
Palladium-Catalyzed Dehydrogenative C-2 Alkenylation of 5-Arylimidazoles and Related Azoles with Styrenes


Abstract
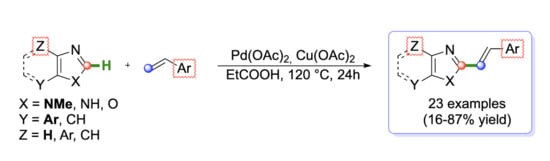
:
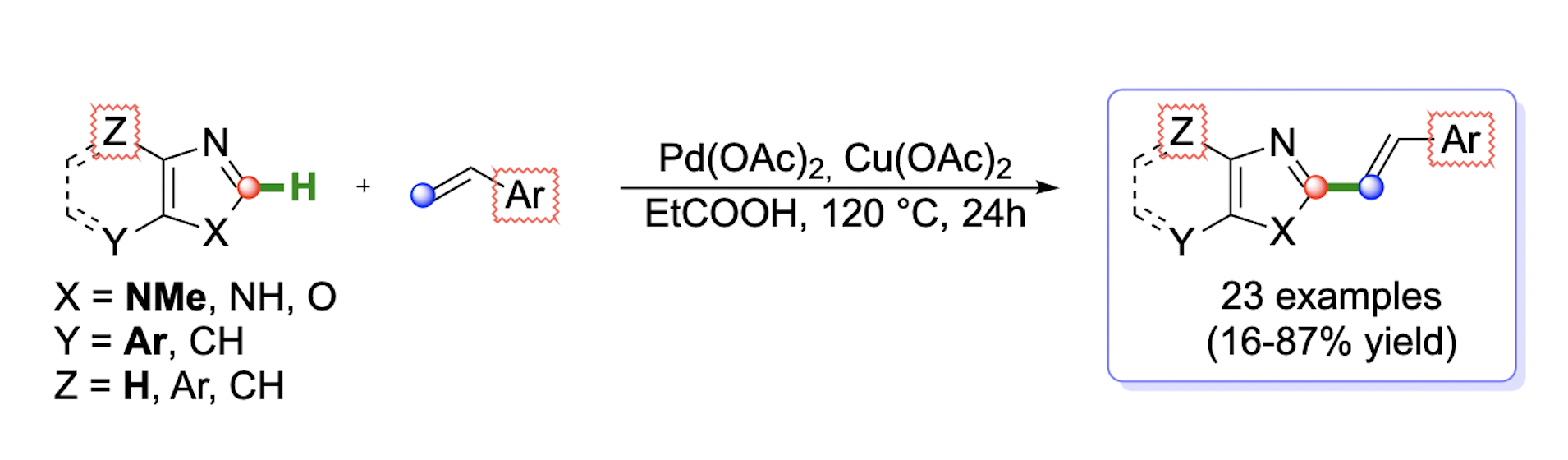
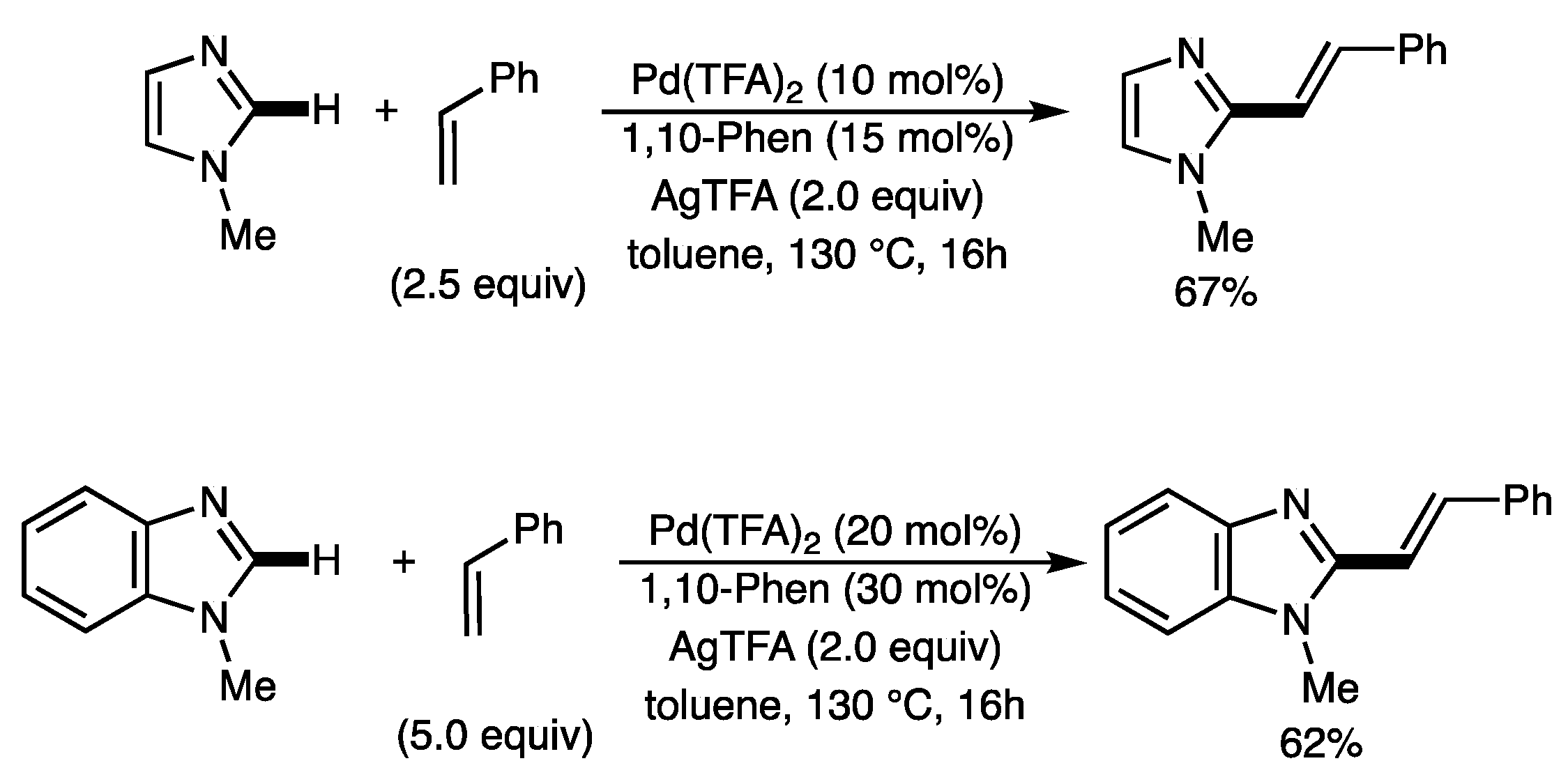
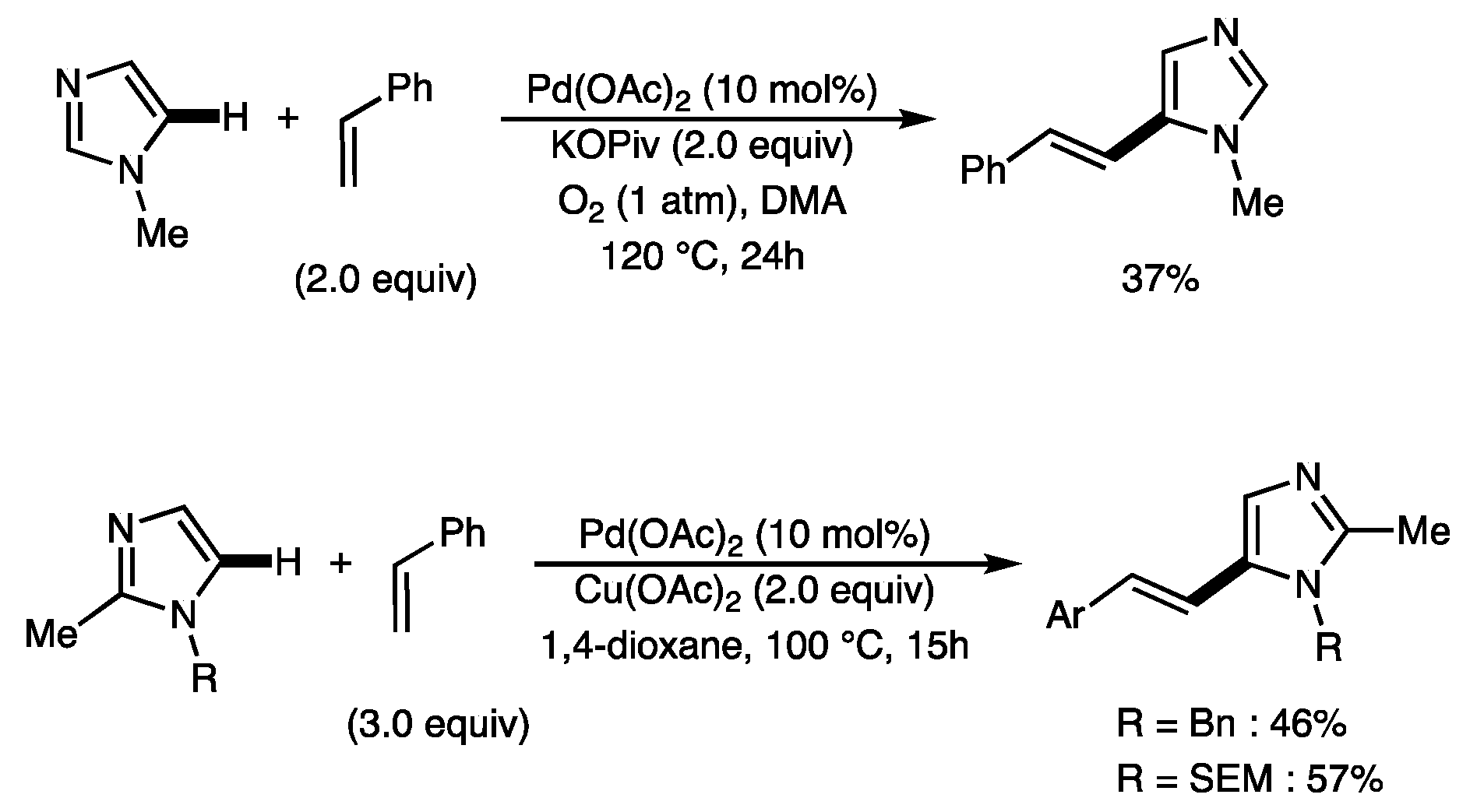
1. Introduction
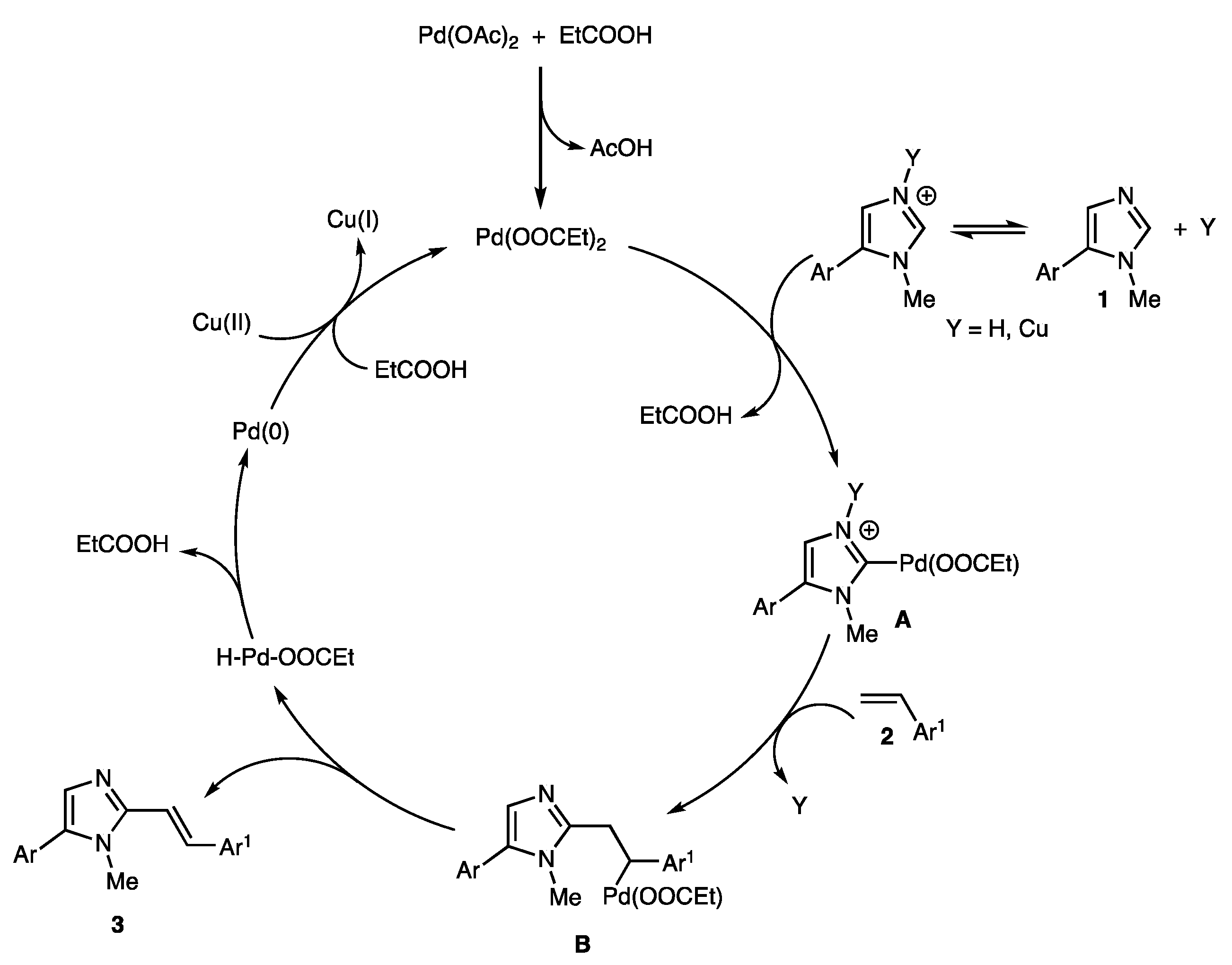
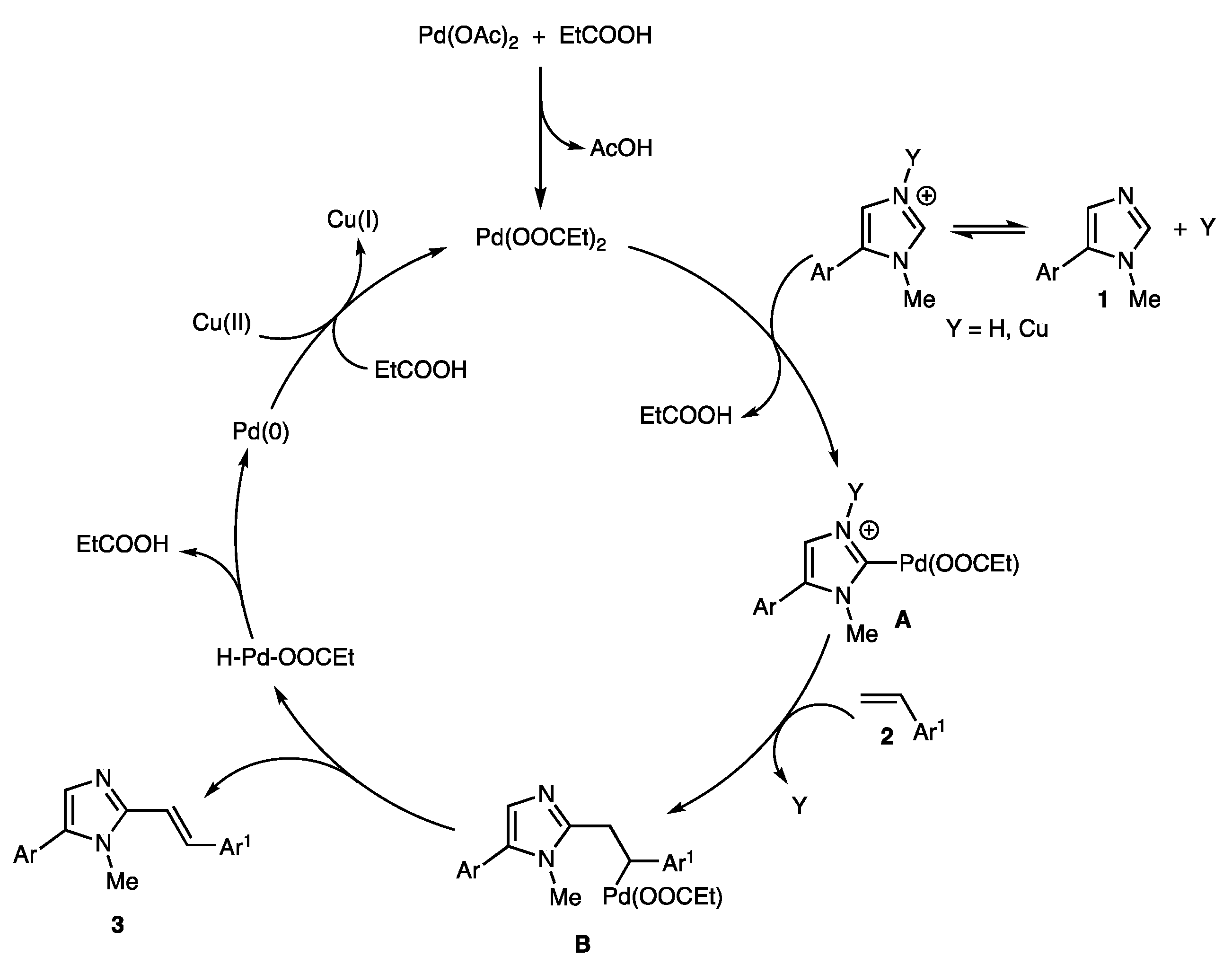
2. Results and Discussion
2.1. Screening of the Reaction Conditions
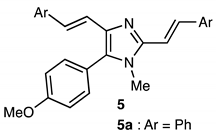
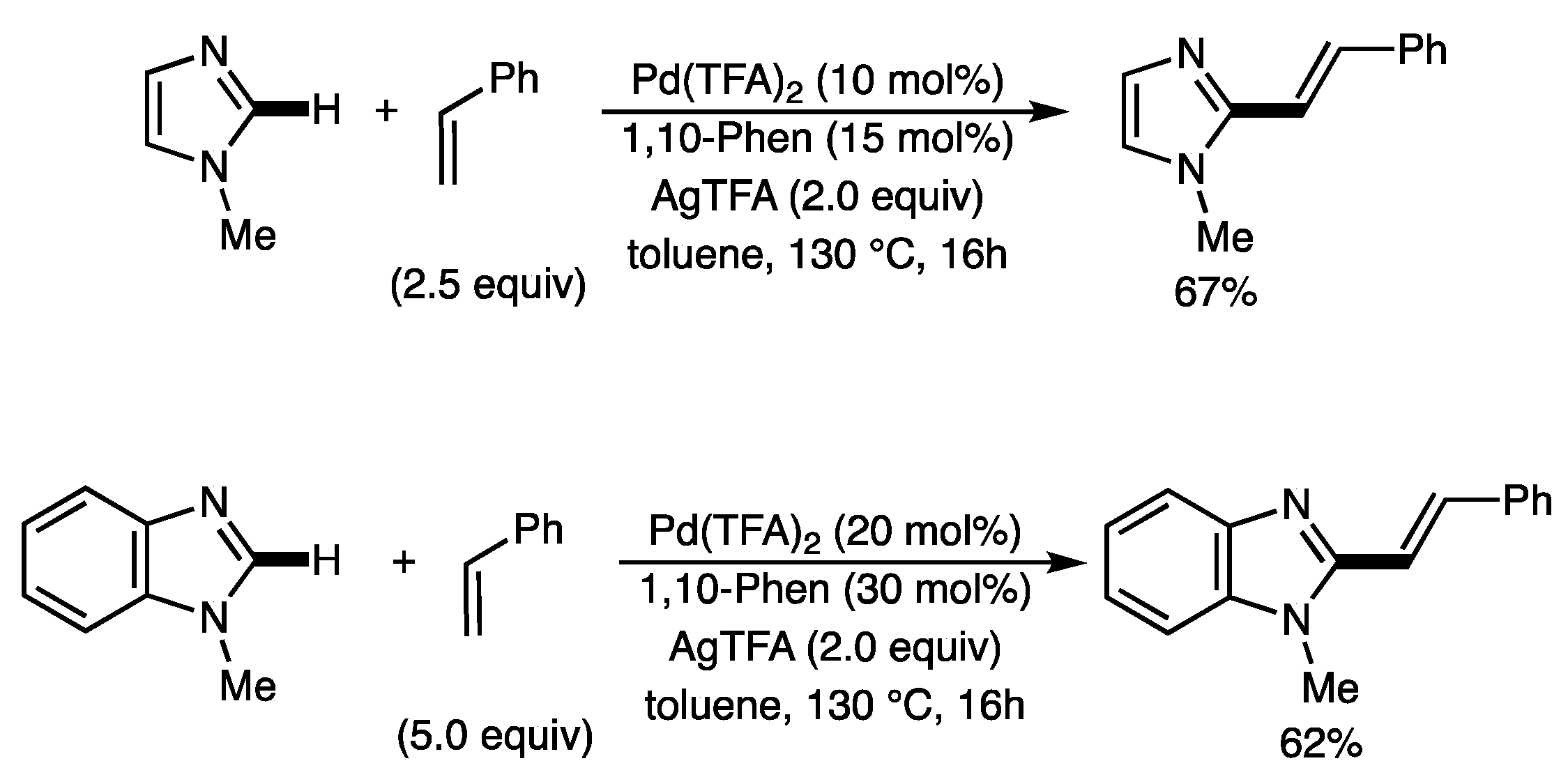
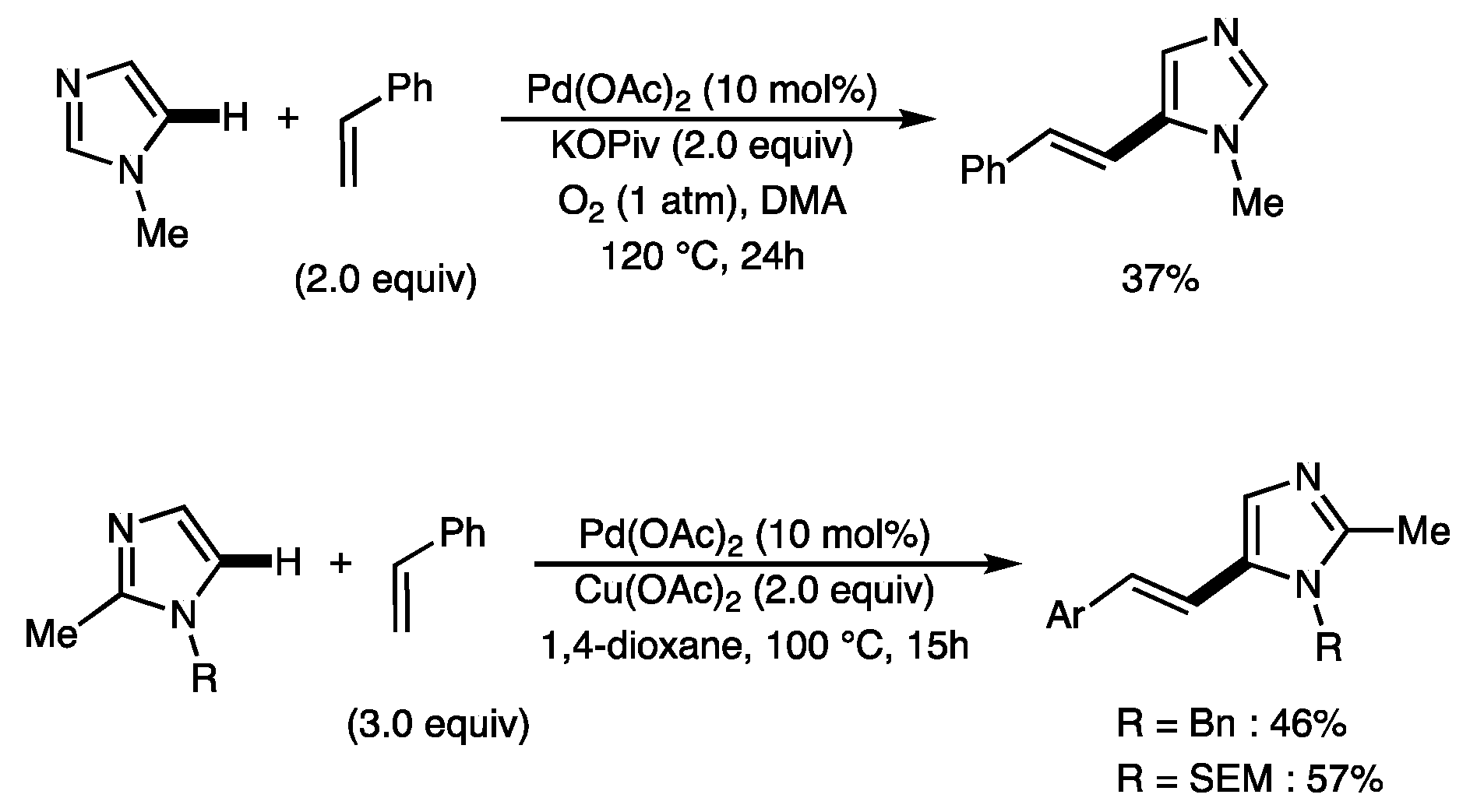
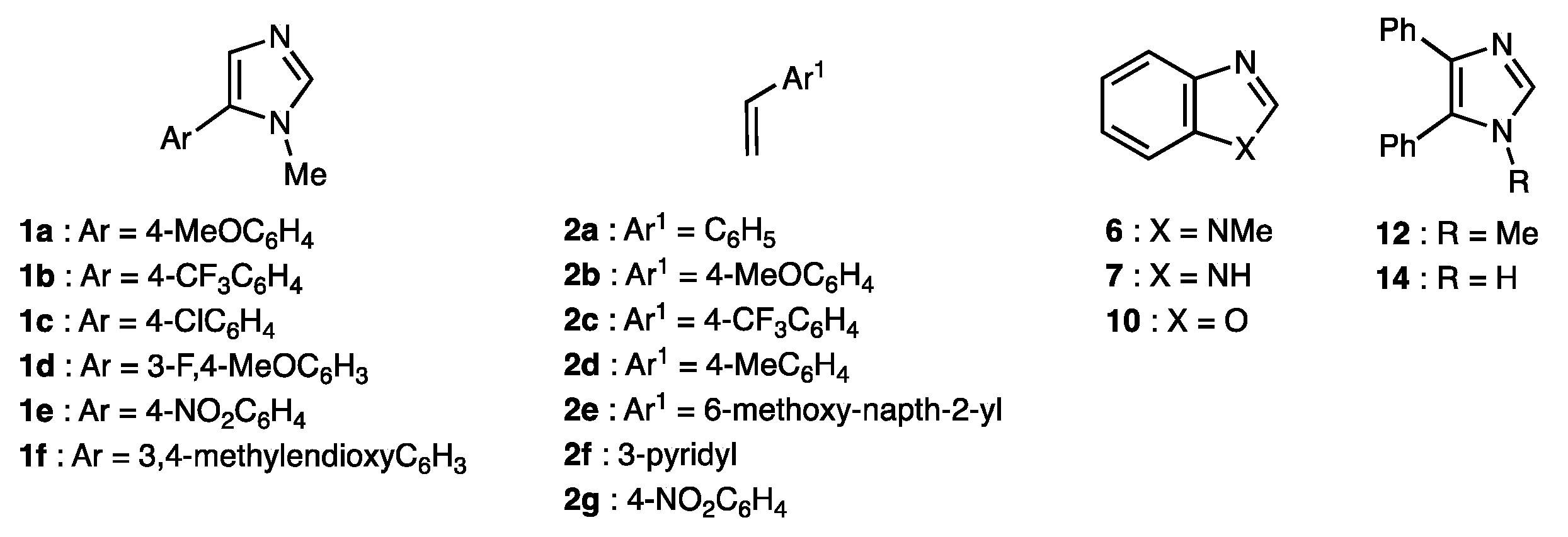
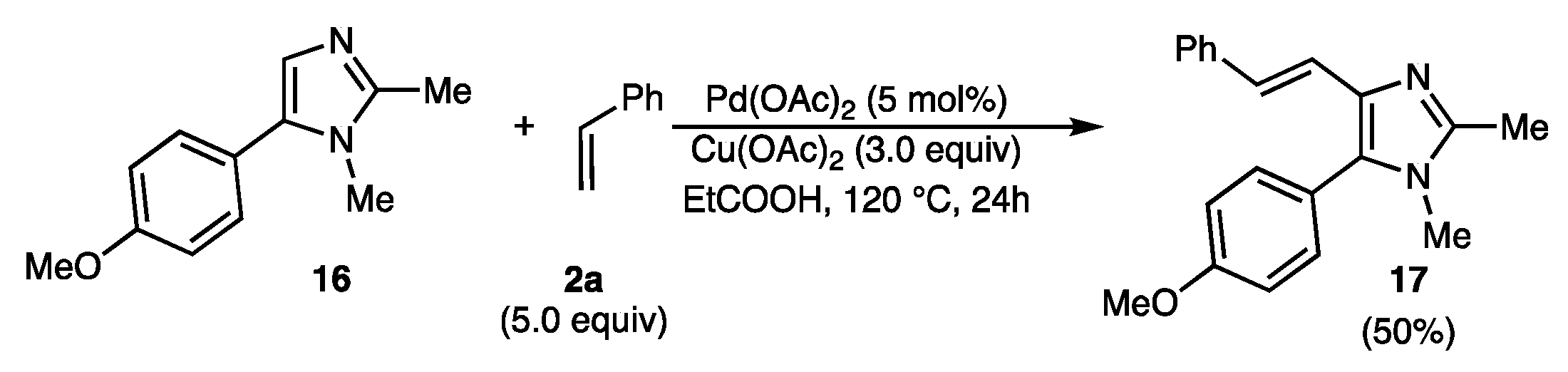
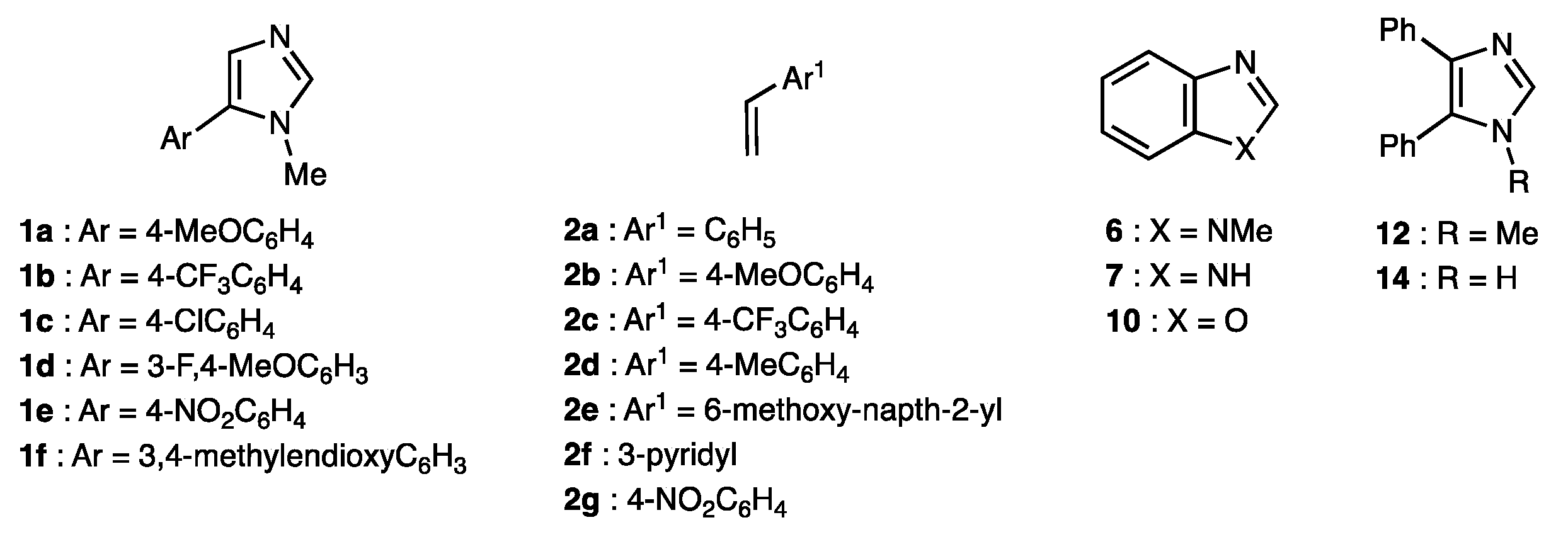
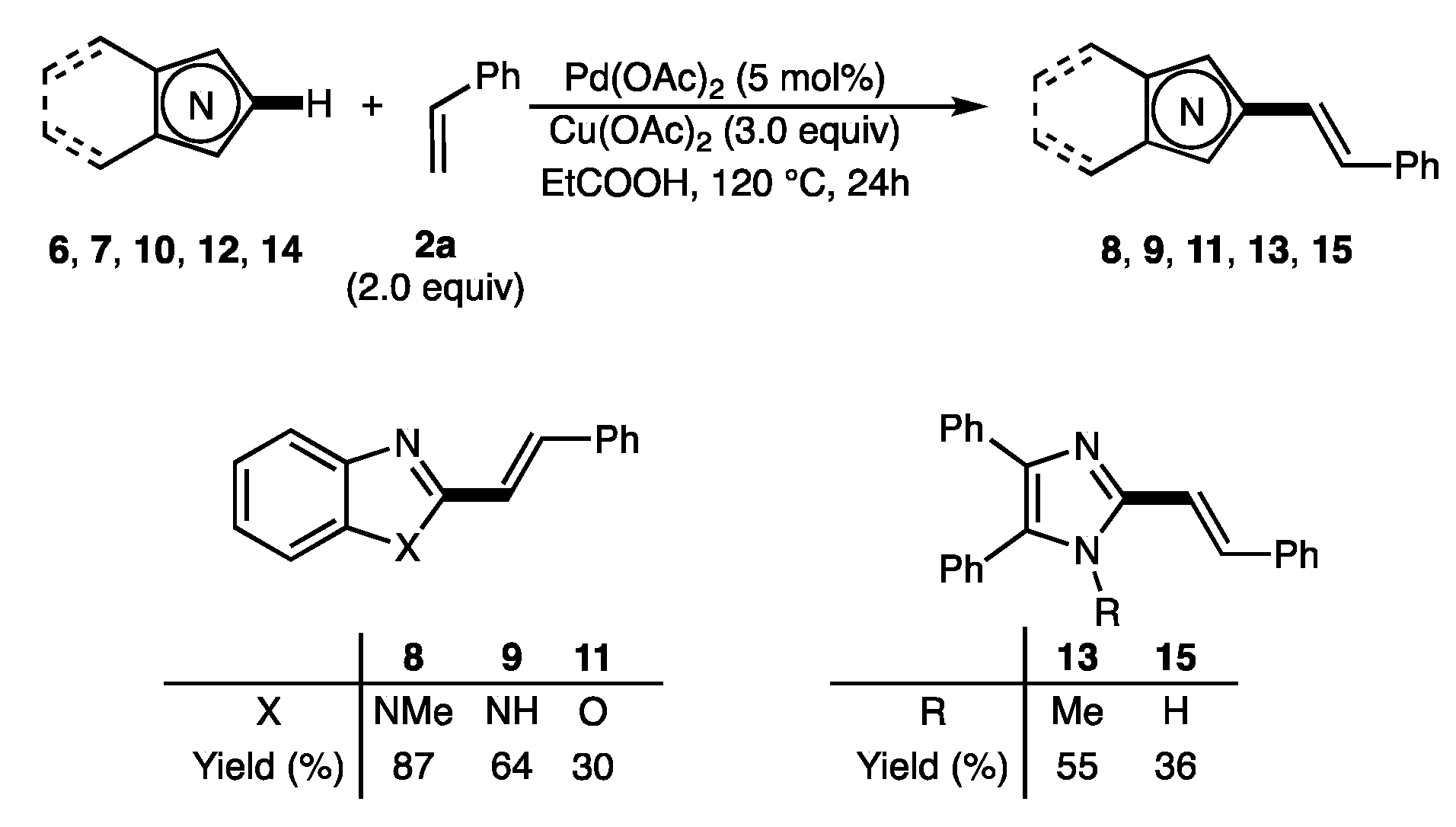
2.2. Scope of the Pd-Catalyzed Dehydrogenative Alkenylation of Imidazoles and Related Azoles
3. Materials and Methods
3.1. (E)-5-(4-Methoxyphenyl)-1-methyl-2-styryl-1H-imidazole (3a) and 5-(4-methoxyphenyl)-1-methyl-2-(1-phenylethyl)-1H-imidazole (4)
3.2. Procedure for the Screening of the Reaction Conditions for the Pd-Catalyzed Dehydrogenative C2-Alkenylation of 5-(4-Methoxyphenyl)-1-Methyl-1H-Imidazole (1a) with Styrene (2a) Using Carboxylic Acids as Reaction Solvents
(E)-5-(4-methoxyphenyl)-1-methyl-2-styryl-1H-imidazole (3a) and 5-(4-methoxyphenyl)-1-methyl-2,4-di((E)-styryl)-1H-imidazole (5a)
3.3. General Procedure for the Pd(II)/Cu(II)-Promoted Dehydrogenative Alkenylation of Azoles with Styrenes
3.3.1. (E)-5-(4-Methoxyphenyl)-2-(4-methoxystyryl)-1-methyl-1H-imidazole (3b)
3.3.2. (E)-5-(4-Methoxyphenyl)-1-methyl-2-(4-(trifluoromethyl)styryl)-1H-imidazole (3c)
3.3.3. (E)-5-(4-Methoxyphenyl)-1-methyl-2-(4-methylstyryl)-1H-imidazole (3d)
3.3.4. (E)-2-(2-(6-Methoxynaphthalen-2-yl)vinyl)-5-(4-methoxyphenyl)-1-methyl-1H-imidazole (3e)
3.3.5. (E)-5-(4-Methoxyphenyl)-1-methyl-2-(4-nitrostyryl)-1H-imidazole (3f)
3.3.6. (E)-1-Methyl-2-styryl-5-(4-(trifluoromethyl)phenyl)-1H-imidazole (3g)
3.3.7. (E)-2-(4-Methoxystyryl)-1-methyl-5-(4-(trifluoro-methyl)phenyl)-1H-imidazole (3h)
3.3.8. (E)-1-Methyl-2-(4-methylstyryl)-5-(4-(trifluoro-methyl)phenyl)-1H-imidazole (3i)
3.3.9. (E)-1-methyl-5-(4-Trifluoromethyl)phenyl)-2-(4-(trifluoromethyl)-styryl)-1H-imidazole (3j)
3.3.10. (E)-5-(4-Chlorophenyl)-1-methyl-2-styryl-1H-imidazole (3k)
