
Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method

 ,
,  ,
,  , ,
, ,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
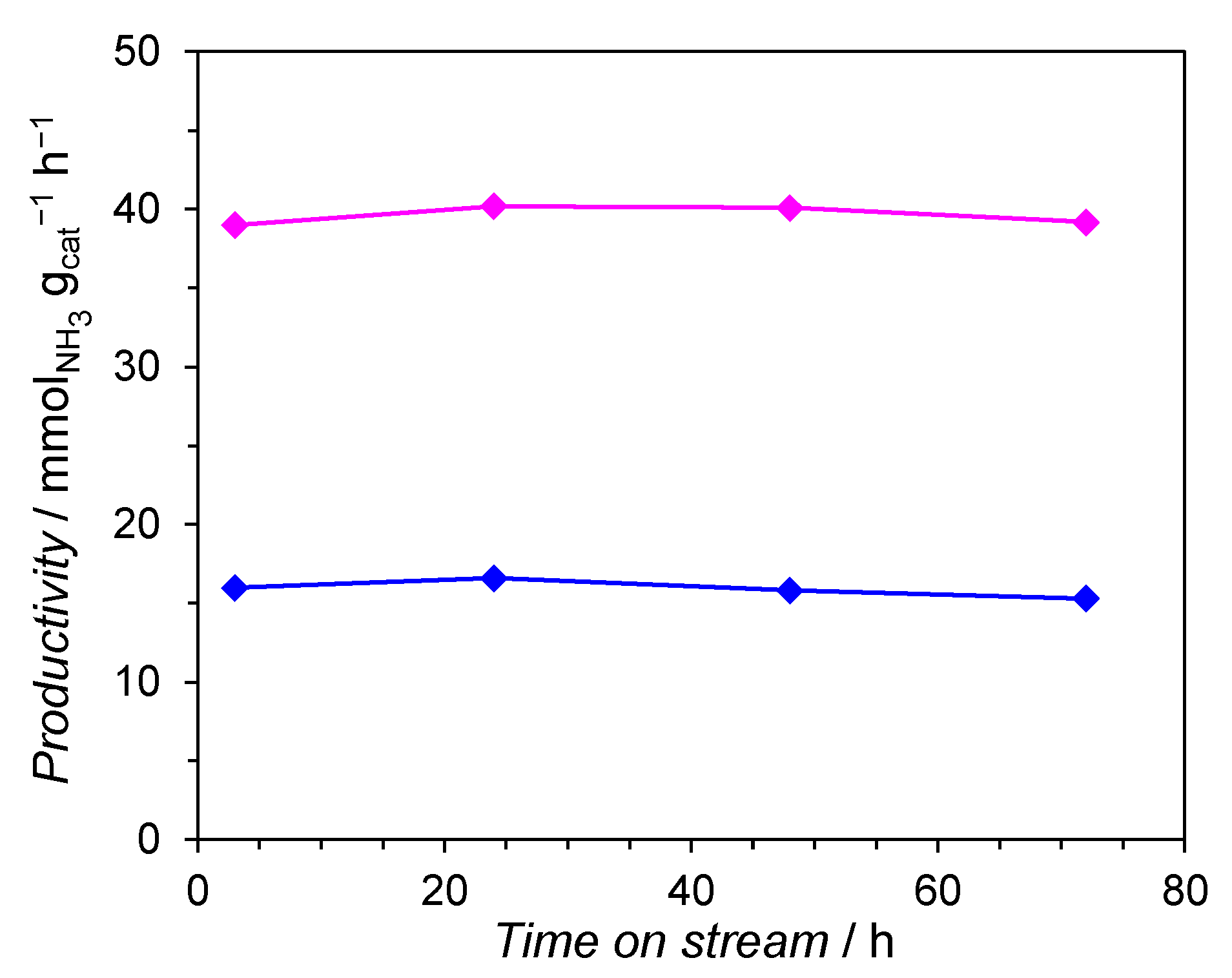
2.1. Evaluation of the Catalytic Performance
2.2. N2 Physisorption
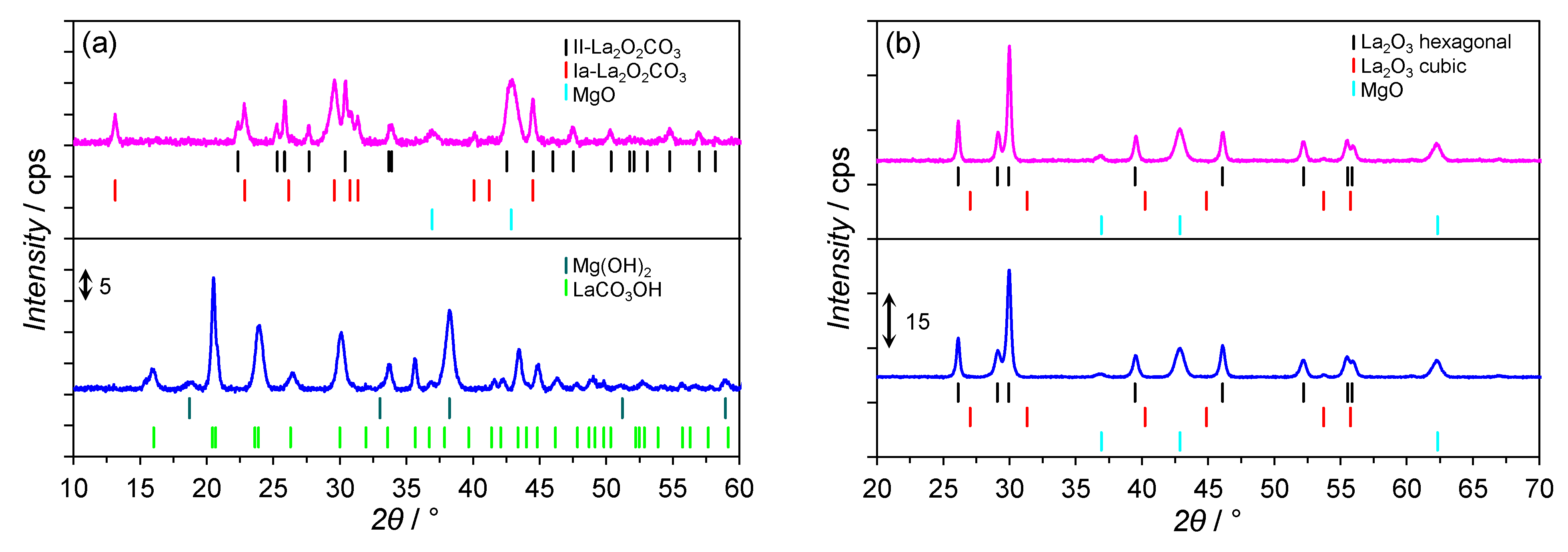
2.3. XRPD
2.4. DRIFTS
