
Synthesis of n-Alkoxy Derivatives of Layered Perovskite-Like Niobate HCa2Nb3O10 and Study of Their Photocatalytic Activity for Hydrogen Production from an Aqueous Solution of Methanol

 , , , ,
, , , , 
Abstract
1. Introduction
2. Results and Discussion
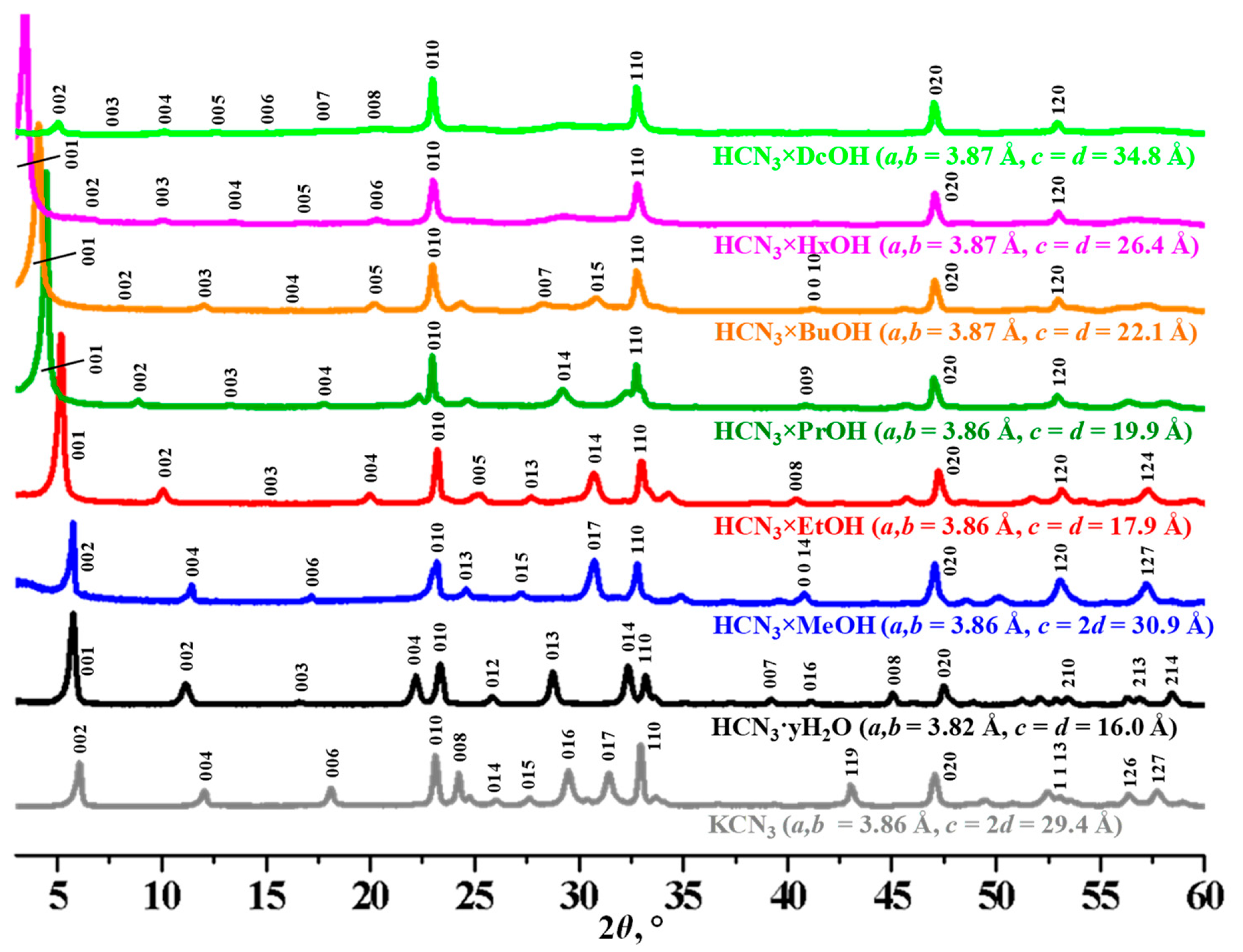
2.1. Identification of the Initial and Protonated Niobates
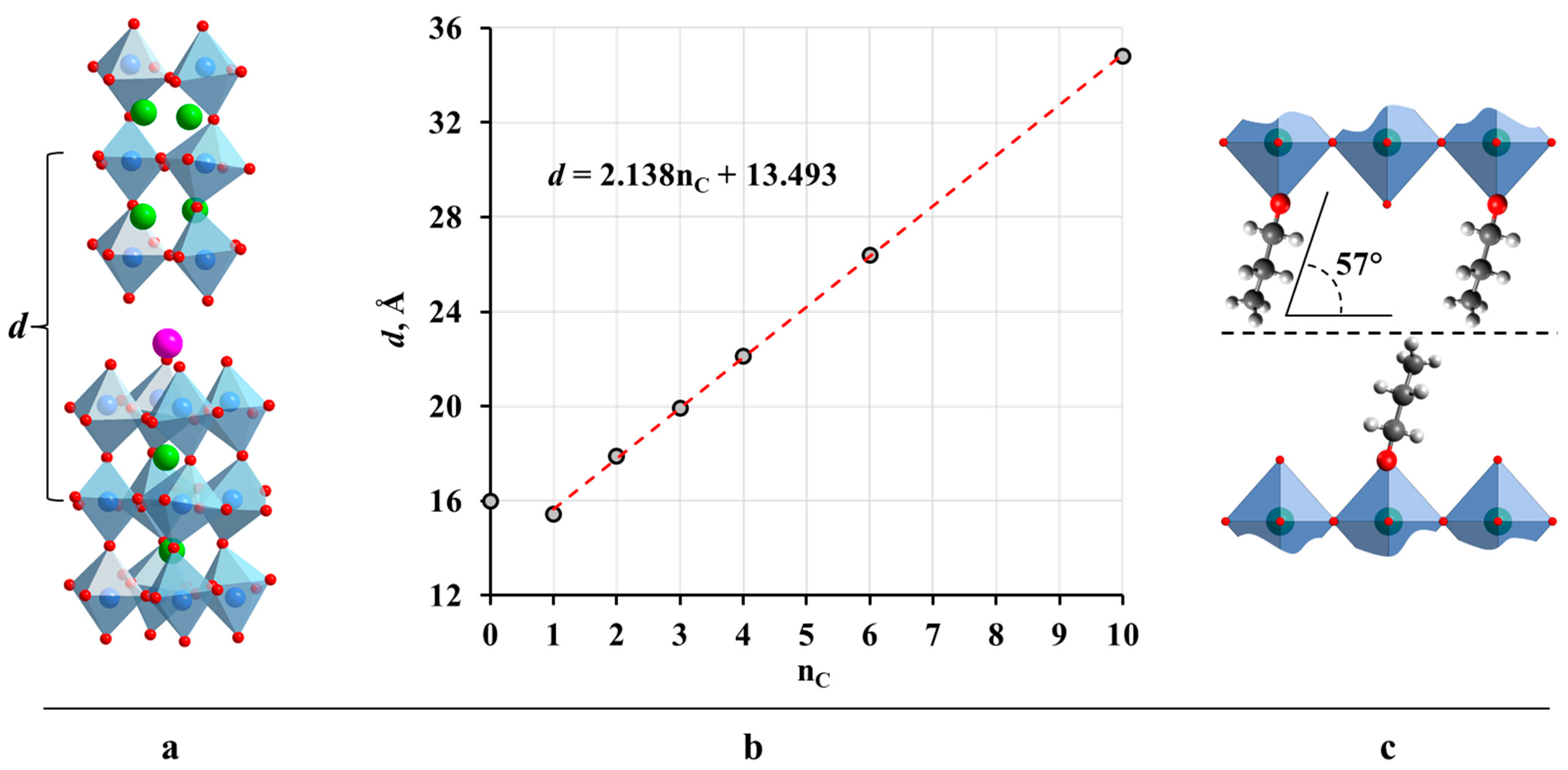
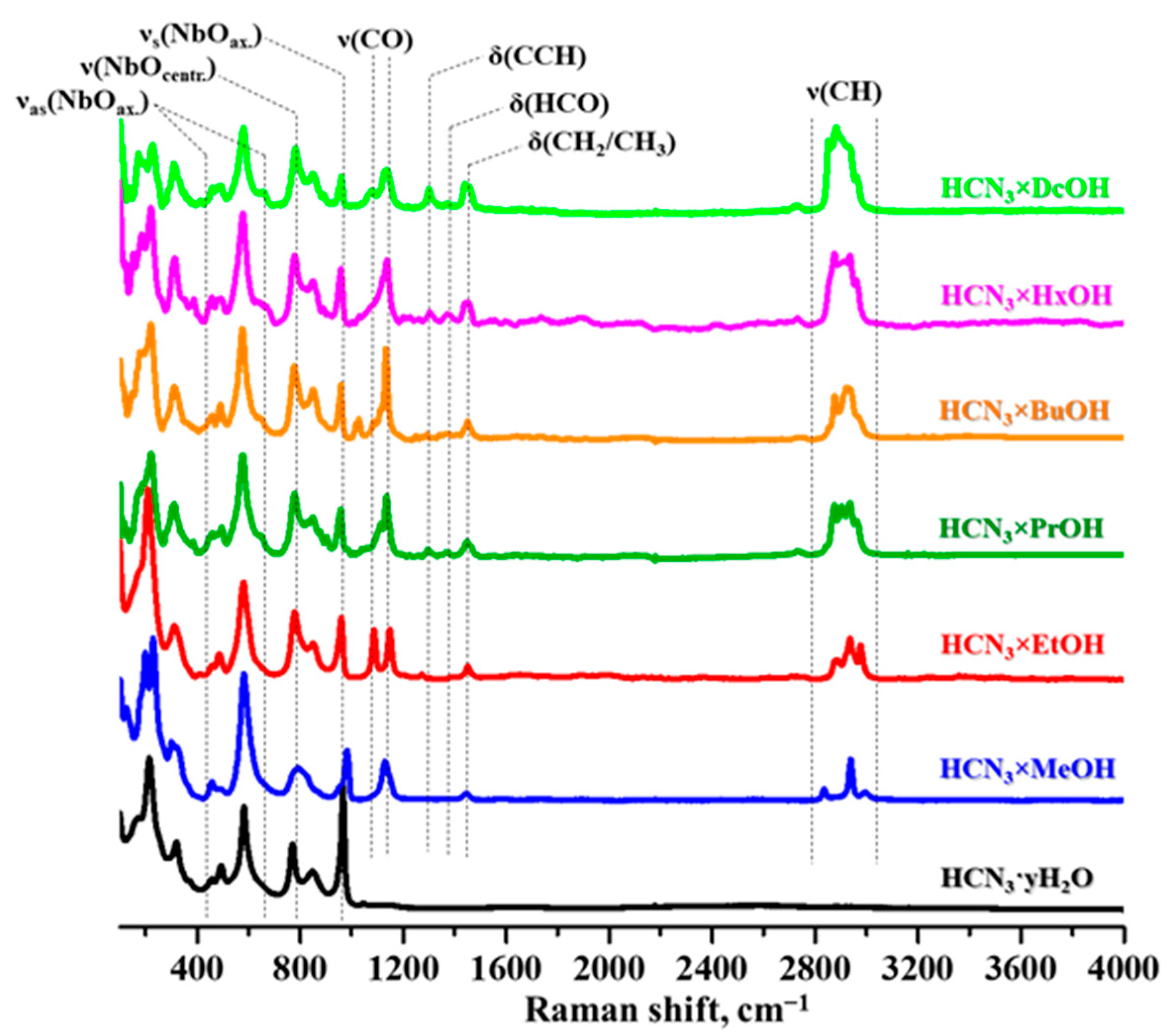
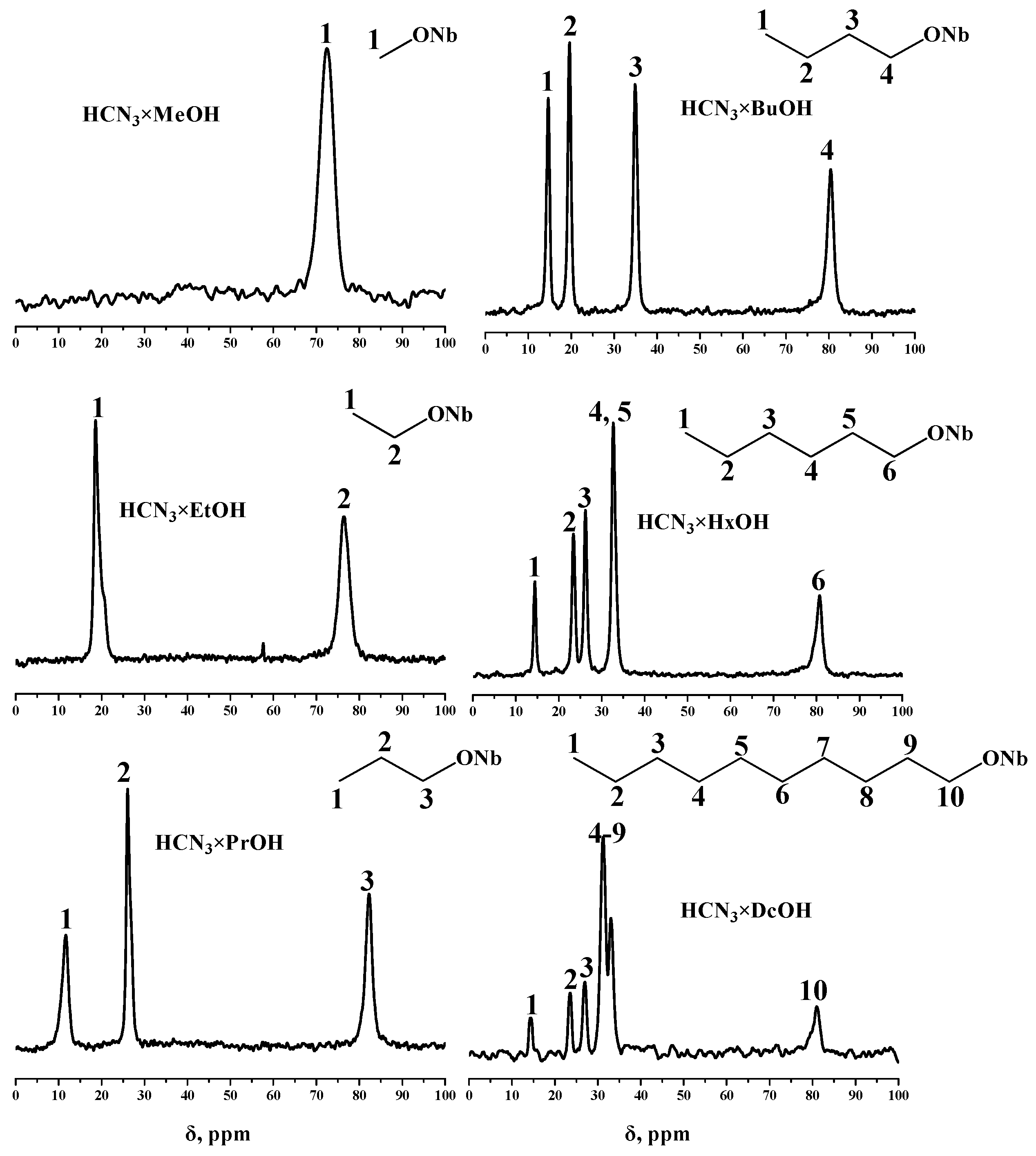
2.2. Analysis of the Inorganic–Organic Compounds
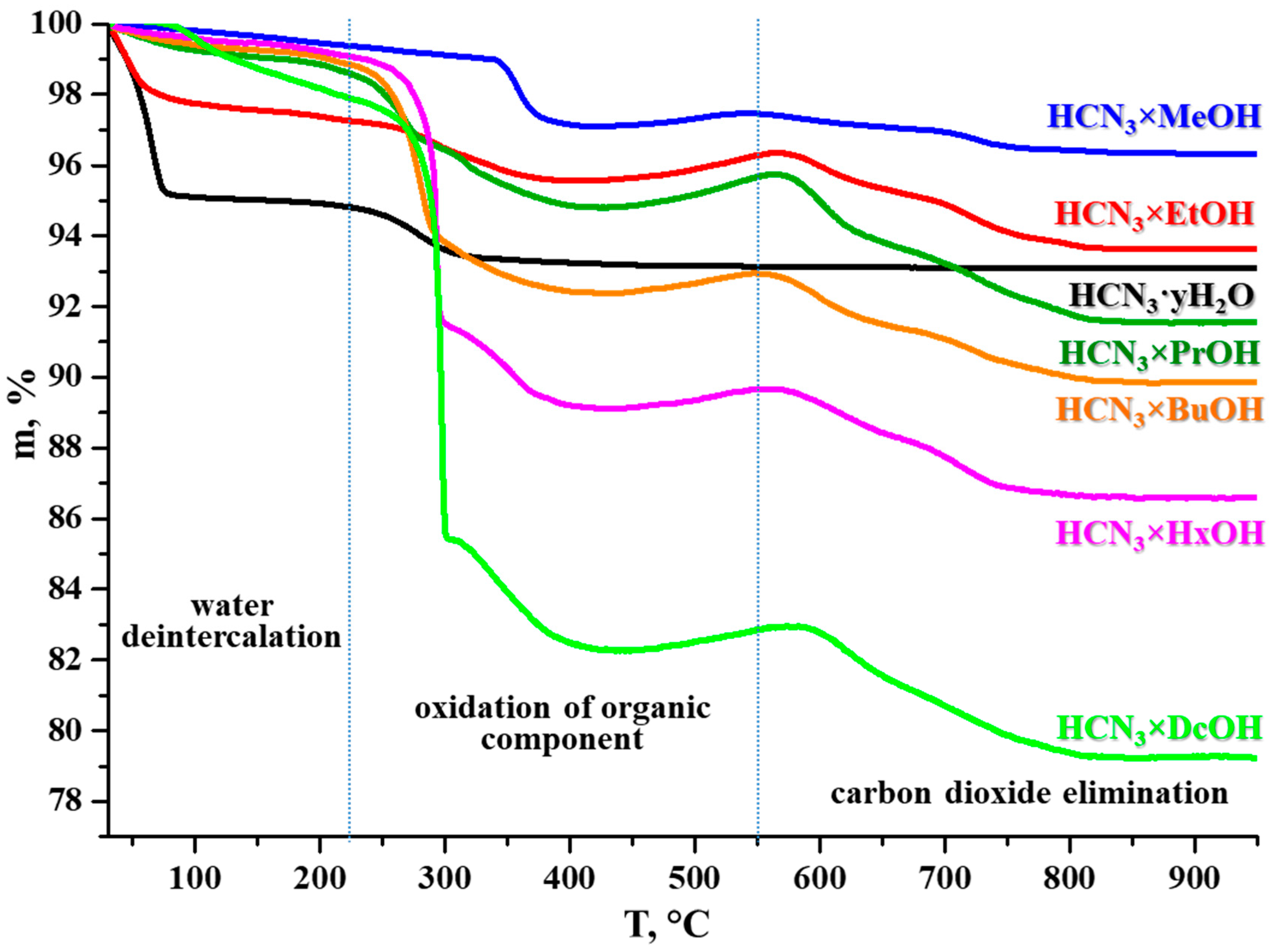
2.3. Thermal, Vacuum, and Hydrolytic Stability of the Inorganic–Organic Hybrids
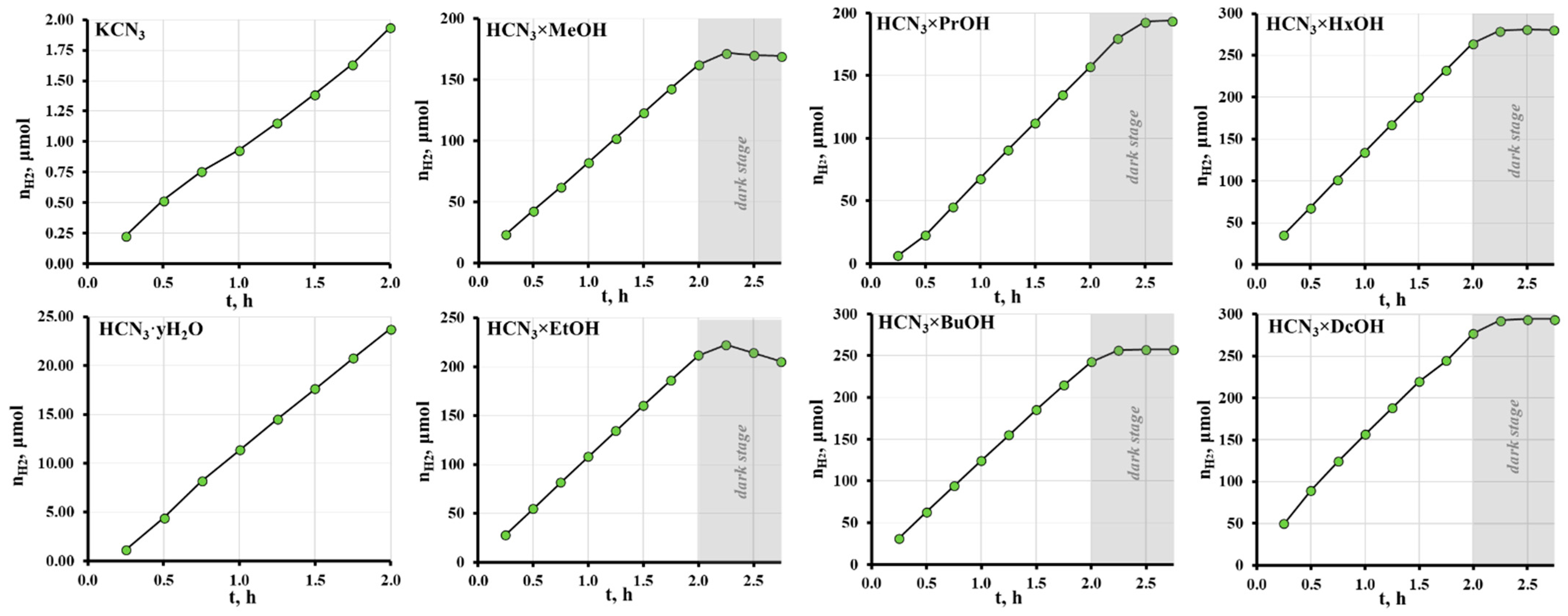
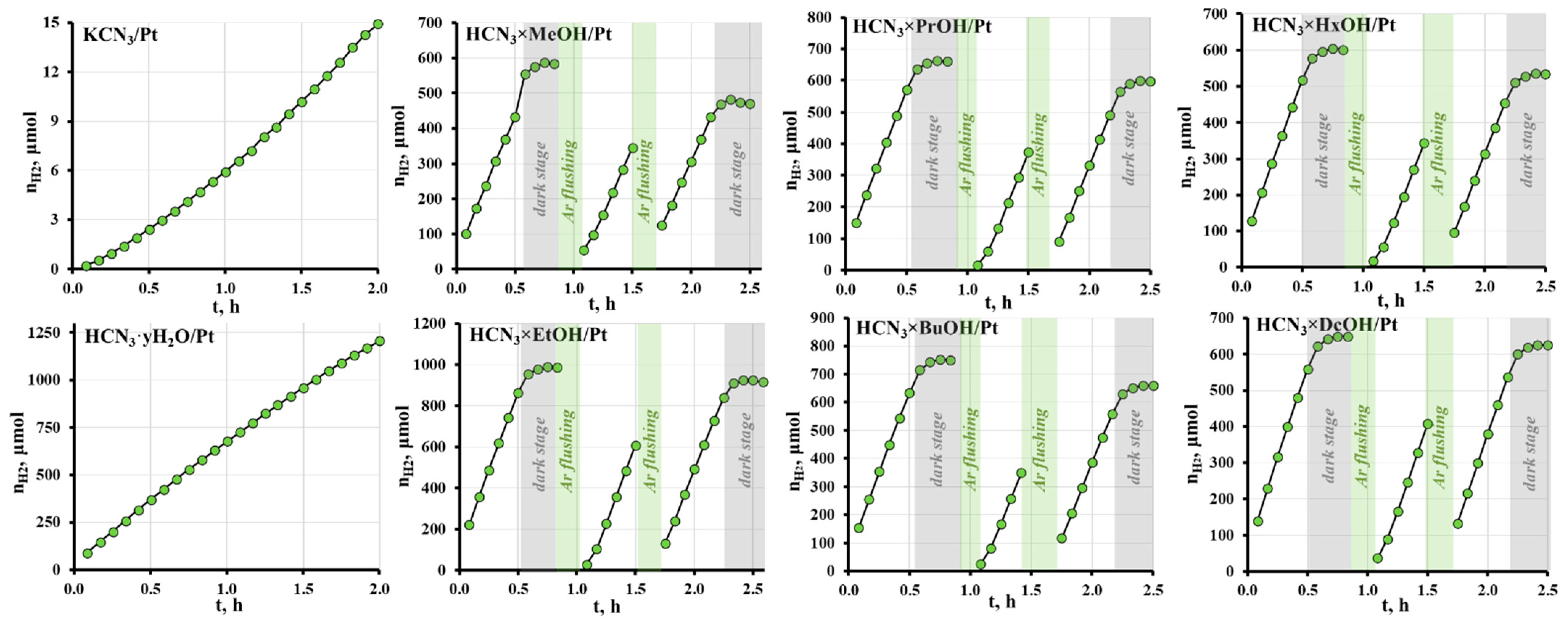
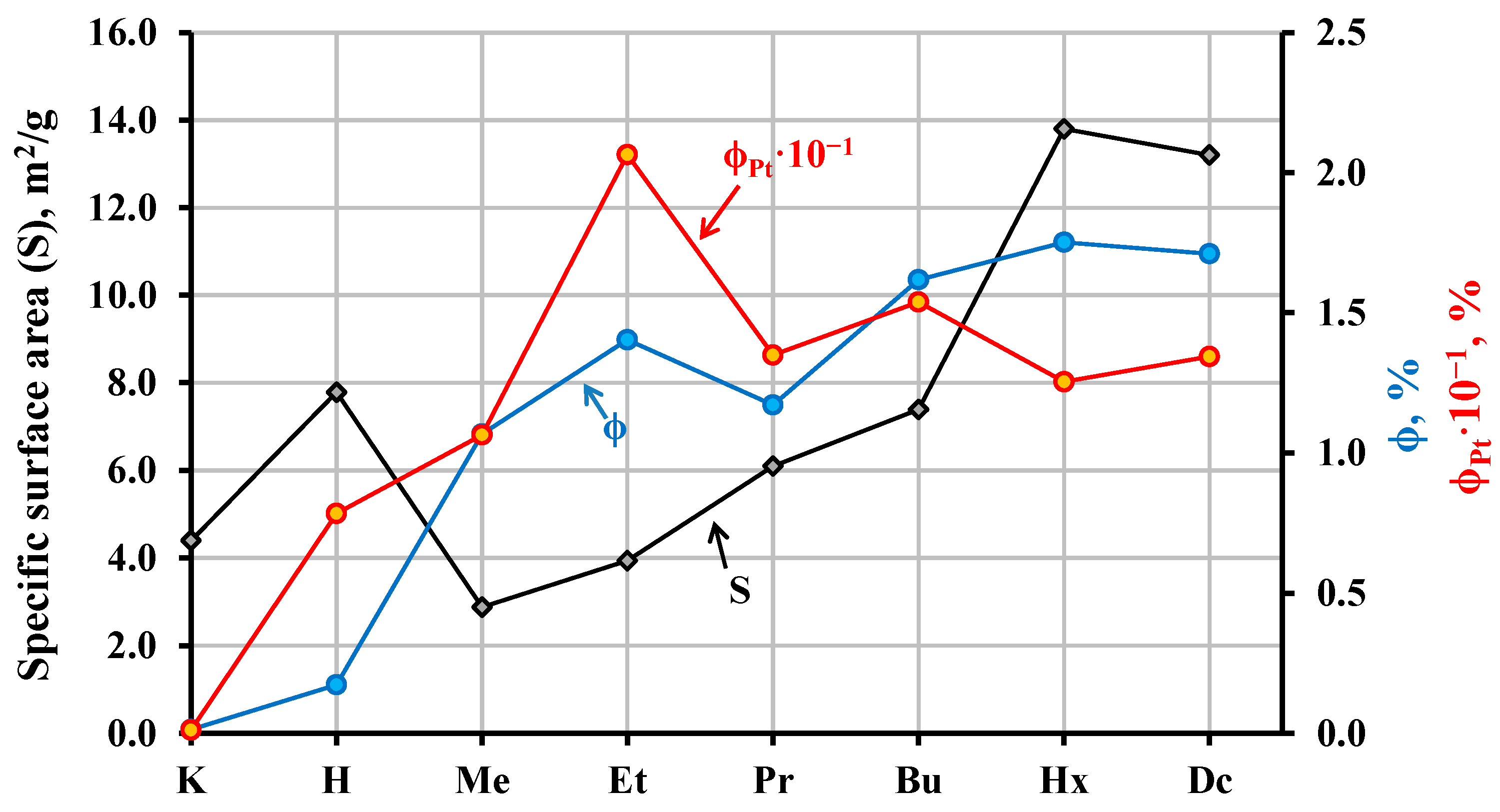
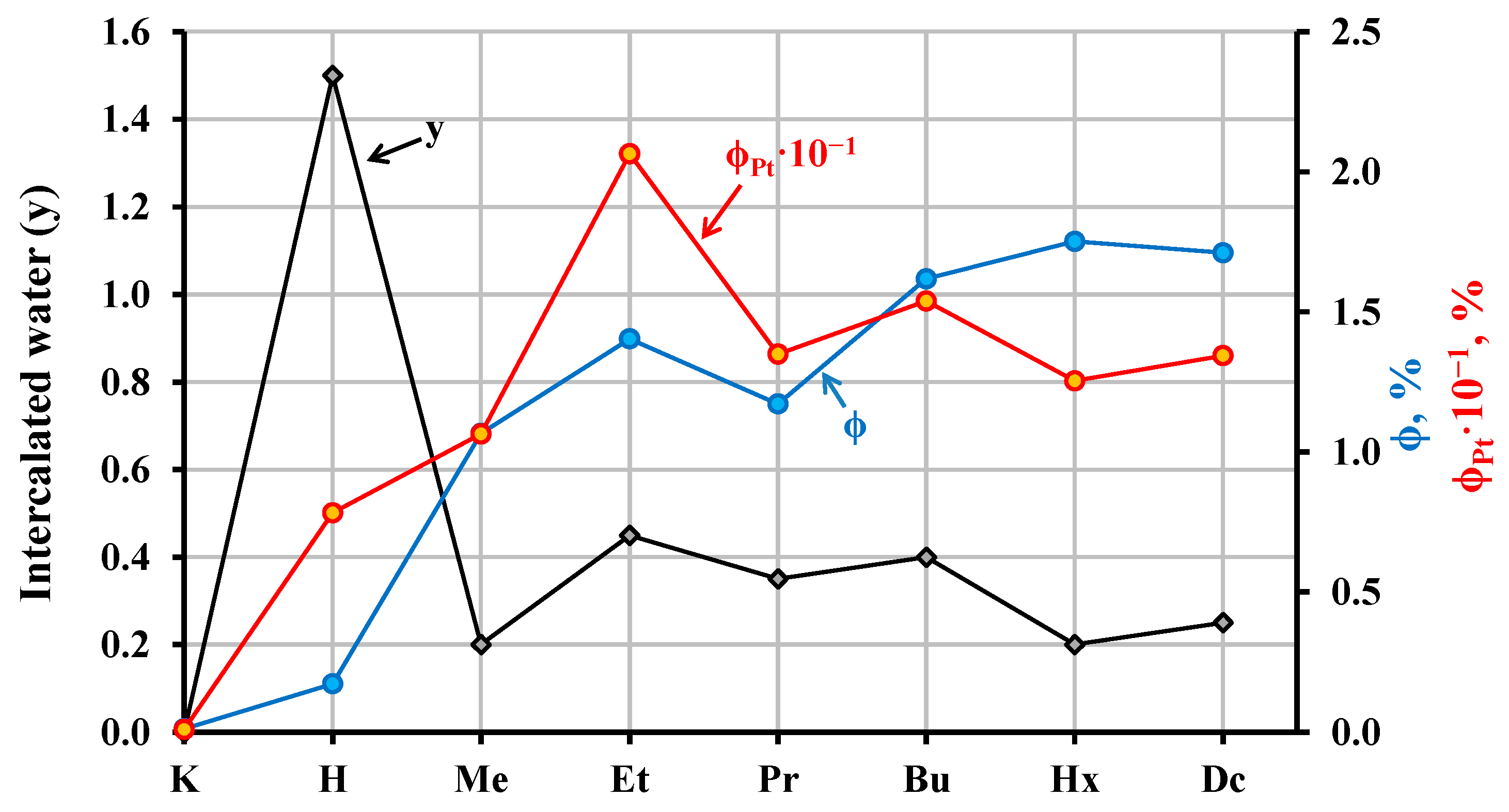
2.4. Photocatalytic Activity of the Hybrid Inorganic–Organic Niobates
2.5. Analysis of the Hybrid Inorganic–Organic Niobates after the Photocatalytic Experiment
2.6. Possible Explanations of Photocatalytic Properties of the Hybrid Inorganic–Organic Niobates
3. Materials and Methods
3.1. Synthesis of the Initial Protonated Niobate
3.2. Synthesis of n-Alkoxy Derivatives
3.3. Investigation of Vacuum and Hydrolytic Stability
3.4. Investigation of Photocatalytic Activity
3.5. Instrumentation
3.5.1. XRD
3.5.2. Raman Spectroscopy
3.5.3. NMR Spectroscopy
3.5.4. DRS
3.5.5. CHN Analysis
3.5.6. TG
3.5.7. SEM
3.5.8. BET
3.5.9. ICP-AES
3.5.10. Spectrophotometry
