Chemistry of the higher fungi. Part IX. Polyacetylenic metabolites from Coprinus quadrifidus. J. Chem. Soc. 1959, 2197–2203. [Google Scholar] [CrossRef]
- Bohlmann, F.; Herbst, P.; Gleinig, H. Synthese von natürlich vorkommenden polyacetylenverbindungen mit endständigen dreifachbindungen. Chem. Ber. 1961, 94, 948–957. [Google Scholar] [CrossRef]
- Jones, E.R.H.; Stephenson, J.S.; Turner, W.B.; Whiting, M.C. Chemistry of the higher fungi. Part XIII. Synthesis of (a) a C9 triacetylenic epoxy-alcohol, a Coprinus quadrifidus metabolite and (b) a C9 triacetylenic 1,2-diol. The structure of biformyne 1. J. Chem. Soc. 1963, 2048–2055. [Google Scholar] [CrossRef]
- Cambie, R.C.; Hirschberg, A.; Jones, E.R.H.; Lowe, G. Chemistry of the higher fungi. Part XVI. Polyacetylenic metabolites from Aleurodiscus roseus. J. Chem. Soc. 1963, 4120–4130. [Google Scholar] [CrossRef]
- Jones, E.R.H.; Lowe, G.; Shannon, P.V.R. Natural acetylenes. Part XX. Tetra-acetylenic and other metabolites from Fistulina hepatica (Huds) Fr. J. Chem. Soc. C 1966, 139–144. [Google Scholar] [CrossRef]
- Miao, Z.; Xu, M.; Hoffmann, B.; Bernet, B.; Vasella, A. Functionalised bicyclic exo-glycals by alkynol cycloisomerisation of hydroxy 1,3-diynes and hydroxy haloalkynes. Helv. Chim. Acta 2005, 88, 1885–1912. [Google Scholar] [CrossRef]