2.5. H2-TPR
- (1)
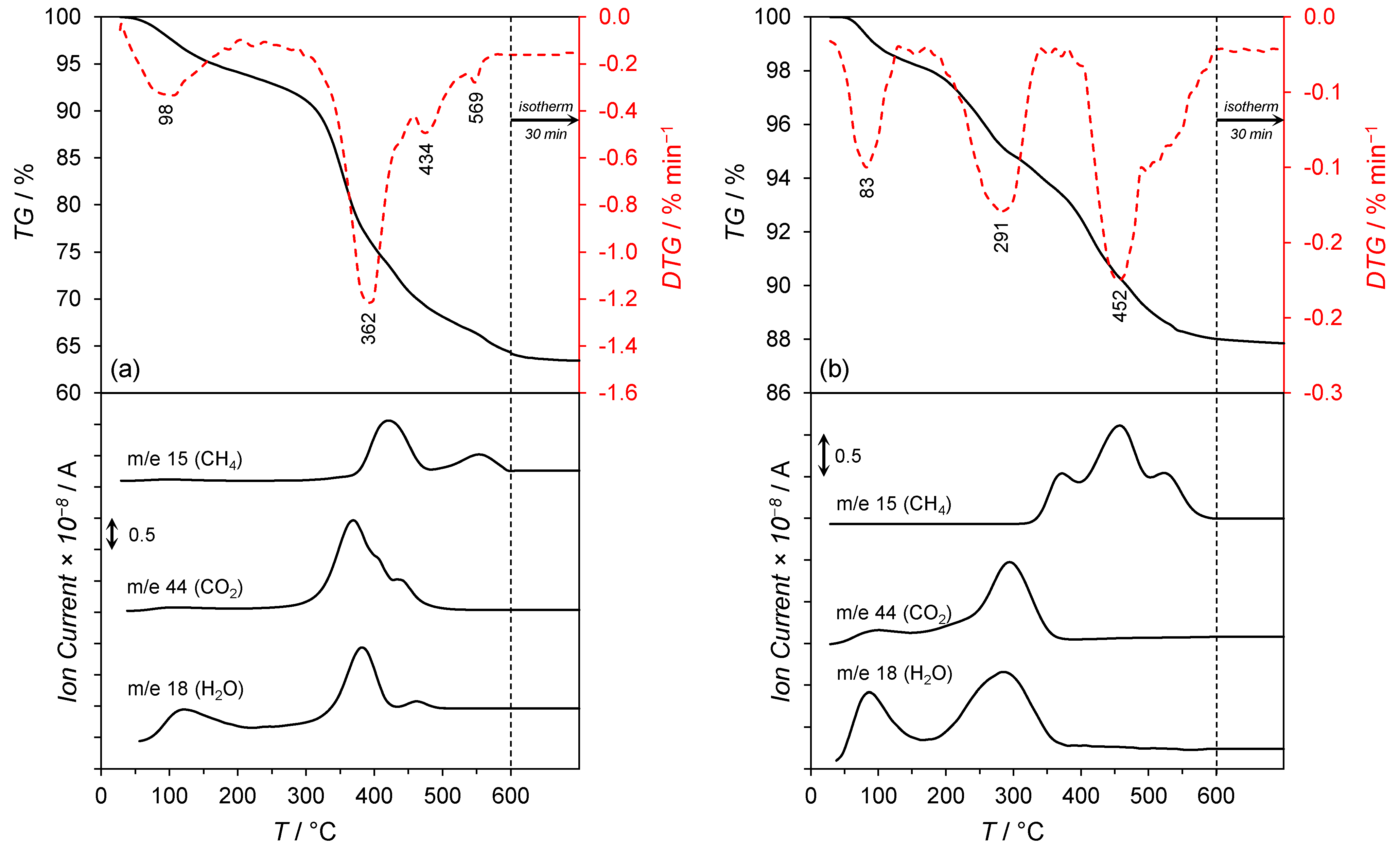
- 30–150 °C, a slight mass loss (ca. 5%) with a maximum rate of mass loss at 98 °C was observed. This was associated with the desorption of physisorbed water and carbon dioxide, as confirmed by the signals at m/e = 18 and m/e = 44 in the mass spectra.
