
CO2 Hydrogenation on NixMg1−xAl2O4: A Comparative Study of MgAl2O4 and NiAl2O4

Abstract
:1. Introduction
2. Experimental Section
2.1. Synthesis of NixMg1−xAl2O4 Catalysts
2.2. Catalytic Activity Tests
2.3. Characterization of Catalysts
2.4. Computation Details
3. Experimental Results
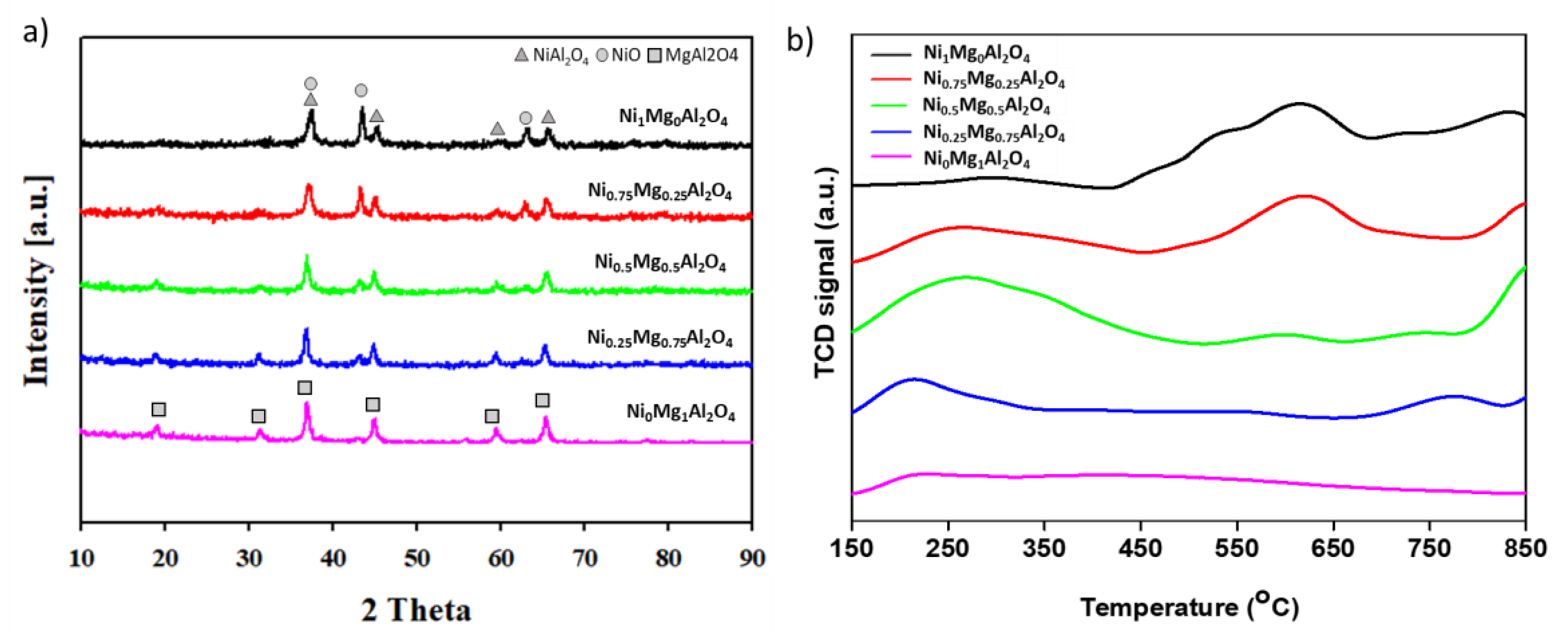
3.1. Characterization of Catalysts
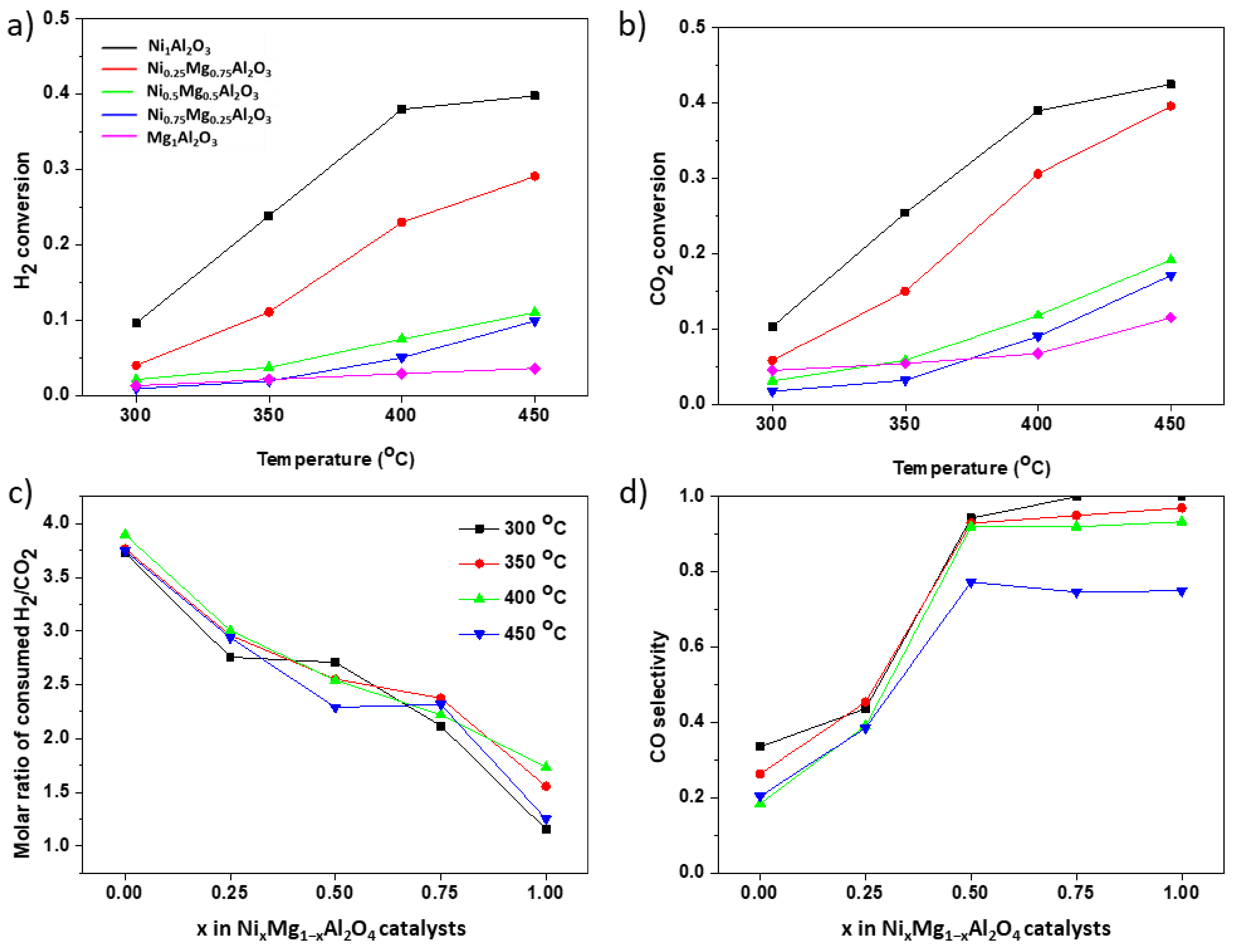
3.2. CO2 Hydrogenation Activity
4. Computational Results
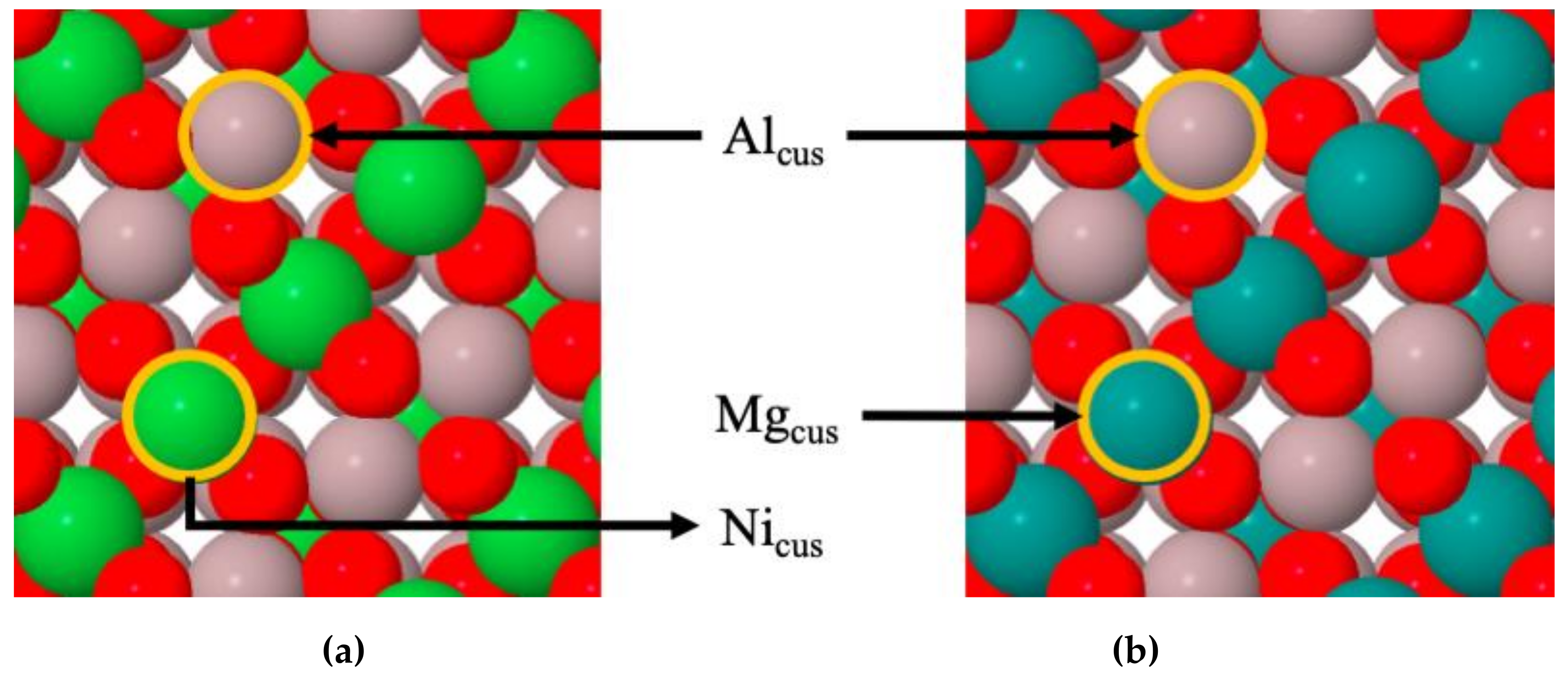
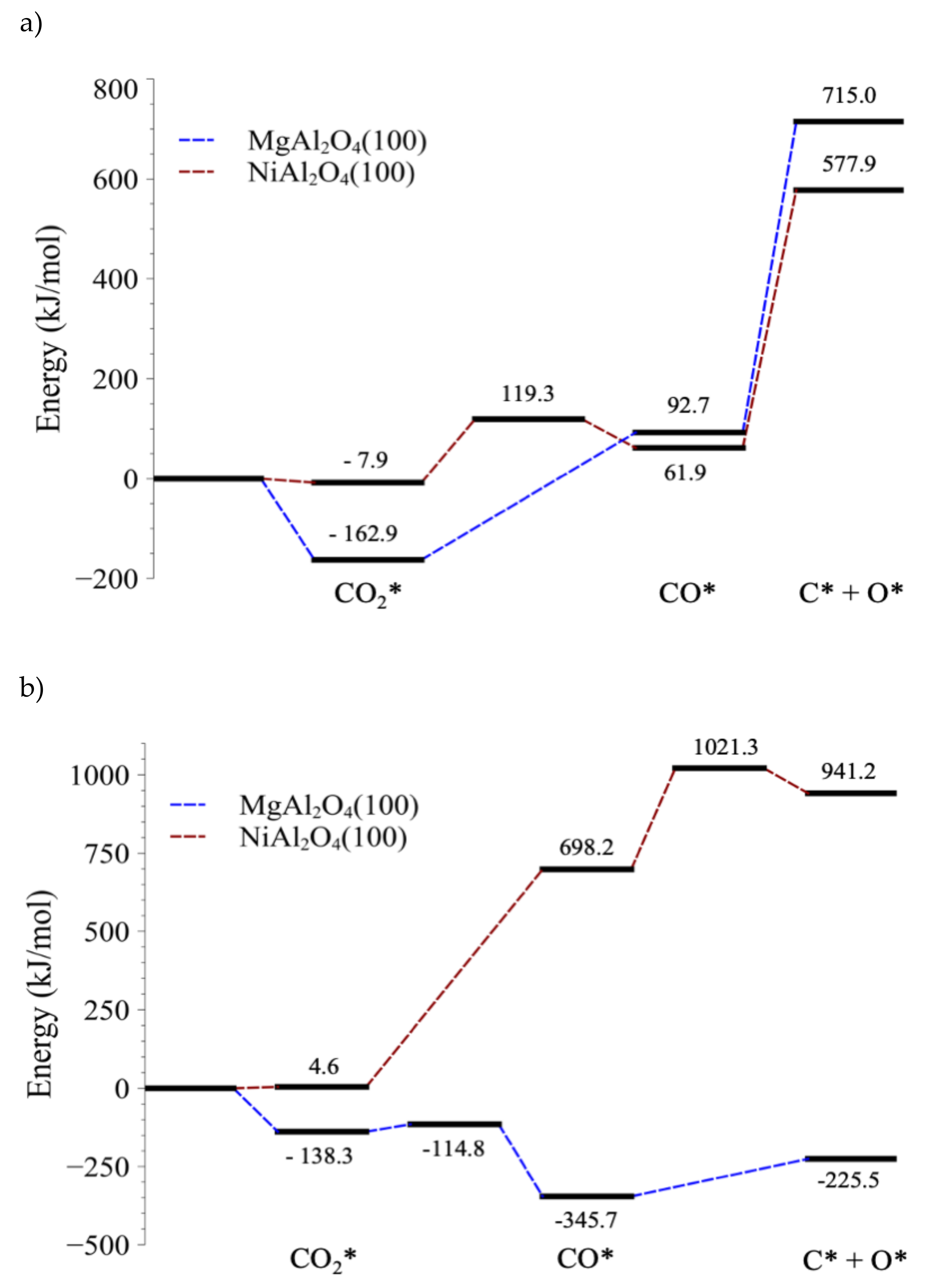
4.1. Stabilities of CO2 and CO on MgAl2O4(100) and NiAl2O4(100)
4.2. Electronic Analysis of the Stability of Adsorbed CO
4.3. C–O Bond-Breaking Kinetics
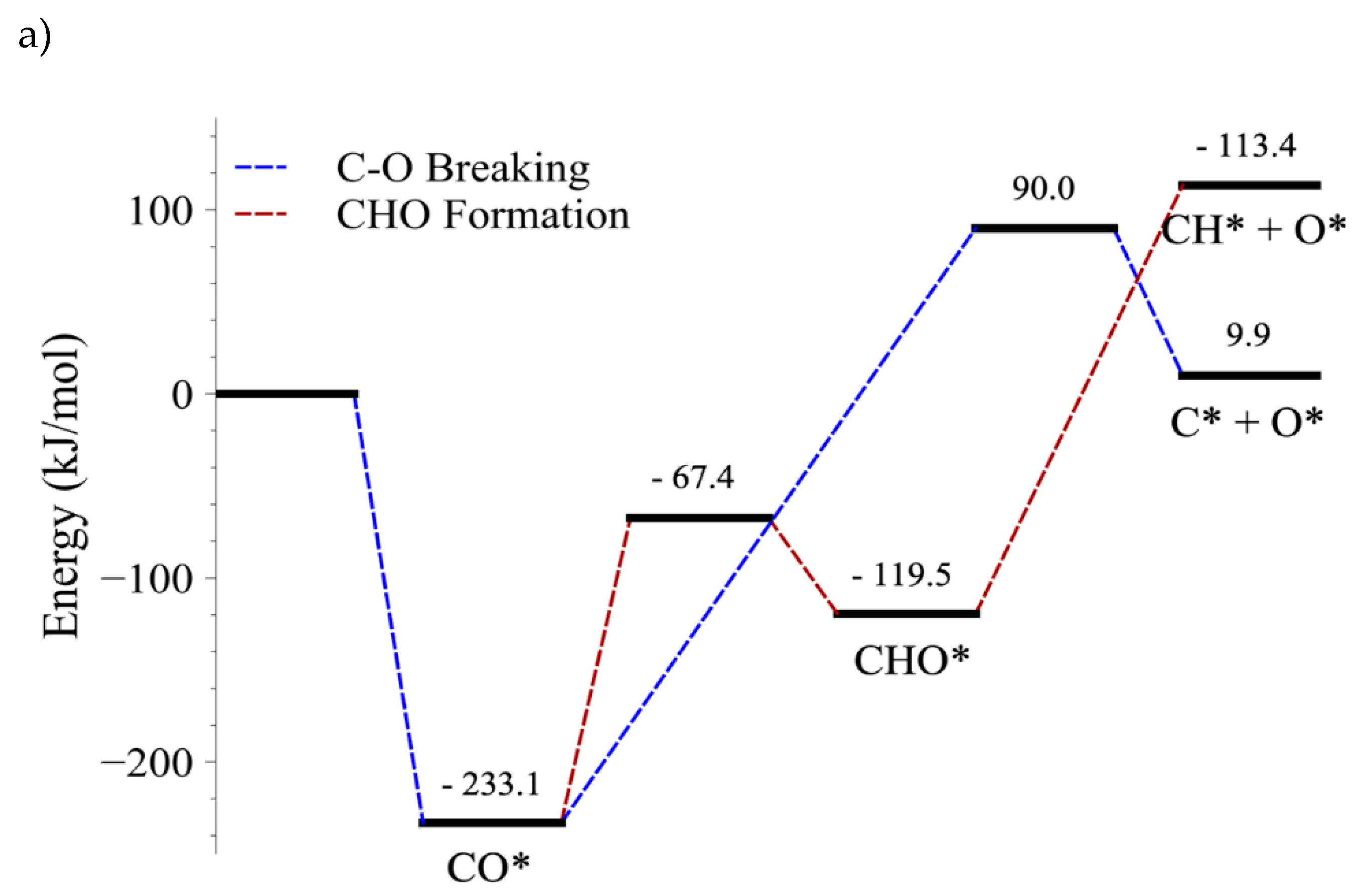
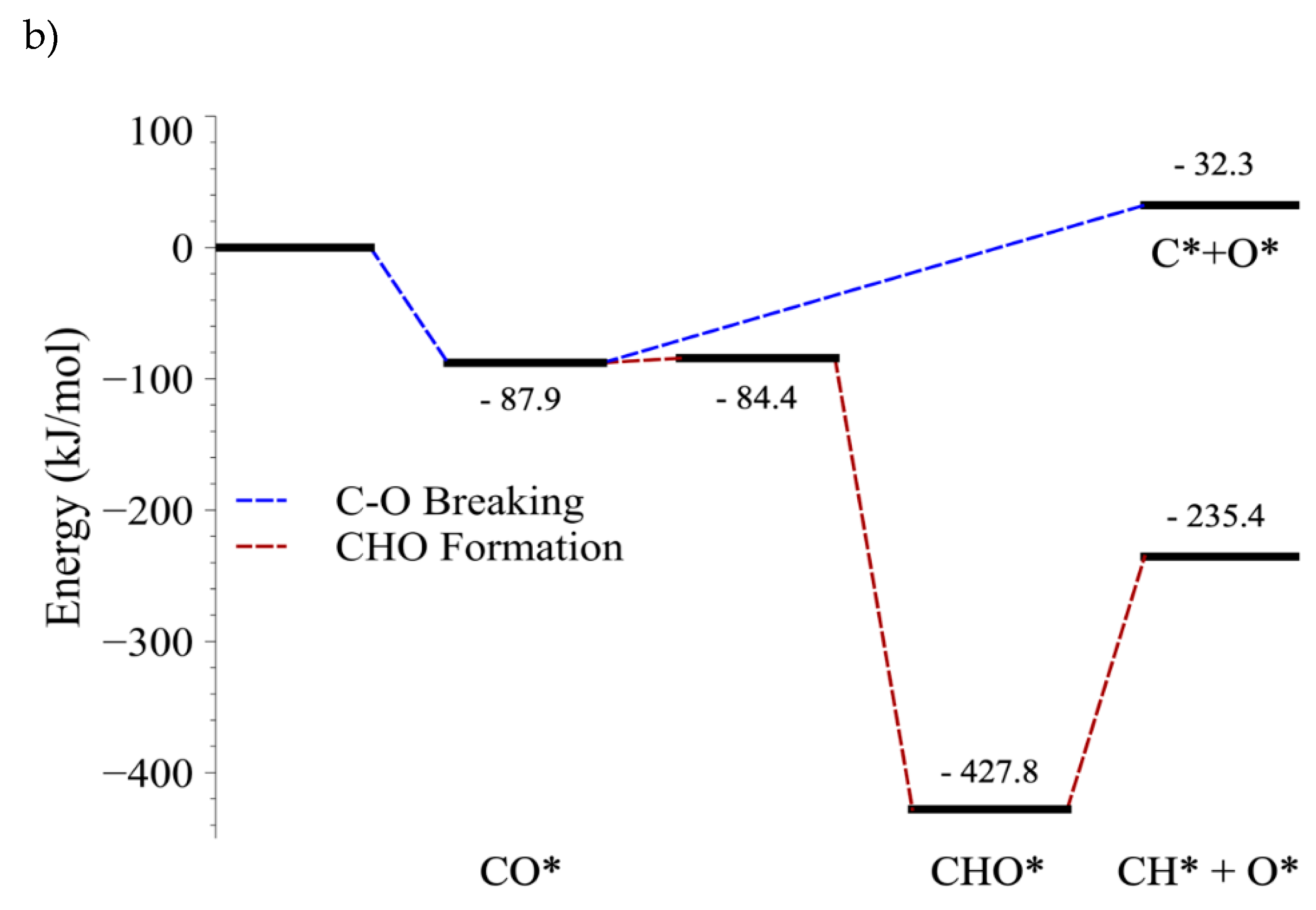
4.4. C–O Bond Cleavage of CO vs. C–O Bond Cleavage of CHO
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strateg. Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Shen, W.; Chen, X.; Qiu, J.; Hayward, J.A.; Sayeef, S.; Osman, P.; Meng, K.; Dong, Z.Y. A comprehensive review of variable renewable energy levelized cost of electricity. Renew. Sustain. Energy Rev. 2020, 133, 110301. [Google Scholar] [CrossRef]
