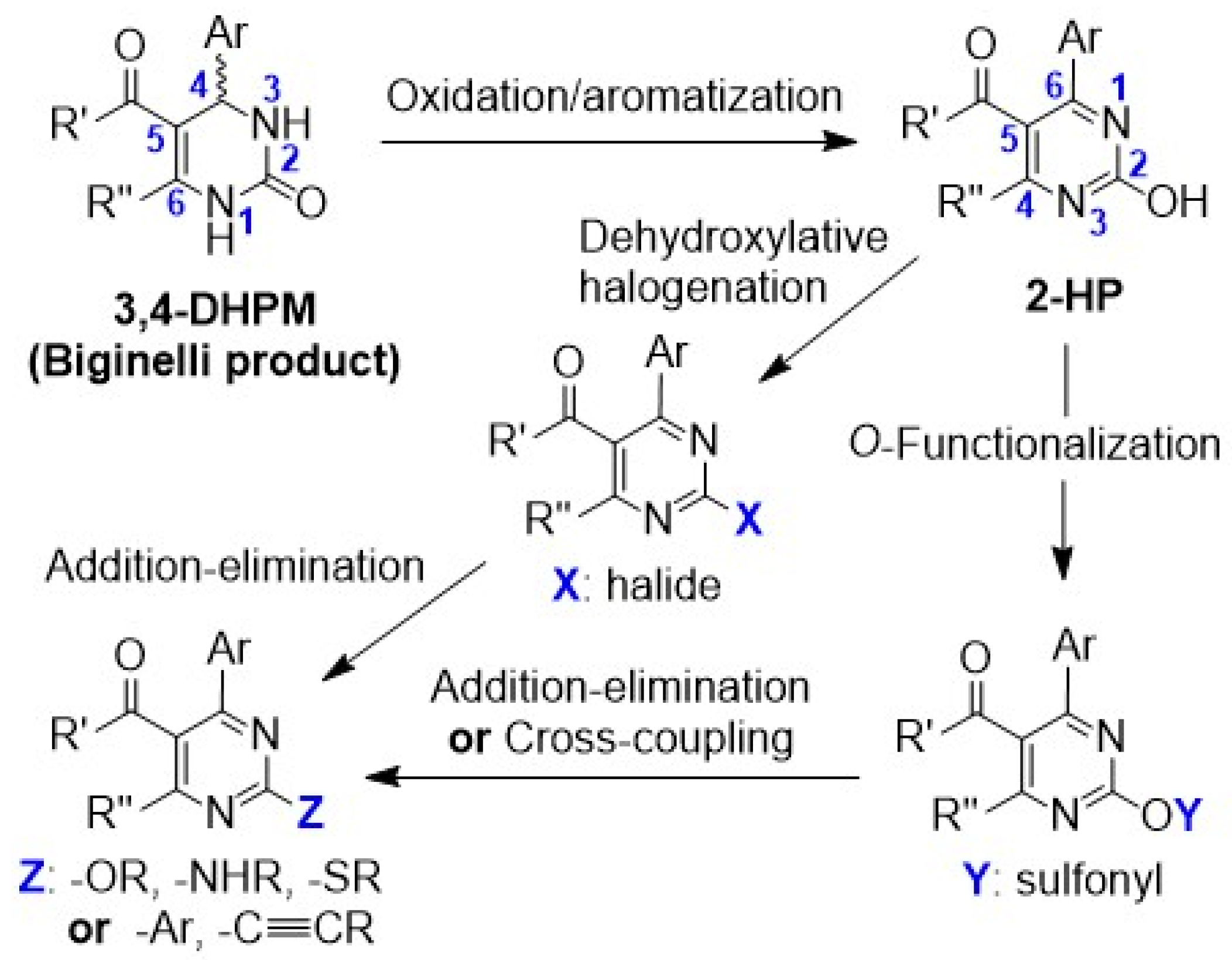
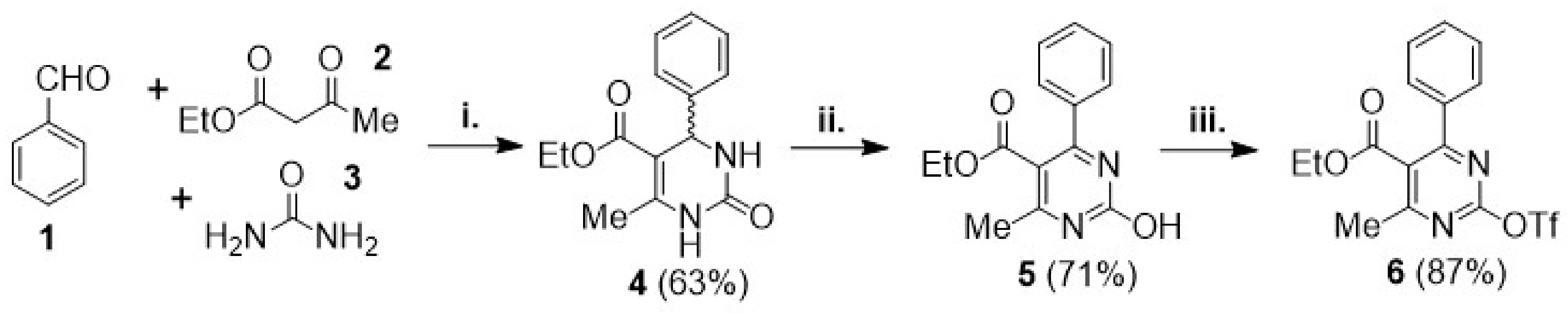
3.2. Synthetic Methods and Characterization Data
Ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4): Urea (3) (2.70 g, 45 mmol, 1.5 equiv) and SnCl2.½H2O (0.30 g, 1.5 mmol, 0.05 equiv) were transferred to a round-bottom flask, which was fitted with a vertical condenser and set under nitrogen atmosphere. Methoxyethanol (30 mL) was added, followed by benzaldehyde (1) (3.0 mL, 30 mmol, 1 equiv) and ethyl acetoacetate (2) (3.8 mL, 30 mmol, 1 equiv), and the mixture was refluxed at 125 °C for 48 h. It was then cooled down and the solvent was removed under vacuum. The resulting crude solid was suspended in CH3CN and collected by filtration, washed with cold CH3CN, and dried under house vacuum overnight. Overall, 4.90 g (18.8 mmol, 63%) of compound 4 were isolated as white powder. It was shown to be pure by 1H and 13C NMR and was progressed to the next step without further purification. 1H NMR (DMSO-d6), δ (ppm): 1.09 (3H, t, J = 7.1 Hz), 2.24 (3H, s), 3.98 (2H, q, J = 7.1 Hz), 5.14 (1H, d, J = 3.3 Hz), 7.21–7.27 (3H, m, signals overlapping), 7.32 (2H, app. t, J = 7.6 Hz), 7.72 (1H, bs), 9.17 (1H, s). 13C NMR (DMSO-d6), δ (ppm): 14.07, 17.77, 53.95, 59.17, 99.24, 126.23, 127.25, 128.38, 144.86, 148.36, 152.11, 165.33. MS (ES-API), m/z: calcd for C14H16N2O3: 260.12; found 261.10 [M + H+]. m.p. 178 °C.
Ethyl 2-hydroxy-4-methyl-6-phenylpyrimidine-5-carboxylate (5): Compound 4 (2.60 g, 10 mmol, 1 equiv), CuCl2 (0.134 g, 1 mmol, 0.1 equiv), and K2CO3 (0.691 g, 5 mmol, 0.5 equiv) were transferred to a round-bottom flask, and the flask was fitted with a vertical condenser and set under nitrogen atmosphere. DCM (30 mL) was added, and the mixture was heated at 35–40 °C. This was followed by slow dropwise addition of TBHP 70% wt solution (6.9 mL solution, 50 mmol, 5 equiv) under vigorous stirring, and the reaction continued for 24 h, maintaining the same temperature. The mixture was then cooled down to r.t. Aqueous sodium thiosulfate 0.5 M solution and aqueous NH4Cl 25% wt solution were added, and the mixture was stirred at r.t. for 1 h. The pH was checked to be around 7–8 at the end of the stirring time. The mixture was then transferred to a separatory funnel and extracted. The organic layer was collected and dried over Na2SO4. After filtering out the drying agent, the solvent was concentrated under vacuum. The crude sample was applied to a silica column for flash chromatography and eluted with hexane-ethyl acetate step gradient (from 1:2 to 1:10) to afford 1.83 g (7.1 mmol, 71%) of compound 5 as a pale yellow powder. 1H NMR (CDCl3), δ (ppm): 0.93 (3H, t, J = 7.1 Hz), 2.62 (3H, s), 4.05 (2H, q, J = 7.1 Hz), 7.43 (2H, app. t, J = 7.5 Hz), 7.48 (1H, tt, J1 = 7.2 Hz, J2 = 1.3 Hz), 7.60 (2H, d, J = 7.8 Hz). 13C NMR (CDCl3), δ (ppm): 13.44, 19.32, 61.62, 111.50, 128.01, 128.38, 130.83, 158.23, 166.08 (three signals overlapping with other peaks). MS (ES-API), m/z: calcd for C14H14N2O3: 258.10; found 259.00 [M + H+]. m.p. 174 °C.
Ethyl 4-methyl-6-phenyl-2-(((trifluoromethyl)sulfonyl)oxy)pyrimidine-5-carboxylate (6): Compound 5 (0.493 g, 1.9 mmol, 1 equiv) was transferred to a round-bottom flask, and the flask was sealed and set under nitrogen atmosphere. Anhydrous DCM (5 mL) was added, followed by Et3N (0.66 mL, 4.75 mmol, 2.5 equiv), and the mixture was stirred and cooled at 0 °C. A solution of triflic anhydride (Tf2O) (0.48 mL, 2.85 mmol, 1.5 equiv) in DCM (2 mL) was added dropwise and the mixture was vigorously stirred at the same temperature for 1 h. The reaction mixture was then brought up to r.t., and the stirring continued for 12 h. The mixture was subsequently diluted with DCM and washed with aqueous NaCl saturated solution and water. The organic layer was collected and dried over Na2SO4. After filtering out the drying agent, the solvent was concentrated under vacuum. The crude sample was applied to a silica column for flash chromatography and eluted with hexane-ethyl acetate step gradient (from 10:1 to 6:1), to afford 0.644 g (1.65 mmol, 87%) of compound 6 as a colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.12 (3H, t, J = 7.1 Hz), 2.67 (3H, s), 4.26 (2H, q, J = 7.1 Hz), 7.48 (2H, app. t, J = 7.5 Hz), 7.54 (1H, t, J = 7.3 Hz), 7.69 (2H, d, J = 7.7 Hz). 13C NMR (CDCl3), δ (ppm): 13.61, 22.62, 62.51, 118.55 (CF3, q, J = 321.1 Hz), 125.51, 128.58, 128.80, 131.34, 135.55, 157.27, 166.66, 167.44, 170.69. MS (ES-API), m/z: calcd for C15H13F3N2O5S: 390.05; found 391.00 [M + H+].