3.5.11. pH Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moiseev, I.I. Green chemistry: Development trajectory. Russ. Chem. Rev. 2013, 82, 616–623. [Google Scholar] [CrossRef]
- Kozlova, E.A.; Parmon, V.N. Heterogeneous semiconductor photocatalysts for hydrogen production from aqueous solutions of electron donors. Russ. Chem. Rev. 2017, 86, 870–906. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Energy Mater. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Ahmad, H.; Kamarudin, S.K.; Minggu, L.J.; Kassim, M. Hydrogen from photo-catalytic water splitting process: A review. Renew. Sustain. Energy Rev. 2015, 43, 599–610. [Google Scholar] [CrossRef]
- Takanabe, K. Photocatalytic Water Splitting: Quantitative Approaches toward Photocatalyst by Design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Rozhkova, E.; Ariga, K. From Molecules to Materials Pathways to Artificial Photosynthesis; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Christoforidis, K.C.; Fornasiero, P. Photocatalytic Hydrogen Production: A Rift into the Future Energy Supply. ChemCatChem 2017, 9, 1523–1544. [Google Scholar] [CrossRef]
- Puga, A.V. Photocatalytic production of hydrogen from biomass-derived feedstocks. Coord. Chem. Rev. 2016, 315, 1–66. [Google Scholar] [CrossRef]
- Liu, R.; Yoshida, H.; ichiro Fujita, S.; Arai, M. Photocatalytic hydrogen production from glycerol and water with NiOx/TiO2 catalysts. Appl. Catal. B Environ. 2014, 144, 41–45. [Google Scholar] [CrossRef]