3.3.11. (E)-5-(4-Chlorophenyl)-2-(4-methoxystyryl)-1-methyl-1H-imidazole (3l)
3.3.12. (E)-5-(4-Chlorophenyl)-1-methyl-2-(4-(trifluoromethyl)styryl)-1H-imidazole (3m)
3.3.13. (E)-4-(2-(5-(4-Chlorophenyl)-1-methyl-1H-imidazol-2-yl)vinyl)-pyridine (3n)
3.3.14. (E)-5-(4-Chlorophenyl)-1-methyl-2-(4-nitrostyryl)-1H-imidazole (3o)
3.3.15. (E)-5-(3-Fluoro-4-methoxyphenyl)-2-(4-methoxystyryl)-1-methyl-1H-imidazole (3p)
3.3.16. (E)-2-(4-Methoxystyryl)-1-methyl-5-(4-nitrophenyl)-1H-imidazole (3q)
3.3.17. (E)-5-(Benzo[d][1,3]dioxol-5-yl)-2-(4-methoxystyryl)-1-methyl-1H-imidazole (3r)
3.3.18. (E)-1-Methyl-2-styryl-1H-benzo[d]imidazole (8)
3.3.19. (E)-2-Styryl-1H-benzo[d]imidazole (9)
3.3.20. (E)-2-Styrylbenzo[d]oxazole (11)
3.3.21. (E)-1-Methyl-4,5-diphenyl-2-styryl-1H-imidazole (13)
3.3.22. (E)-4,5-Diphenyl-2-styryl-1H-imidazole (15)
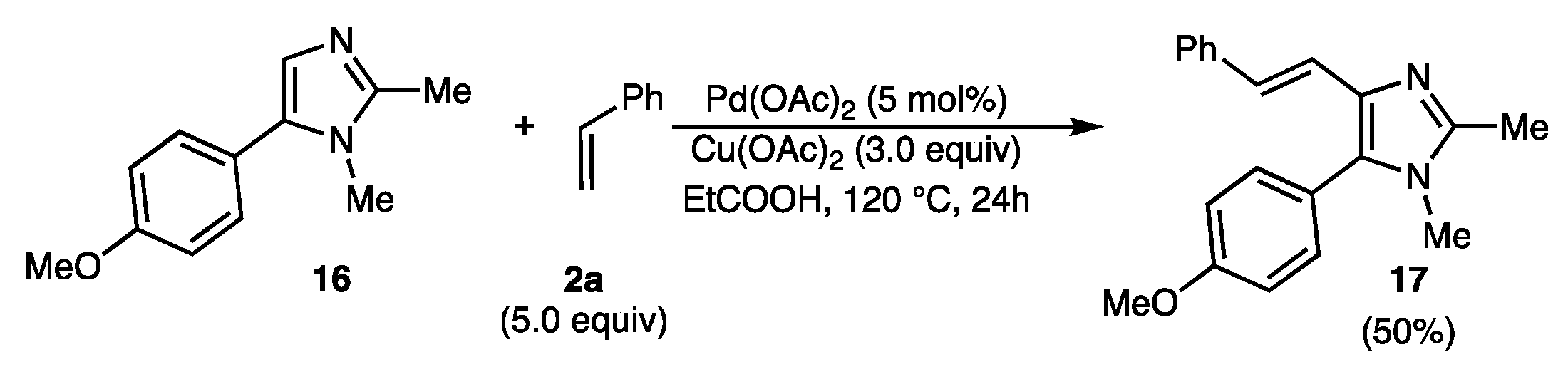
3.3.23. (E)-5-(4-Methoxyphenyl)-1,2-dimethyl-4-styryl-1H-imidazole (17)
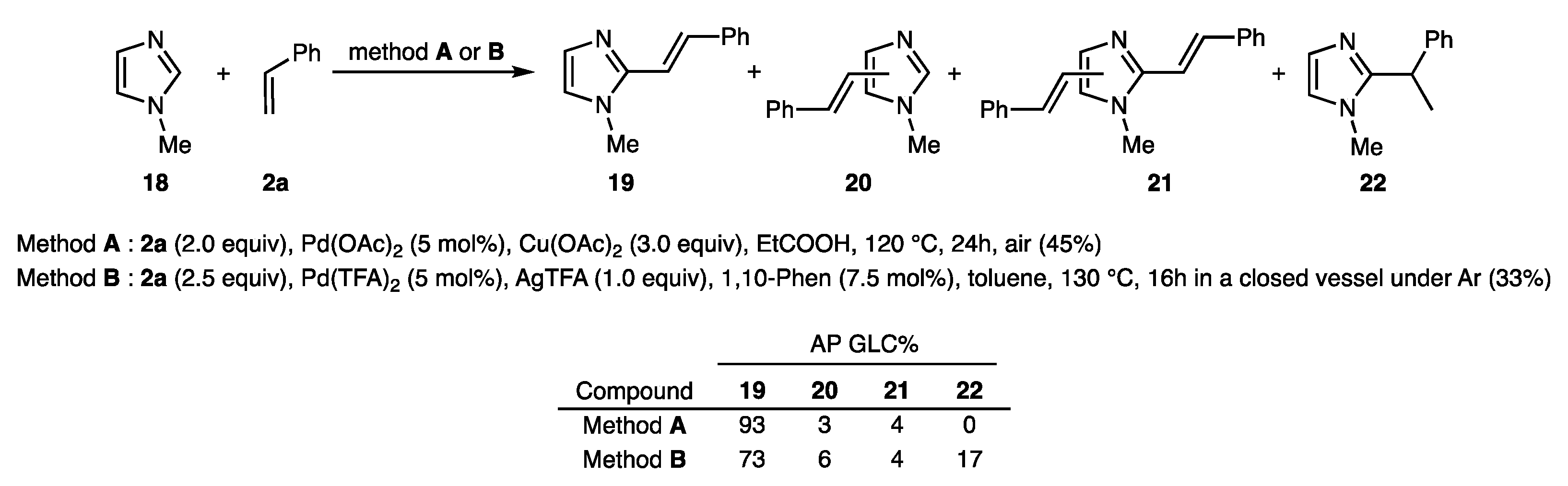
3.3.24. (E)-1-Methyl-2-styryl-1H-imidazole (19)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bansal, S.; Shabade, A.B.; Punji, B. Advances in C(sp2)-H/C(sp2)-H Oxidative Coupling of (Hetero)arenes Using 3d Transition Metal Catalysts. Adv. Synth. Catal. 2021, 363, 1998–2022. [Google Scholar] [CrossRef]
- Bellina, F.; Perego, L.A. 12 Aerobic Oxidative Intermolecular Cross-Coupling and Heck Reactions; Georg Thieme Verlag: Stuttgart, Germany, 2017. [Google Scholar]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C-H bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, J.; Muzart, J. Intermolecular dehydrogenative Heck reactions. Chem. Rev. 2011, 111, 1170–1214. [Google Scholar] [CrossRef]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative Coupling between Two Hydrocarbons: An Update of Recent C-H Functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef]
- Maes, J.; Maes, B.U.W. A Journey through Metal-Catalyzed CH Functionalization of Heterocycles. In Heterocyclic Chemistry in the 21st Century—A Tribute to Alan Katritzky; Academic Press: Cambridge, MA, USA, 2016; pp. 137–194. [Google Scholar]
- Varun, B.V.; Dhineshkumar, J.; Bettadapur, K.R.; Siddaraju, Y.; Alagiri, K.; Prabhu, K.R. Recent advancements in dehydrogenative cross coupling reactions for CC bond formation. Tetrahedron Lett. 2017, 58, 803–824. [Google Scholar] [CrossRef]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef]
