
Atoms vs. Ions: Intermediates in Reversible Electrochemical Hydrogen Evolution Reaction



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanisms of H and H2 Evolution/Oxidation Reactions
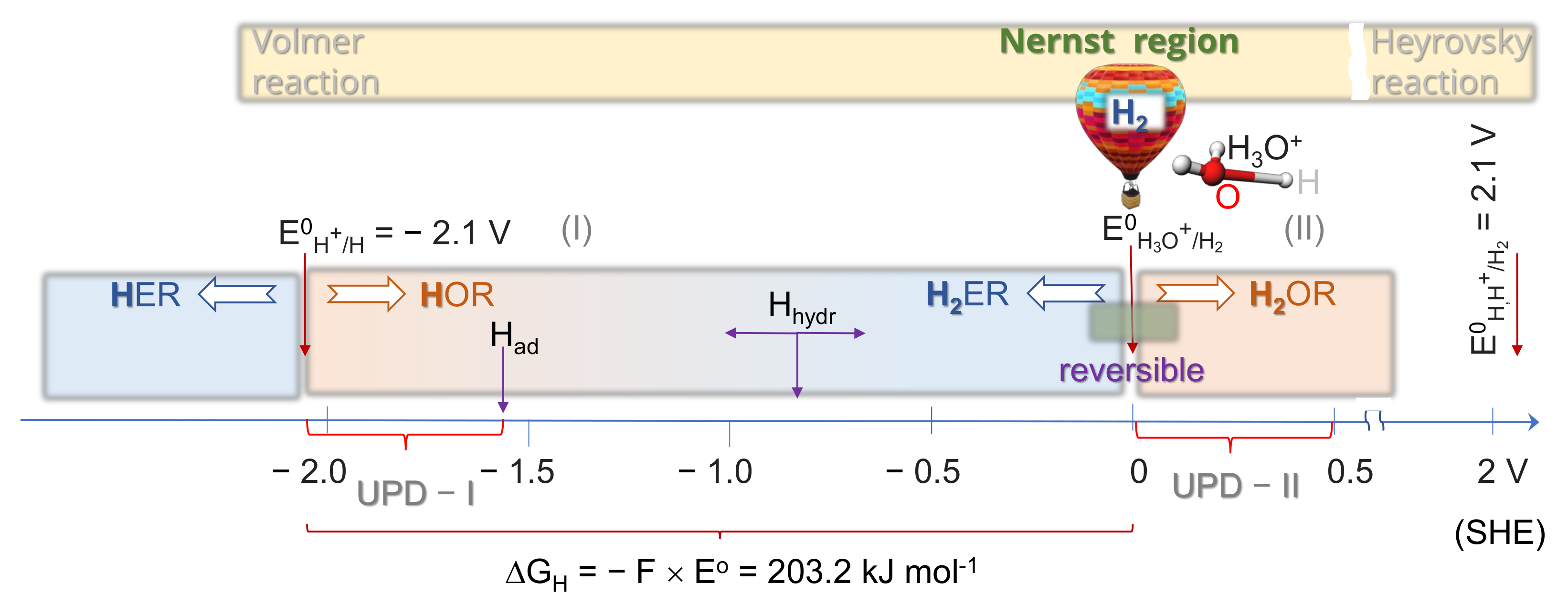
2.1. Thermodynamics of H Atom Formation
2.2. Energetics of Bond Breaking in H2 and H
2.3. Depolarization of H2 Evolution Due to Hydrogen Molecular Ion H2+ Formation
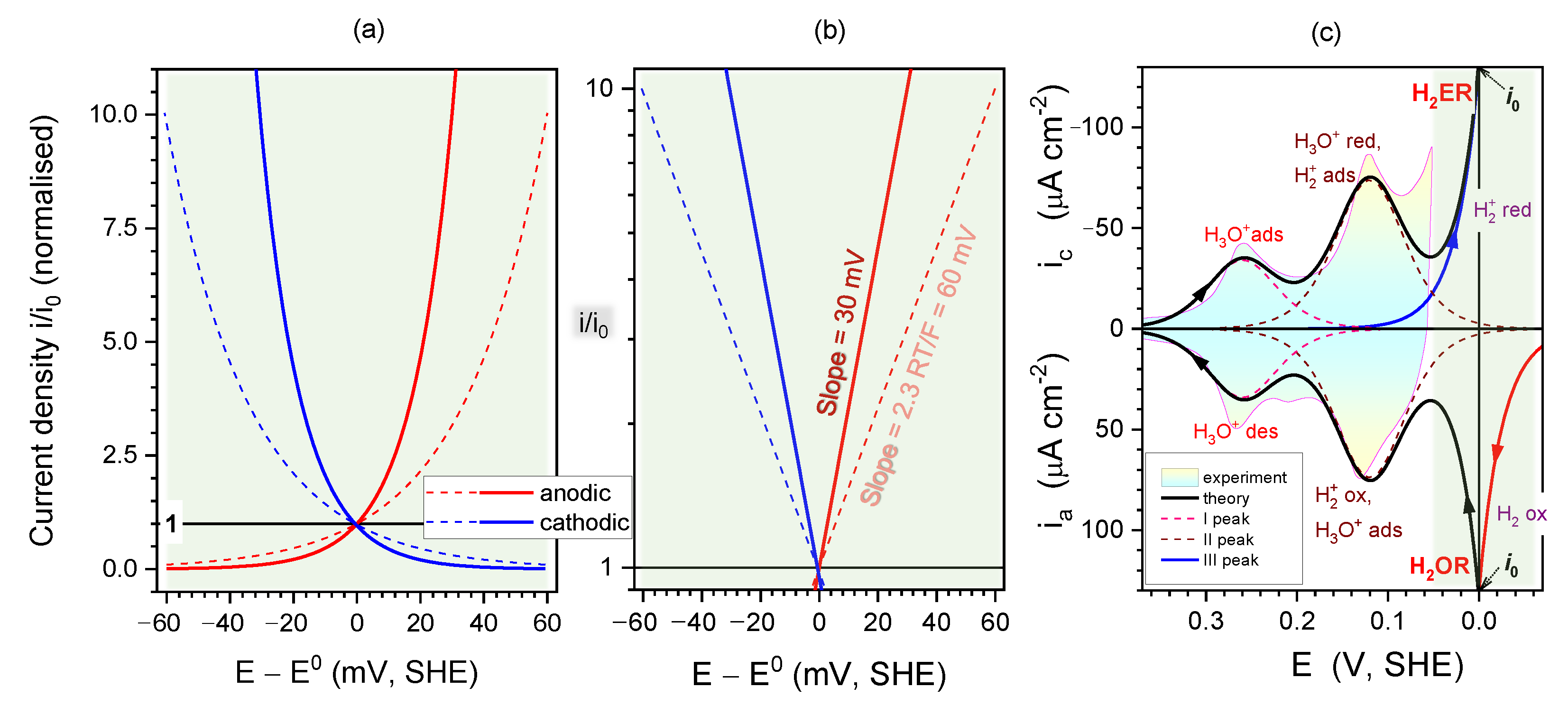
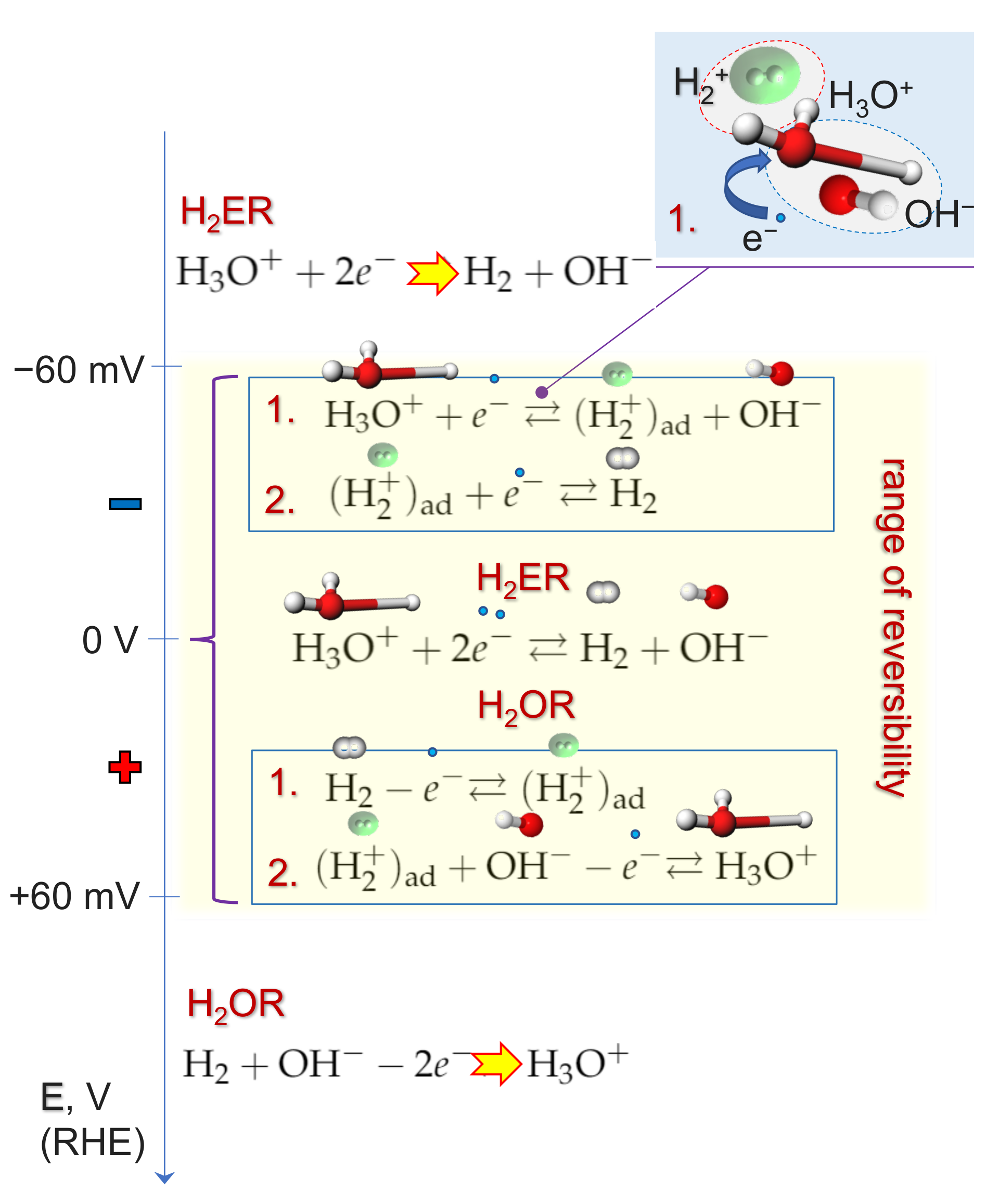
2.4. Kinetics and pH-Dependence of E in the Case of Reversible H2 Evolution/Oxidation Processes
2.5. Reversibility: Thermodynamics at Work for SHE Electrode
3. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Juodkazis, K.; Juodkazytė, J.; Jelmakas, E.; Kalinauskas, P.; Valsiūnas, I.; Mečinskas, P.; Juodkazis, S. Photoelectrolysis of Water: Solar Hydrogen-Achievements and Perspectives. Opt. Express Energy Express 2010, 18, A147–A160. [Google Scholar] [CrossRef]
- Conway, B.E.; Tilak, B.V. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochim. Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Dubouis, N.; Grimaud, A. The hydrogen evolution reaction: From material to interfacial descriptors. Chem. Sci. 2019, 10, 9165–9181. [Google Scholar] [CrossRef] [PubMed]
- Lasia, A. Handbook of Fuel Cells; John Wiley & Son, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Lasia, A. On the mechanism of the hydrogen absorption reaction. J. Electroanal. Chem. 2006, 593, 159–166. [Google Scholar] [CrossRef]
- Losiewicz, B.; Jurczakowski, R.; Lasia, A. Kinetics of hydrogen underpotential deposition at polycrystalline platinum in acidic solutions. Electroch. Acta 2012, 80, 292–301. [Google Scholar] [CrossRef]
- Elayappana, V.; Shanmugamb, R.; Chinnusamyc, S.; Yoob, D.J.; Mayakrishnane, G.; Kima, K.; Noha, H.S.; Kima, M.K.; Lee, H. Three-dimensional bimetal TMO supported carbon based electrocatalyst developed via dry synthesis for hydrogen and oxygen evolution. Appl. Surf. Sci. 2020, 505, 144642. [Google Scholar] [CrossRef]