- MacDowell, N.; Florin, N.; Buchard, A.; Hallett, J.; Galindo, A.; Jackson, G.; Adjiman, C.S.; Williams, C.K.; Shah, N.; Fennell, P. An overview of CO2 capture technologies. Energy Environ. Sci. 2010, 3, 1645–1669. [Google Scholar] [CrossRef] [Green Version]
- Tapia, J.F.D.; Lee, J.-Y.; Ooi, R.E.H.; Foo, D.C.Y.; Tan, R.R. A review of optimization and decision-making models for the planning of CO2 capture, utilization and storage (CCUS) systems. Sustain. Prod. Consum. 2018, 13, 1–15. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Hernandez Lalinde, J.A.; Roongruangsree, P.; Ilsemann, J.; Bäumer, M.; Kopyscinski, J. CO2 methanation and reverse water gas shift reaction. Kinetic study based on in situ spatially-resolved measurements. Chem. Eng. J. 2020, 390, 124629. [Google Scholar] [CrossRef]
- Mills, G.A.; Steffgen, F.W. Catalytic Methanation. Catal. Rev. 1974, 8, 159–210. [Google Scholar] [CrossRef]
- Kirchner, J.; Baysal, Z.; Kureti, S. Activity and Structural Changes of Fe-based Catalysts during CO2 Hydrogenation towards CH4—A Mini Review. ChemCatChem 2020, 12, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Chen, C.-S.; Budi, C.S.; Wu, H.-C.; Saikia, D.; Kao, H.-M. Size-Tunable Ni Nanoparticles Supported on Surface-Modified, Cage-Type Mesoporous Silica as Highly Active Catalysts for CO2 Hydrogenation. ACS Catal. 