- Yang, Y.; Lan, J.; You, J. Oxidative C-H/C-H Coupling Reactions between Two (Hetero)arenes. Chem. Rev. 2017, 117, 8787–8863. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- Bras, J.L.; Muzart, J. Pd-Catalyzed Intermolecular Dehydrogenative Heck Reactions of Five-Membered Heteroarenes. Catalysts 2020, 10, 571. [Google Scholar] [CrossRef]
- Moritanl, I.; Fujiwara, Y. Aromatic substitution of styrene-palladium chloride complex. Tetrahedron Lett. 1967, 8, 1119–1122. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Moritani, I.; Danno, S.; Asano, R.; Teranishi, S. Aromatic substitution of olefins. VI. Arylation of olefins with palladium(II) acetate. J. Am. Chem. Soc. 1969, 91, 7166–7169. [Google Scholar] [CrossRef]
- Moritani, I.; Danno, S.; Fujiwara, Y.; Teranishi, S. Aromatic Substitution of Olefins XV. The Steric Course of the Reaction. Bull. Chem. Soc. Jpn. 1971, 44, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, J.; Mao, F.; Kwong, F.Y. Palladium-catalyzed cross-dehydrogenative functionalization of C(sp2)-H Bonds. Chem. Asian J. 2014, 9, 26–47. [Google Scholar] [CrossRef] [PubMed]
- Broggini, G.; Beccalli, E.M.; Fasana, A.; Gazzola, S. Palladium-catalyzed dual C-H or N-H functionalization of unfunctionalized indole derivatives with alkenes and arenes. Beilstein J. Org. Chem. 2012, 8, 1730–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Gunnoe, T.B. Advances in Group 10 Transition-Metal-Catalyzed Arene Alkylation and Alkenylation. J. Am. Chem. Soc. 2021, 143, 6746–6766. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J.P. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 2002, 37, 2320–2322. [Google Scholar] [CrossRef]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of Olefin with Aryl Iodide Catalyzed by Palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Besselievre, F.; Piguel, S.; Mahuteau-Betzer, F.; Grierson, D.S. Stereoselective direct copper-catalyzed alkenylation of oxazoles with bromoalkenes. Org. Lett. 2008, 10, 4029–4032. [Google Scholar] [CrossRef]