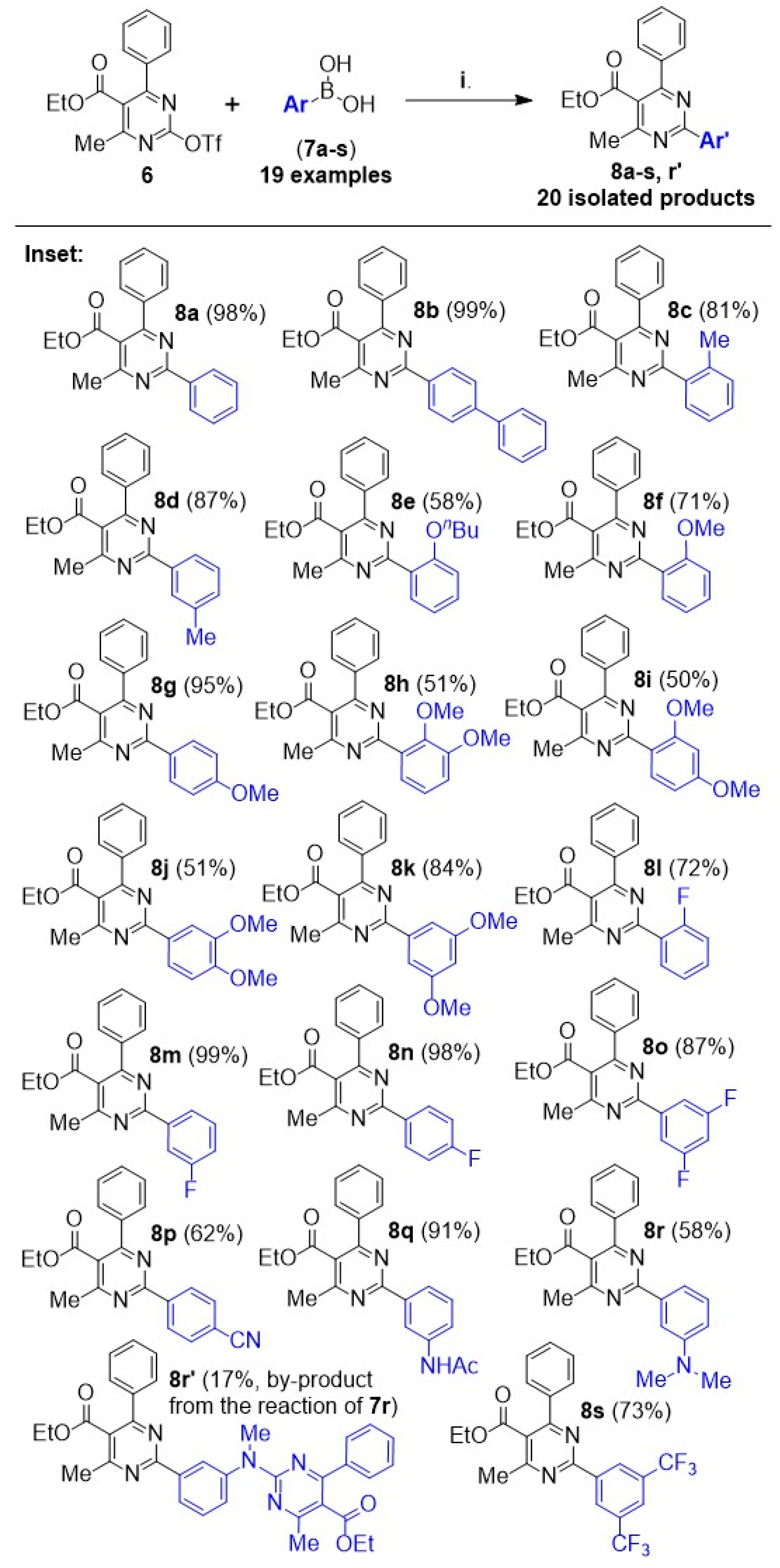
General method for the synthesis of ethyl 4-methyl-2,6-diphenylpyrimidine-5-carboxylates via Suzuki–Miyaura C–C cross-coupling: Boronic acid 7 (0.75 mmol, 1.5 equiv), Pd(OAc)2 (0.006 g, 0.025 mmol, 0.05 equiv), PPh3 (0.026 g, 0.1 mmol, 0.2 equiv), and K3PO4 (0.265 g, 1.25 mmol, 2.5 equiv) were transferred to a round-bottom flask, and the flask was fitted with a vertical condenser and set under nitrogen atmosphere. A solution of compound 6 (0.195 g, 0.5 mmol, 1 equiv) in anhydrous 1,4-dioxane (3.5 mL) was syringed in, and the resulting mixture was refluxed at 110 °C for 16 h. It was then cooled down to r.t., quenched with aqueous NH4Cl 25% wt solution, and extracted three times with diethyl ether. The combined organic phase was washed with aqueous Na2CO3 and NaCl solution and dried over Na2SO4. After filtering out the drying agent, the solvent was removed under vacuum. The crude sample was re-dissolved in DCM, applied to a silica column for flash chromatography, and eluted first with hexane and then with hexane-ethyl acetate step gradient (the end-ratio of solvents was different in each case, depending on product polarity).
● Ethyl 4-methyl-2,6-diphenylpyrimidine-5-carboxylate (8a): Yield: 98%; white solid. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.1 Hz), 2.70 (3H, s), 4.22 (2H, q, J = 7.1 Hz), 7.45–7.53 (6H, m, signals overlapping), 7.76 (2H, m), 8.56 (2H, m). 13C NMR (CDCl3), δ (ppm): 13.66, 22.87, 61.76, 123.35, 128.45, 128.47, 128.50, 128.63, 129.96, 131.03, 137.16, 138.23, 163.57, 163.70, 165.39, 168.46. MS (ES-API), m/z: calcd for C20H18N2O2: 318.14; found 319.10 [M + H+]. m.p. 64 °C.
● Ethyl 2-([1,1’-biphenyl]-4-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8b): Yield: 99%; white solid. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.1 Hz), 2.73 (3H, s), 4.23 (2H, q, J = 7.1 Hz), 7.39 (1H, t, J = 7.5 Hz), 7.47–7.51 (5H, m, signals overlapping), 7.69 (2H, d, J = 7.5 Hz), 7.74 (2H, d, J = 8.5 Hz), 7.79 (2H, dd, J1 = 7.2 Hz, J2 = 2.0 Hz), 8.65 (2H, d, J = 8.5 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 22.89, 61.75, 123.27, 127.17, 127.19, 127.70, 128.44, 128.47, 128.80, 129.10, 129.95, 136.09, 138.25, 140.52, 143.67, 163.44, 163.59, 165.40, 168.45. MS (ES-API), m/z: calcd for C26H22N2O2: 394.17; found 395.10 [M + H+]. m.p. 69 °C.
● Ethyl 4-methyl-6-phenyl-2-(o-tolyl)pyrimidine-5-carboxylate (8c): Yield: 81%; white solid. 1H NMR (CDCl3), δ (ppm): 1.11 (3H, t, J = 7.1 Hz), 2.63 (3H, s), 2.70 (3H, s), 4.24 (2H, q, J = 7.1 Hz), 7.31 (2H, app. t, J = 7.5 Hz), 7.35 (1H, app. dt, J1 = 7.3 Hz, J2 = 1.5 Hz), 7.44–7.50 (3H, m, signals overlapping), 7.73 (2H, m), 7.90 (1H, d, J = 7.3 Hz). 13C NMR (CDCl3), δ (ppm): 13.67, 21.34, 22.78, 61.87, 122.83, 125.95, 128.44, 128.50, 129.68, 129.98, 130.61, 131.35, 137.53, 137.69, 138.02, 163.16, 165.02, 166.80, 168.40. MS (ES-API), m/z: calcd for C21H20N2O2: 332.15; found 333.10 [M + H+]. m.p. 70 °C.
● Ethyl 4-methyl-6-phenyl-2-(m-tolyl)pyrimidine-5-carboxylate (8d): Yield: 87%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.1 Hz), 2.46 (3H, s), 2.70 (3H, s), 4.22 (2H, q, J = 7.1 Hz), 7.32 (1H, d, J = 7.6 Hz), 7.39 (1H, app. t, J = 7.8 Hz), 7.47–7.51 (3H, m, signals overlapping), 7.75 (2H, m), 8.35 (1H, d, J = 8.0 Hz), 8.36 (1H, s). 13C NMR (CDCl3), δ (ppm): 13.65, 21.48, 22.86, 61.74, 123.28, 125.85, 128.43, 128.44, 128.46, 129.12, 129.94, 131.86, 137.09, 138.16, 138.28, 163.56, 163.87, 165.33, 168.47. MS (ES-API), m/z: calcd for C21H20N2O2: 332.15; found 333.10 [M + H+].
● Ethyl 2-(2-butoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8e): Yield: 58%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 0.90 (3H, t, J = 7.3 Hz), 1.10 (3H, t, J = 7.1 Hz), 1.45 (2H, app. hex, J = 7.5 Hz), 1.75 (2H, app. quint, J = 7.1 Hz), 2.69 (3H, s), 4.05 (2H, t, 6.5 Hz), 4.22 (2H, q, J = 7.1 Hz), 7.03 (2H, m, signals overlapping), 7.39 (1H, dt, J1 = 7.9 Hz, J2 = 1.8 Hz), 7.43–7.47 (3H, m, signals overlapping), 7.71 (2H, m), 7.78 (1H, dd, J1 = 7.6 Hz, J2 = 1.8 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 13.84, 19.26, 22.69, 31.40, 61.77, 68.46, 113.17, 120.48, 122.95, 128.22, 128.38, 128.40, 129.78, 131.12, 131.66, 138.16, 157.50, 163.33, 164.74, 165.27, 168.41. MS (ES-API), m/z: calcd for C24H26N2O3: 390.19; found 391.10 [M + H+].
● Ethyl 2-(2-methoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8f): Yield: 71%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.1 Hz), 2.70 (3H, s), 3.89 (3H, s), 4.22 (2H, q, J = 7.1 Hz), 7.03 (1H, d, J = 8.4 Hz), 7.06 (1H, app. dt, J1 = 7.5 Hz, J2 = 0.7 Hz), 7.42 (1H, m), 7.43–7.47 (3H, m, signals overlapping), 7.73 (3H, m, signals overlapping). 13C NMR (CDCl3), δ (ppm): 13.64, 22.75, 56.01, 61.78, 112.18, 120.68, 123.15, 128.33, 128.45, 128.46, 129.86, 131.09, 131.52, 138.06, 157.74, 163.34, 164.89, 165.27, 168.30. MS (ES-API), m/z: calcd for C21H20N2O3: 348.15; found 349.10 [M + H+].
