
Sustainable Electrochemical NO Capture and Storage System Based on the Reversible Fe2+/Fe3+-EDTA Redox Reaction

 ,
,  ,
, 
Abstract
1. Introduction
2. Experiment
2.1. Materials
2.2. Preparation of Fe-Complex Solutions
2.3. Characterizations
2.4. Electrochemical Measurement
3. Result and Discussions
3.1. Electrochemical Redox Property of Fe-EDTA Solution
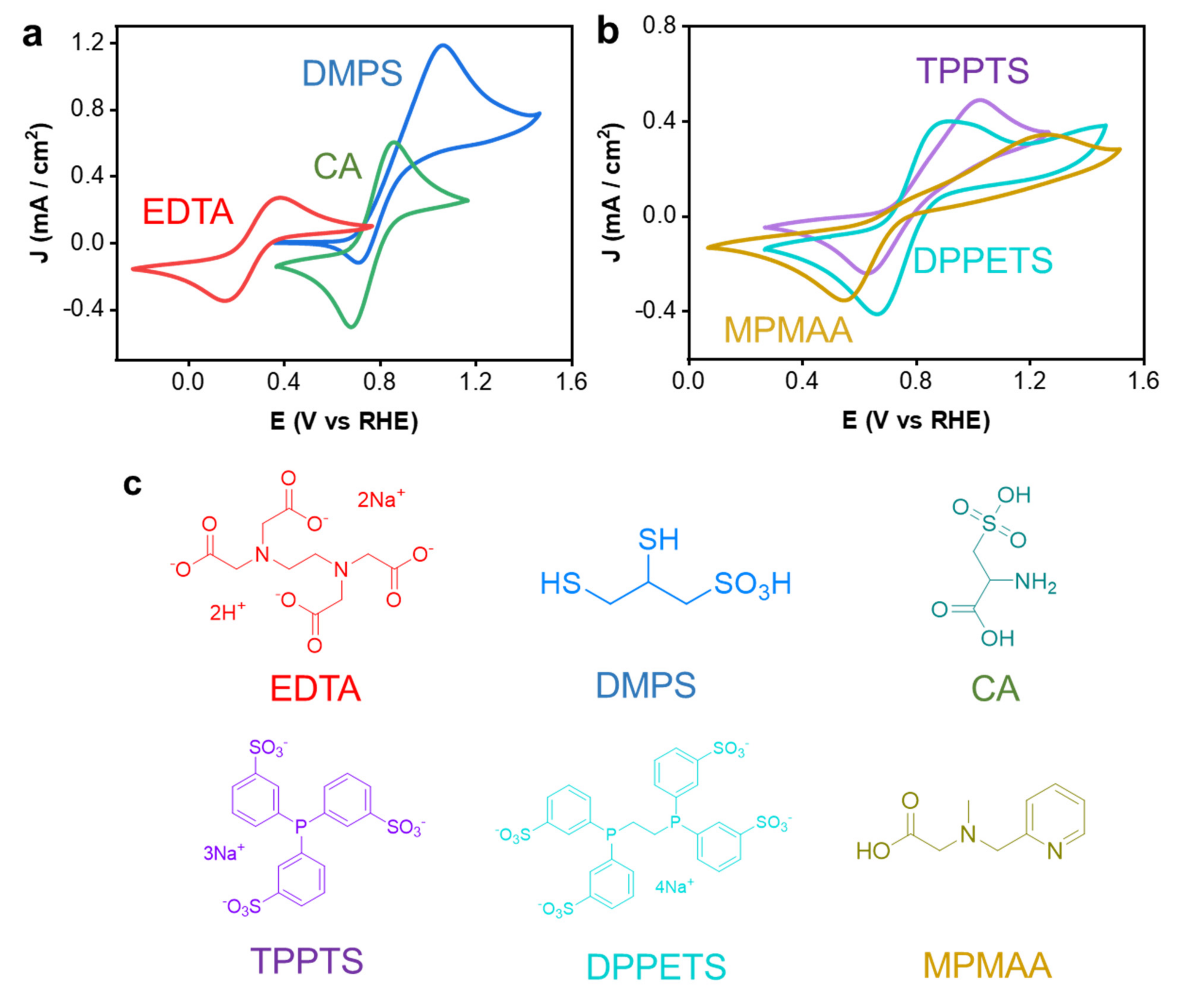
3.2. NO Absorption Capacity and Electrochemical Redox Property of Diverse Fe-Complexes
3.3. Fe-EDTA Conversion Using a Half-Cell Reaction System
3.4. NO Absorption and Emission Using Fe-EDTA Solution in a Full-Cell System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sun, Y.; Zwolińska, E.; Chmielewski, A.G. Abatement technologies for high concentrations of NOx and SO2 removal from exhaust gases: A review. Crit. Rev. Environ. Sci. Technol. 2015, 46, 119–142. [Google Scholar] [CrossRef]
- Long, X.-L.; Xin, Z.-L.; Chen, M.-B.; Li, W.; Xiao, W.-D.; Yuan, W.-K. Kinetics for the simultaneous removal of NO and SO2 with cobalt ethylenediamine solution. Sep. Purif. Technol. 2008, 58, 328–334. [Google Scholar] [CrossRef]
- Chock, D.P.; Dunker, A.M.; Kumar, S.; Sloane, C.S. Effect of nitrogen oxides (NOx) emission rates on smog formation in the California South Coast Air Basin. Environ. Sci. Technol. 1981, 15, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Portmann, R.W.; Garcia, R.R.; Thomason, L.W.; Poole, L.R.; McCormick, M.P. The role of aerosol variations in anthropogenic ozone depletion at northern midlatitudes. J. Geophys. Res. Atmos. 1996, 101, 6713–6727. [Google Scholar] [CrossRef]
- Su, I.H.; Wu, J.C.S. Photo selective catalytic reduction of nitric oxide with propane at room temperature. Catal. Commun. 2009, 10, 1534–1537. [Google Scholar] [CrossRef]
- Zeng, Z.; Lu, P.; Li, C.; Mai, L.; Li, Z.; Zhang, Y. Removal of NO by carbonaceous materials at room temperature: A review. Catal. Sci. Technol. 2012, 2, 2188–2199. [Google Scholar] [CrossRef]
- Guo, L.; Shu, Y.; Gao, J. Present and Future Development of Flue Gas Control Technology of DeNO_X in the World. Energy Procedia 2012, 17, 397–403. [Google Scholar] [CrossRef]
- Bae, S.W.; Roh, S.A.; Kim, S.D. NO removal by reducing agents and additives in the selective non-catalytic reduction (SNCR) process. Chemosphere 2006, 65, 170–175. [Google Scholar] [CrossRef]
- Yang, S.; Fu, Y.; Liao, Y.; Xiong, S.; Qu, Z.; Yan, N.; Li, J. Competition of selective catalytic reduction and non selective catalytic reduction over MnOx/TiO2 for NO removal: The relationship between gaseous NO concentration and N2O selectivity. Catal. Sci. Technol. 2014, 4, 224–232. [Google Scholar] [CrossRef]
- Choi, S.-W.; Choi, S.-K.; Bae, H.-K. Hybrid selective noncatalytic reduction (SNCR)/selective catalytic reduction (SCR) for NOx removal using low-temperature SCR with Mn-V2O5/TiO2 catalyst. J. Air Waste Manag. Assoc. 2015, 65, 485–491. [Google Scholar] [CrossRef]