- Gottumukkala, A.L.; Derridj, F.; Djebbar, S.; Doucet, H. Alkenyl bromides: Useful coupling partners for the palladium-catalysed coupling with heteroaromatics via a C–H bond activation. Tetrahedron Lett. 2008, 49, 2926–2930. [Google Scholar] [CrossRef]
- Verrier, C.; Hoarau, C.; Marsais, F. Direct palladium-catalyzed alkenylation, benzylation and alkylation of ethyl oxazole-4-carboxylate with alkenyl-, benzyl- and alkyl halides. Org. Biomol. Chem. 2009, 7, 647–650. [Google Scholar] [CrossRef]
- Sahnoun, S.; Messaoudi, S.; Brion, J.-D.; Alami, M. Pd/Cu-Catalyzed Direct Alkenylation of Azole Heterocycles with Alkenyl Halides. Eur. J. Org. Chem. 2010, 2010, 6097–6102. [Google Scholar] [CrossRef]
- Vabre, R.; Chevot, F.; Legraverend, M.; Piguel, S. Microwave-assisted Pd/Cu-catalyzed C-8 direct alkenylation of purines and related azoles: An alternative access to 6,8,9-trisubstituted purines. J. Org. Chem. 2011, 76, 9542–9547. [Google Scholar] [CrossRef]
- Yao, Y.-X.; Fang, D.-M.; Gao, F.; Liang, X.-X. Room-temperature palladium-catalyzed direct 2-alkenylation of azole derivatives with alkenyl bromides. Tetrahedron Lett. 2019, 60, 68–71. [Google Scholar] [CrossRef]
- Mahuteau-Betzer, F.; Piguel, S. Synthesis and evaluation of photophysical properties of Series of π-conjugated oxazole dyes. Tetrahedron Lett. 2013, 54, 3188–3193. [Google Scholar] [CrossRef]
- Waldvogel, S.R.; Lips, S.; Selt, M.; Riehl, B.; Kampf, C.J. Electrochemical Arylation Reaction. Chem. Rev. 2018, 118, 6706–6765. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lv, S.; Tian, S. Electrochemical Transition-Metal-Catalyzed C-H Bond Functionalization: Electricity as Clean Surrogates of Chemical Oxidants. ChemSusChem 2019, 12, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Lei, A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. Acc. Chem. Res. 2019, 52, 3309–3324. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, J.C. From Bench to Plant: An Opportunity for Transition Metal Paired Electrocatalysis. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Lessi, M.; Manzini, C.; Minei, P.; Perego, L.A.; Bloino, J.; Egidi, F.; Barone, V.; Pucci, A.; Bellina, F. Synthesis and Optical Properties of Imidazole-Based Fluorophores having High Quantum Yields. ChemPlusChem 2014, 79, 366–370. [Google Scholar] [CrossRef]