● Ethyl 2-(4-methoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8g): Yield: 95%; white wax. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.1 Hz), 2.68 (3H, s), 3.89 (3H, s), 4.20 (2H, q, J = 7.1 Hz), 7.00 (2H, d, J = 9.1 Hz), 7.45–7.51 (3H, m, signals overlapping), 7.74 (2H, m), 8.52 (2H, d, J = 9.1 Hz). 13C NMR (CDCl3), δ (ppm): 13.67, 22.88, 55.37, 61.69, 113.83, 122.60, 128.41, 128.42, 128.43, 129.86, 130.36, 138.43, 162.15, 163.44, 163.57, 165.28, 168.60. MS (ES-API), m/z: calcd for C21H20N2O3: 348.15; found 349.10 [M + H+].
● Ethyl 2-(2,3-dimethoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8h): Yield: 51%; white wax. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.2 Hz), 2.69 (3H, s), 3.91 (3H, s), 3.96 (3H, s), 4.22 (2H, q, J = 7.2 Hz), 7.03 (1H, dd, J1 = 8.2 Hz, J2 = 1.3 Hz), 7.15 (1H, app. t, J = 8.0 Hz), 7.37 (1H, dd, J1 = 7.8 Hz, J2 = 1.3 Hz), 7.42–7.48 (3H, m, signals overlapping), 7.72 (2H, m). 13C NMR (CDCl3), δ (ppm): 13.66, 22.78, 56.05, 61.69, 61.82, 113.97, 122.91, 123.36, 123.99, 128.44, 128.49, 129.88, 133.77, 138.10, 147.99, 153.51, 163.38, 164.91, 165.01, 168.35. MS (ES-API), m/z: calcd for C22H22N2O4: 378.16; found 379.10 [M + H+].
● Ethyl 2-(2,4-dimethoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8i): Yield: 50%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.2 Hz), 2.68 (3H, s), 3.86 (3H, s), 3.89 (3H, s), 4.20 (2H, q, J = 7.2 Hz), 6.57 (1H, d, J = 2.3 Hz), 6.60 (1H, dd, J1 = 8.5 Hz, J2 = 2.3 Hz), 7.42–7.48 (3H, m, signals overlapping), 7.71 (2H, m), 7.82 (1H, d, J = 8.5 Hz). 13C NMR (CDCl3), δ (ppm): 13.65, 22.78, 55.46, 56.08, 61.70, 99.56, 105.04, 121.16, 122.52, 128.43, 128.48, 129.79, 133.08, 138.29, 159.47, 162.46, 163.31, 164.81, 164.86, 168.44. MS (ES-API), m/z: calcd for C22H22N2O4: 378.16; found 379.10 [M + H+].
● Ethyl 2-(3,4-dimethoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8j): Yield: 51%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.07 (3H, t, J = 7.1 Hz), 2.68 (3H, s), 3.95 (3H, s), 4.01 (3H, s), 4.19 (2H, q, J = 7.1 Hz), 6.96 (1H, d, J = 8.5 Hz), 7.45–7.51 (3H, m, signals overlapping), 7.74 (2H, m), 8.11 (1H, d, J = 2.0 Hz), 8.20 (1H, dd, J1 = 8.5 Hz, J2 = 2.0 Hz). 13C NMR (CDCl3), δ (ppm): 13.63, 22.86, 55.91, 55.96, 61.64, 110.65, 111.12, 122.23, 122.66, 128.38, 128.41, 129.83, 129.99, 138.37, 148.85, 151.66, 163.31, 163.53, 165.24, 168.51. MS (ES-API), m/z: calcd for C22H22N2O4: 378.16; found 379.10 [M + H+].
● Ethyl 2-(3,5-dimethoxyphenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8k): Yield: 84%; white wax. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.2 Hz), 2.69 (3H, s), 3.89 (6H, s), 4.21 (2H, q, J = 7.2 Hz), 6.62 (1H, t, J = 2.2 Hz), 7.45–7.51 (3H, m, signals overlapping), 7.72–7.78 (4H, m, signals overlapping). 13C NMR (CDCl3), δ (ppm): 13.65, 22.83, 55.56, 61.79, 103.76, 106.44, 123.53, 128.45, 128.49, 129.98, 138.15, 139.26, 160.94, 163.31, 163.47, 165.33, 168.43. MS (ES-API), m/z: calcd for C22H22N2O4: 378.16; found 379.10 [M + H+].
● Ethyl 2-(2-fluorophenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8l): Yield: 72%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.71 (3H, s), 4.23 (2H, q, J = 7.2 Hz), 7.19 (1H, app. t, J = 9.5 Hz), 7.26 (1H, app. t, J = 7.5 Hz), 7.42–7.50 (4H, m, signals overlapping), 7.75 (2H, m), 8.11 (1H, app. t, J = 7.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.62, 22.77, 61.87, 116.84 (d, J = 22.2 Hz), 123.44, 124.06 (d, J = 3.5 Hz), 126.31 (d, J = 10.1 Hz), 128.46, 128.52, 130.07, 131.91, 131.92 (d, J = 8.6 Hz), 137.77, 161.27 (d, J = 256.0 Hz), 162.65 (d, J = 4.5 Hz), 163.48, 165.36, 168.11. MS (ES-API), m/z: calcd for C20H17FN2O2: 336.13; found 337.10 [M + H+].
● Ethyl 2-(3-fluorophenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8m): Yield: 99%; white solid. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.2 Hz), 2.70 (3H, s), 4.22 (2H, q, J = 7.2 Hz), 7.19 (1H, ddt, J1 = 8.2 Hz, J2 = 2.7 Hz, J3 = 1.1 Hz), 7.46 (1H, m, J1 = 7.8 Hz, J2 = 5.7 Hz), 7.47–7.51 (3H, m, signals overlapping), 7.75 (2H, dd, J1 = 7.8 Hz, J2 = 2.0 Hz), 8.26 (1H, ddd, J1 = 10.3 Hz, J2 = 2.7 Hz, J3 = 1.5 Hz), 8.35 (1H, app. td, J1 = 7.8 Hz, J2 = 1.1 Hz). 13C NMR (CDCl3), δ (ppm): 13.65, 22.83, 61.86, 115.42 (d, J = 23.9 Hz), 117.90 (d, J = 20.9 Hz), 123.78, 124.24 (d, J = 2.8 Hz), 128.44, 128.52, 129.97 (d, J = 7.8 Hz), 130.11, 137.97, 139.54 (d, J = 7.7 Hz), 162.51 (d, J = 3.3 Hz), 163.13 (d, J = 244.8 Hz), 163.62, 165.56, 168.27. MS (ES-API), m/z: calcd for C20H17FN2O2: 336.13; found 337.10 [M + H+]. m.p. 101 °C.
● Ethyl 2-(4-fluorophenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8n): Yield: 98%; white wax. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.2 Hz), 2.69 (3H, s), 4.21 (2H, q, J = 7.2 Hz), 7.16 (2H, app. t, J = 8.7 Hz), 7.45–7.53 (3H, m, signals overlapping), 7.74 (2H, dd, J1 = 7.6 Hz, J2 = 2.2 Hz), 8.57 (2H, dd, J1 = 8.9 Hz, J2 = 5.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.64, 22.86, 61.79, 115.46 (d, J = 21.7 Hz), 123.26, 128.41, 128.49, 130.02, 130.80 (d, J = 8.7 Hz), 133.33 (d, J = 3.3 Hz), 138.14, 162.73, 163.63, 164.91 (d, J = 250.7 Hz), 165.48, 168.38. MS (ES-API), m/z: calcd for C20H17FN2O2: 336.13; found 337.10 [M + H+].
● Ethyl 2-(3,5-difluorophenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8o): Yield: 87%; white solid. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.69 (3H, s), 4.23 (2H, q, J = 7.2 Hz), 6.94 (1H, tt, J1 = 8.6 Hz, J2 = 2.4 Hz), 7.47–7.53 (3H, m, signals overlapping), 7.74 (2H, dd, J1 = 7.8 Hz, J2 = 2.0 Hz), 8.10 (2H, m). 13C NMR (CDCl3), δ (ppm): 13.67, 22.79, 61.94, 106.19 (t, J = 25.9 Hz), 111.39 (dd, J1 = 19.9 Hz, J2 = 6.4 Hz), 124.22, 128.43, 128.57, 130.24, 137.75, 140.69 (t, J = 9.6 Hz), 161.39 (t, J = 3.7 Hz), 163.19 (dd, J1 = 248.7 Hz, J2 = 12.6 Hz), 163.68, 165.72, 168.07. MS (ES-API), m/z: calcd for C20H16F2N2O2: 354.12; found 355.10 [M + H+]. m.p. 124 °C.
● Ethyl 2-(4-cyanophenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8p): Yield: 62%; white solid. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.71 (3H, s), 4.23 (2H, q, J = 7.2 Hz), 7.46–7.54 (3H, m, signals overlapping), 7.75 (2H, m), 7.79 (2H, d, J = 8.6Hz), 8.68 (2H, d, J = 8.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.65, 22.81, 61.99, 114.25, 118.77, 124.29, 128.42, 128.60, 129.06, 130.29, 132.30, 137.70, 141.16, 161.77, 163.78, 165.79, 168.01. MS (ES-API), m/z: calcd for C21H17N3O2: 343.13; found 344.10 [M + H+]. m.p. 101 °C.
● Ethyl 2-(3-acetamidophenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8q): Yield: 91%; beige solid. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.2 Hz), 2.20 (3H, s), 2.69 (3H, s), 4.21 (2H, q, J = 7.2 Hz), 7.36 (1H, bs), 7.44–7.52 (4H, m, signals overlapping), 7.74 (2H, m), 7.99 (1H, dd, J1 = 7.7 Hz, J2 = 1.4 Hz), 8.30 (1H, d, J = 7.9 Hz), 8.37 (1H, app. t, J = 1.7 Hz). 13C NMR (CDCl3), δ (ppm): 13.61, 22.75, 24.41, 61.82, 119.79, 122.78, 123.45, 124.40, 128.34, 128.40, 129.16, 129.94, 137.66, 138.01, 138.31, 163.03, 163.50, 165.31, 168.36, 168.86. MS (ES-API), m/z: calcd for C22H21N3O3: 375.16; found 376.10 [M + H+], 398.10 [M + Na+]. m.p. 153 °C.
● Ethyl 2-(3-(dimethylamino)phenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8r): The general method was modified in this case to include 3.5 equiv (instead of 2.5 equiv) of K3PO4 due to the fact that the boronic acid 7r was in the form of HCl salt. Yield: 58%; pale yellow solid. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.1 Hz), 2.69 (3H, s), 3.05 (6H, s), 4.21 (2H, q, J = 7.1 Hz), 6.91 (1H, d, J = 7.0 Hz), 7.36 (1H, app. t, J = 8.0 Hz), 7.46–7.50 (3H, m, signals overlapping), 7.75 (2H, m), 7.92 (1H, d, J = 7.5 Hz), 7.96 (1H, bs). 13C NMR (CDCl3), δ (ppm): 13.69, 22.95, 40.83, 61.75, 112.68, 115.47, 117.36, 123.20, 128.42, 128.48, 129.20, 129.84, 137.89, 138.40, 150.85, 163.40, 164.29, 165.21, 168.60. MS (ES-API), m/z: calcd for C22H23N3O2: 361.18; found 362.10 [M + H+]. m.p. 78 °C.