2017, 7, 8367–8381. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Mukti, R.R.; Taufiq-Yap, Y.H.; Sazegar, M.R. Highly active Ni-promoted mesostructured silica nanoparticles for CO2 methanation. Appl. Catal. B Environ. 2014, 147, 359–368. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdöhelyi, A.; Bánsági, T. Methanation of CO2 on supported rhodium catalyst. J. Catal. 1981, 68, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on group VIII metals: II. Kinetics and mechanism of CO2 hydrogenation on nickel. J. Catal. 1982, 77, 460–472. [Google Scholar] [CrossRef]
- Yang Lim, J.; McGregor, J.; Sederman, A.J.; Dennis, J.S. Kinetic studies of CO2 methanation over a Ni/γ-Al2O3 catalyst using a batch reactor. Chem. Eng. Sci. 2016, 141, 28–45. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.C.; Park, N.-K.; Kang, M.; Kang, D.; Seo, M.W.; Lee, D.; Jeon, S.G.; Ryu, H.-J. CO2 hydrogenation activity of Ni-Mg-Al2O3 catalysts: Reaction behavior on NiAl2O4 and MgAl2O4. Korean J. Chem. Eng. 2021, 38, 1188–1196. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 Methanation: The Effect of Catalysts and Reaction Conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Karam, L.; Bacariza, M.C.; Lopes, J.M.; Henriques, C.; Massiani, P.; El Hassan, N. Assessing the potential of xNi-yMg-Al2O3 catalysts prepared by EISA-one-pot synthesis towards CO2 methanation: An overall study. Int. J. Hydrogen Energy 2020, 45, 28626–28639. [Google Scholar] [CrossRef]
- Galhardo, T.S.; Braga, A.H.; Arpini, B.H.; Szanyi, J.; Gonçalves, R.V.; Zornio, B.F.; Miranda, C.R.; Rossi, L.M. Optimizing Active Sites for High CO Selectivity during CO2 Hydrogenation over Supported Nickel Catalysts. J. Am. Chem. Soc. 2021, 143, 4268–4280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ying, M.; Yu, J.; Zhan, W.; Wang, L.; Guo, Y.; Guo, Y. NixAl1O2-δ mesoporous catalysts for dry reforming of methane: The special role of NiAl2O4 spinel phase and its reaction mechanism. Appl. Catal. B Environ. 2021, 291, 120074. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.M.; Dholabhai, P.P.; Castro, R.H.R.; Uberuaga, B.P. Stabilization of MgAl2O4 spinel surfaces via doping. Surf. Sci. 2016, 649, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P.E. Crystal orbital hamilton populations (COHP). Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [Green Version]