- Barone, V.; Bellina, F.; Biczysko, M.; Bloino, J.; Fornaro, T.; Latouche, C.; Lessi, M.; Marianetti, G.; Minei, P.; Panattoni, A.; et al. Toward the design of alkynylimidazole fluorophores: Computational and experimental characterization of spectroscopic features in solution and in poly(methyl methacrylate). Phys. Chem. Chem. Phys. 2015, 17, 26710–26723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellina, F.; Manzini, C.; Marianetti, G.; Pezzetta, C.; Fanizza, E.; Lessi, M.; Minei, P.; Barone, V.; Pucci, A. Colourless p-phenylene-spaced bis-azoles for luminescent concentrators. Dyes Pigment. 2016, 134, 118–128. [Google Scholar] [CrossRef]
- Lessi, M.; Panzetta, G.; Marianetti, G.; Bellina, F. Improved Synthesis of Symmetrical 2,5-Diarylimidazoles by One-Pot Palladium-Catalyzed Direct Arylation Tailored on the Electronic Features of the Aryl Halide. Synthesis 2017, 49, 4676–4686. [Google Scholar] [CrossRef] [Green Version]
- Marianetti, G.; Lessi, M.; Barone, V.; Bellina, F.; Pucci, A.; Minei, P. Solar collectors based on luminescent 2,5-diarylimidazoles. Dyes Pigment. 2018, 157, 334–341. [Google Scholar] [CrossRef]
- Lessi, M.; Lucci, A.; Cuzzola, A.; Bellina, F. Imidazo-Fused Isoindoles by Pd(II)/Ag(I)-Promoted Intramolecular Dehydrogenative Coupling. Eur. J. Org. Chem. 2020, 2020, 796–802. [Google Scholar] [CrossRef]
- Lee, W.C.; Wang, T.H.; Ong, T.G. Ligand promoted Pd-catalyzed dehydrogenative alkenylation of hetereoarenes. Chem. Commun. 2014, 50, 3671–3673. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hwang, Y.J.; Han, I.; Joo, J.M. Regioselective C-H alkenylation of imidazoles and its application to the synthesis of unsymmetrically substituted benzimidazoles. Chem. Commun. 2018, 54, 6879–6882. [Google Scholar] [CrossRef]
- Miyasaka, M.; Hirano, K.; Satoh, T.; Miura, M. Palladium-catalyzed direct oxidative alkenylation of azoles. J. Org. Chem. 2010, 75, 5421–5424. [Google Scholar] [CrossRef]
- Vana, J.; Bartacek, J.; Hanusek, J.; Roithova, J.; Sedlak, M. C-H Functionalizations by Palladium Carboxylates: The Acid Effect. J. Org. Chem. 2019, 84, 12746–12754. [Google Scholar] [CrossRef]
- Schubert, W.M.; Keeffe, J.R. Acid-catalyzed hydration of styrenes. J. Am. Chem. Soc. 1972, 94, 559–566. [Google Scholar] [CrossRef]