● Ethyl 2-(3-((5-(ethoxycarbonyl)-4-methyl-6-phenylpyrimidin-2-yl)(methyl)amino) phenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8r’): This compound occurred as a by-product alongside the main product, 8r, in the reaction of compound 6 with boronic acid 7r. Yield: 17%; colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 0.99 (3H, t, J = 7.1 Hz), 1.09 (3H, t, J = 7.1 Hz), 2.49 (3H, s), 2.70 (3H, s), 3.69 (3H, s), 4.09 (2H, q, J =7.1 Hz), 4.22 (2H, q, J = 7.1 Hz), 7.32 (2H, t, J = 7.5 Hz), 7.37 (1H, t, J = 7.1 Hz), 7.45–7.49 (3H, m, signals overlapping), 7.51 (2H, m), 7.59 (2H, d, J = 7.8 Hz), 7.75 (2H, m), 8.42 (1H, dt, J1 = 4.0 Hz, J2 = 1.8 Hz), 8.62 (1H, bs). 13C NMR (CDCl3), δ (ppm): 13.60, 13.67, 22.85, 23.11, 38.50, 61.07, 61.79, 115.45, 123.43, 125.76, 126.54, 128.09, 128.33, 128.47, 128.48, 128.83, 128.93, 129.45, 129.97, 137.89, 138.18, 139.06, 145.33, 160.48, 163.37, 163.58, 165.13, 165.41, 166.80, 168.48, 169.25. MS (ES-API), m/z: calcd for C35H33N5O4: 587.25; found 588.20 [M + H+].
● Ethyl 2-(3,5-bis(trifluoromethyl)phenyl)-4-methyl-6-phenylpyrimidine-5-carboxylate (8s): Yield: 73%; white solid. 1H NMR (CDCl3), δ (ppm): 1.11 (3H, t, J = 7.1 Hz), 2.73 (3H, s), 4.24 (2H, q, J = 7.1 Hz), 7.52 (3H, m, signals overlapping), 7.76 (2H, m), 8.00 (1H, s), 9.04 (2H, s). 13C NMR (CDCl3), δ (ppm): 13.65, 22.79, 62.05, 123.35 (q, J = 272.8 Hz), 124.30 (hep, J = 3.7 Hz), 124.68, 128.46, 128.63 (q, J = 3.3 Hz), 128.67, 130.40, 131.95 (q, J = 33.4 Hz), 137.52, 139.22, 160.73, 163.97, 166.03, 167.90. MS (ES-API), m/z: calcd for C22H16F6N2O2: 454.11; found 455.10 [M + H+]. m.p. 92 °C.
Phenyldiazonium tetrafluoroborate (9): In a round-bottom flask open to air, 9.13 mL (0.1 mol, 1 equiv) of aniline were dissolved in a mixture of water (40 mL) and 50% aqueous tetrafluoroboric acid solution (35.12 mL solution, 17.56 g HBF4, 0.2 mol, 2 equiv). The resulting solution was cooled at 0 °C, followed by dropwise addition of a solution of sodium nitrite (7.59 g, 0.11 mol, 1.1 equiv) in water (15 mL), while the temperature was maintained at 0–5 °C. Stirring was continued at the same temperature for 2 more hours. The crude solid product was collected by filtration and washed with cold water. It was then dissolved in acetone and precipitated again by addition of diethyl ether. The solid was collected by filtration and dried under house vacuum. In total, 14 g (0.073 mol, 73%) of product 9 were isolated. It was stored in small portions in a fridge until use.
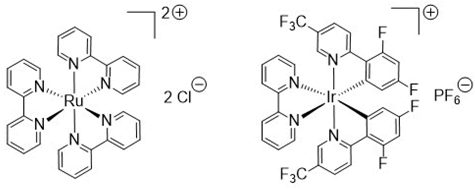
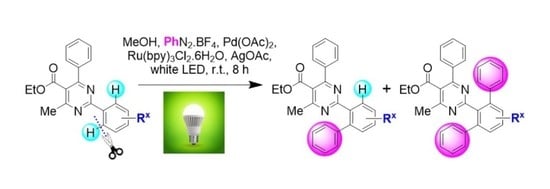
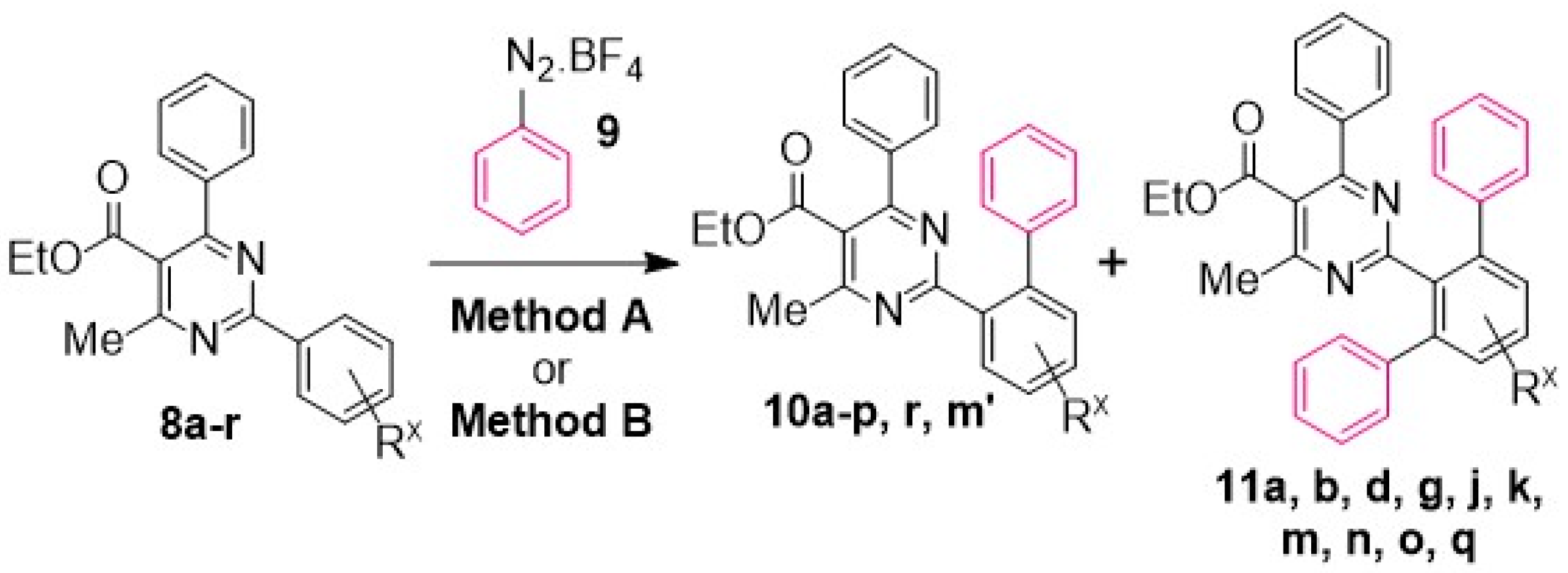
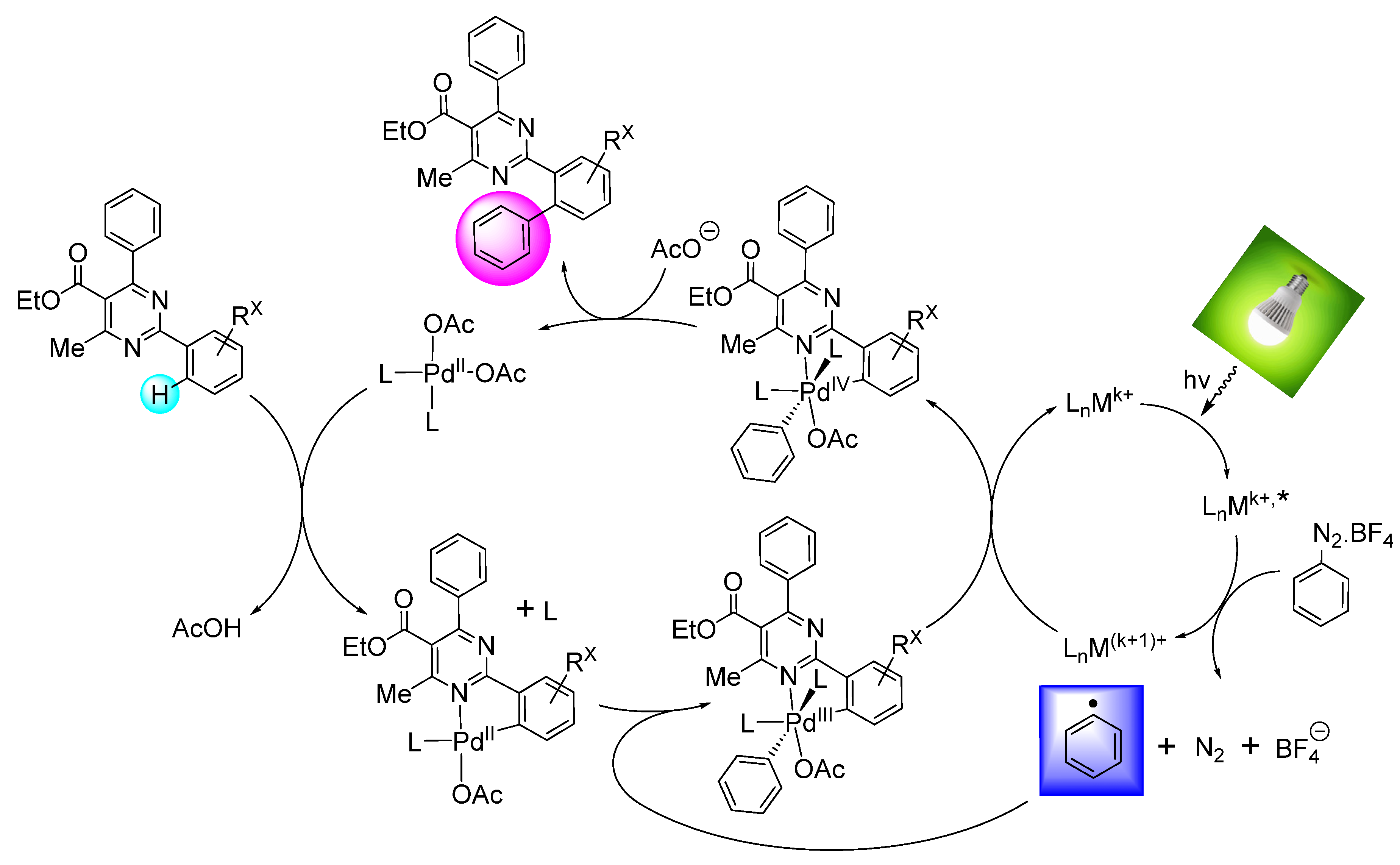
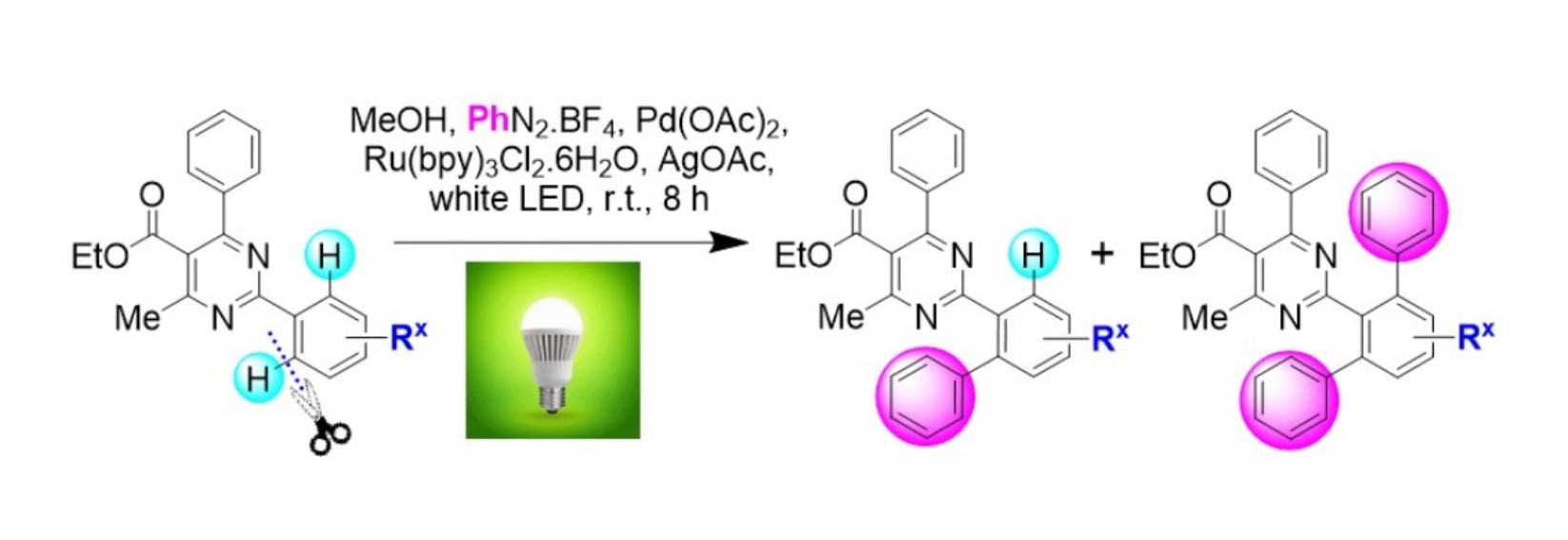
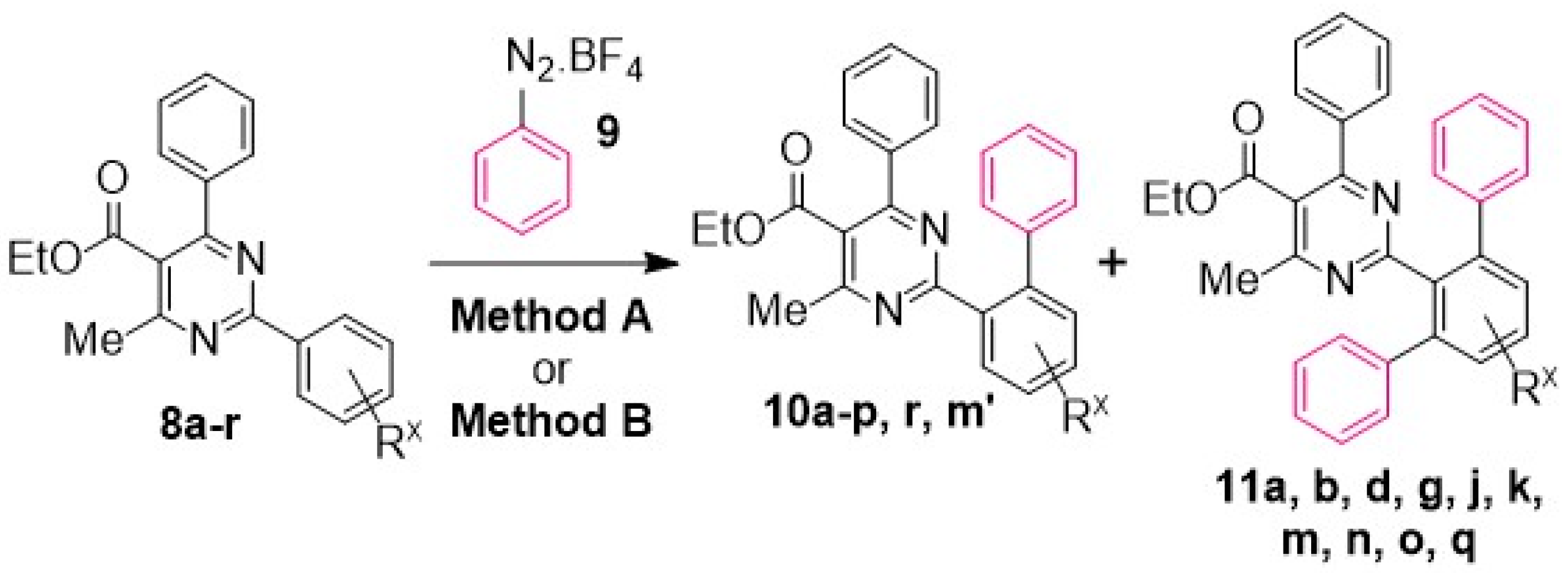
General Method A for Pd(II)-catalyzed, Ru(II)-photoinitiated (mono- and bis-) C–H phenylation of ethyl 4-methyl-2,6-diphenylpyrimidine-5-carboxylates: Phenyldiazonium tetrafluoroborate (9) (0.115 g, 0.6 mmol, 4 equiv), Pd(OAc)2 (0.003 g, 0.015 mmol, 0.1 equiv), Ru(bpy)3Cl2.6H2O (0.006 g, 0.0075 mmol, 0.05 equiv) and AgOAc (0.050 g, 0.3 mmol, 2 equiv) were transferred to a small round-bottom flask and the flask was sealed and set under nitrogen atmosphere. Anhydrous MeOH (1 mL) was added, and the mixture was vigorously stirred for 5 min. The substrate ethyl 4-methyl-2,6-diphenylpyrimidine-5-carboxylate of type 8 (0.15 mmol, 1 equiv), dissolved in MeOH (1 mL), was syringed in, and the flask was submitted to irradiation with