- Taboada, E.; Angurell, I.; Llorca, J. Hydrogen photoproduction from bio-derived alcohols in an optical fiber honeycomb reactor loaded with Au/TiO2. J. Photochem. Photobiol. A Chem. 2014, 281, 35–39. [Google Scholar] [CrossRef]
- Al-Azri, Z.H.N.; Chen, W.T.; Chan, A.; Jovic, V.; Ina, T.; Idriss, H.; Waterhouse, G.I.N. The roles of metal co-catalysts and reaction media in photocatalytic hydrogen production: Performance evaluation of M/TiO2 photocatalysts (M = Pd, Pt, Au) in different alcohol-water mixtures. J. Catal. 2015, 329, 355–367. [Google Scholar] [CrossRef]
- Dosado, A.G.; Chen, W.T.; Chan, A.; Sun-Waterhouse, D.; Waterhouse, G.I.N. Novel Au/TiO2 photocatalysts for hydrogen production in alcohol-water mixtures based on hydrogen titanate nanotube precursors. J. Catal. 2015, 330, 238–254. [Google Scholar] [CrossRef]
- Wang, X.; Dong, H.; Hu, Z.; Qi, Z.; Li, L. Fabrication of a Cu2O/Au/TiO2 composite film for efficient photocatalytic hydrogen production from aqueous solution of methanol and glucose. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2017, 219, 10–19. [Google Scholar] [CrossRef]
- Li, C.; Wang, H.; Ming, J.; Liu, M.; Fang, P. Hydrogen generation by photocatalytic reforming of glucose with heterostructured CdS/MoS2 composites under visible light irradiation. Int. J. Hydrogen Energy 2017, 42, 16968–16978. [Google Scholar] [CrossRef]
- Jaswal, R.; Shende, R.; Nan, W.; Shende, A. Photocatalytic reforming of pinewood (Pinus ponderosa) acid hydrolysate for hydrogen generation. Int. J. Hydrogen Energy 2017, 42, 2839–2848. [Google Scholar] [CrossRef]