- Ashok, J.; Raju, G.; Reddy, P.S.; Subrahmanyam, M.; Venugopal, A. Catalytic decomposition of CH4 over NiO–Al2O3–SiO2 catalysts: Influence of catalyst preparation conditions on the production of H2. Int. J. Hydrogen Energy 2008, 33, 4809–4818. [Google Scholar] [CrossRef]
- Vesselli, E.; Schweicher, J.; Bundhoo, A.; Frennet, A.; Kruse, N. Catalytic CO2 Hydrogenation on Nickel: Novel Insight by Chemical Transient Kinetics. J. Phys. Chem. C 2011, 115, 1255–1260. [Google Scholar] [CrossRef]
- Hinojosa, J.A.; Antony, A.; Hakanoglu, C.; Asthagiri, A.; Weaver, J.F. Adsorption of CO2 on a PdO(101) Thin Film. J. Phys. Chem. C 2012, 116, 3007–3016. [Google Scholar] [CrossRef]
- Thompson, T.L.; Diwald, O.; Yates, J.T. CO2 as a Probe for Monitoring the Surface Defects on TiO2(110)Temperature-Programmed Desorption. J. Phys. Chem. B 2003, 107, 11700–11704. [Google Scholar] [CrossRef]
- Henderson, M.A.; Epling, W.S.; Perkins, C.L.; Peden, C.H.F.; Diebold, U. Interaction of Molecular Oxygen with the Vacuum-Annealed TiO2(110) Surface: Molecular and Dissociative Channels. J. Phys. Chem. B 1999, 103, 5328–5337. [Google Scholar] [CrossRef]
- Wang, Y.; Lafosse, A.; Jacobi, K. Adsorption and Reaction of CO2 on the RuO2(110) Surface. J. Phys. Chem. B 2002, 106, 5476–5482. [Google Scholar] [CrossRef]