- Xu, M.; Miao, Z.; Bernet, B.; Vasella, A. Functionalised monocyclic five- to seven-membered exo-glycals by alkynol cycloisomerisation of hydroxyl buta-1,3-diynes and 1-haloalkynes. Helv. Chim. Acta 2005, 88, 2918–2937. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, H.; Zhu, L.-L.; Li, X.X.; Chen, Z. Base-catalyzed cascade 1,3-H shift/cyclization reaction to construct polyaromatic furans. Adv. Synth. Catal. 2011, 353, 707–712. [Google Scholar] [CrossRef]
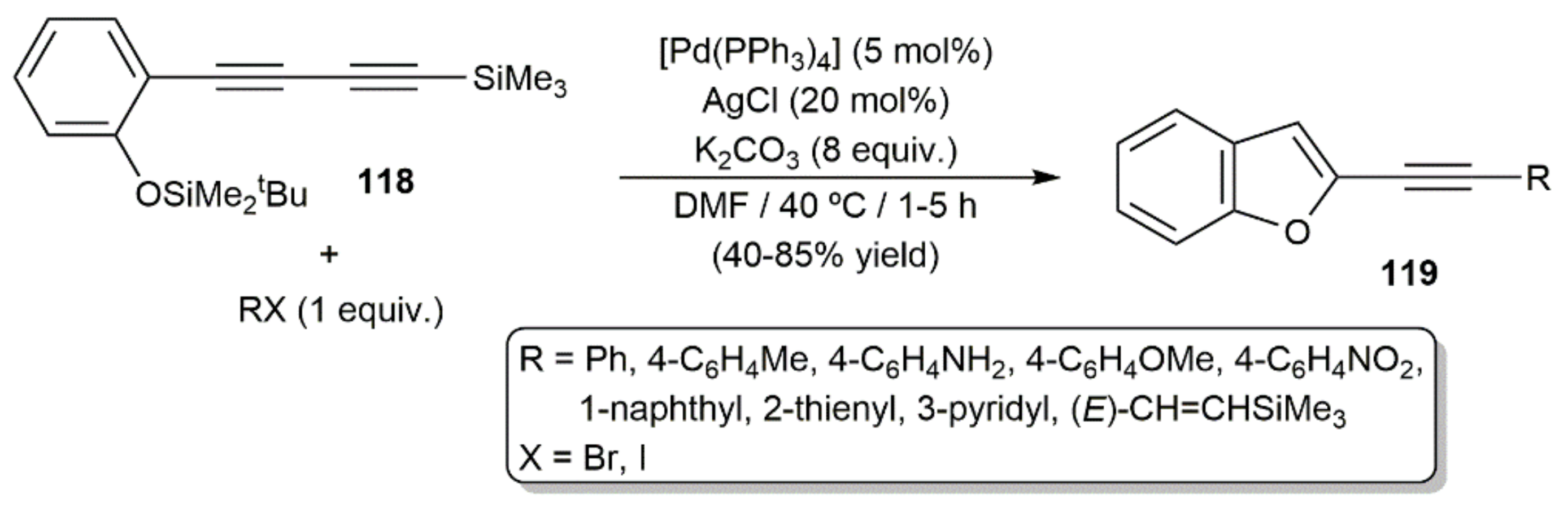
- Fiandanese, V.; Bottalico, D.; Marchese, G.; Punzi, A. A straightforward synthesis of indole and benzofuran derivatives. Tetrahedron 2008, 64, 53–60. [Google Scholar] [CrossRef]
- Gooβen, L.J.; Rodríguez, N.; Gooβen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar]
- Francos, J.; Cadierno, V. Metal-catalyzed intra- and intermolecular addition of carboxylic acids to alkynes in aqueous media: A review. Catalysts 2017, 7, 328. [Google Scholar] [CrossRef] [Green Version]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. The intermolecular hydro-oxycarbonylation of internal alkynes: Current state of the art. Arkivoc 2018, 2, 17–39. [Google Scholar] [CrossRef] [Green Version]