- Udom, I.; Myers, P.; Ram, M.K.; Hepp, A.F.; Archibong, E.; Stefanakos, E.K.; Goswami, D.Y. Optimization of Photocatalytic Degradation of Phenol Using Simple Photocatalytic Reactor. Am. J. Anal. Chem. 2014, 5, 743–750. [Google Scholar] [CrossRef]
- Long, Z.; Li, Q.; Wei, T.; Zhang, G.; Ren, Z. Historical development and prospects of photocatalysts for pollutant removal in water. J. Hazard. Mater. 2020, 395, 122599. [Google Scholar] [CrossRef]
- Rueda-Marquez, J.J.; Levchuk, I.; Fernandez-Ibañez, P.; Sillanpää, M. A critical review on application of photocatalysis for toxicity reduction of real wastewaters. J. Clean. Prod. 2020, 258, 120694. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, X.; Meng, Y.; Pan, G.; Ni, Z.; Xia, S. Layered double hydroxides-based photocatalysts and visible-light driven photodegradation of organic pollutants: A review. Chem. Eng. J. 2020, 392, 123684. [Google Scholar] [CrossRef]
- Karthikeyan, C.; Arunachalam, P.; Ramachandran, K.; Al-Mayouf, A.M.; Karuppuchamy, S. Recent advances in semiconductor metal oxides with enhanced methods for solar photocatalytic applications. J. Alloys Compd. 2020, 828, 154281. [Google Scholar] [CrossRef]
- Teixeira, I.; Quiroz, J.; Homsi, M.; Camargo, P. An overview of the photocatalytic H2 evolution by semiconductor-based materials for nonspecialists. J. Braz. Chem. Soc. 2020, 31, 211–229. [Google Scholar] [CrossRef]
- Schaak, R.E.; Mallouk, T. Perovskites by Design: A Toolbox of Solid-State Reactions. Chem. Mater. 2002, 14, 1455–1471. [Google Scholar] [CrossRef]