- (2)
- 150–500 °C, a significant mass loss (ca. 25%) with the maximum rates of mass loss at 362 and 434 °C were recorded and associated with an intense water and carbon dioxide release (the signals at m/e = 18 and m/e = 44 in the mass spectrum). This was attributed to the thermal decomposition of the co-precipitated cobalt hydroxy carbonate to cobalt(II,III) oxide and lanthanum hydroxy carbonate to lanthanum oxide carbonate. The signal at m/e = 18 in the mass spectrum could also be attributable to the dehydroxylation of Mg(OH)2 occurring at ca. 350 °C [30] and the reduction of Co3O4 to metallic Co, occurring typically in the temperature range of 250–450 °C [31]. Moreover, the hydrogenation of CO2 (released upon the decarboxylation of lanthanum oxide carbonate) to methane occurred. The confirmation was the appearance of the signal at m/e = 15 in the mass spectrum with the maximum at 425 °C.
- (3)
- 500–600 °C, a slight mass loss (ca. 3%) related to the thermal decomposition of La2O2CO3 with subsequent hydrogenation of CO2 to CH4 was detected. The confirmation was the signal at m/e = 15 in the mass spectrum with the maximum at 551 °C.
- (1)
- 30–150 °C, a slight mass loss (ca. 2%) with a maximum rate of mass loss at 83 °C associated with the desorption of physisorbed water and carbon dioxide was observed. The confirmation was the signals at m/e = 18 and m/e = 44 in the mass spectra.
- (2)
- 150–400 °C, a mass loss of ca. 4% related to the reduction of Co3O4 to metallic Co and the thermal decomposition of La2O2CO3 to La2O3 was detected. This was confirmed by the signals at m/e = 18 and m/e = 44 in the mass spectrum, respectively. The appearance of the signal at m/e = 15 in the mass spectra was due to the hydrogenation of carbon dioxide (released upon the decarboxylation of La2O2CO3) to methane.
- (3)
- 400–600 °C, a mass loss of ca. 6% associated with the continuous hydrogenation of carbon dioxide to methane was observable, as confirmed by the signal at m/e = 15 in the mass spectrum with the maximum at 452 °C.
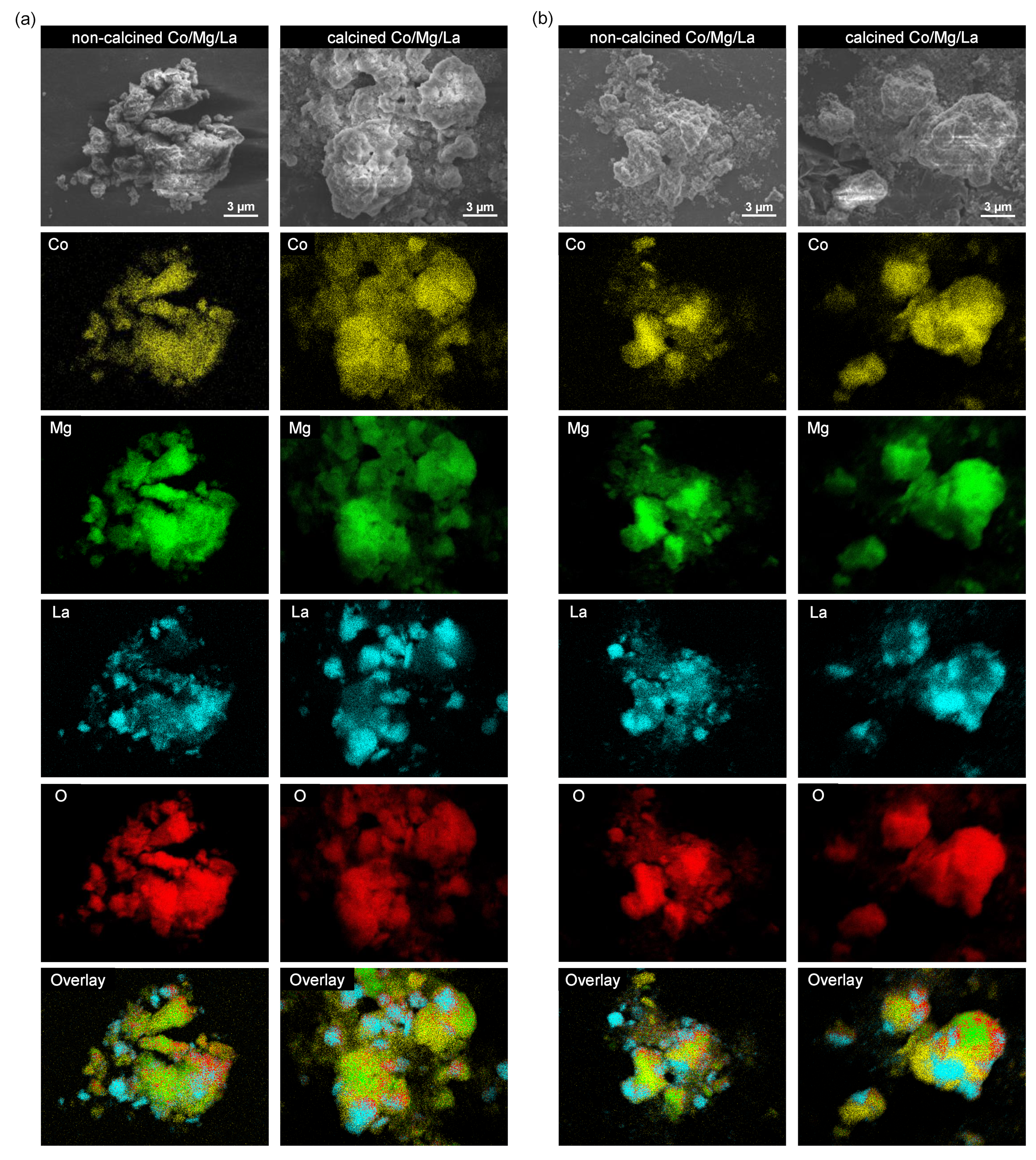
2.6. SEM and TEM
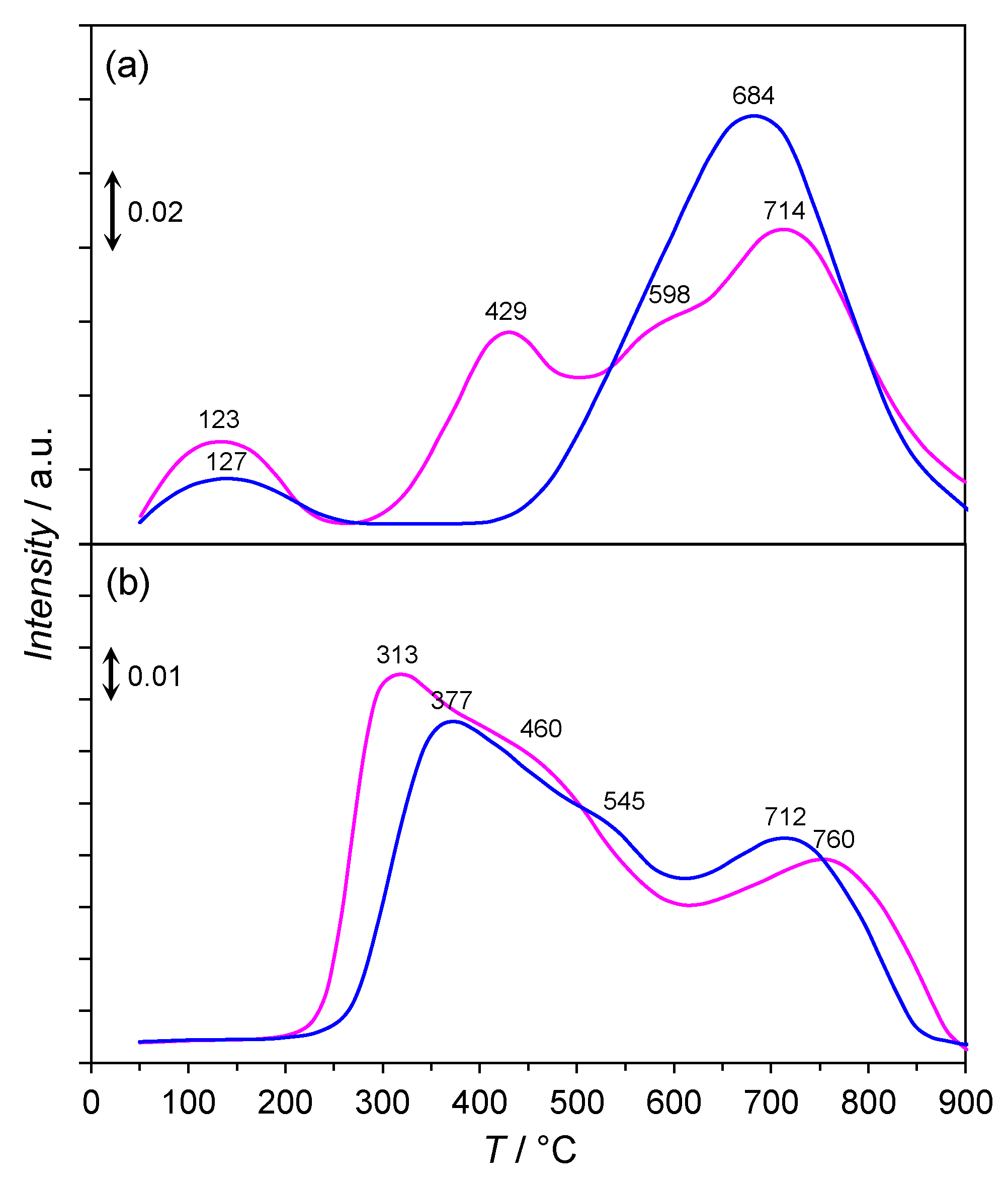
2.7. CO2-TPD
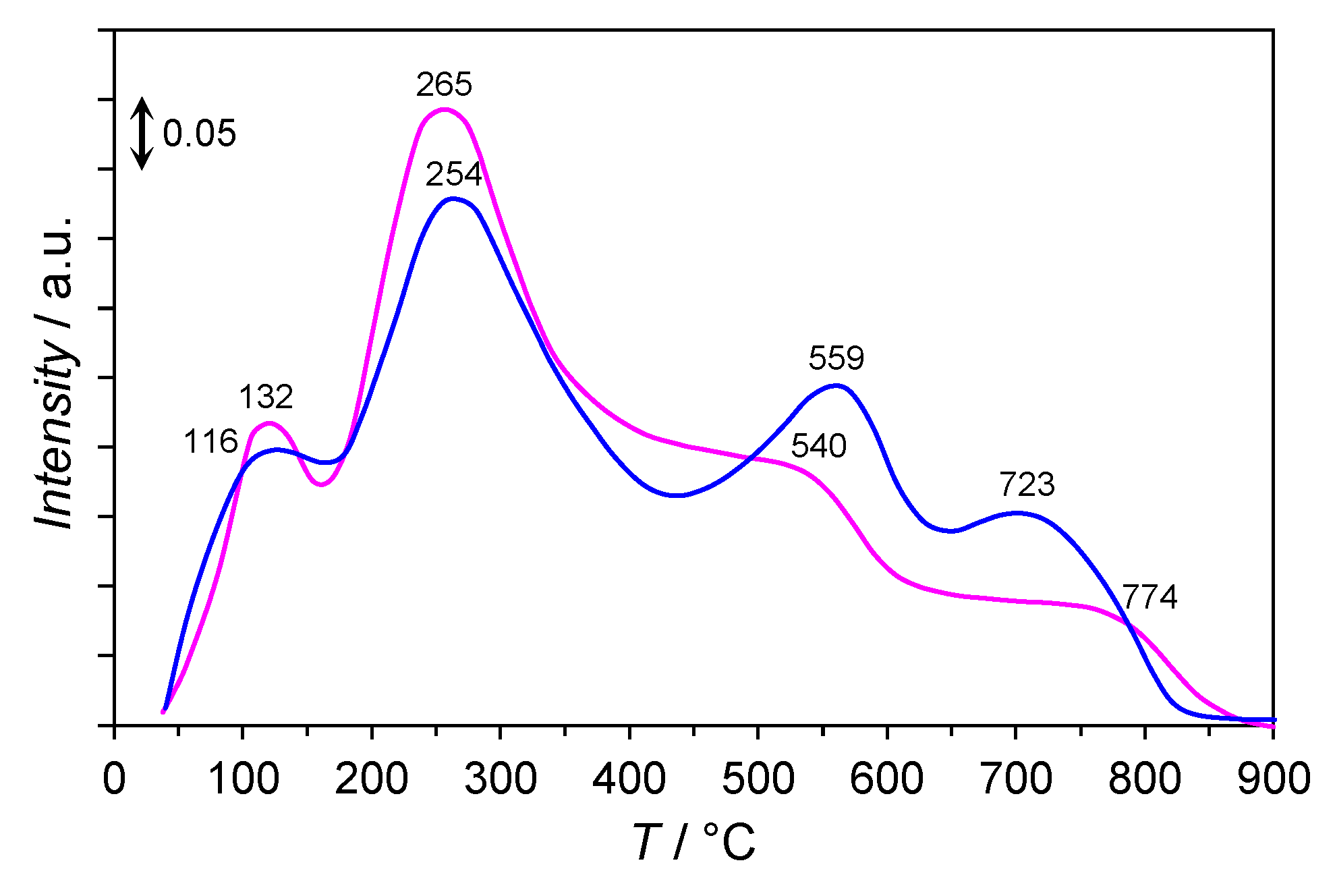
2.8. H2-TPD and N2-TPD
3. Materials and Methods
3.1. Catalysts Synthesis
3.2. Characterization Studies
3.3. Catalytic Activity Studies
4. Conclusions
- (1)
- Strong basicity,
- (2)
- More availability of the Co active sites under the reaction conditions, and
- (3)
- Competitive adsorption between the H2 and N2 molecules.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Keane, M.A. Ceramics for catalysis. J. Mater. Sci. 2003, 38, 4661–4675. [Google Scholar] [CrossRef]