- Logeshwaran, N.; Ramakrishnan, S.; Chandrasekaran, S.S.; Vinothkannan, M.; Kim, A.R.; Sengodan, S.; Velusamy, D.B.; Varadhan, P.; He, J.H.; Yoo, D.J. An efficient and durable trifunctional electrocatalyst for zinc–air batteries driven overall water splitting. Appl. Catal. B Environ. 2021, 297, 120405. [Google Scholar] [CrossRef]
- Sui, Y.; Ji, X. Anticatalytic Strategies to Suppress Water Electrolysis in Aqueous Batteries. Chem. Rev. 2021, 121, 6654–6695. [Google Scholar] [CrossRef] [PubMed]
- Vetter, K.J. Elektrochemische Kinetik; Springer: Berlin, Germany, 1961. [Google Scholar]
- Will, F.G.; Knorr, C.A. Untersuchung von Adsorptionserscheinungen an Rhodium, Iridium, Palladium und Gold mit der potentiostatischen Dreieckmethode. Z. Electrochem. 1960, 64, 270–275. [Google Scholar]
- Biegler, T.; Rand, D.A.J.; Woods, R. Limiting oxygen coverage on platinized platinum; Relevance to determination of real platinum area by hydrogen adsorption. J. Electroanal. Chem. 1971, 29, 269–277. [Google Scholar] [CrossRef]
- Gomez, R.; Orts, J.M.; Alvarez-Ruiz, B.; Feliu, J.M. Effect of Temperature on Hydrogen Adsorption on Pt(111), Pt(110), and Pt(100) Electrodes in 0.1 M HClO4. J. Phys. Chem. B 2004, 108, 228–238. [Google Scholar] [CrossRef]
- Sudhagar, P.; Roy, N.; Vedarajan, R.; Devadoss, A.; Terashima, C.; Nakata, K.; Fujishima, A. Photoelectrochemical Solar Fuel Production From Basic Principles to Advanced Devicess; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Pourbaix, M. Atlas D’équilibres Électrochimiques; Gauthier-Villars: Paris, France, 1963. [Google Scholar]
- Jerkiewicz, G. Standard and Reversible Hydrogen Electrodes: Theory, Design, Operation and Applications. ACS Catal. 2020, 10, 8409–8417. [Google Scholar] [CrossRef]