- Pham, E.K.; Chang, S.-G. Removal of NO from flue gases by absorption to an iron(ii) thiochelate complex and subsequent reduction to ammonia. Nature 1994, 369, 139–141. [Google Scholar] [CrossRef]
- Juzeliúnas, E.; Jüttner, K. Electrochemical Study of NO Conversion from Fe(II)-EDTA-NO Complex on Pt Electrodes. J. Electrochem. Soc. 1998, 145, 53–58. [Google Scholar] [CrossRef]
- Demmink, J.F.; van Gils, I.C.F.; Beenackers, A.A.C.M. Absorption of Nitric Oxide into Aqueous Solutions of Ferrous Chelates Accompanied by Instantaneous Reaction. Ind. Eng. Chem. Res. 1997, 36, 4914–4927. [Google Scholar] [CrossRef]
- Mendelsohn, M.H.; Harkness, J.B.L. Enhanced flue-gas denitrification using ferrous.cntdot.EDTA and a polyphenolic compound in an aqueous scrubber system. Energy Fuels 1991, 5, 244–248. [Google Scholar] [CrossRef]
- Gambardella, F.; Sanchez, L.; Ganzeveld, K.; Winkelman, J.; Heeres, H. Reactive NO absorption in aqueous FeII(EDTA) solutions in the presence of denitrifying micro-organisms. Chem. Eng. J. 2006, 116, 67–75. [Google Scholar] [CrossRef][Green Version]
- Jin, Y.; Veiga, M.a.C.; Kennes, C. Bioprocesses for the removal of nitrogen oxides from polluted air. J. Chem. Technol. Biotechnol. 2005, 80, 483–494. [Google Scholar] [CrossRef]
- Xia, Y.; Zhao, J.; Li, M.; Zhang, S.; Li, S.; Li, W. Bioelectrochemical Reduction of Fe(II)EDTA–NO in a Biofilm Electrode Reactor: Performance, Mechanism, and Kinetics. Environ. Sci. Technol. 2016, 50, 3846–3851. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Han, J.-I. An innovative dual fuel cell to capture and collect pure NO X from flue gases. J. Appl. Electrochem. 2013, 43, 1011–1016. [Google Scholar] [CrossRef]
- Garedew, M.; Lin, F.; Song, B.; DeWinter, T.M.; Jackson, J.E.; Saffron, C.M.; Lam, C.H.; Anastas, P.T. Greener Routes to Biomass Waste Valorization: Lignin Transformation Through Electrocatalysis for Renewable Chemicals and Fuels Production. ChemSusChem 2020, 13, 4214–4237. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- Zhu, K.; Zhu, H.; Feng, S.; Fu, J.; Guo, D.; Sun, Q.; Huang, L.; Hao, X. Electrochemical degradation of chemical wastewater by anodic oxidation process. IOP Conf. Ser. Earth Environ. Sci. 2019, 371, 032018. [Google Scholar] [CrossRef]
- Cai, J.; Zhou, M.; Pan, Y.; Du, X.; Lu, X. Extremely efficient electrochemical degradation of organic pollutants with co-generation of hydroxyl and sulfate radicals on Blue-TiO2 nanotubes anode. Appl. Catal. B Environ. 2019, 257, 117902. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, Q.; Song, X.; Wang, H.; Bian, Z. Recent electrochemical methods in electrochemical degradation of halogenated organics: A review. Environ. Sci. Pollut. Res. 2019, 26, 10457–10486. [Google Scholar] [CrossRef]