- Ogura, Y.; Sato, K.; Miyahara, S.I.; Kawano, Y.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Hosokawa, S.; Nagaoka, K. Efficient ammonia synthesis over a Ru/La0.5Ce0.5O1.75 catalyst pre-reduced at high temperature. Chem. Sci. 2018, 9, 2230–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, Y.; Tsujimaru, K.; Sato, K.; Miyahara, S.I.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. Ru/La0.5Pr0.5O1.75 Catalyst for Low-Temperature Ammonia Synthesis. ACS Sustain. Chem. Eng. 2018, 6, 17258–17266. [Google Scholar] [CrossRef]
- Gao, W.; Wang, P.; Guo, J.; Chang, F.; He, T.; Wang, Q.; Wu, G.; Chen, P. Barium Hydride-Mediated Nitrogen Transfer and Hydrogenation for Ammonia Synthesis: A Case Study of Cobalt. ACS Catal. 2017, 7, 3654–3661. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Zhang, T.; Lin, B.; Ni, J.; Au, C.T.; Jiang, L. Strong metal-support interactions of Co-based catalysts facilitated by dopamine for highly efficient ammonia synthesis: In situ XPS and XAFS spectroscopy coupled with TPD studies. Chem. Commun. 2019, 55, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, Z.; Jodzis, S.; Sentek, J. Studies on kinetics of ammonia synthesis over ruthenium catalyst supported on active carbon. Appl. Catal. A Gen. 1996, 138, 83–91. [Google Scholar] [CrossRef]
- Kowalczyk, Z.; Sentek, J.; Jodzis, S.; Muhler, M.; Hinrichsen, O. Effect of potassium on the kinetics of ammonia synthesis and decomposition over fused iron catalyst at atmospheric pressure. J. Catal. 1997, 169, 407–414. [Google Scholar] [CrossRef]
- Kowalczyk, Z. Effect of potassium on the high pressure kinetics of ammonia synthesis over fused iron catalyst. Catal. Lett. 1996, 37, 173–179. [Google Scholar] [CrossRef]
- Jacobsen, C.J.H. Boron nitride: A novel support for ruthenium-based ammonia synthesis catalysts. J. Catal. 2001, 200, 1–3. [Google Scholar] [CrossRef]
- Imamura, K.; Miyahara, S.; Kawano, Y.; Sato, K.; Nakasaka, Y.; Nagaoka, K. Kinetics of ammonia synthesis over Ru/Pr2O3. J. Taiwan Inst. Chem. Eng. 2019, 105, 50–56. [Google Scholar] [CrossRef]
- Li, W.; Wang, S.; Li, J. Highly Effective Ru/BaCeO3 Catalysts on Supports with Strong Basic Sites for Ammonia Synthesis. Chem. Asian J. 2019, 14, 2815–2821. [Google Scholar]
- Cai, J.; Wang, C.; Liu, Y.; Ni, J.; Lin, B.; Wang, X.; Lin, J.; Jiang, L. Effect of pore-size distribution on Ru/ZSM-5 catalyst for enhanced N2 activation to ammonia via dissociative mechanism. J. Rare Earths 2020, 38, 873–882. [Google Scholar] [CrossRef]
- Miyahara, S.; Sato, K.; Kawano, Y.; Imamura, K.; Ogura, Y.; Tsujimaru, K.; Nagaoka, K. Ammonia synthesis over lanthanoid oxide–supported ruthenium catalysts. Catal. Today 2021, 376, 36–40. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, J.; Ni, J.; Wang, R.; Wei, K. Highly efficient Ru/Sm2O3-CeO2 catalyst for ammonia synthesis. Catal. Commun. 2011, 15, 23–26. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Ma, Y.C.; Lin, J.X. Ruthenium catalyst supported on high-surface-area basic ZrO2 for ammonia synthesis. J. Mol. Catal. A Chem. 2013, 378, 307–313. [Google Scholar] [CrossRef]