- Juodkazis, K.; Juodkazytė, J.; Grigucevičienė, A.; Juodkazis, S. Hydrogen species within the metals: Role of molecular hydrogen ion . Appl. Surf. Sci. 2011, 258, 743–747. [Google Scholar] [CrossRef]
- Kriek, R.J.; Mogwase, B.M.S.; Vorster, S. Relation of the electrochemical interplay between H2PtCl6 and H2O/H3O+/H2+ and the hydrogen-evolution reaction. Electrochem. Sci. Adv. 2021, e2100041. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.; Liu, H.; Suen, N.T.; Yu, X.; Feng, L. Electrochemical Hydrogen Evolution Reaction Efficiently Catalyzed by Ru 2 P Nanoparticles. ChemSusChem 2018, 11, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Peter, S.C. An overview on Pd-based electrocatalysts for the hydrogen evolution reaction. Inorg. Chem. Front. 2018, 5, 2060–2080. [Google Scholar] [CrossRef]
- Fan, X.; Du, P.; Ma, X.; Wang, R.; Ma, J.; Wang, Y.; Fan, D.; Long, Y.; Deng, B.; Huang, K.; et al. Mechanochemical Synthesis of Pt/Nb2CTx MXene Composites for Enhanced Electrocatalytic Hydrogen Evolution. Materials 2021, 14, 2426. [Google Scholar] [CrossRef]
- Emsley, J. The Elements, 2nd ed.; Clarendon Press: Oxford, UK, 1991. [Google Scholar]
- Markovic, M.M.; Ross, P.N., Jr. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Nobuhara, K.; Nakanishi, H.; Kasai, H.; Okiji, A. Interactions of atomic hydrogen with Cu(111), Pt(111), and Pd(111). J. Appl. Phys. 2000, 88, 6897–6901. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Hydrogen sorption AT/IN electrodes. Prog. Surf. Sci. 1998, 57, 137–186. [Google Scholar] [CrossRef]
- Jerkiewicz, G.; Zolfaghari, A. Comparison of Hydrogen Electroadsorption from the Electrolyte with Hydrogen Adsorption from the Gas Phase. J. Electrochem. Soc. 1996, 143, 1240–1248. [Google Scholar] [CrossRef]
- Ooka, H.; Wintzer, M.E.; Nakamura, R. Non-Zero Binding Enhances Kinetics of Catalysis: Machine Learning Analysis on the Experimental Hydrogen Binding Energy of Platinum. ACS Catal. 2021, 11, 6298–6303. [Google Scholar] [CrossRef]