- Cadierno, V. Recent advances in the transition metal-catalyzed addition of carboxylic acids to alkynes. Curr. Org. Chem. 2021, 25, 2260–2303. [Google Scholar] [CrossRef]
- Cadierno, V. Gold-catalyzed addition of carboxylic acids to alkynes and allenes: Valuable tools for organic synthesis. Catalysts 2020, 10, 1206. [Google Scholar] [CrossRef]
- León, F.; Francos, J.; López-Serrano, J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Double asymmetric hydrogenation of conjugated dienes: A self-breeding chirality route for C2 symmetric 1,4-diols. Chem. Commun. 2019, 55, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.E.; Reish, J.; Hörner, L. Thiophen-derivate aus polyacetylenen. Angew. Chem. 1960, 72, 920. [Google Scholar] [CrossRef]
- Tang, J.; Zhao, X. Synthesis of 2,5-disubstituted thiophenes via metal-free sulfur heterocyclization of 1,3-diynes with sodium hydrosulfide. RSC Adv. 2012, 2, 5488–5490. [Google Scholar] [CrossRef]
- Zhang, G.; Yi, H.; Chen, H.; Bian, C.; Liu, C.; Lei, A. Trisulfur radical anion as the key intermediate for the synthesis of thiophene via the interaction between elemental sulfur and NaOtBu. Org. Lett. 2014, 16, 6156–6159. [Google Scholar] [CrossRef]