- Luo, X.; Wang, R.; Ni, J.; Lin, J.; Lin, B.; Xu, X.; Wei, K. Effect of La2O3 on Ru/CeO2-La2O3 catalyst for ammonia synthesis. Catal. Lett. 2009, 133, 382–387. [Google Scholar] [CrossRef]
- Javaid, R.; Aoki, Y.; Nanba, T. Highly efficient Ru/MgO–Er2O3 catalysts for ammonia synthesis. J. Phys. Chem. Solids 2020, 146, 109570. [Google Scholar] [CrossRef]
- Li, W.; Liu, P.; Niu, R.; Li, J.; Wang, S. Influence of CeO2 supports prepared with different precipitants over Ru/CeO2 catalysts for ammonia synthesis. Solid State Sci. 2020, 99, 105983. [Google Scholar] [CrossRef]
- Lin, B.; Liu, Y.; Heng, L.; Ni, J.; Lin, J.; Jiang, L. Effect of ceria morphology on the catalytic activity of Co/CeO2 catalyst for ammonia synthesis. Catal. Commun. 2017, 101, 15–19. [Google Scholar] [CrossRef]
- Wang, X.; Ni, J.; Lin, B.; Wang, R.; Lin, J.; Wei, K. Highly efficient Ru/MgO-CeO2 catalyst for ammonia synthesis. Catal. Commun. 2010, 12, 251–254. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Tarka, A.; Ostrowski, A.; Raróg-Pilecka, W. Kinetic studies of ammonia synthesis over a barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide. J. Taiwan Inst. Chem. Eng. 2020, 114, 241–248. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Tarka, A.; Ostrowski, A.; Jodłowski, P.; Szymański, D.; Kępiński, L.; Raróg-Pilecka, W. A high performance barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide for ammonia synthesis. RSC Adv. 2021, 11, 14218–14228. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Tarka, A.; Jodłowski, P.; Kępiński, L.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Tuning the catalytic performance of Co/Mg-La system for ammonia synthesis via the active phase precursor introduction method. Appl. Catal. A Gen. 2020, 598, 117553. [Google Scholar] [CrossRef]
- Zybert, M.; Wyszyńska, M.; Tarka, A.; Patkowski, W.; Ronduda, H.; Mierzwa, B.; Kępiński, L.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Surface enrichment phenomenon in the Ba-doped cobalt catalyst for ammonia synthesis. Vacuum 2019, 168, 108831. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Gan, Y.M.; Huang, C.; Lu, Z.H.; Wang, X.; Zhang, R.; Feng, G. Ni/La2O3 and Ni/MgO–La2O3 catalysts for the decomposition of NH3 into hydrogen. Int. J. Hydrog. Energy 2020, 45, 16528–16539. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Shamsuzzaman Microwave-assisted MgO NP catalyzed one-pot multicomponent synthesis of polysubstituted steroidal pyridines. New J. Chem. 2018, 42, 184–197. [Google Scholar] [CrossRef]
- Ni, J.; Chen, L.; Lin, J.; Schreyer, M.K.; Wang, Z.; Kawi, S. High performance of Mg-La mixed oxides supported Ni catalysts for dry reforming of methane: The effect of crystal structure. Int. J. Hydrog. Energy 2013, 38, 13631–13642. [Google Scholar] [CrossRef]
- Turcotte, R.P.; Sawyer, J.O.; Eyring, L. On the rare earth dioxymonocarbonates and their decomposition. Inorg. Chem. 1969, 8, 238–246. [Google Scholar] [CrossRef]