- Uppuluri, R.; Gupta, A.S.; Rosas, A.S.; Mallouk, T.E. Soft chemistry of ion-exchangeable layered metal oxides. Chem. Soc. Rev. 2018, 47, 2401–2430. [Google Scholar] [CrossRef]
- Tani, S.; Komori, Y.; Hayashi, S.; Sugahara, Y. Local environments and dynamics of hydrogen atoms in protonated forms of ion-exchangeable layered perovskites estimated by solid-state 1H NMR. J. Solid State Chem. 2006, 179, 3357–3364. [Google Scholar] [CrossRef]
- Utkina, T.; Chislov, M.; Silyukov, O.; Burovikhina, A.; Zvereva, I.A. TG and DSC investigation of water intercalation and protonation processes in perovskite-like layered structure of titanate K2Nd2Ti3O10. J. Therm. Anal. Calorim. 2016, 125, 281–287. [Google Scholar] [CrossRef]
- Silyukov, O.; Chislov, M.; Burovikhina, A.; Utkina, T.; Zvereva, I. Thermogravimetry study of ion exchange and hydration in layered oxide materials. J. Therm. Anal. Calorim. 2012, 110, 187–192. [Google Scholar] [CrossRef]
- Jacobson, A.; Lewandowski, J.; Johnson, J.W. Ion exchange of the layered perovskite KCa2Nb3O10 by protons. J. Less Common Met. 1986, 116, 137–146. [Google Scholar] [CrossRef]
- Kumada, N.; Fukazawa, Y.; Yonesaki, Y.; Takei, T.; Kinomura, N. Ion-exchange Reaction of Protonated Layer Compounds with Bi3+ Ion. Clay Sci. 2006, 12, 331–335. Available online: https://www.jstage.jst.go.jp/article/jcssjclayscience1960/12/Supplement2/12_Supplement2_331/_pdf/-char/ja (accessed on 1 June 2021).