two white household LED lamps (Phillips, 12.5 W, 1521 lumen) placed on opposite sides of the sample at 1–2 cm distance from the flask at ambient temperature for 8 h. Subsequently, the reaction mixture was diluted with diethyl ether and washed first with aqueous NH4Cl 25% wt solution. The aqueous phase was back-extracted 2 more times with diethyl ether. The combined organic phase was then washed with aqueous Na2CO3 10% wt and aqueous NaCl saturated solution and dried over Na2SO4. The drying agent was removed by filtration and the solvent was removed under vacuum. The sample was re-dissolved in DCM and applied to a silica column prepared with hexane. Elution took place first with hexane and then with hexane-ethyl acetate step gradient (the end-ratio of solvents was different in each case depending on product polarity), leading to isolation of mono- and bis- (where applicable) CH-arylation products of types 10 and 11, respectively.
General Method B for Pd(II)-catalyzed, Ir(III)-photoinitiated (mono- and bis-) C–H phenylation of ethyl 4-methyl-2,6-diphenylpyrimidine-5-carboxylates: Phenyldiazonium tetrafluoroborate (9) (0.115 g, 0.6 mmol, 4 equiv), Pd(OAc)2 (0.003 g, 0.015 mmol, 0.1 equiv), and (2,2’-bipyridine)bis[3,5-difluoro-2-[5-(trifluoro-methyl)-2-pyridinyl-kN][phenyl-kC]iridium(III) hexa-fluorophosphate (0.008 g, 0.0075 mmol, 0.05 equiv) were transferred to a small round-bottom flask, and the flask was sealed and set under nitrogen atmosphere. Anhydrous MeOH (1 mL) was added, and the mixture was vigorously stirred for 5 min. The substrate ethyl 4-methyl-2,6-diphenylpyrimidine-5-carboxylate of type 8 (0.15 mmol, 1 equiv), dissolved in MeOH (1 mL), was syringed in, and the flask was submitted to irradiation with two white household LED lamps (Phillips, 12.5 W, 1521 lumen) placed on opposite sides of the sample at 1–2 cm distance from the flask at ambient temperature for 8 h. Subsequently, the reaction mixture was diluted with diethyl ether and washed first with aqueous NH4Cl 25% wt solution. The aqueous phase was back-extracted 2 more times with diethyl ether. The combined organic phase was then washed with aqueous Na2CO3 10% wt and aqueous NaCl saturated solution and dried over Na2SO4. The drying agent was removed by filtration and the solvent was removed under vacuum. The sample was re-dissolved in DCM and applied to a silica column prepared with hexane. Elution took place first with hexane and then with hexane-ethyl acetate step gradient (the end-ratio of solvents was different in each case depending on product polarity), leading to isolation of mono- and bis- (where applicable) CH-arylation products of types 10 and 11, respectively.
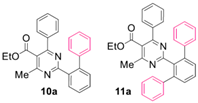
Reaction of substrate 8a: Method A; conversion 59%; ratio of 10a/11a = 1.24:1.
● Ethyl 2-([1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10a): White solid. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.59 (3H, s), 4.21 (2H, q, J = 7.2 Hz), 7.08 (2H, d, J = 7.8 Hz), 7.23 (2H, d, J= 7.2 Hz), 7.28 (2H, app. t, J = 7.7 Hz), 7.31–7.39 (4H, m, signals overlapping), 7.45–7.55 (3H, m, signals overlapping), 7.98 (1H, d, J = 7.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.67, 22.67, 61.82, 122.52, 126.40, 127.52, 128.11, 128.18, 128.32, 129.30, 129.71, 129.79, 130.79, 130.94, 137.39, 137.53, 142.04, 142.29, 162.70, 165.12, 166.59, 168.22. MS (ES-API), m/z: calcd for C26H22N2O2: 394.17; found 395.10 [M + H+]. m.p. 116 °C.