- Rao, M.L.N.; Islam, S.K.; Dasgupta, P. Rapid access to unsymmetrical 1,3-diynes and 2,5-disubstituted thiophenes under ligand and Pd/Ni-free Cu-catalysis. RSC Adv. 2015, 5, 78090–78098. [Google Scholar] [CrossRef]
- Talbi, I.; Alayrac, C.; Lohier, J.-F.; Touil, S.; Witulski, B. Application of ynamides in the synthesis of 2-(tosylamido)- and 2,5-bis(tosylamido)thiophenes. Org. Lett. 2016, 18, 2656–2659. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.B.; Carvalho, D.B.; Trfzger, O.S.; Kassab, N.M.; Guerrero, P.G., Jr.; Barbosa, S.L.; Shiguemoto, C.Y.K.; Baroni, A.C.M. One-pot synthesis of unsymmetrical 1,3-butadiyne derivatives and their application in the synthesis of unsymmetrical 2,5-diarylthiophenes. Eur. J. Org. Chem. 2019, 2019, 696–704. [Google Scholar] [CrossRef]
- Curtis, R.F.; Hasnain, S.N.; Taylor, J.A. Selenophens from diacetylenes. Chem. Commun. 1968, 365a. [Google Scholar] [CrossRef]
- Barancelli, D.A.; Acker, C.I.; Menezes, P.H.; Zeni, G. Selective base-promoted synthesis of substituted selenophenes by carbocyclization of (Z)-benzylselenoenynes. Org. Biomol. Chem. 2011, 9, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Mack, W. Synthesis of tellurophene and its 2,5-disubstituted derivatives. Angew. Chem. Int. Ed. 1966, 5, 896. [Google Scholar] [CrossRef]