- Sutradhar, N.; Sinhamahapatra, A.; Pahari, S.K.; Pal, P.; Bajaj, H.C.; Mukhopadhyay, I.; Panda, A.B. Controlled synthesis of different morphologies of MgO and their use as solid base catalysts. J. Phys. Chem. C 2011, 115, 12308–12316. [Google Scholar] [CrossRef]
- Tomić-Tucaković, B.; Majstorović, D.; Jelić, D.; Mentus, S. Thermogravimetric study of the kinetics of Co3O4 reduction by hydrogen. Thermochim. Acta 2012, 541, 15–24. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, B.; Lin, J. Highly effective perovskite-type BaZrO3 supported Ru catalyst for ammonia synthesis. Appl. Catal. A Gen. 2013, 458, 130–136. [Google Scholar] [CrossRef]
- Sato, K.; Imamura, K.; Kawano, Y.; Miyahara, S.-i.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. A low-crystalline ruthenium nano-layer supported on praseodymium oxide as an active catalyst for ammonia synthesis. Chem. Sci. 2016, 8, 674–679. [Google Scholar] [CrossRef] [Green Version]
- Cornu, D.; Guesmi, H.; Krafft, J.M.; Lauron-Pernot, H. Lewis acido-basic interactions between CO2 and MgO surface: DFT and DRIFT approaches. J. Phys. Chem. C 2012, 116, 6645–6654. [Google Scholar] [CrossRef] [Green Version]
- Rosowski, F.; Hornung, A.; Hinrichsen, O.; Herein, D.; Muhler, M.; Ertl, G. Ruthenium catalysts for ammonia synthesis at high pressures: Preparation, characterization, and power-law kinetics. Appl. Catal. A Gen. 1997, 151, 443–460. [Google Scholar] [CrossRef]
- Urabe, K.; Aika, K.; Ozaki, A. Activation of nitrogen by alkali metal-promoted transition metal: VI Hydrogen effect on isotopic equilibration of nitrogen and rate-determining step of ammonia synthesis on potassium-promoted ruthenium catalysts. J. Catal. 1976, 42, 197–204. [Google Scholar] [CrossRef]
- Muhler, M.; Rosowski, F.; Hinrichsen, O.; Hornung, A.; Ertl, G. Ruthenium as catalyst for ammonia synthesis. Stud. Surf. Sci. Catal. 1996, 101, 317–326. [Google Scholar]
- Hinrichsen, O.; Rosowski, F.; Hornung, A.; Muhler, M.; Ertl, G. The Kinetics of Ammonia Synthesis over Ru-Based Catalysts. J. Catal. 1997, 165, 33–44. [Google Scholar] [CrossRef]
- Hinrichsen, O.; Rosowski, F.; Muhler, M.; Ertl, G. The microkinetics of ammonia synthesis catalyzed by cesium-promoted supported ruthenium. Chem. Eng. Sci. 1996, 51, 1683–1690. [Google Scholar] [CrossRef]
- Zhang, B.Y.; Chen, P.P.; Liu, J.X.; Su, H.Y.; Li, W.X. Influence of Cobalt Crystal Structures on Activation of Nitrogen Molecule: A First-Principles Study. J. Phys. Chem. C 2019, 123, 10956–10966. [Google Scholar] [CrossRef]
- Hagen, S.; Barfod, R.; Fehrmann, R.; Jacobsen, C.J.H.; Teunissen, H.T.; Chorkendorff, I. Ammonia synthesis with barium-promoted iron-cobalt alloys supported on carbon. J. Catal. 2003, 214, 327–335. [Google Scholar] [CrossRef]
- Kojima, R.; Enomoto, H.; Muhler, M.; Aika, K.I. Cesium-promoted rhenium catalysts supported on alumina for ammonia synthesis. Appl. Catal. A Gen. 2003, 246, 311–322. [Google Scholar] [CrossRef]