● Ethyl 2-([1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11a): White wax. 1H NMR (CDCl3), δ (ppm): 1.06 (3H, t, J = 7.1 Hz), 2.31 (3H, s), 4.16 (2H, q, J = 7.1 Hz), 7.00 (2H, d, J = 8.0 Hz), 7.20 (4H, dd, J1 = 7.7 Hz, J2 = 2.0 Hz), 7.26–7.30 (8H, m, signals overlapping), 7.37 (1H, tt, J1 = 7.3 Hz, J2 = 1.3 Hz), 7.51 (2H, d, J = 7.7 Hz), 7.57 (1H, t, J = 6.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.63, 22.09, 61.77, 122.26, 126.52, 127.85, 128.12, 128.15, 128.94, 129.31, 129.54, 129.62, 136.98, 137.58, 141.59, 141.95, 162.60, 164.04, 166.96, 167.95. MS (ES-API), m/z: calcd for C32H26N2O2: 470.20; found 471.20 [M + H+].
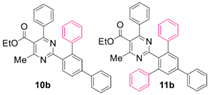
Reaction of substrate 8b: Method A; conversion 58%; ratio of 10b/11b = 1.27:1.
● Ethyl 2-([1,1’:3’,1’’-terphenyl]-4’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10b): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.11 (3H, t, J = 7.1 Hz), 2.60 (3H, s), 4.22 (2H, q, J = 7.1 Hz), 7.11 (2H, d, J = 8.0 Hz), 7.27–7.31 (4H, m, signals overlapping), 7.34–7.40 (5H, m, signals overlapping), 7.47 (2H, app. t, J = 7.6 Hz), 7.68 (2H, d, J = 8.0 Hz), 7.72 (2H, m, signals overlapping), 8.09 (1H, d, J = 8.8 Hz). 13C NMR (CDCl3), δ (ppm): 13.67, 22.67, 61.86, 122.49, 126.16, 126.53, 127.28, 127.73, 128.16, 128.19, 128.37, 128.83, 129.31, 129.82, 129.84, 131.43, 136.21, 137.55, 140.41, 142.38, 142.52, 142.59, 162.72, 165.13, 166.28, 168.25. MS (ES-API), m/z: calcd for C32H26N2O2: 470.20; found 471.10 [M + H+].
● Ethyl 4-methyl-6-phenyl-2-(5’-phenyl-[1,1’:3’,1’’-terphenyl]-2’-yl)pyrimidine-5-carboxylate (11b): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.05 (3H, t, J = 7.1 Hz), 2.30 (3H, s), 4.15 (2H, q, J = 7.1 Hz), 7.00 (2H, d, J = 8.0 Hz), 7.21–7.31 (12H, m, signals overlapping), 7.36 (1H, t, J = 7.6 Hz), 7.38 (1H, t, J = 7.6 Hz), 7.46 (2H, app. t, J = 7.6 Hz), 7.69 (2H, d, J = 8.0 Hz), 7.73 (2H, s). 13C NMR (CDCl3), δ (ppm): 13.62, 22.14, 61.77, 122.27, 126.67, 127.33, 127.72, 127.94, 128.15, 128.17, 128.35, 128.85, 129.35, 129.69, 135.90, 137.58, 140.45, 141.60, 141.80, 142.56, 162.63, 164.12, 166.82, 167.98. MS (ES-API), m/z: calcd for C38H30N2O2: 546.23; found 547.20 [M + H+].
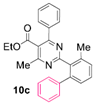
Reaction of substrate 8c: Method B; conversion 52%; only 10c.
● Ethyl 4-methyl-2-(3-methyl-[1,1’-biphenyl]-2-yl)-6-phenylpyrimidine-5-carboxylate (10c): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.01 (3H, t, J = 7.2 Hz), 2.27 (3H, s), 2.49 (3H, s), 4.12 (2H, q, J = 7.2 Hz), 7.06 (2H, m), 7.12 (1H, m), 7.15 (2H, d, J = 7.4 Hz), 7.19 (2H, d, J = 7.3 Hz), 7.22 (1H, d, J = 7.4 Hz), 7.24 (1H, d, J = 6.4 Hz), 7.26 (2H, app. t, J = 7.4 Hz), 7.30 (1H, d, J = 7.4 Hz), 7.31 (1H, t, J = 7.2 Hz). 13C NMR (CDCl3), δ (ppm): 13.63, 20.38, 22.50, 61.84, 122.74, 126.39, 127.79, 127.81, 128.23, 128.28, 128.80, 129.19, 129.65, 129.78, 136.31, 137.61, 137.69, 141.54, 141.84, 162.99, 164.44, 167.26, 168.07. MS (ES-API), m/z: calcd for C27H24N2O2: 408.18; found 409.10 [M + H+].
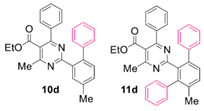
Reaction of substrate 8d: Method A; conversion 42%; ratio of 10d/11d = 3.67:1.
● Ethyl 4-methyl-2-(4-methyl-[1,1’-biphenyl]-2-yl)-6-phenylpyrimidine-5-carboxylate (10d): White solid. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.46 (3H, s), 2.59 (3H, s), 4.21 (2H, q, J = 7.2 Hz), 7.08 (2H, d, J = 8.0 Hz), 7.21 (2H, d, J = 7.5 Hz), 7.28 (2H, app. t, J = 7.3 Hz), 7.30–7.38 (6H, m, signals overlapping), 7.78 (1H, bs). 13C NMR (CDCl3), δ (ppm): 13.65, 21.09, 22.63, 61.78, 122.50, 126.18, 128.03, 128.16, 128.35, 129.34, 129.75, 130.48, 130.89, 131.27, 137.19, 137.23, 137.61, 139.32, 142.30, 162.70, 165.04, 166.76, 168.25. MS (ES-API), m/z: calcd for C27H24N2O2: 408.18; found 409.10 [M + H+]. m.p. 133 °C.
● Ethyl 4-methyl-2-(4’-methyl-[1,1’:3’,1’’-terphenyl]-2’-yl)-6-phenylpyrimidine-5-carboxylate (11d): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.00 (3H, t, J =7.1 Hz), 2.21 (3H, s), 2.26 (3H, s), 4.10 (2H, q, J = 7.1 Hz), 7.03 (2H, d, J = 8.2 Hz), 7.15 (2H, app. t, J = 7.7 Hz), 7.16–7.24 (8H, m, signals overlapping), 7.28 (2H, app. t, J =7.6 Hz), 7.35 (1H, t, J = 7.4 Hz), 7.39 (1H, d, J= 8.0 Hz), 7.41 (1H, d, J = 8.0 Hz). 13C NMR (CDCl3), δ (ppm): 13.56, 20.69, 21.99, 61.63, 121.88, 126.33, 126.40, 127.50, 127.79, 128.09, 128.12, 129.13, 129.35, 129.49, 129.96, 130.58, 135.69, 137.76, 137.85, 139.02, 140.04, 141.11, 141.63, 162.38, 163.67, 167.17, 167.96. MS (ES-API), m/z: calcd for C33H28N2O2: 484.22; found 485.20 [M + H+].
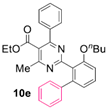
Reaction of substrate 8e: Method A; conversion 30%; only 10e.
● Εthyl 2-(3-butoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10e): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 0.83 (3H, t, J = 7.5 Hz), 1.06 (3H, t, J = 7.2 Hz), 1.32 (2H, hex, J = 7.5 Hz), 1.62 (2H, quint, J = 7.0 Hz), 2.58 (3H, s), 4.00 (2H, t, J = 6.5 Hz), 4.18 (2H, q, J = 7.2 Hz), 6.99 (1H, d, J = 8.4 Hz), 7.06 (1H, d, J = 7.8 Hz), 7.16–7.26 (7H, m, signals overlapping), 7.32 (2H, app. t, J = 7.7 Hz), 7.36 (1H, t, J = 7.2 Hz), 7.40 (1H, app. t, J = 8.1 Hz). 13C NMR (CDCl3), δ (ppm): 13.62, 13.72, 19.14, 22.43, 31.19, 61.73, 68.43, 111.52, 122.29, 122.78, 126.57, 127.80, 127.95, 128.20, 128.22, 129.22, 129.57, 129.90, 137.85, 141.16, 142.70, 156.99, 163.01, 164.11, 165.39, 168.17. MS (ES-API), m/z: calcd for C30H30N2O3: 466.23; found 467.20 [M + H+].
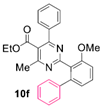
Reaction of substrate 8f: Method B; conversion 31%; only 10f.
● Ethyl 2-(3-methoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10f): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.07 (3H, t, J = 7.1 Hz), 2.59 (3H, s), 3.82 (3H, s), 4.18 (2H, q, J = 7.1 Hz), 7.01 (1H, d, J = 8.2 Hz), 7.07 (1H, dd, J1 = 7.8 Hz, J2 = 0.8 Hz), 7.17 (2H, m), 7.20 (1H, m), 7. 22 (2H, m), 7.25 (2H, m), 7.32 (2H, app. t, J = 7.5 Hz), 7.37 (1H, tt, J1 = 7.3 Hz, J2 = 1.5 Hz), 7.43 (1H, app. t, J = 8.0 Hz). 13C NMR (CDCl3), δ (ppm): 13.65, 22.57, 56.00, 61.76, 110.50, 122.47, 123.02, 126.63, 127.80, 128.25, 128.26, 128.51, 129.20, 129.62, 129.92, 137.79, 141.02, 142.81, 157.32, 163.05, 164.32, 165.34, 168.13. MS (ES-API), m/z: calcd for C27H24N2O3: 424.18; found 425.10 [M + H+].
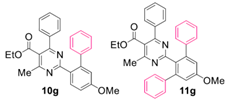
Reaction of substrate 8g: Method A; conversion 33%; ratio of 10g/11g = 1.22:1.
● Ethyl 2-(5-methoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10g): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.2 Hz), 2.55 (3H, s), 3.88 (3H, s), 4.19 (2H, q, J = 7.2 Hz), 6.97 (1H, d, J = 2.6 Hz), 7.00 (1H, dd, J1 = 8.6 Hz, J2 = 2.6 Hz), 7.06 (2H, d, J = 8.2 Hz), 7.25 (2H, dd, J1 = 7.8 Hz, J2 =1.6 Hz), 7.27 (2H, d, J = 8.0 Hz), 7.32–7.38 (4H, m, signals overlapping), 7.99 (1H, d, J = 8.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 22.67, 55.47, 61.74, 113.06, 116.37, 121.97, 126.48, 128.11, 128.12, 128.33, 129.15, 129.70, 130.11, 132.63, 137.67, 142.56, 143.97, 160.61, 162.56, 164.99, 166.12, 168.39. MS (ES-API), m/z: calcd for C27H24N2O3: 424.18; found 425.10 [M + H+].