- Ulman, A.; Manassen, J.; Frolow, F.; Rabinovich, D. Synthesis of new tetraphenylporphyrin molecules containing heteroatoms. III. Tetraphenyl-21-tellura-23-thiaprophyrin: An internally-bridged porphyrin. Tetrahedron Lett. 1978, 19, 1885–1886. [Google Scholar] [CrossRef]
- McCormick, T.M.; Jahnke, A.A.; Lough, A.J.; Seferos, D.S. Tellurophenes with delocalized π-systems and their extended valence adducts. J. Am. Chem. Soc. 2012, 134, 3542–3548. [Google Scholar] [CrossRef] [PubMed]
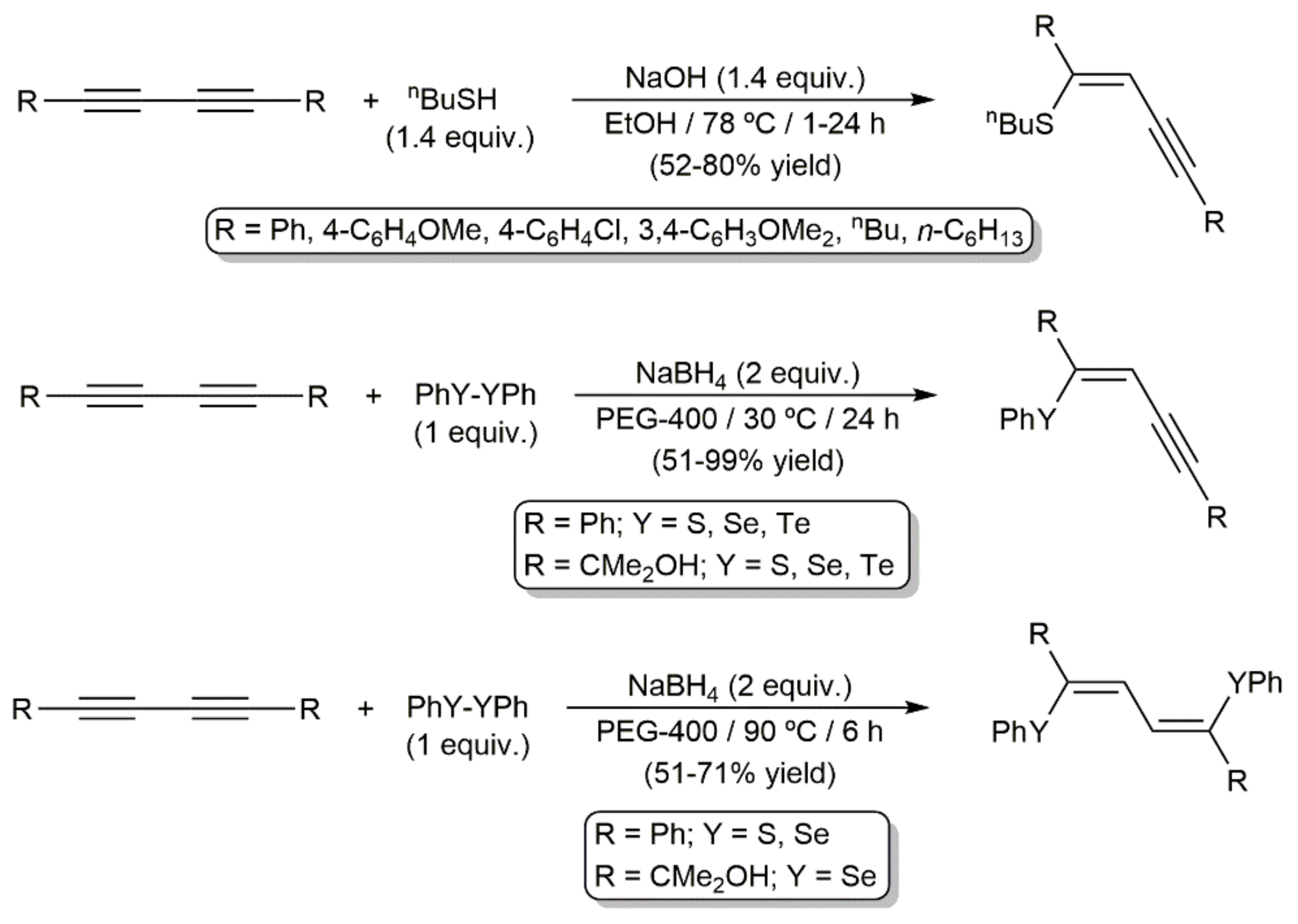
- Alves, D.; Sachini, M.; Jacob, R.G.; Lenardão, E.J.; Contreira, M.E.; Savegnano, L.; Perin, G. Synthesis of (Z)-organylthioenynes using KF/Al2O3/solvent as recyclable system. Tetrahedron Lett. 2011, 52, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Santana, A.S.; Carvalho, D.B.; Casemiro, N.S.; Hurtado, G.R.; Viana, L.H.; Kassab, N.M.; Barbosa, S.L.; Marques, F.A.; Guerrero, P.G., Jr.; Baroni, A.C.M. Improvement in the synthesis of (Z)-organylthioenynes via hydrothiolation of buta-1,3-diynes: A comparative study using NaOH or TBAOH as base. Tetrahedron Lett. 2012, 53, 5733–5738. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, J.; Li, H.; Sun, Q.; Xiong, L.; Yin, G. Highly regio- and stereoselective synthesis of bis-sulfanyl substituted conjugated dienes by copper-palladium cooperative catalysis. Org. Chem. Front. 2021, 8, 628–634. [Google Scholar] [CrossRef]