- Lafosse, A.; Wang, Y.; Jacobi, K. Carbonate formation on the O-enriched RuO2(110) surface. J. Chem. Phys. 2002, 117, 2823–2831. [Google Scholar] [CrossRef]
- Seiferth, O.; Wolter, K.; Dillmann, B.; Klivenyi, G.; Freund, H.J.; Scarano, D.; Zecchina, A. IR investigations of CO2 adsorption on chromia surfaces: Cr2O3 (0001)/Cr(110) versus polycrystalline α-Cr2O3. Surf. Sci. 1999, 421, 176–190. [Google Scholar] [CrossRef]
- Kim, M.; Pan, L.; Weaver, J.F.; Asthagiri, A. Initial Reduction of the PdO(101) Surface: Role of Oxygen Vacancy Formation Kinetics. J. Phys. Chem. C 2018, 122, 26007–26017. [Google Scholar] [CrossRef]
- Zhang, F.; Pan, L.; Li, T.; Diulus, J.T.; Asthagiri, A.; Weaver, J.F. CO Oxidation on PdO(101) during Temperature-Programmed Reaction Spectroscopy: Role of Oxygen Vacancies. J. Phys. Chem. C 2014, 118, 28647–28661. [Google Scholar] [CrossRef]
- Van Den Bossche, M.; Grönbeck, H. Methane Oxidation over PdO(101) Revealed by First-Principles Kinetic Modeling. J. Am. Chem. Soc. 2015, 137, 12035–12044. [Google Scholar] [CrossRef]
- Chin, Y.C.; Garcia-Dieguez, M.; Iglesia, E. Dynamics and Thermodynamics of Pd−PdO Phase Transitions: Effects of Pd Cluster Size and Kinetic Implications for Catalytic Methane Combustion. J. Phys. Chem. C 2016, 120, 1446–1460. [Google Scholar] [CrossRef]