● Ethyl 2-(5’-methoxy-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11g): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.03 (3H, t, J = 7.2 Hz), 2.26 (3H, s), 3.90 (3H, s), 4.13 (2H, q, J = 7.2 Hz), 6.93 (2H, d, J = 8.0 Hz), 7.02 (2H, s), 7.18 (4H, dd, J1 = 7.6 Hz, J2 = 2.1 Hz), 7.22–7.28 (7H, m, signals overlapping), 7.34 (2H, t, J = 7.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.60, 22.08, 55.54, 61.70, 115.05, 122.62, 127.86, 128.10, 128.14, 129.15, 129.23, 129.55, 132.64, 137.67, 141.73, 143.75, 159.49, 162.49, 163.96, 166.80, 168.06. MS (ES-API), m/z: calcd for C33H28N2O3: 500.21; found 501.10 [M + H+].
Reaction of substrate 8h: Method B; conversion 42%; only 10h.
● Ethyl 2-(3,4-dimethoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10h): White wax. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.1 Hz), 2.58 (3H, s), 3.88 (3H, s), 3.94 (3H, s), 4.20 (2H, q, J = 7.1 Hz), 7.06 (1H, d, J = 8.5 Hz), 7.12 (2H, m), 7.18–7.22 (4H, m, signals overlapping), 7.29 (2H, d, J = 8.3 Hz), 7.33 (2H, app. t, J = 7.6 Hz), 7.39 (1H, t, J1 = 7.3 Hz). 13C NMR (CDCl3), δ (ppm): 13.65, 22.45, 56.09, 61.57, 61.83, 113.22, 125.68, 126.32, 127.02, 127.84, 128.28, 128.29, 129.25, 129.76, 133.35, 134.61, 137.69, 140.69, 147.28, 152.37, 163.00, 164.32, 164.93, 168.04. MS (ES-API), m/z: calcd for C28H26N2O4: 454.19; found 455.10 [M + H+].
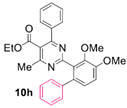
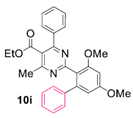
Reaction of substrate 8i: Method B; conversion 30%; only 10i.
● Ethyl 2-(3,5-dimethoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10i): White wax. 1H NMR (CDCl3), δ (ppm): 1.07 (3H, t, J = 7.1 Hz), 2.58 (3H, s), 3.80 (3H, s), 3.86 (3H, s), 4.18 (2H, q, J = 7.1 Hz), 6.57 (1H, d, J = 2.2 Hz), 6.60 (1H, d, J = 2.2 Hz), 7.17 (2H, m), 7.19–7.25 (5H, m, signals overlapping), 7.31 (2H, app. t, J = 7.4 Hz), 7.36 (1H, t, J = 7.3 Hz). 13C NMR (CDCl3), δ (ppm): 13.64, 22.55, 55.52, 55.96, 61.72, 98.15, 106.68, 121.13, 122.69, 126.74, 127.83, 128.19, 128.23, 129.10, 129.56, 137.81, 141.32, 143.90, 158.63, 160.91, 162.94, 164.21, 165.20, 168.19. MS (ES-API), m/z: calcd for C28H26N2O4: 454.19; found 455.10 [M + H+].
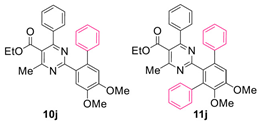
Reaction of substrate 8j: Method A; conversion 47%; ratio of 10j/11j = 1.85:1.
● Ethyl 2-(4,5-dimethoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10j): Colorless wax. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.2 Hz), 2.58 (3H, s), 3.95 (3H, s), 4.01 (3H, s), 4.20 (2H, q, J = 7.2 Hz), 6.95 (1H, s), 7.05 (2H, d, J = 7.8 Hz), 7.24 (2H, d, J = 7.6 Hz), 7.27 (2H, app. t, J = 7.8 Hz), 7.31–7.39 (4H, m, signals overlapping), 7.58 (1H, s). 13C NMR (CDCl3), δ (ppm): 13.67, 22.68, 56.08, 56.15, 61.77, 113.64, 113.98, 122.13, 126.23, 128.12, 128.13, 128.34, 129.37, 129.60, 129.74, 135.85, 137.62, 142.55, 148.23, 150.03, 162.63, 165.04, 166.05, 168.31. MS (ES-API), m/z: calcd for C28H26N2O4: 454.19; found 455.10 [M + H+].
● Ethyl 2-(4’,5’-dimethoxy-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11j): Colorless wax. 1H NMR (CDCl3), δ (ppm): 1.01 (3H, t, J = 7.2 Hz), 2.24 (3H, s), 3.57 (3H, s), 3.96 (3H, s), 4.11 (2H, q, J = 7.2 Hz), 6.97 (2H, d, J = 7.7 Hz), 7.05 (1H, s), 7.17–7.29 (12H, m, signals overlapping), 7.34 (1H, t, J = 7.4 Hz). 13C NMR (CDCl3), δ (ppm): 13.59, 21.99, 56.10, 60.69, 61.68, 113.71, 121.85, 126.57, 127.36, 127.89, 128.08, 128.09, 128.13, 129.33, 129.55, 130.33, 131.17, 136.49, 136.76, 137.67, 138.01, 141.61, 146.16, 153.16, 162.49, 163.82, 166.38, 167.96. MS (ES-API), m/z: calcd for C34H30N2O4: 530.22; found 531.20 [M + H+].
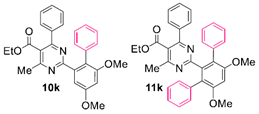
Reaction of substrate 8k: Method A; conversion 44%; ratio of 10k/11k = 1.20:1.
● Ethyl 2-(4,6-dimethoxy-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10k): White wax. 1H NMR (CDCl3), δ (ppm): 1.07 (3H, t, J = 7.2 Hz), 2.55 (3H, s), 3.76 (3H, s), 3.90 (3H, s), 4.18 (2H, q, J = 7.2 Hz), 6.66 (1H, d, J =2.5 Hz), 7.05 (1H, d, J = 2.5 Hz), 7.11 (2H, m), 7.19 (2H, m), 7.24 (1H, tt, J1 = 7.3 Hz, J2 = 1.8 Hz), 7.27–7.31 (4H, m, signals overlapping), 7.37 (1H, tt, J1 = 7.4 Hz, J2 = 1.6 Hz). 13C NMR (CDCl3), δ (ppm): 13.64, 22.57, 55.55, 56.03, 61.77, 100.51, 106.32, 122.56, 123.87, 126.12, 127.54, 128.12, 128.40, 129.75, 131.09, 137.20, 137.58, 139.98, 158.24, 159.82, 162.75, 164.88, 166.66, 168.16. MS (ES-API), m/z: calcd for C28H26N2O4: 454.19; found 455.20 [M + H+].
● Ethyl 2-(4’,6’-dimethoxy-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11k): White wax. 1H NMR (CDCl3), δ (ppm): 0.99 (3H, t, J = 7.2 Hz), 2.23 (3H, s), 3.83 (6H, s), 4.08 (2H, q, J = 7.2 Hz), 6.75 (1H, s), 7.02 (2H, m), 7.12–7.20 (10H, m, signals overlapping), 7.28 (2H, app. t, J = 7.5 Hz), 7.35 (1H, tt, J1 = 7.5 Hz, J2 = 1.3 Hz). 13C NMR (CDCl3), δ (ppm): 13.53, 21.91, 56.25, 61.60, 96.89, 121.88, 123.14, 126.23, 127.29, 128.06, 128.11, 129.46, 130.95, 136.65, 137.73, 140.63, 157.23, 162.37, 163.57, 166.42, 167.86. MS (ES-API), m/z: calcd for C34H30N2O4: 530.22; found 531.20 [M + H+]. m.p. 145 °C.
Reaction of substrate 8l: Method A; conversion 20%; only 10l.
● Ethyl 2-(3-fluoro-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10l): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.59 (3H, s), 4.21 (2H, q, J = 7.2 Hz), 7.18 (2H, m, signals overlapping), 7.22 (2H, d, J = 7.2 Hz), 7.26–7.30 (5H, m, signals overlapping), 7.33 (2H, app. t, J = 8.0 Hz), 7.39 (1H, tt, J1 = 7.3 Hz, J2 = 1.5 Hz), 7.46 (1H, app. dt, J1 = 8.0 Hz, J2 = 5.7 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 22.60, 61,91, 114.99 (d, J = 22.2 Hz), 123.40, 125.91 (d, J = 3.1 Hz), 126.43 (d, J = 14.5 Hz), 127.06, 128.09, 128.25, 128.35, 129.20, 129.90, 130.45 (d, J = 9.2 Hz), 137.38, 140.15 (d, J = 2.1 Hz), 143.69 (d, J = 2.7 Hz), 160.56 (d, J = 248.8 Hz), 162.93, 163.17, 164.86, 167.83. MS (ES-API), m/z: calcd for C26H21FN2O2: 412.16; found 413.10 [M + H+].
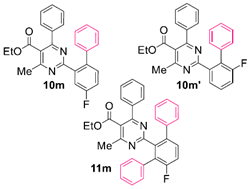
Reaction of substrate 8m: Method A; conversion 28%; ratio of 10m/10m’/11m = 3.33:1:1.
● Ethyl 2-(4-fluoro-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10m): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.1 Hz), 2.58 (3H, s), 4.21 (2H, q, J = 7.1 Hz), 7.09 (2H, d, J = 7.5 Hz), 7.21 (3H, m, signals overlapping), 7.29 (2H, app. t, J = 7.5 Hz), 7.31–7.35 (3H, m, signals overlapping), 7.38 (1H, t, J = 7.1 Hz), 7.43 (1H, dd, J1 = 9.4 Hz, J2 = 5.6 Hz), 7.72 (1H, dd, J1 = 9.4 Hz, J2 = 3.0 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 22.61, 61.91, 116.57 (d, J = 21.4 Hz), 117.58 (d, J = 22.9 Hz), 122.91, 126.52, 128.14, 128.23, 128.33, 129.31, 129.92, 132.64 (d, J = 8.1 Hz), 137.37, 138.23 (d, J = 3.7 Hz), 139.07 (d, J = 7.4 Hz), 141.44, 162.07 (d, J = 247.2 Hz), 162.85, 165.31, 165.35, 168.05. MS (ES-API), m/z: calcd for C26H21FN2O2: 412.16; found 413.10 [M + H+].
● Ethyl 2-(6-fluoro-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10m’): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.08 (3H, t, J = 7.1 Hz), 2.56 (3H, s), 4.20 (2H, q, J = 7.1 Hz), 7.08 (2H, m), 7.25 (2H, m), 7.27 (1H, m), 7.30 (2H, app. t, J = 7.5 Hz), 7.34–7.40 (4H, m, signals overlapping), 7.45 (1H, app. dt, J1 = 7.9 Hz, J2 = 5.3 Hz), 7.79 (1H, d, J = 7.5 Hz). 13C NMR (CDCl3), δ (ppm): 13.65, 22.57, 61.89, 117.08 (d, J = 23.6 Hz), 122.79, 126.45 (d, J = 3.2 Hz), 127.07, 127.98, 127.99, 128.22, 128.34, 128.86 (d, J = 8.9 Hz), 129.93, 130.35, 134.82, 137.35, 139.82, 160.07 (d, J = 245.8 Hz), 162.85, 165.13, 165.37, 168.02. MS (ES-API), m/z: calcd for C26H21FN2O2: 412.16; found 413.10 [M + H+].