- Dinega, D.P.; Bawendi, M.G. A solution-phase chemical approach to a new crystal structure of cobalt. Angew. Chem. Int. Ed. 1999, 38, 1788–1791. [Google Scholar] [CrossRef]
- Sato, H.; Kitakami, O.; Sakurai, T.; Shimada, Y.; Otani, Y.; Fukamichi, K. Structure and magnetism of hcp-Co fine particles. J. Appl. Phys. 1997, 81, 1858–1862. [Google Scholar] [CrossRef]
- Kitakami, O.; Sato, H.; Shimada, Y.; Sato, F.; Tanaka, M. Size effect on the crystal phase of cobalt fine particles. Phys. Rev. B 1997, 56, 13849–13854. [Google Scholar] [CrossRef]
- Liu, J.X.; Wang, P.; Xu, W.; Hensen, E.J.M. Particle Size and Crystal Phase Effects in Fischer-Tropsch Catalysts. Engineering 2017, 3, 467–476. [Google Scholar] [CrossRef]
- Mehrbod, M.; Martinelli, M.; Martino, A.G.; Cronauer, D.C.; Jeremy Kropf, A.; Marshall, C.L.; Jacobs, G. Fischer-Tropsch synthesis: Direct cobalt nitrate reduction of promoted Co/TiO2 catalysts. Fuel 2019, 245, 488–504. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; Van Diilen, A.J.; De Jong, K.P. Cobalt particle size effects in the Fischer-Tropsch reaction studied with carbon nanofiber supported catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef] [Green Version]
- Rambeau, G.; Jorti, A.; Amariglio, H. Catalytic activity of a cobalt powder in NH3 synthesis in relation with the allotropic transformation of the metal. J. Catal. 1985, 94, 155–165. [Google Scholar] [CrossRef]
- Liu, J.X.; Su, H.Y.; Sun, D.P.; Zhang, B.Y.; Li, W.X. Crystallographic dependence of CO activation on cobalt catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. [Google Scholar] [CrossRef]
- Yu, M.; Liu, L.; Wang, Q.; Jia, L.; Hou, B.; Si, Y.; Li, D.; Zhao, Y. High coverage H2 adsorption and dissociation on fcc Co surfaces from DFT and thermodynamics. Int. J. Hydrog. Energy 2018, 43, 5576–5590. [Google Scholar] [CrossRef]
- Chen, Q.; Svenum, I.H.; Gavrilovic, L.; Chen, D.; Blekkan, E.A. Effect of trace potassium on hydrogen adsorption and dissociation on hcp cobalt: A density functional theory study. Surf. Sci. 2019, 681, 24–31. [Google Scholar] [CrossRef]
- Lisowski, W. Kinetics and thermodynamics of hydrogen interaction with thin cobalt films. Appl. Surf. Sci. 1989, 35, 399–408. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, R.; Jia, L.; Hou, B.; Li, D.; Wang, B. Insight into the effect of surface coverage and structure over different Co surfaces on the behaviors of H2 adsorption and activation. Int. J. Hydrog. Energy 2016, 41, 23022–23032. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Mahmoodinia, M.; Farstad, M.H.; Svenum, I.H.; Strømsheim, M.D.; Niemantsverdriet, J.W.; Venvik, H.J. Interaction of hydrogen with flat (0001) and corrugated (11–20) and (10–12) cobalt surfaces: Insights from experiment and theory. Catal. Today 2020, 342, 124–130. [Google Scholar] [CrossRef]