- Schilling, A.C.; Groden, K.; Simonovis, J.P.; Hunt, A.; Hannagan, R.T.; Çlnar, V.; McEwen, J.S.; Sykes, E.C.H.; Waluyo, I. Accelerated Cu2O Reduction by Single Pt Atoms at the Metal-Oxide Interface. ACS Catal. 2020, 10, 4215–4226. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Sterk, E.B.; Palle, J.; Melcherts, A.E.M.; Zijlstra, B.; Groeneveld, E.; Berben, P.H.; Boereboom, J.M.; Hensen, E.J.M.; et al. Understanding carbon dioxide activation and carbon–carbon coupling over nickel. Nat. Commun. 2019, 10, 5330. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Surface Area (m2/g) | Total Pore Volume (mL/g) | Average Pore Size (nm) |
|---|---|---|---|
| Ni1Mg0Al2O4 | 38.0 | 0.17 | 10.8 |
| Ni0.75Mg0.25Al2O4 | 64.2 | 0.26 | 11.4 |
| Ni0.5Mg0.5Al2O4 | 38.1 | 0.13 | 9.4 |
| Ni0.25Mg0.75Al2O4 | 32.8 | 0.10 | 8.4 |
| Ni0Mg1Al2O4 | 32.1 | 0.10 | 8.2 |
| Adsorbed Molecule | MgAl2O4 | NiAl2O4 | ||||||
|---|---|---|---|---|---|---|---|---|
| Alcus | Mgcus | Alcus | Nicus | |||||
| 0 Ov | 1 Ov | 0 Ov | 1 Ov | 0 Ov | 1 Ov | 0 Ov | 1 Ov | |
| CO | 30.8 | 87.9 | 59.7 | 57.8 | 16.9 | - | 179.8 | 233.1 |
| CO2 | 75.3 | 187.1 | 162.9 | 138.3 | 7.9 | 68.3 | 36.5 | −4.6 |