- Lee, D.K.; Choi, K.-S. Enhancing long-term photostability of BiVO4 photoanodes for solar water splitting by tuning electrolyte composition. Nature Energy 2018, 3, 53–60. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; Arquer, F.P.G.d.; Kiani, A.; Edwards, J.P.; Luna, P.D.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef]
- Verma, S.; Lu, S.; Kenis, P.J.A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nature Energy 2019, 4, 466–474. [Google Scholar] [CrossRef]
- Allcorn, E.; Nagasubramanian, G.; Pratt, H.D.; Spoerke, E.; Ingersoll, D. Elimination of active species crossover in a room temperature, neutral pH, aqueous flow battery using a ceramic NaSICON membrane. J. Power Sources 2018, 378, 353–361. [Google Scholar] [CrossRef]
- Guo, Q.; Sun, T.; Wang, Y.; He, Y.; Jia, J. Spray absorption and electrochemical reduction of nitrogen oxides from flue gas. Environ. Sci Technol. 2013, 47, 9514–9522. [Google Scholar] [CrossRef]
- Shimizu, K.; Hutcheson, R.; Engelmann, M.D.; Francis Cheng, I. Cyclic voltammetric and aqueous equilibria model study of the pH dependant iron(II/III)ethylenediamminetetraacetate complex reduction potential. J. Electroanal. Chem. 2007, 603, 44–50. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe-Complexes | NO Absorption Capacity (Equivalent) | Remained Fe2+ Ratio after Exposed in Air for 24 h (%) | Note |
|---|---|---|---|
| Fe2+-EDTA | 1 | 0 | Poor oxidation resistance |
| Fe2+-(DMPS)2 | 2 | 97 | Asymmetric reactivity between oxidation and reduction reaction |
| Fe2+-(CA)2 | >2 | 96 | Fe precipitation after NO absorption |
| Fe2+-MPMAA | 2 | 97 | Poor electrochemical reactivity, side reactions with electrode |
| Fe2+-(TPPTS)2 | 0 | 98 | Poor NO absorption, side reactions with electrode |
| Fe2+-(DPPETS)2 | 2 | 97 | Side reactions with electrode |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eum, H.; Cheong, S.; Kim, J.; Han, S.-J.; Cheong, M.; Lee, H.; Lee, H.-S.; Lee, D.K. Sustainable Electrochemical NO Capture and Storage System Based on the Reversible Fe2+/Fe3+-EDTA Redox Reaction. Catalysts 2022, 12, 79. https://doi.org/10.3390/catal12010079
Eum H, Cheong S, Kim J, Han S-J, Cheong M, Lee H, Lee H-S, Lee DK. Sustainable Electrochemical NO Capture and Storage System Based on the Reversible Fe2+/Fe3+-EDTA Redox Reaction. Catalysts. 2022; 12(1):79. https://doi.org/10.3390/catal12010079
Chicago/Turabian StyleEum, Heesung, Seokhyeon Cheong, Jiyun Kim, Seo-Jung Han, Minserk Cheong, Hyunjoo Lee, Hae-Seok Lee, and Dong Ki Lee. 2022. "Sustainable Electrochemical NO Capture and Storage System Based on the Reversible Fe2+/Fe3+-EDTA Redox Reaction" Catalysts 12, no. 1: 79. https://doi.org/10.3390/catal12010079
APA StyleEum, H., Cheong, S., Kim, J., Han, S.-J., Cheong, M., Lee, H., Lee, H.-S., & Lee, D. K. (2022). Sustainable Electrochemical NO Capture and Storage System Based on the Reversible Fe2+/Fe3+-EDTA Redox Reaction. Catalysts, 12(1), 79. https://doi.org/10.3390/catal12010079