● Ethyl 2-(4’-fluoro-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11m): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.02 (3H, t, J = 7.1 Hz), 2.27 (3H, s), 4.12 (2H, q, J = 7.1 Hz), 6.98 (2H, d, J = 8.0 Hz), 7.15 (2H, dd, J1 = 7.5 Hz, J2 = 2.2 Hz), 7.20–7.24 (6H, m, signals overlapping), 7.26–7.30 (4H, m, signals overlapping), 7.32 (1H, app. t, J = 8.8 Hz), 7.36 (1H, t, J = 7.4 Hz), 7.46 (1H, dd, J1 = 8.6 Hz, J2 = 5.2 Hz). 13C NMR (CDCl3), δ (ppm): 13.58, 22.04, 61.79, 116.23 (d, J = 22.6 Hz), 122.47, 126.67, 127.23, 127.73, 127.94, 128.11, 128.19, 129.32, 129.72, 130.30, 130.32, 130.89 (d, J = 8.7 Hz), 134.26, 137.46, 137.92, 139.29, 140.77, 159.26 (d, J = 246.8 Hz), 162.68, 164.13, 165.69, 167.75. MS (ES-API), m/z: calcd for C32H25FN2O2: 488.19; found 489.20 [M + H+].
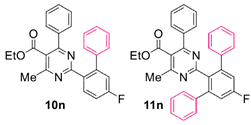
Reaction of substrate 8n: Method A; conversion 24%; ratio of 10n/11n = 4.05:1.
● Ethyl 2-(5-fluoro-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10n): White solid. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.2 Hz), 2.57 (3H, s), 4.21 (2H, q, J = 7.2 Hz), 7.07 (2H, d, J = 7.9 Hz), 7.18 (2H, m, signals overlapping), 7.22 (2H, dd, J1 = 7.3 Hz, J2 = 2.5 Hz), 7.28 (2H, app. t, J = 7.8 Hz), 7.36 (4H, m, signals overlapping), 8.00 (1H, m). 13C NMR (CDCl3), δ (ppm): 13.66, 22.64, 61.86, 114.39 (d, J = 21.8 Hz), 117.76 (d, J = 21.8 Hz), 122.56, 126.91, 128.22, 128.26, 128.28, 129.07, 129.88, 133.01 (d, J = 8.8 Hz), 133.59 (d, J = 2.9 Hz), 137.42, 141.28 (d, J = 2.0 Hz), 144.58 (d, J = 8.4 Hz), 162.76, 163.31 (d, J = 249.3 Hz), 165.21, 165.65, 168.13. MS (ES-API), m/z: calcd for C26H21FN2O2: 412.16; found 413.10 [M + H+]. m.p. 118 °C.
● Ethyl 2-(5’-fluoro-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11n): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.03 (3H, t, J = 7.1 Hz), 2.28 (3H, s), 4.14 (2H, q, J = 7.1 Hz), 6.96 (2H, d, J = 7.5 Hz), 7.16 (4H, m), 7.20 (2H, d, J = 9.1 Hz), 7.23–7.29 (8H, m, signals overlapping), 7.35 (1H, t, J = 7.2 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 22.10, 61.87, 116.21 (d, J = 21.7 Hz), 122.38, 127.03, 128.01, 128.10, 128.20, 129.11, 129.72, 133.41, 137.46, 140.60 (d, J = 2.0 Hz), 144.47 (d, J = 9.1 Hz), 162.28 (d, J = 246.2 Hz), 162.70, 165.00, 166.19, 167.86. MS (ES-API), m/z: calcd for C32H25FN2O2: 488.19; found 489.10 [M + H+].
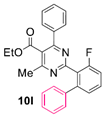
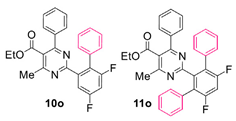
Reaction of substrate 8o: Method A; conversion 26%; ratio of 10o/11o = 1.60:1.
● Ethyl 2-(4,6-difluoro-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10o): Colorless wax. 1H NMR (CDCl3), δ (ppm): 1.09 (3H, t, J = 7.1 Hz), 2.55 (3H, s), 4.20 (2H, q, J = 7.1), 7.02 (1H, app. dt, J1 = 9.0 Hz, J2 = 2.8 Hz), 7.09 (2H, d, J = 8.0 Hz), 7.22 (2H, d, J = 7.0 Hz), 7.30 (2H, app. t, J = 7.7 Hz), 7.33–7.37 (3H, m, signals overlapping), 7.39 (1H, t, J = 7.4 Hz), 7.57 (1H, ddd, J1 = 9.0 Hz, J2 = 2.8 Hz, J3 = 1.3 Hz). 13C NMR (CDCl3), δ (ppm): 13.66, 22.57, 61.94, 105.25 (dd, J1 = 27.8 Hz, J2 = 25.5 Hz), 113.64 (dd, J1 = 23.0 Hz, J2 = 3.6 Hz), 123.15, 125.88, 127.19, 128.06, 128.26, 128.33, 130.03, 130.39, 134.15, 137.17, 140.76 (dd, J1 = 9.0 Hz, J2 = 4.5 Hz), 160.31 (dd, J1 = 246.5 Hz, J2 = 12.0 Hz), 161.78 (dd, J1 = 250.2 Hz, J2 = 13.0 Hz), 162.96, 164.34, 165.33, 167.88. MS (ES-API), m/z: calcd for C26H20F2N2O2: 430.15; found 431.10 [M + H+].
● Ethyl 2-(4’,6’-difluoro-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11o): Colorless viscous oil. 1H NMR (CDCl3), δ (ppm): 1.01 (3H, t, J = 7.1 Hz), 2.26 (3H, s), 4.11 (2H, q, J = 7.1 Hz), 7.00 (2H, d, J = 7.9 Hz), 7.12 (1H, t, J = 9.2 Hz), 7.19 (4H, dd, J1 = 7.3 Hz, J2 = 2.0 Hz), 7.23–7.27 (6H, m, peaks overlapping), 7.29 (2H, t, J = 7.9 Hz), 7.37 (1H, t, J = 7.5 Hz). 13C NMR (CDCl3), δ (ppm): 13.63, 22.00, 61.82, 104.74 (t, J = 26.6 Hz), 122.58, 127.38, 127.70, 127.86, 128.06, 128.22, 129.83, 130.40, 133.50, 137.29, 141.20 (dd, J1 = 25.3 Hz, J2 = 11.0 Hz), 159.12 (dd, J1 = 248.6 Hz, J2 = 12.7 Hz), 162.74, 164.18, 164.51, 167.59. MS (ES-API), m/z: calcd for C32H24F2N2O2: 506.18; found 507.10 [M + H+].
Reaction of substrate 8p: Method A; conversion 58%; only 10p.
● Ethyl 2-(5-cyano-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10p): White wax. 1H NMR (CDCl3), δ (ppm): 1.10 (3H, t, J = 7.1 Hz), 2.58 (3H, s), 4.22 (2H, q, J = 7.1 Hz), 7.09 (2H, m), 7.20 (2H, m), 7.30 (2H, app. t, J = 7.6 Hz), 7.34–7.42 (4H, m, signals overlapping), 7.76 (1H, dd, J1 = 7.7 Hz, J2 = 1.6 Hz), 7.77 (1H, s), 8.10 (1H, d, J1 = 7.7 Hz). 13C NMR (CDCl3), δ (ppm): 13.67, 22.61, 62.04, 113.34, 118.48, 123.31, 127.39, 128.27, 128.33, 128.46, 129.05, 130.12, 130.84, 131.62, 134.49, 137.10, 140.11, 141.46, 143.20, 162.96, 164.85, 165.54, 167.84. MS (ES-API), m/z: calcd for C27H21N3O2: 419.16; found 420.10 [M + H+].
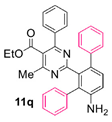
Reaction of substrate 8q: Method A; conversion 33%; only 11q.
● Ethyl 2-(4’-amino-[1,1’:3’,1’’-terphenyl]-2’-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (11q): Brown wax. 1H NMR (CDCl3), δ (ppm): 1.01 (3H, t, J =7.1 Hz), 2.26 (3H, s), 4.10 (2H, q, J = 7.1 Hz), 6.93 (1H, d, J = 8.2 Hz), 7.01 (2H, d, J = 8.0 Hz), 7.12 (2H, d, J = 7.6 Hz), 7.16 (1H, t, J = 7.0 Hz), 7.19 (2H, d, J = 7.3 Hz), 7.21 (1H, t, J = 7.0 Hz), 7.22–7.30 (6H, m, signals overlapping), 7.31 (1H, d, J =8.2 Hz), 7.34 (1H, t, J = 7.3 Hz). 13C NMR (CDCl3), δ (ppm): 13.57, 22.01, 61.64, 116.08, 121.94, 125.81, 127.00, 127.75, 128.08, 128.12, 128.33, 129.33, 129.49, 130.37, 130.40, 130.41, 132.08, 137.37, 137.72, 138.23, 141.79, 143.36, 162.38, 163.73, 167.02, 167.98. MS (ES-API), m/z: calcd for C32H27N3O2: 485.21; found 486.20 [M + H+].
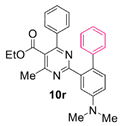
Reaction of substrate 8r: Method A; conversion 64%; only 10r.
● Ethyl 2-(4-(dimethylamino)-[1,1’-biphenyl]-2-yl)-4-methyl-6-phenylpyrimidine-5-carboxylate (10r): Orange film. 1H NMR (CDCl3), δ (ppm): 1.11 (3H, t, J = 7.1 Hz), 2.71 (3H, s), 3.12 (6H, s), 4.24 (2H, q, J = 7.1 Hz), 6.85 (1H, dd, J1 = 9.1 Hz, J2 = 2.9 Hz), 7.13 (1H, d, J = 2.9 Hz), 7.33 (1H, t, J = 7.1 Hz), 7.37 (4H, m, signals overlapping), 7.42 (1H, t, J = 7.3 Hz), 7.67 (4H, m, signals overlapping), 7.88 (1H, d, J = 9.1 Hz). 13C NMR (CDCl3), δ (ppm): 13.71, 22.77, 40.39, 61.86, 112.75, 113.16, 118.66, 122.55, 123.18, 128.39, 128.51, 128.72, 129.31, 129.80, 137.99, 139.68, 141.66, 151.94, 153.31, 162.90, 164.50, 166.56, 168.41. MS (ES-API), m/z: calcd for C28H27N3O2: 437.21; found 467.2 [M + Li+ + Na+], 543.2 [M + Pd2+].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}