- Zvereva, I.A.; Silyukov, O.; Chislov, M. Ion-exchange reactions in the structure of perovskite-like layered oxides: I. Protonation of NaNdTiO4 complex oxide. Russ. J. Gen. Chem. 2011, 81, 1434–1441. [Google Scholar] [CrossRef]
- Yip, T.W.S.; Cussen, E.J. Ion Exchange and Structural Aging in the Layered Perovskite Phases. Inorg. Chem. 2013, 52, 6985–6993. [Google Scholar] [CrossRef]
- Yafarova, L.V.; Silyukov, O.I.; Myshkovskaya, T.D.; Minich, I.A.; Zvereva, I.A. New data on protonation and hydration of perovskite-type layered oxide KCa2Nb3O10. J. Therm. Anal. Calorim. 2020, 1, 87–93. [Google Scholar] [CrossRef]
- Silyukov, O.I.; Minich, I.A.; Zvereva, I.A. Synthesis of Protonated Derivatives of Layered Perovskite-Like Bismuth Titanates. Glass Phys. Chem. 2018, 44, 115–119. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Silyukov, O.I.; Utkina, T.D.; Chislov, M.V.; Sokolova, Y.P.; Zvereva, I.A. Photocatalytic properties and hydration of perovskite-type layered titanates A2Ln2Ti3O10 (A = Li, Na, K.; Ln = La, Nd). Russ. J. Gen. Chem. 2012, 82, 1191–1196. [Google Scholar] [CrossRef]
- Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Preparation of Porous Particles of Layered Perovskite-Like Titanate HLaTiO4. Glass Phys. Chem. 2020, 46, 272–276. [Google Scholar] [CrossRef]
- Cattaneo, A.S.; Ferrara, C.; Marculescu, A.M.; Giannici, F.; Tealdi, C.; Martorana, A.; Mustarelli, P. Solid-state NMR characterization of the structure and thermal stability of hybrid organic–inorganic compounds based on a HLaNb2O7 Dion–Jacobson layered perovskite. Phys. Chem. Chem. Phys. 2016, 18, 21903–21912. [Google Scholar] [CrossRef]
- Wang, B.; Dong, X.; Pan, Q.; Cheng, Z.; Yang, Y. Intercalation behavior of n-alkylamines into an A-site defective layered perovskite H2W2O7. J. Solid State Chem. 2007, 180, 1125–1129. [Google Scholar] [CrossRef]
- Jacobson, A.J.; Johnson, J.W.; Lewandowski, J. Intercalation of the layered solid acid HCa2Nb3O10 by organic amines. Mater. Res. Bull. 1987, 22, 45–51. [Google Scholar] [CrossRef]
- Takata, T.; Furumi, Y.; Shinohara, K.; Tanaka, A.; Hara, M.; Kondo, J.; Domen, K. Photocatalytic Decomposition of Water on Spontaneously Hydrated Layered Perovskites. Chem. Mater. 1997, 9, 1063–1064. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Zvereva, I.A. Photocatalytic activity of layered perovskite-like oxides in practically valuable chemical reactions. Russ. Chem. Rev. 2016, 85, 248–279. [Google Scholar] [CrossRef]
- Cui, W.; Liu, L.; Ma, S.; Liang, Y.; Zhang, Z. CdS-sensitized K2La2Ti3O10 composite: A new photocatalyst for hydrogen evolution under visible light irradiation. Catal. Today 2013, 207, 44–49. [Google Scholar] [CrossRef]
- Cui, W.; Liu, L.; Ma, S.; Liang, Y.; Zhang, Z. Substitution effects of In3+ by Fe3+ on photocatalytic and structural properties of Bi 2InNbO7 photocatalysts. J. Mol. Catal. 2001, 168, 289–297. [Google Scholar]
- Reddy, V.; Hwang, D.; Lee, J. Effect of Zr substitution for Ti in KLaTiO4 for photocatalytic water splitting. Catal. Lett. 2003, 90, 39–44. [Google Scholar] [CrossRef]
- Kumar, V.; Govind; Uma, S. Investigation of cation (Sn2+) and anion (N3-) substitution in favor of visible light photocatalytic activity in the layered perovskite K2La2Ti3O10. J. Hazard. Mater. 2011, 189, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wen, T.; Guo, Y.; Yang, B.; Wang, Y. Controllable doping of nitrogen and tetravalent niobium affords yellow and black calcium niobate nanosheets for enhanced photocatalytic hydrogen evolution. RSC Adv. 2016, 6, 64930–64936. [Google Scholar] [CrossRef]
- Kawashima, K.; Hojamberdiev, M.; Chen, S.; Yubuta, K.; Wagata, H.; Domen, K.; Teshima, K. Understanding the effect of partial N3−-to-O2− substitution and H+-to-K+ exchange on photocatalytic water reduction activity of Ruddlesden–Popper layered perovskite KLaTiO4. Mol. Catal. 2017, 432, 250–258. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Wei, Y.; Li, Y.; Lin, J.; Wu, J. Fabrication and photocatalytic property of Pt-intercalated layered perovskite niobates H1−xLaNb2−xMoxO7 (x = 0–0.15). J. Hazard. Mater. 2009, 166, 103–108. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Wei, Y.; Huang, M.; Wu, J. Photocatalytic property of partially substituted Pt-intercalated layered perovskite, ASr2TaxNb3−xO10 (A = K, H; x = 0, 1, 1.5, 2 and 3). Sol. Energy Mater. Sol. Cells 2011, 95, 1019–1027. [Google Scholar] [CrossRef]
- Oshima, T.; Wang, Y.; Lu, D.; Yokoi, T.; Maeda, K. Photocatalytic overall water splitting on Pt nanocluster-intercalated, restacked KCa2Nb3O10 nanosheets: The promotional effect of co-existing ions. Nanoscale Adv. 2019, 1, 189–194. [Google Scholar] [CrossRef]
- Cui, W.; Qi, Y.; Liu, L.; Rana, D.; Hu, J.; Liang, Y. Synthesis of PbS–K2La2Ti3O10 composite and its photocatalytic activity for hydrogen production. Prog. Nat. Sci. Mater. Int. 2012, 22, 120–125. [Google Scholar] [CrossRef][Green Version]
- Cui, W.; Guo, D.; Liu, L.; Hu, J.; Rana, D.; Liang, Y. Preparation of ZnIn2S4/K2La2Ti3O10 composites and their photocatalytic H2 evolution from aqueous Na2S/Na2SO3 under visible light irradiation. Catal. Commun. 2014, 48, 55–59. [Google Scholar] [CrossRef]
- Saito, K.; Kozeni, M.; Sohmiya, M.; Komaguchi, K.; Ogawa, M.; Sugahara, Y.; Ide, Y. Unprecedentedly enhanced solar photocatalytic activity of a layered titanate simply integrated with TiO2 nanoparticles. Phys. Chem. Chem. Phys. 2016, 18, 30920–30925. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Lv, C.; Zhang, C.; Jin, X.; Meng, Q.; Chen, G. Construction of 2D-composite HCa2Nb3O10/CaNb2O6 heterostructured photocatalysts with enhanced hydrogen production performance. New J. Chem. 2018, 42, 681–687. [Google Scholar] [CrossRef]