- Van Helden, P.; Van Den Berg, J.A.; Weststrate, C.J. Hydrogen adsorption on co surfaces: A density functional theory and temperature programmed desorption study. ACS Catal. 2012, 2, 1097–1107. [Google Scholar] [CrossRef]
- Nakhaei Pour, A.; Keyvanloo, Z.; Izadyar, M.; Modaresi, S.M. Dissociative hydrogen adsorption on the cubic cobalt surfaces: A DFT study. Int. J. Hydrog. Energy 2015, 40, 7064–7071. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, C.; Peng, X.; Zhang, T.; Wang, X.; Jiang, Y.; Qi, H.; Zheng, L.; Lin, J.; Jiang, L. Boosting Efficient Ammonia Synthesis over Atomically Dispersed Co-Based Catalyst via the Modulation of Geometric and Electronic Structures. CCS Chem. 2021, 3, 1881–1892. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63–77. [Google Scholar] [CrossRef]
- Borodziński, A.; Bonarowska, M. Relation between crystallite size and dispersion on supported metal catalysts. Langmuir 1997, 13, 5613–5620. [Google Scholar] [CrossRef]
- Huang, C.; Yu, Y.; Tang, X.; Liu, Z.; Zhang, J.; Ye, C.; Ye, Y.; Zhang, R. Hydrogen generation by ammonia decomposition over Co/CeO2 catalyst: Influence of support morphologies. Appl. Surf. Sci. 2020, 532, 147335. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Co Content 1 (wt%) | SBET 2 (m2 g−1) | Vp 3 (cm3 g−1) | Φ 4 (nm) | Number of Basic Sites (μmol g−1) | Density of Basic Sites 6 (μmol m−2) |
|---|---|---|---|---|---|---|
| non-calcined Co/Mg/La | 9.9 | 30.7 (60.4) 5 | 0.11 (0.31) 5 | 7.4 (14.6) 5 | 346 | 5.7 |
| calcined Co/Mg/La | 10.2 | 31.4 (29.1) 5 | 0.20 (0.27) 5 | 18.2 (27.2) 5 | 352 | 12.1 |
| Catalyst | H2 Desorbed 1 (μmol g−1) | MT/HT 2 (—) | FE 3 (%) | dH2 4 (nm) | |||
|---|---|---|---|---|---|---|---|
| LT | MT | HT | Total | ||||
| non-calcined Co/Mg/La | 7.2 | 17.4 | 59.3 | 83.9 | 0.29 | 16.2 | 7.8 |
| calcined Co/Mg/La | 11.9 | 27.1 | 52.2 | 91.1 | 0.52 | 11.7 | 10.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronduda, H.; Zybert, M.; Patkowski, W.; Ostrowski, A.; Jodłowski, P.; Szymański, D.; Kępiński, L.; Raróg-Pilecka, W. Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method. Catalysts 2021, 11, 941. https://doi.org/10.3390/catal11080941
Ronduda H, Zybert M, Patkowski W, Ostrowski A, Jodłowski P, Szymański D, Kępiński L, Raróg-Pilecka W. Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method. Catalysts. 2021; 11(8):941. https://doi.org/10.3390/catal11080941
Chicago/Turabian StyleRonduda, Hubert, Magdalena Zybert, Wojciech Patkowski, Andrzej Ostrowski, Przemysław Jodłowski, Damian Szymański, Leszek Kępiński, and Wioletta Raróg-Pilecka. 2021. "Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method" Catalysts 11, no. 8: 941. https://doi.org/10.3390/catal11080941
APA StyleRonduda, H., Zybert, M., Patkowski, W., Ostrowski, A., Jodłowski, P., Szymański, D., Kępiński, L., & Raróg-Pilecka, W. (2021). Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method. Catalysts, 11(8), 941. https://doi.org/10.3390/catal11080941