- Grimster, N.P.; Gauntlett, C.; Godfrey, C.R.; Gaunt, M.J. Palladium-catalyzed intermolecular alkenylation of indoles by solvent-controlled regioselective C-H functionalization. Angew. Chem. Int. Ed. 2005, 44, 3125–3129. [Google Scholar] [CrossRef]
- Djakovitch, L.; Rouge, P. New homogeneously and heterogeneously [Pd/Cu]-catalysed C3-alkenylation of free NH-indoles. J. Mol. Catal. A Chem. 2007, 273, 230–239. [Google Scholar] [CrossRef]
- Bellina, F.; Cauteruccio, S.; Mannina, L.; Rossi, R.; Viel, S. Regiocontrolled Synthesis of 1,2-Diaryl-1H-imidazoles by Palladium- and Copper-Mediated Direct Coupling of 1-Aryl-1H-imidazoles with Aryl Halides under Ligandless Conditions. Eur. J. Org. Chem. 2006, 693–703. [Google Scholar] [CrossRef]
- Storr, T.E.; Baumann, C.G.; Thatcher, R.J.; De Ornellas, S.; Whitwood, A.C.; Fairlamb, I.J. Pd(0)/Cu(I)-mediated direct arylation of 2’-deoxyadenosines: Mechanistic role of Cu(I) and reactivity comparisons with related purine nucleosides. J. Org. Chem. 2009, 74, 5810–5821. [Google Scholar] [CrossRef]
- Gorelsky, S.I. Tuning the Regioselectivity of Palladium-Catalyzed Direct Arylation of Azoles by Metal Coordination. Organometallics 2012, 31, 794–797. [Google Scholar] [CrossRef]
- Bellina, F.; Cauteruccio, S.; Di Fiore, A.; Rossi, R. Regioselective Synthesis of 4,5-Diaryl-1-methyl-1H-imidazoles Including Highly Cytotoxic Derivatives by Pd-Catalyzed Direct C-5 Arylation of 1-Methyl-1H-imidazole with Aryl Bromides. Eur. J. Org. Chem. 2008, 2008, 5436–5445. [Google Scholar] [CrossRef]
- Kang, P.; Foote, C.S. Photosensitized oxidation of 13C,15N-labeled imidazole derivatives. J. Am. Chem. Soc. 2002, 124, 9629–9638. [Google Scholar] [CrossRef] [PubMed]
- Baghbanzadeh, M.; Pilger, C.; Kappe, C.O. Palladium-catalyzed direct arylation of heteroaromatic compounds: Improved conditions utilizing controlled microwave heating. J. Org. Chem. 2011, 76, 8138–8142. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Brown, A.R. Suzuki-Miyaura cross-coupling reactions of potassium vinyltrifluoroborate with aryl and heteroaryl electrophiles. J. Org. Chem. 2006, 71, 9681–9686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petzer, J.P.; Steyn, S.; Castagnoli, K.P.; Chen, J.-F.; Schwarzschild, M.A.; Van der Schyf, C.J.; Castagnoli, N. Inhibition of monoamine oxidase B by selective adenosine A2A receptor antagonists. Biorg. Med. Chem. 2003, 11, 1299–1310. [Google Scholar] [CrossRef]