- Dabdoub, M.J.; Baroni, A.C.M.; Lenardão, E.J.; Gianeti, T.R.; Hurtado, G.R. Synthesis of (Z)-1-phenylseleno-1,4-diorganyl-1-buten-3-ynes: Hydroselenation of symmetrical and unsymmetrical 1,4-diorganyl-1,3-butadiynes. Tetrahedron 2001, 57, 4271–4276. [Google Scholar] [CrossRef]
- Alves, D.; Luchese, C.; Nogueira, C.W.; Zeni, G. Electrophilic cyclization of (Z)-selenoenynes: Synthesis and reactivity of 3-iodoselenophenes. J. Org. Chem. 2007, 72, 6726–6734. [Google Scholar] [CrossRef]
- Dabdoub, M.J.; Dabdoub, V.B.; Lenardão, E.J.; Hurtado, G.R.; Barbosa, S.L.; Guerrero, P.G., Jr.; Nazário, C.E.D.; Viana, L.H.; Santana, A.S.; Baroni, A.C.M. Synthesis of (Z)-1-organylthiobut-1-en-3-ynes: Hydrothiolation of symmetrical and unsymmetrical buta-1,3-diynes. Synlett 2009, 986–990. [Google Scholar] [CrossRef]
- Ishii, A.; Annaka, T.; Nakata, N. Convenient synthesis and photophysical properties of 1-thio- and 1-seleno-1,3-butadiene fluorophores in rigid dibenzobarrelene and benzobarrelene skeletons. Chem. Eur. J. 2012, 18, 6428–6432. [Google Scholar] [CrossRef] [PubMed]
