
Light-Driven Hydrogen Evolution Assisted by Covalent Organic Frameworks



Abstract
:
{kind=link}
{kind=link}
{kind=link}
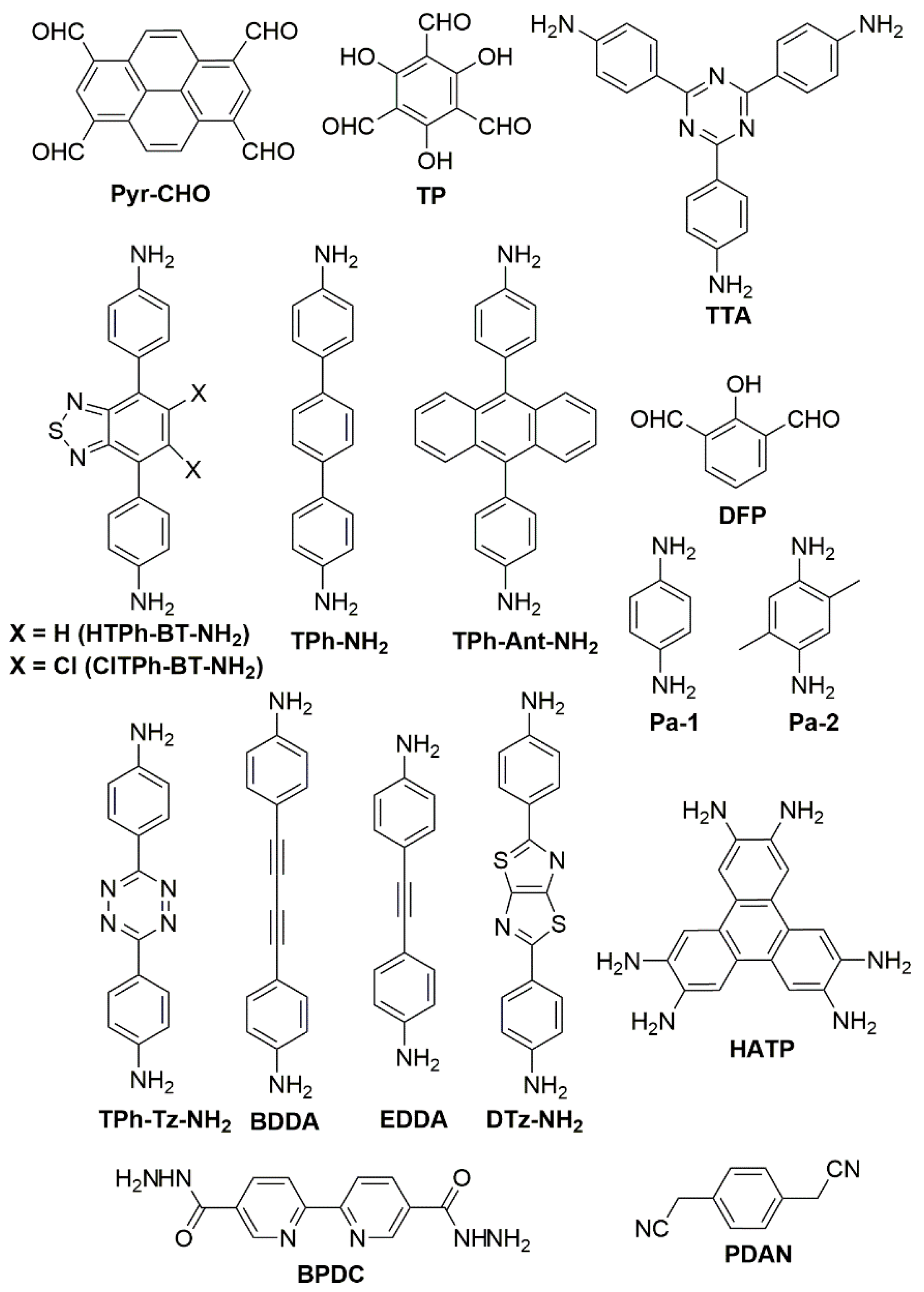{kind=link}