- Moorthy, J.N.; Neogi, I. IBX-mediated one-pot synthesis of benzimidazoles from primary alcohols and arylmethyl bromides. Tetrahedron Lett. 2011, 52, 3868–3871. [Google Scholar] [CrossRef]
- Kidwai, M.; Bansal, V.; Saxena, A.; Aerry, S.; Mozumdar, S. Cu-Nanoparticles: Efficient catalysts for the oxidative cyclization of Schiffs’ bases. Tetrahedron Lett. 2006, 47, 8049–8053. [Google Scholar] [CrossRef]
- Moore, T.W.; Sana, K.; Yan, D.; Krumm, S.A.; Thepchatri, P.; Snyder, J.P.; Marengo, J.; Arrendale, R.F.; Prussia, A.J.; Natchus, M.G.; et al. Synthesis and Metabolic Studies of Host Directed Inhibitors for Antiviral Therapy. ACS Med. Chem. Lett. 2013, 4, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S. Synthesis and anthelmintic activity of some novel 2-substituted-4,5-diphenyl imidazoles. Acta Pharm. 2010, 60, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, R.; Baldwin, S.; Flechtner, T.; Kalicky, P.; Liu, S.; Washburn, W. Remote oxidation of steroids by photolysis of attached benzophenone groups. J. Am. Chem. Soc. 1973, 95, 3251–3262. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Glorius, F. C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. [Google Scholar] [CrossRef]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
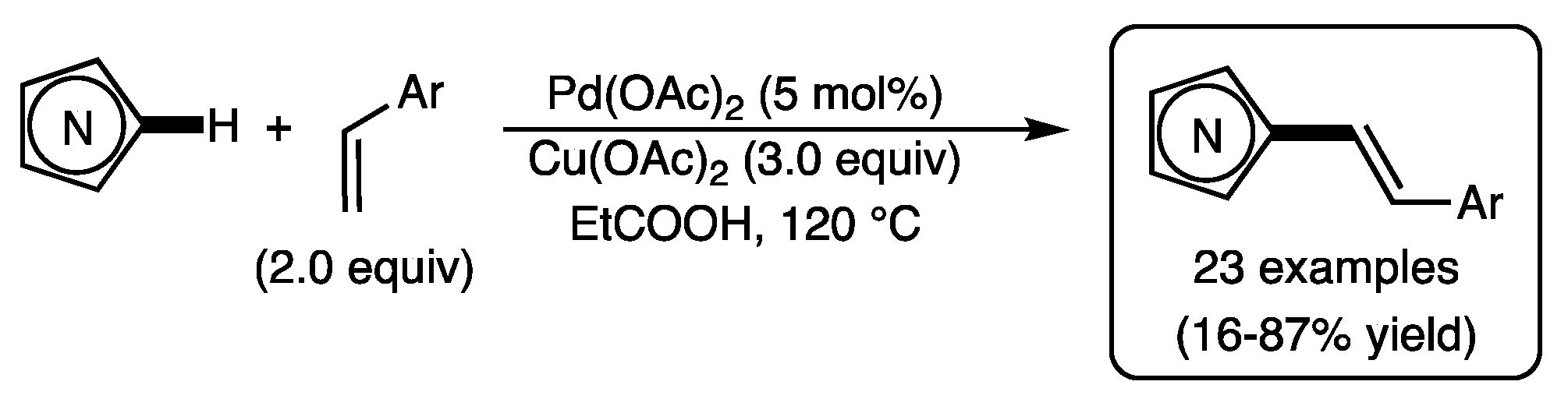




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
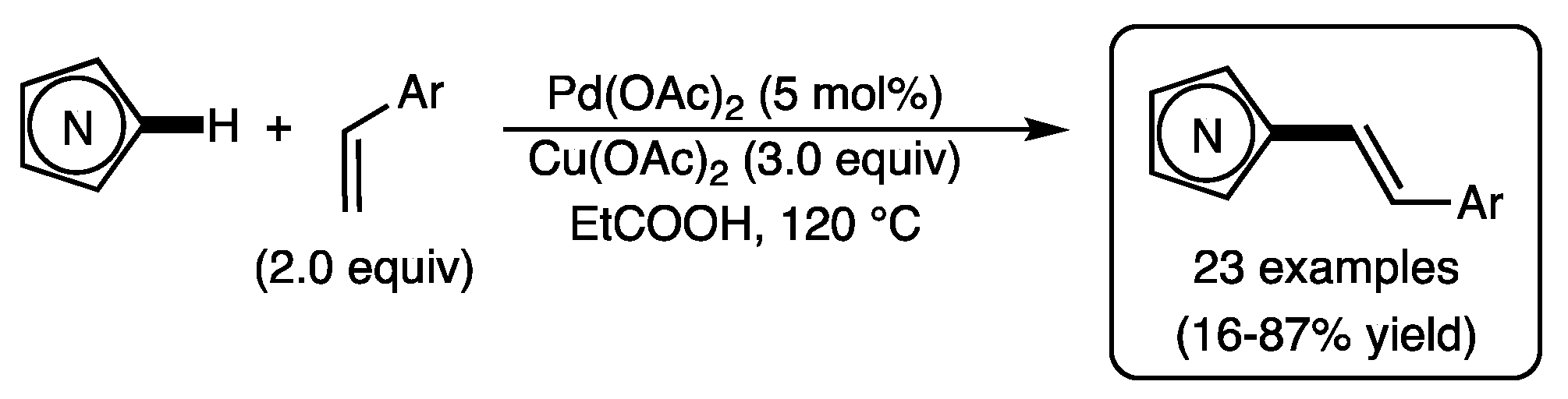{kind=link}
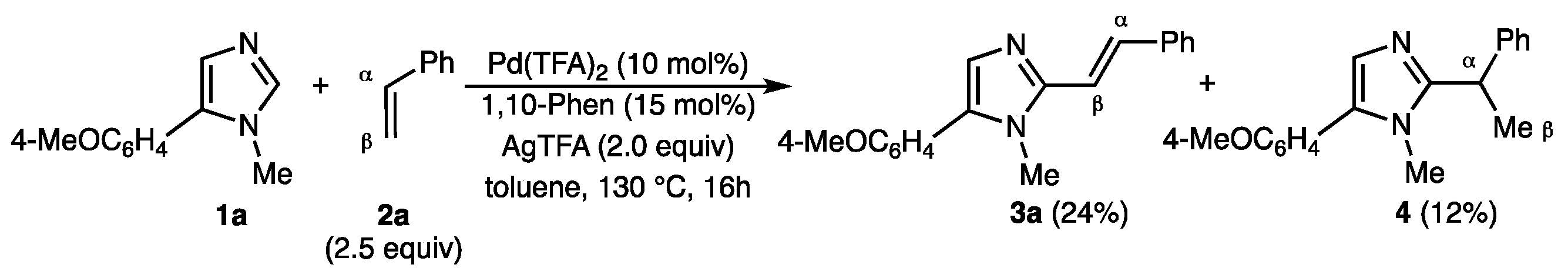{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry 1 | Oxidant | Solvent | 1a Conversion (GLC %) | Yield of 3a (%) 2 | 3a:5a Ratio (AP%) 3 |
|---|---|---|---|---|---|
| 1 | AgOAc | EtCOOH | 90 | 33 | 68:32 |
| 2 | AgOAc | AcOH | 69 | 24 | 65:35 |
| 3 | AgOAc | PivOH | 64 | 23 | 60:40 |
| 4 | Ag2cO3 | EtCOOH | 93 | 32 | 61:39 |
| 5 | Ag2O | EtCOOH | 79 | 38 | 70:30 |
| 6 | AgTFA | EtCOOH | 75 | 35 | 66:35 |
| 7 | Cu(OAc)2 | EtCOOH | >95 | 61(56) 4 | 76:24 |
| 8 5 | Cu(OAc)2 | EtCOOH | 81 | 39 | 82:18 |
| 9 6 | Cu(OAc)2 | EtCOOH | <5 | traces | – |
| 10 7 | Cu(OAc)2 | EtCOOH | <5 | traces | – |
| 11 | CuO | EtCOOH | >95 | 31 | 52:48 |
| 12 | CuCl2 | EtCOOH | <5 | traces | – |
| 13 | PhI(OAc)2 | EtCOOH | <5 | traces | – |
| 14 | NMO | EtCOOH | <5 | traces | – |