- Venkateswarlu, C.; Chandrasekaran, S. Hydrochalcogenation of symmetrical and unsymmetrical buta-1,3-diynes with diaryl dichalcogenides: Facile entry to (Z)-1-(organylchalcogeno)but-1-en-3-yne derivatives. Synthesis 2015, 47, 395–410. [Google Scholar] [CrossRef]
- Lopes, E.F.; Gonçalves, L.C.; Vinueza, J.C.G.; Jacob, R.G.; Perin, G.; Santi, C.; Lenardão, E.J. DES as a green solvent to prepare 1,2-bis-organylseleno alkenes. Scope and limitations. Tetrahedron Lett. 2015, 56, 6890–6895. [Google Scholar] [CrossRef]
- Lara, R.G.; Soares, L.K.; Jacob, R.G.; Schumacher, R.F.; Perin, G. Selective synthesis of (Z)-chalcogenoenynes and (Z,Z)-1,4-bis-chalcogenbuta-1,3-dienes using PEG-400. J. Braz. Chem. Soc. 2016, 27, 2046–2054. [Google Scholar] [CrossRef]
- Profir´eva, Y.I.; Vasil´eva, L.A.; Turbanova, E.S.; Petrov, A.A. Electrophilic and radical addition of hydrogen halides to diacetylene and its homologs. Russ. J. Org. Chem. 1969, 5, 591–600. [Google Scholar]
- Radchenko, S.I. Reaction of alkylthioalkyldiacetylenes with hydrogen halides and alkyllithiums. Russ. J. Org. Chem. 1976, 12, 229–230. [Google Scholar]
- Radchenko, S.I.; Komarov, V.Y.; Ionin, B.I. Regioselectivity of addition of electrophilic reagents to conjugated acetylenic sulfides and selenides. Russ. J. Org. Chem. 1985, 21, 244–249. [Google Scholar]