- Chen, X.; Shen, S.; Guo, L.; Mao, S. Semiconductor-based photocatalytic hydrogen generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wong, K.-H.; Chen, Z.; Yu, J.; Zhao, J.; Hu, C.; Chan, C.-Y.; Wong, P.-K. AgBr-Ag-Bi2WO6 nanojunction system: A novel and efficient photocatalyst with double visible-light active components. Appl. Catal. A Gen. 2009, 363, 221–229. [Google Scholar] [CrossRef]
- Kim, H.G.; Jeong, E.D.; Borse, P.H.; Jeon, S.; Yong, K.; Lee, J.S.; Li, W.; Oh, S.H. Photocatalytic Ohmic layered nanocomposite for efficient utilization of visible light photons. Appl. Phys. Lett. 2006, 89, 2012–2015. [Google Scholar] [CrossRef]
- Kim, H.G.; Borse, P.H.; Choi, W.; Lee, J.S. Photocatalytic Nanodiodes for Visible-Light Photocatalysis. Angew. Chem. 2005, 117, 4661–4665. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, G.; Xiong, Z.; Tang, H.; Jiang, C. Fabrication of flower-like direct Z-scheme β-Bi2O3/g-C3N4 photocatalyst with enhanced visible light photoactivity for Rhodamine B degradation. Appl. Surf. Sci. 2018, 436, 162–171. [Google Scholar] [CrossRef]
- Youngblood, W.J.; Lee, S.-H.A.; Maeda, K.; Mallouk, T. Visible light water splitting using dye-sensitized oxide semiconductors. Acc. Chem. Res. 2009, 42, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cai, F.; Chen, T.; Fan, M.; Song, C.; Yan, X.; Shi, W. Enhanced Photocatalystic Activity for Hydrogen Production under Visible Light Irradiation. ACS Appl. Mater. Interfaces 2015, 7, 33. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, J.; Zhang, Q.; Yu, H.; Ding, F.; Xu, B.; Sun, Y.; Xu, Z. Facile synthesis of heterostructured YVO4/g-C3N4/Ag photocatalysts with enhanced visible-light photocatalytic performance. Appl. Catal. B Environ. 2018, 224, 586–593. [Google Scholar] [CrossRef]
- Cui, Z.; Yang, H.; Zhao, X. Enhanced photocatalytic performance of g-C3N4/Bi4Ti3O12 heterojunction nanocomposites. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2018, 229, 160–172. [Google Scholar] [CrossRef]
- Guo, Y.; Li, J.; Gao, Z.; Zhu, X.; Liu, Y.; Wei, Z.; Zhao, W.; Sun, C. A simple and effective method for fabricating novel p-n heterojunction photocatalyst g-C3N4/Bi4Ti3O12 and its photocatalytic performances. Appl. Catal. B Environ. 2016, 192, 57–71. [Google Scholar] [CrossRef]
- Maeda, K.; Mallouk, T.E. Two-Dimensional Metal Oxide Nanosheets as Building Blocks for Artificial Photosynthetic Assemblies. Bull. Chem. Soc. Jpn. 2018, 92, 38–54. [Google Scholar] [CrossRef]
- Gómez-Romero, P.; Sanchez, C. Functional Hybrid Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Sanchez, C.; Soler-Illia, G.; Ribot, F.; Lalot, T.; Mayer, C.R.; Cabuil, V. Designed hybrid organic-inorganic nanocomposites from functional nanobuilding blocks. Chem. Mater. 2001, 13, 3061–3083. [Google Scholar] [CrossRef]
- Mir, S.H.; Nagahara, L.A.; Thundat, T.; Tabari, P.M.; Furukawa, H.; Khosla, A. Review—Organic-Inorganic Hybrid Functional Materials: An Integrated Platform for Applied Technologies. J. Electrochem. Soc. 2018, 165, B3137–B3156. [Google Scholar] [CrossRef]
- Tahara, S.; Ichikawa, T.; Kajiwara, G.; Sugahara, Y. Reactivity of the Ruddlesden−Popper Phase H2La2Ti3O10 with Organic Compounds: Intercalation and Grafting Reactions. Chem. Mater. 2007, 19, 2352–2358. [Google Scholar] [CrossRef]
- Silyukov, O.I.; Kurnosenko, S.A.; Zvereva, I.A. Intercalation of Methylamine into the Protonated Forms of Layered Perovskite-Like Oxides HLnTiO4 (Ln = La and Nd). Glass Phys. Chem. 2018, 44, 428–432. [Google Scholar] [CrossRef]
- Minich, I.A.; Silyukov, O.I.; Gak, V.V.; Borisov, E.V.; Zvereva, I.A. Synthesis of Organic-Inorganic Hybrids Based on Perovskite-like Bismuth Titanate H2K0.5Bi2.5Ti4O13·H2O and n-Alkylamines. ACS Omega 2020, 5, 8158–8168. [Google Scholar] [CrossRef]
- Kurnosenko, S.A.; Silyukov, O.I.; Minich, I.A.; Zvereva, I.A. Exfoliation of methylamine and n-butylamine derivatives of layered perovskite-like oxides HLnTiO4 and H2Ln2Ti3O10 (Ln = La, Nd) into nanolayers. Glass Phys. Chem. 2021, 44, 428–432. [Google Scholar]
- Kurnosenko, S.A.; Silyukov, O.I.; Mazur, A.S.; Zvereva, I.A. Synthesis and thermal stability of new inorganic-organic perovskite-like hybrids based on layered titanates HLnTiO4 (Ln = La, Nd). Ceram. Int. 2019, 46, 5058–5068. [Google Scholar] [CrossRef]
- Silyukov, O.I.; Khramova, A.D.; Zvereva, I.A. Synthesis of Organic-Inorganic Derivatives of Perovskite-Like Layered HCa2Nb3O10 Oxide with Monoethanolamine and Glycine. Glass Phys. Chem. 2020, 46, 256–259. [Google Scholar] [CrossRef]
- Han, Y.-S.; Park, I.; Choy, J.-H. Exfoliation of layered perovskite, KCa2Nb3O10, into colloidal nanosheets by a novel chemical process. J. Mater. Chem. 2001, 11, 1277–1282. [Google Scholar] [CrossRef]
- Tahara, S.; Sugahara, Y. Interlayer Surface Modification of the Protonated Triple-Layered Perovskite HCa2Nb3O10·xH2O with n-Alcohols. Langmuir 2003, 19, 9473–9478. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Lushpinskaya, I.P.; Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Identification of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10 by Multinuclear NMR. Russ. J. Gen. Chem. 2020, 90, 760–761. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Silyukov, O.I.; Lushpinskaia, I.P.; Kurnosenko, S.A.; Mazur, A.S.; Shenderovich, I.G.; Zvereva, I.A. NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10. Molecules 2020, 25, 5229. [Google Scholar] [CrossRef] [PubMed]
- Minich, I.A.; Silyukov, O.I.; Mazur, A.S.; Zvereva, I.A. Grafting reactions of perovskite-like bismuth titanate H2K0.5Bi2.5Ti4O13·H2O with n-alcohols. Ceram. Int. 2020, 46, 29373–29381. [Google Scholar] [CrossRef]