- Yan, L.; Sun, Y.; Yamamoto, Y.; Kasamatsu, S.; Hamada, I.; Sugino, O. Hydrogen adsorption on Pt(111) revisited from random phase approximation. J. Chem. Phys. 2018, 149, 164702. [Google Scholar] [CrossRef] [PubMed]
- Nordholm, S.; Bacskay, G.B. The Basics of Covalent Bonding in Terms of Energy and Dynamics. Molecules 2020, 25, 2667. [Google Scholar] [CrossRef]
- Lopez, G.V.; Fournier, M.; Jankunas, J.; Spiliotis, A.K.; Rakitzis, T.P.; Chandler, D.W. Alignment of the hydrogen molecule under intense laser fields. J. Chem. Phys. 2017, 147, 013948. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merényi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueous solution: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Huber, K.P.; Herzberg, G. Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules; Springer: Boston, MA, USA, 1979. [Google Scholar]
- Pavičić, D.; Kiess, A.; Hānsch, T.W.; Figger, H. Intense-Laser-Field Ionization of the Hydrogen Molecular Ions and at Critical Internuclear Distances. Phys. Rev. Lett. 2005, 94, 163002. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, Z.; He, F.; Wang, X.; Atia-Tul-Noor, A.; Kielpinski, D.; Sang, R.T.; Litvinyuk, I.V. Observing electron localization in a dissociating H2+ molecule in real time. Nat. Commun. 2017, 8, 15849. [Google Scholar] [CrossRef][Green Version]
- Sun, Z.; Yao, H.; Ren, X.; Liu, Y.; Wang, D.; Zhao, W.; Wang, C.; Yang, C. Imaging of electron transition and bond breaking in the photodissociation of via ultrafast X-ray photoelectron diffraction. Opt. Express 2021, 29, 10893–10902. [Google Scholar] [CrossRef]
- Palascak, M.W.; Shields, G.C. Accurate Experimental Values for the Free Energies of Hydration of H+, OH-, and H3O+. J. Phys. Chem. A 2004, 108, 3692–3694. [Google Scholar] [CrossRef]
- Juodkazis, K.; Juodkazytė, J.; Šebeka, B.; Juodkazis, S. Reversible hydrogen evolution and oxidation on Pt electrode mediated by molecular ion. Appl. Surf. Sci. 2014, 290, 13–17. [Google Scholar] [CrossRef]