| Bonding Types | MgAl2O4 | NiAl2O4 | ||||
|---|---|---|---|---|---|---|
| Mgcus | Nicus | |||||
| s, p | d | s, p, d | s, p | d | s, p, d | |
| Bonding | −2.08 | - | −2.08 | −2.68 | −3.58 | −6.26 |
| Anti-Bonding | 0.36 | - | 0.36 | 0.31 | 1.11 | 1.42 |
| Total Bonding | −1.72 | - | −1.72 | −2.37 | −2.47 | −4.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, B.; Ko, E.H.; Boo, J.; Kim, M.; Kang, D.; Park, N.-K. CO2 Hydrogenation on NixMg1−xAl2O4: A Comparative Study of MgAl2O4 and NiAl2O4. Catalysts 2021, 11, 1026. https://doi.org/10.3390/catal11091026
Seo B, Ko EH, Boo J, Kim M, Kang D, Park N-K. CO2 Hydrogenation on NixMg1−xAl2O4: A Comparative Study of MgAl2O4 and NiAl2O4. Catalysts. 2021; 11(9):1026. https://doi.org/10.3390/catal11091026
Chicago/Turabian StyleSeo, Boseok, Eun Hee Ko, Jinho Boo, Minkyu Kim, Dohyung Kang, and No-Kuk Park. 2021. "CO2 Hydrogenation on NixMg1−xAl2O4: A Comparative Study of MgAl2O4 and NiAl2O4" Catalysts 11, no. 9: 1026. https://doi.org/10.3390/catal11091026
APA StyleSeo, B., Ko, E. H., Boo, J., Kim, M., Kang, D., & Park, N.-K. (2021). CO2 Hydrogenation on NixMg1−xAl2O4: A Comparative Study of MgAl2O4 and NiAl2O4. Catalysts, 11(9), 1026. https://doi.org/10.3390/catal11091026