| 15 8 | Cu(OAc)2 | EtCOOH | >95 | 50(42) | 74:26 |
| 16 9 | Cu(OAc)2 | EtCOOH | 94 | 37 | 69:31 |
| 17 | Cu(OAc)2 | EtCOOH/DMF | 91 | 44 | 61:39 |
| 18 | Cu(OAc)2 | EtCOOH/NMP | 94 | 45 | 56:44 |
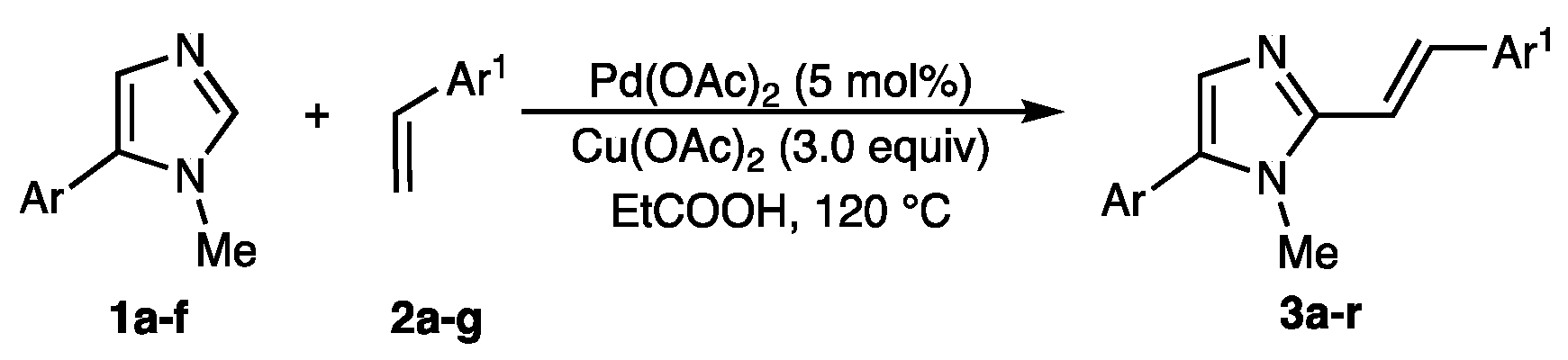
| Entry 1 | Product 3 | Ar | Ar 1 | Yield of 3 (%) 2,3 | 3:5 Ratio (AP%) 4 |
|---|---|---|---|---|---|
| 1 | a | 4-MeOC6H4 | C6H5 | 56 | 76:24 |
| 2 | b | 4-MeOC6H4 | 4-MeOC6H4 | 41 | 77:23 |
| 3 | c | 4-MeOC6H4 | 4-CF3C6H4 | 61 | 85:15 |
| 4 | d | 4-MeOC6H4 | 4-MeC6H4 | 49 | 79:21 |
| 5 | e | 4-MeOC6H4 | 6-MeO-naphth-2-yl | 51 | 77:23 |
| 6 | f | 4-MeOC6H4 | 4-NO2C6H4 | 44 | 82:18 |
| 7 | g | 4-CF3C6H4 | C6H5 | 43 | 78:22 |
| 8 | h | 4-CF3C6H4 | 4-MeOC6H4 | 41 | 75:25 |
| 9 | i | 4-CF3C6H4 | 4-MeC6H4 | 44 | 75:25 |
| 10 | j | 4-CF3C6H4 | 4-CF3C6H4 | 50 | 79:21 |
| 11 | k | 4-ClC6H4 | C6H5 | 45 | 78:22 |
| 12 | l | 4-ClC6H4 | 4-MeOC6H4 | 28 5 | nd |
| 13 | m | 4-ClC6H4 | 4-CF3C6H4 | 46 | 93:7 |
| 14 | n | 4-ClC6H4 | 4-pyridyl | 16 6,7 | nd |
| 15 | o | 4-ClC6H4 | 4-NO2C6H4 | 27 6,8 | nd |
| 16 | p | 3-F,4-MeOC6H3 | 4-MeOC6H4 | 56 | 78:22 |
| 17 | q | 4-NO2C6H4 | 4-MeOC6H4 | 30 9 | nd |
| 18 | r | 3,4-MethylendioxyC6H3 | 4-MeOC6H4 | 52 | 76:24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lessi, M.; Nania, A.; Pittari, M.; Lodone, L.; Cuzzola, A.; Bellina, F. Palladium-Catalyzed Dehydrogenative C-2 Alkenylation of 5-Arylimidazoles and Related Azoles with Styrenes. Catalysts 2021, 11, 762. https://doi.org/10.3390/catal11070762
Lessi M, Nania A, Pittari M, Lodone L, Cuzzola A, Bellina F. Palladium-Catalyzed Dehydrogenative C-2 Alkenylation of 5-Arylimidazoles and Related Azoles with Styrenes. Catalysts. 2021; 11(7):762. https://doi.org/10.3390/catal11070762
Chicago/Turabian StyleLessi, Marco, Attilio Nania, Melania Pittari, Laura Lodone, Angela Cuzzola, and Fabio Bellina. 2021. "Palladium-Catalyzed Dehydrogenative C-2 Alkenylation of 5-Arylimidazoles and Related Azoles with Styrenes" Catalysts 11, no. 7: 762. https://doi.org/10.3390/catal11070762
APA StyleLessi, M., Nania, A., Pittari, M., Lodone, L., Cuzzola, A., & Bellina, F. (2021). Palladium-Catalyzed Dehydrogenative C-2 Alkenylation of 5-Arylimidazoles and Related Azoles with Styrenes. Catalysts, 11(7), 762. https://doi.org/10.3390/catal11070762