- Rabinovich, V.A.; Khavin, Z.Y. Kratkiy Khimicheskiy Spravochnik; Khimiya: Leningrad, Russia, 1977. [Google Scholar]
- Jerkiewicz, G.; Vatankhan, G.; Lessard, J.; Soriaga, M.P.; Park, Y.S. Surface-oxide growth at platinum electrodes in aqueous H2SO4: Reexamination of its mechanism through combined cyclic-voltammetry, electrochemical quartz-crystal nanobalance, and Auger electron spectroscopy measurements. Electroch. Acta 2004, 49, 1451–1459. [Google Scholar] [CrossRef]
- Juodkazytė, J.; Juodkazis, K.; Matulaitienė, I.; Šebeka, B.; Savickaja, I.; Balčytis, A.; Nishijima, Y.; Niaura, G.; Juodkazis, S. Hydrogen Evolution on Nano-Structured CuO/Pd Electrode: Raman Scattering Study. Appl. Sci. 2019, 9, 5301. [Google Scholar] [CrossRef]
- Tafel, J. Über die Polarisation bei kathodischer Wasserstoffentwicklung. Z. Für Phys. Chem. 1905, 50U, 641–712. [Google Scholar] [CrossRef]
- Butler, J.A.V. Studies in heterogeneous equilibria. Part II. The kinetic interpretation of the Nernst theory of electromotive force. Trans. Faraday Soc. 1924, 19, 729–733. [Google Scholar] [CrossRef]
- Erdey-Gruz, T.; Volmer, M. Zur theorie der wasserstoffüberspannung. Z. Phys. Chem. 1930, 150, 203–213. [Google Scholar] [CrossRef]
- Bazant, M.Z. Theory of Chemical Kinetics and Charge Transfer based on Nonequilibrium Thermodynamics. Acc. Chem. Res. 2013, 46, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Santra, L.Z.B.; Ko, H.Y.; Di Stasio, R.A.; Klein, M.L.; Car, R.; Wu, X. Hydroxide diffuses slower than hydronium in water because its solvated structure inhibits correlated proton transfer. Nat. Chem. 2018, 10, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, K.; Yang, J.; Zheng, Y.; Hübner, R.; Ou, Z.; Dong, X.; He, L.; Wang, H.; Li, J.; et al. Tungsten Oxide/Reduced Graphene Oxide Aerogel with Low-Content Platinum as High-Performance Electrocatalyst for Hydrogen Evolution Reaction. Small 2021, 17, 2102159. [Google Scholar] [CrossRef]