- Ide, M.; Ohashi, K.; Mihara, S.; Iwasawa, T. Regio- and stereoselective hydrohalogenation of ynamide components in 1,3-butadiynes with in situ generated HX. Tetrahedron Lett. 2014, 55, 2130–2133. [Google Scholar] [CrossRef]








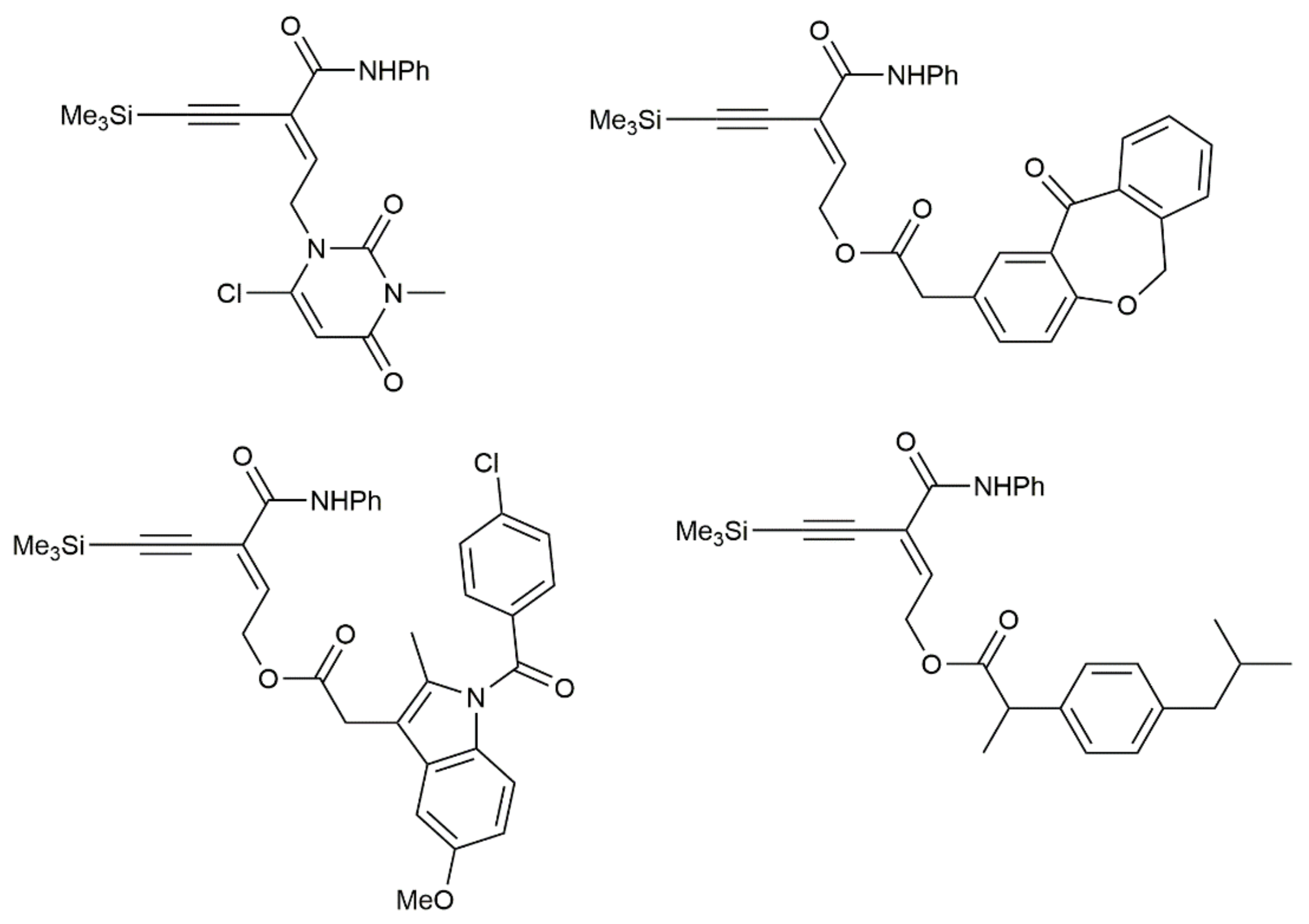

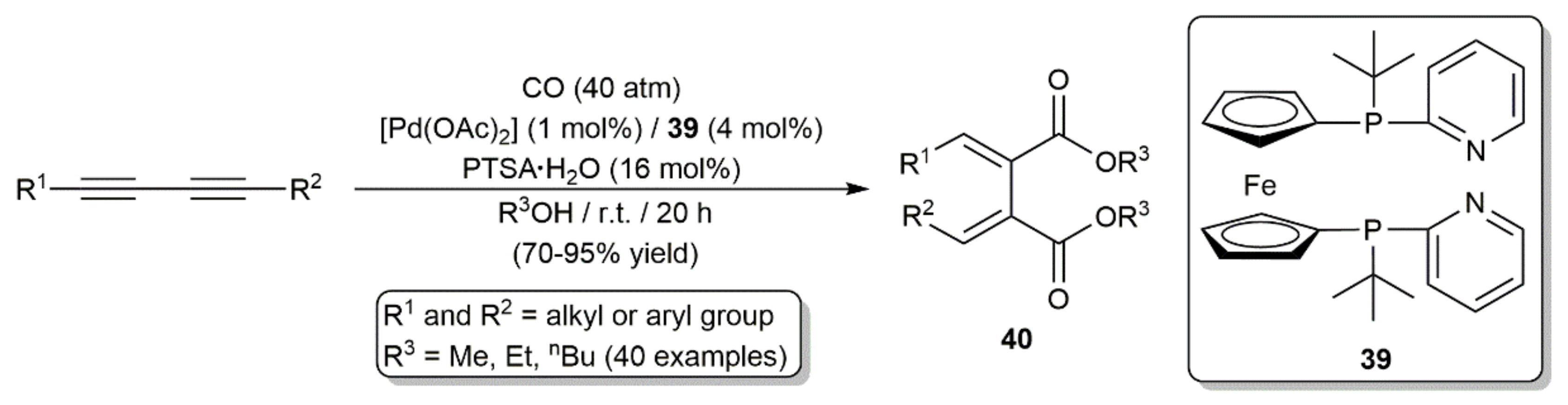
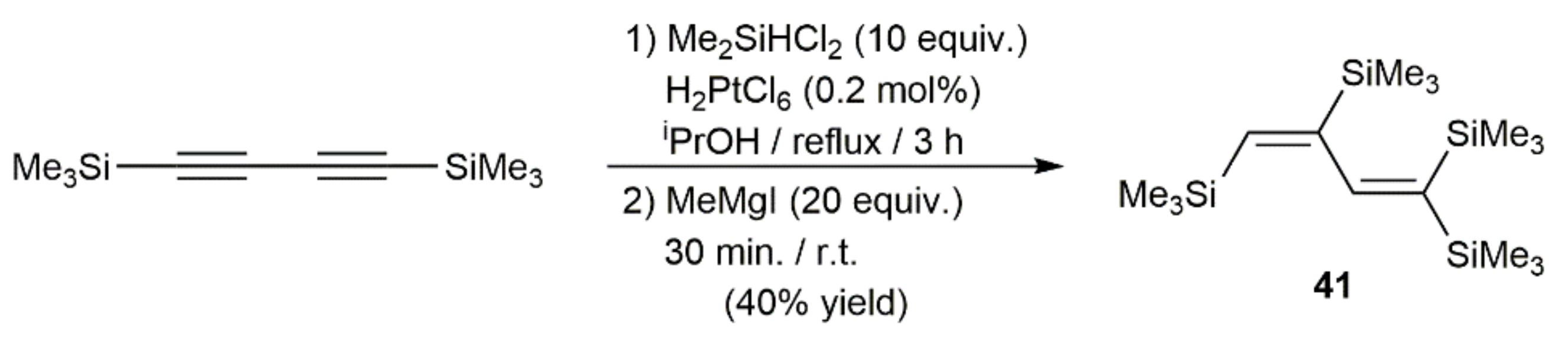
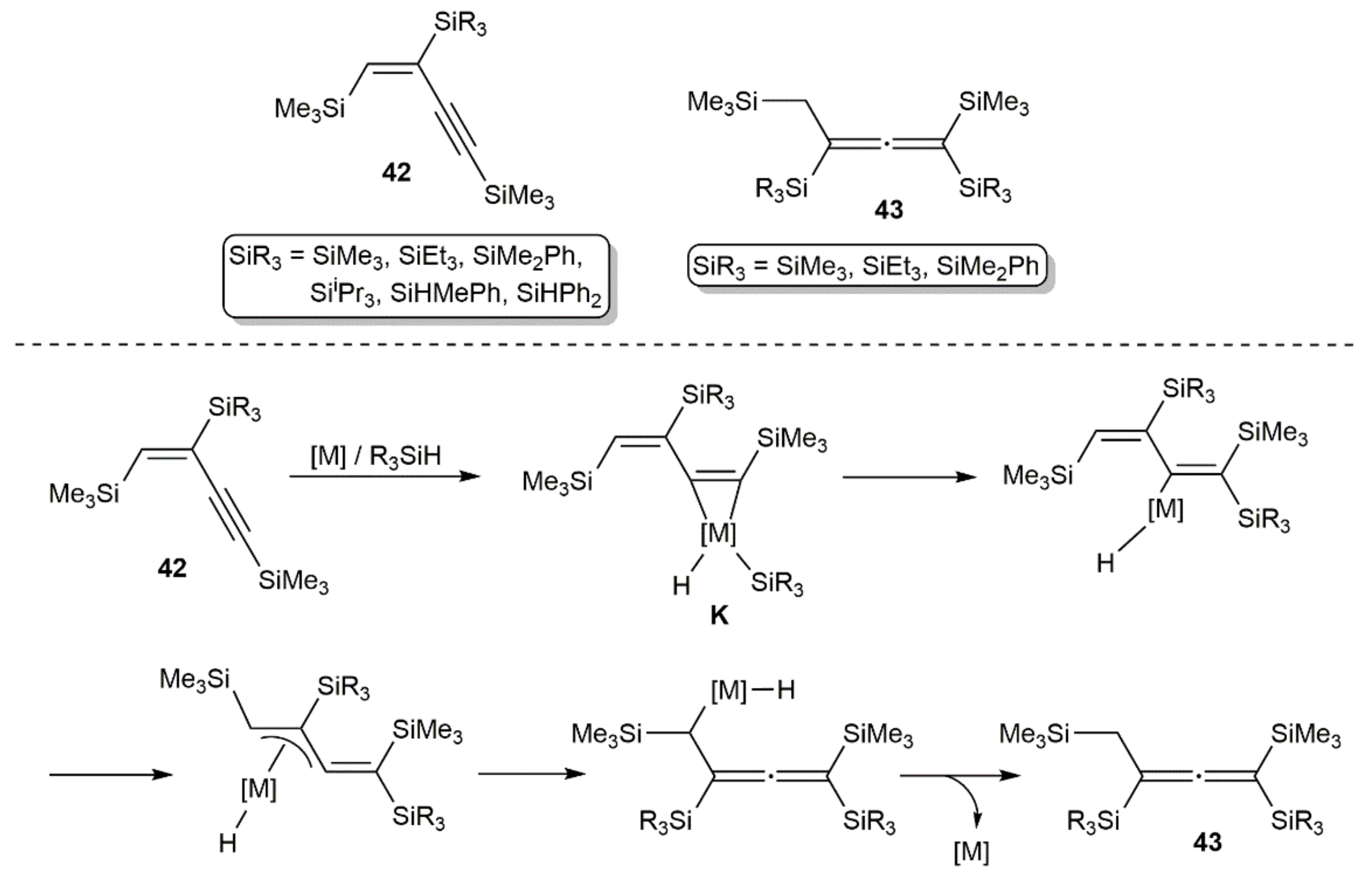

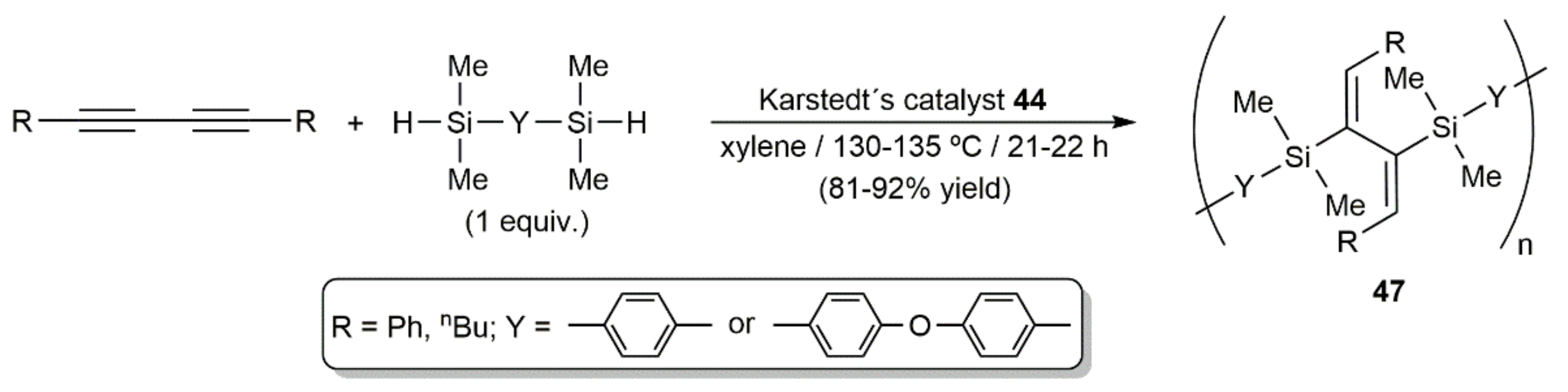
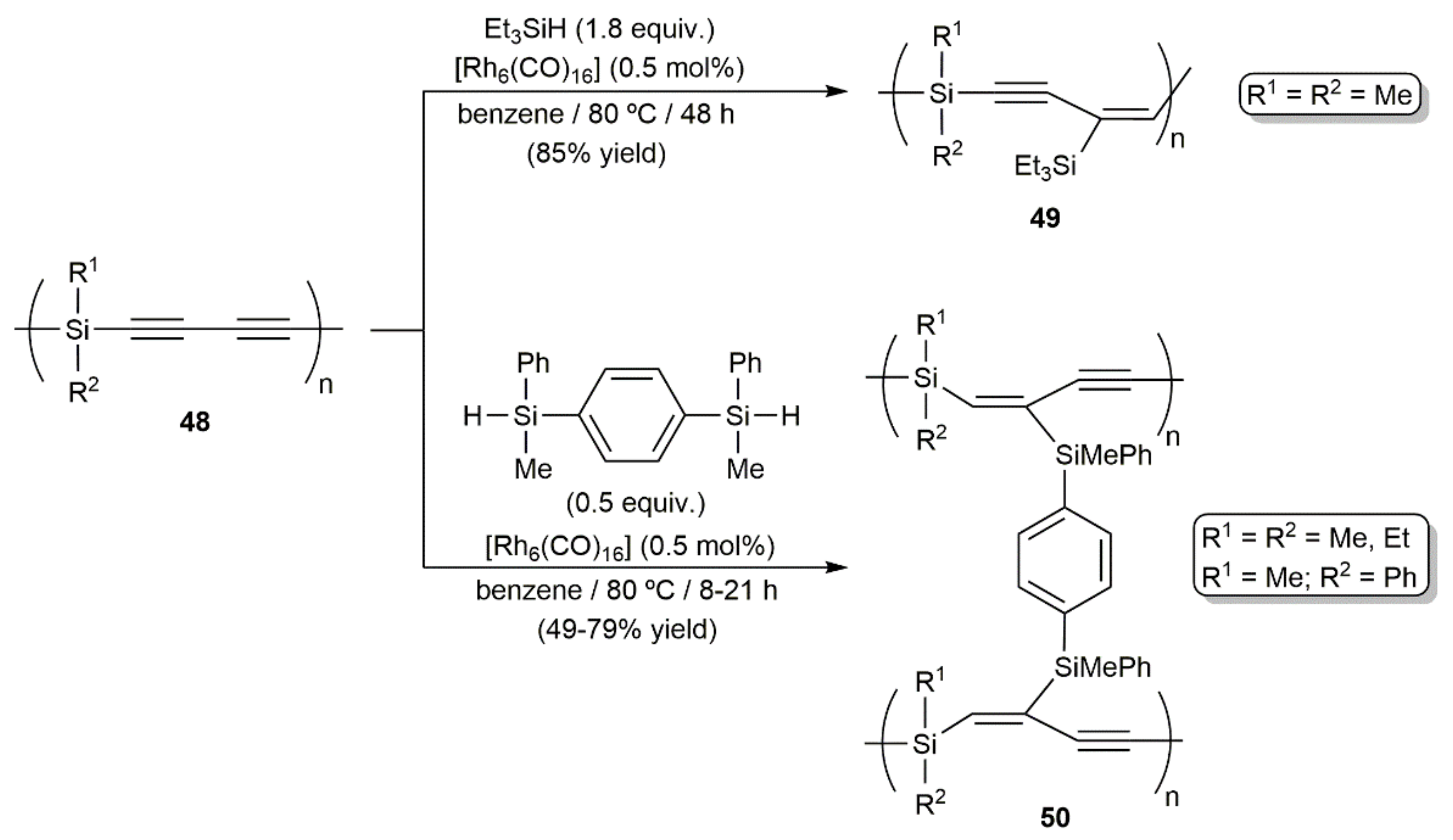
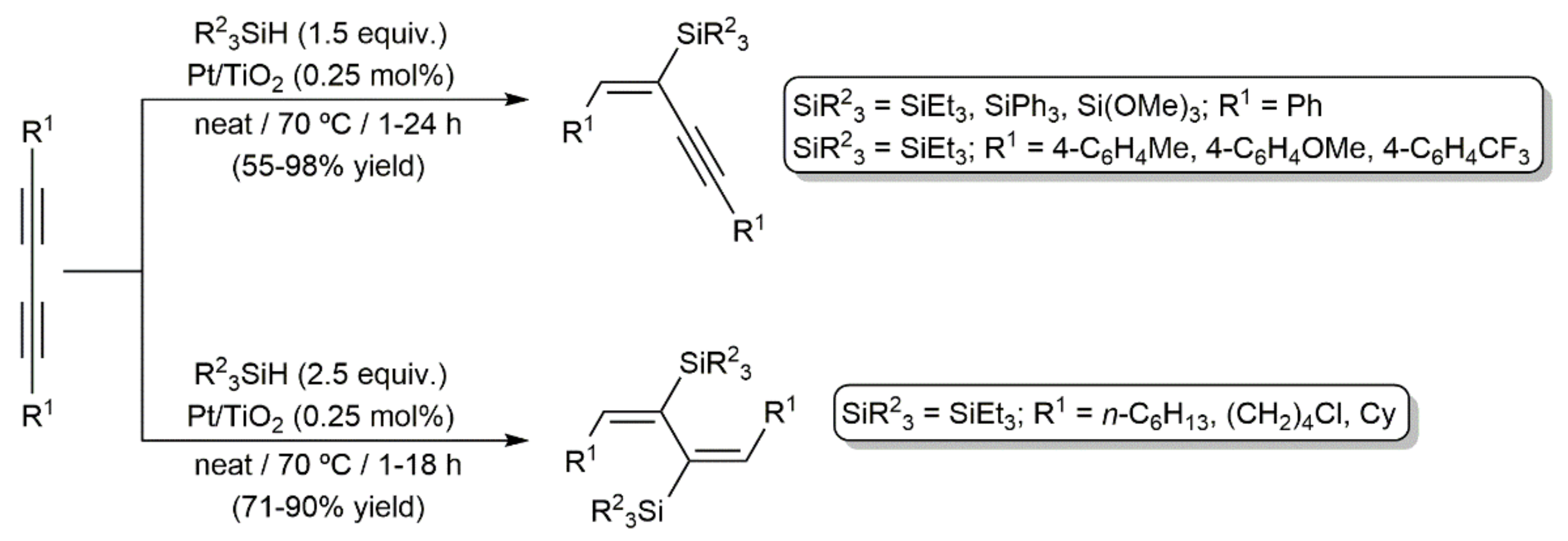
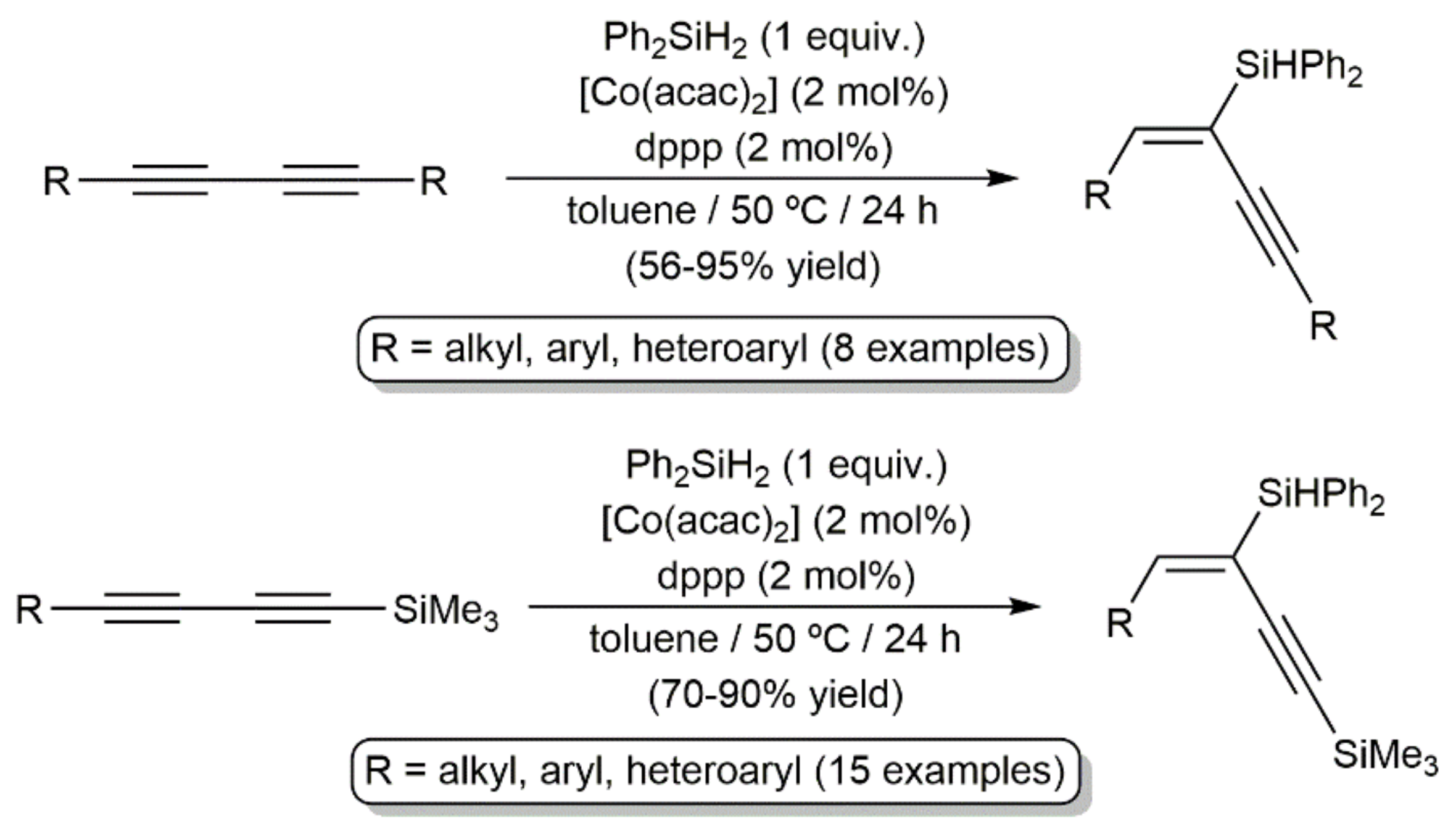












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadierno, V. Catalytic Hydrofunctionalization Reactions of 1,3-Diynes. Catalysts 2022, 12, 89. https://doi.org/10.3390/catal12010089
Cadierno V. Catalytic Hydrofunctionalization Reactions of 1,3-Diynes. Catalysts. 2022; 12(1):89. https://doi.org/10.3390/catal12010089
Chicago/Turabian StyleCadierno, Victorio. 2022. "Catalytic Hydrofunctionalization Reactions of 1,3-Diynes" Catalysts 12, no. 1: 89. https://doi.org/10.3390/catal12010089
APA StyleCadierno, V. (2022). Catalytic Hydrofunctionalization Reactions of 1,3-Diynes. Catalysts, 12(1), 89. https://doi.org/10.3390/catal12010089