- Wang, C.; Tang, K.; Wang, D.; Liu, Z.; Wang, L.; Zhu, Y.; Qian, Y. A new carbon intercalated compound of Dion-Jacobson phase HLaNb2O7. J. Mater. Chem. 2012, 22, 11086–11092. [Google Scholar] [CrossRef]
- Aznar, A.J.; Sanz, J.; Ruiz-Hitzky, E. Mechanism of the grafting of organosilanes on mineral surfaces. IV. Phenylderivatives of sepiolite and poly (organosiloxanes). Colloid Polym. Sci. 1992, 270, 165–176. [Google Scholar] [CrossRef]
- Shimada, A.; Yoneyama, Y.; Tahara, S.; Mutin, H.; Sugahara, Y. Interlayer surface modification of the protonated ion-exchangeable layered perovskite HLaNb2O7??xH2O with organophosphonic acids. Chem. Mater. 2009, 21, 4155–4162. [Google Scholar] [CrossRef]
- Machida, M.; Mitsuyama, T.; Ikeue, K.; Matsushima, S.; Arai, M. Photocatalytic property and electronic structure of triple-layered perovskite tantalates, MCa2Ta3O10 (M = Cs, Na, H, and C6H13NH3). J. Phys. Chem. B 2005, 109, 7801–7806. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Wang, L.; Hao, Q.; Zhu, X.; Chen, X.; Tang, K. Preparation of interlayer surface tailored protonated double-layered perovskite H 2 CaTa 2 O 7 with n-alcohols, and their photocatalytic activity. RSC Adv. 2014, 4, 4047–4054. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Maksimova, E.A.; Pozhidaev, A.Y.; Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Layered Titanate H2Nd2Ti3O10 Intercalated With n-Butylamine: A New Highly Efficient Hybrid Photocatalyst for Hydrogen Production From Aqueous Solutions of Alcohols. Front. Chem. 2019, 7, 863. [Google Scholar] [CrossRef]
- Voytovich, V.V.; Kurnosenko, S.A.; Silyukov, O.I.; Rodionov, I.A.; Minich, I.A.; Zvereva, I.A. Study of n-alkylamine Intercalated Layered Perovskite-Like Niobates HCa2Nb3O10 as Photocatalysts for Hydrogen Production From an Aqueous Solution of Methanol. Front. Chem. 2020, 8, 300. [Google Scholar] [CrossRef]
- Fukuoka, H.; Isami, T.; Yamanaka, S. Crystal Structure of a Layered Perovskite Niobate KCa2Nb3O10. J. Solid State Chem. 2000, 151, 40–45. [Google Scholar] [CrossRef]
- Jehng, J.M.; Wachs, I.E. Structural Chemistry and Raman Spectra of Niobium Oxides. Chem. Mater. 1991, 3, 100–107. [Google Scholar] [CrossRef]
- Kim, H.J.; Byeon, S.H.; Yun, H. Raman spectra of the solid-solution between Rb2La2Ti3O10 and RbCa2Nb3O10. Bull. Korean Chem. Soc. 2001, 22, 298–302. [Google Scholar]
- Rodionov, I.A.; Sokolova, I.P.; Silyukov, O.I.; Burovikhina, A.A.; Fateev, S.A.; Zvereva, I.A. Protonation and Photocatalytic Activity of the Rb2La2Ti3O10 Layered Oxide in the Reaction of Hydrogen Production. Int. J. Photoenergy 2017, 2017, 9628146. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition | Light Absorption | Specific Surface, m2/g | |||
|---|---|---|---|---|---|---|
| x | y | Eg, eV | λmax, nm | N2 | Kr | |
| KCN3 | − | − | 3.54 | 350 | 3.8 | 4.4 |
| HCN3·yH2O | − | 1.5 | 3.50 | 354 | 5.6 | 7.6 |
| HCN3×MeOH | 1.00 | 0.20 | 3.45 | 359 | 2.4 | 2.9 |
| HCN3×EtOH | 0.90 | 0.45 | 3.50 | 354 | 4.7 | 3.9 |
| HCN3×PrOH | 0.85 | 0.35 | 3.53 | 351 | 6.4 | 6.1 |
| HCN3×BuOH | 0.85 | 0.40 | 3.50 | 354 | 7.3 | 7.4 |
| HCN3×HxOH | 0.85 | 0.20 | 3.47 | 357 | 10.7 | 13.8 |
| HCN3×DcOH | 0.90 | 0.25 | 3.51 | 353 | 10.2 | 13.2 |
| Compound | Bare | Pt-Loaded | kPt | ||
|---|---|---|---|---|---|
| ω, μmol/h | ϕ, % | ω, μmol/h | ϕ, % | ||
| KCN3 | 0.94 | 0.013 | 7.8 | 0.11 | 8.40 |
| HCN3·yH2O | 13 | 0.17 | 586 | 7.8 | 45.4 |
| HCN3×MeOH | 80 | 1.1 | 796 | 10.6 | 10.0 |
| HCN3×EtOH | 105 | 1.4 | 1544 | 20.6 | 14.7 |
| HCN3×PrOH | 88 | 1.2 | 1009 | 13.5 | 11.5 |
| HCN3×BuOH | 121 | 1.6 | 1151 | 15.4 | 9.50 |
| HCN3×HxOH | 131 | 1.8 | 938 | 12.5 | 7.20 |
| HCN3×DcOH | 128 | 1.7 | 1005 | 13.4 | 7.90 |
| R | Precursors | Alcohol Concentration, % | Temperature, °C | Flushing | Approximate Yield, % |
|---|---|---|---|---|---|
| Me | HCN3·yH2O | 90 (in water) | 100 | acetone | 85 |
| Et | 96 (in water) | ||||
| Pr | HCN3×EtOH | 100 | 150 | 75 | |
| Bu | |||||
| Hx | n-hexane | ||||
| Dc | HCN3×PrOH | 65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voytovich, V.V.; Kurnosenko, S.A.; Silyukov, O.I.; Rodionov, I.A.; Bugrov, A.N.; Minich, I.A.; Malygina, E.N.; Zvereva, I.A. Synthesis of n-Alkoxy Derivatives of Layered Perovskite-Like Niobate HCa2Nb3O10 and Study of Their Photocatalytic Activity for Hydrogen Production from an Aqueous Solution of Methanol. Catalysts 2021, 11, 897. https://doi.org/10.3390/catal11080897
Voytovich VV, Kurnosenko SA, Silyukov OI, Rodionov IA, Bugrov AN, Minich IA, Malygina EN, Zvereva IA. Synthesis of n-Alkoxy Derivatives of Layered Perovskite-Like Niobate HCa2Nb3O10 and Study of Their Photocatalytic Activity for Hydrogen Production from an Aqueous Solution of Methanol. Catalysts. 2021; 11(8):897. https://doi.org/10.3390/catal11080897
Chicago/Turabian StyleVoytovich, Vladimir V., Sergey A. Kurnosenko, Oleg I. Silyukov, Ivan A. Rodionov, Alexander N. Bugrov, Iana A. Minich, Ekaterina N. Malygina, and Irina A. Zvereva. 2021. "Synthesis of n-Alkoxy Derivatives of Layered Perovskite-Like Niobate HCa2Nb3O10 and Study of Their Photocatalytic Activity for Hydrogen Production from an Aqueous Solution of Methanol" Catalysts 11, no. 8: 897. https://doi.org/10.3390/catal11080897
APA StyleVoytovich, V. V., Kurnosenko, S. A., Silyukov, O. I., Rodionov, I. A., Bugrov, A. N., Minich, I. A., Malygina, E. N., & Zvereva, I. A. (2021). Synthesis of n-Alkoxy Derivatives of Layered Perovskite-Like Niobate HCa2Nb3O10 and Study of Their Photocatalytic Activity for Hydrogen Production from an Aqueous Solution of Methanol. Catalysts, 11(8), 897. https://doi.org/10.3390/catal11080897