- Takenaka, M.; Hashimoto, Y.; Iwasa, T.; Taketsugu, T.; Seniutinas, G.; Balcytis, A.; Juodkazis, S.; Nishijima, Y. First Principles Calculations Toward Understanding SERS of 2,2’-Bipyridyl Adsorbed on Au, Ag, and Au–Ag Nanoalloy. J. Comput. Chem. 2004, 40, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Kronberg, R.; Laasonen, K. Reconciling the Experimental and Computational Hydrogen Evolution Activities of Pt(111) through DFT-Based Constrained MD Simulations. ACS Catal. 2021, 11, 8062–8078. [Google Scholar] [CrossRef]
- Lindgren, P.; Kastlunger, G.; Peterson, A.A. A Challenge to the G∼ 0 Interpretation of Hydrogen Evolution. ACS Catal. 2020, 10, 121–128. [Google Scholar] [CrossRef]
- Nishijima, Y.; Shimizu, S.; Kurihara, K.; Hashimoto, Y.; Takahashi, H.; Balcytis, A.; Seniutinas, G.; Okazaki, S.; Juodkazyte, J.; Iwasa, T.; et al. Optical readout of hydrogen storage in films of Au and Pd. Opt. Express 2017, 20, 24081–24092. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juodkazytė, J.; Juodkazis, K.; Juodkazis, S. Atoms vs. Ions: Intermediates in Reversible Electrochemical Hydrogen Evolution Reaction. Catalysts 2021, 11, 1135. https://doi.org/10.3390/catal11091135
Juodkazytė J, Juodkazis K, Juodkazis S. Atoms vs. Ions: Intermediates in Reversible Electrochemical Hydrogen Evolution Reaction. Catalysts. 2021; 11(9):1135. https://doi.org/10.3390/catal11091135
Chicago/Turabian StyleJuodkazytė, Jurga, Kȩstutis Juodkazis, and Saulius Juodkazis. 2021. "Atoms vs. Ions: Intermediates in Reversible Electrochemical Hydrogen Evolution Reaction" Catalysts 11, no. 9: 1135. https://doi.org/10.3390/catal11091135
APA StyleJuodkazytė, J., Juodkazis, K., & Juodkazis, S. (2021). Atoms vs. Ions: Intermediates in Reversible Electrochemical Hydrogen Evolution Reaction. Catalysts, 11(9), 1135. https://doi.org/10.3390/catal11091135