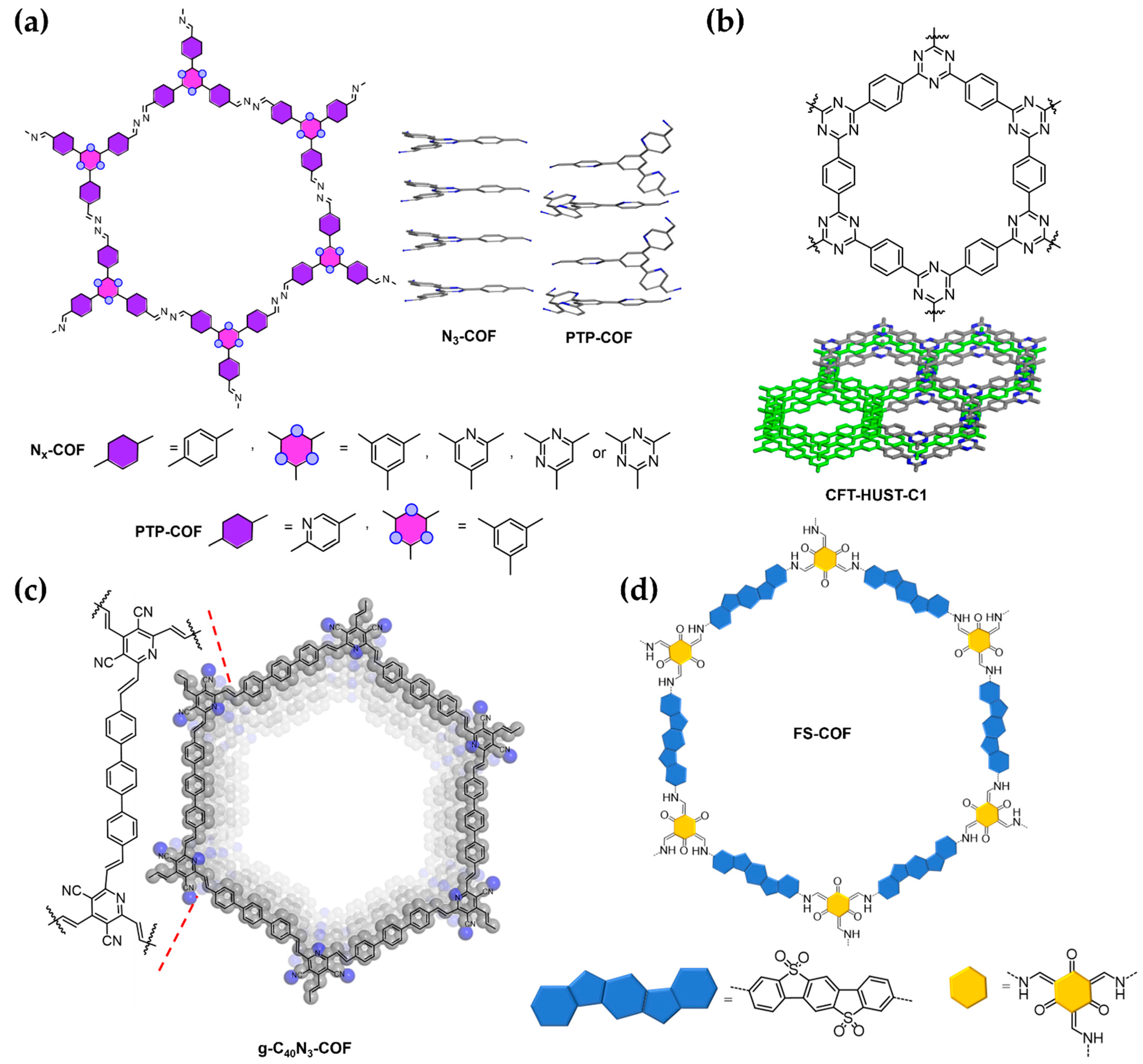{kind=link}
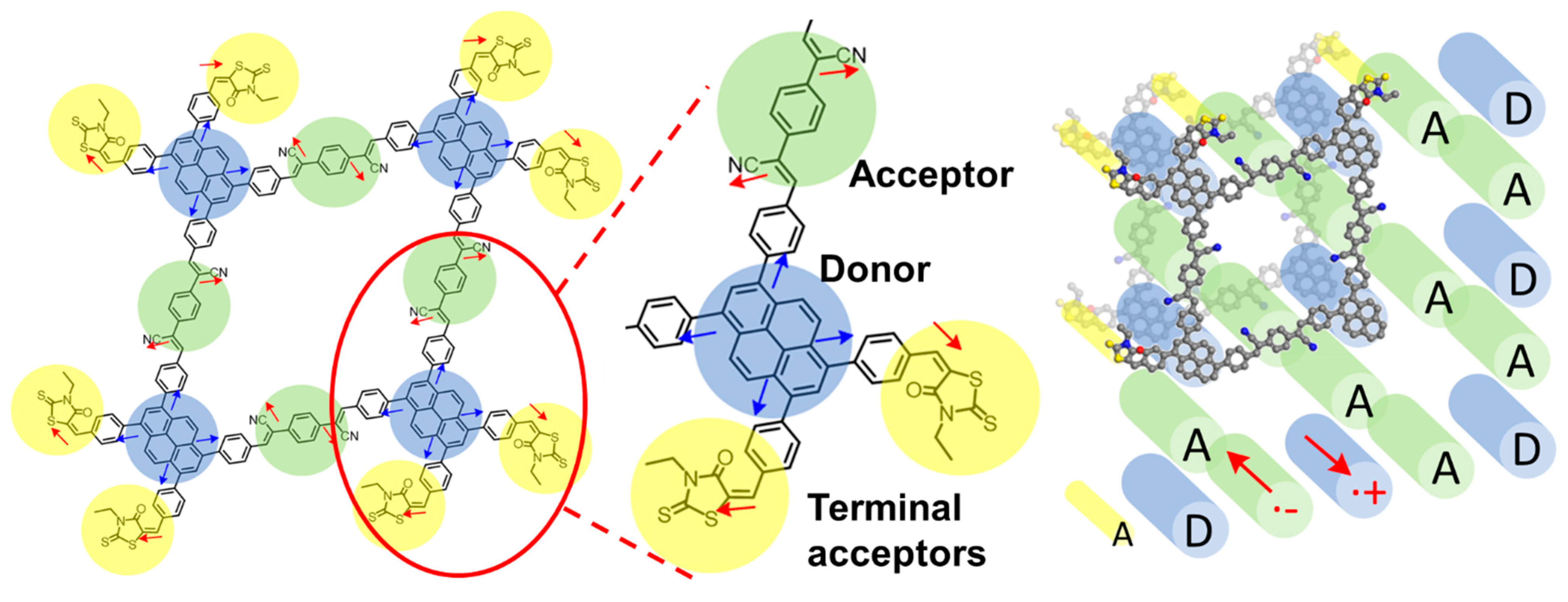{kind=link}
1. Introduction
2. Crucial Aspects Determining the Photocatalytic Hydrogen Evolution Performance of COF-Mediated Systems
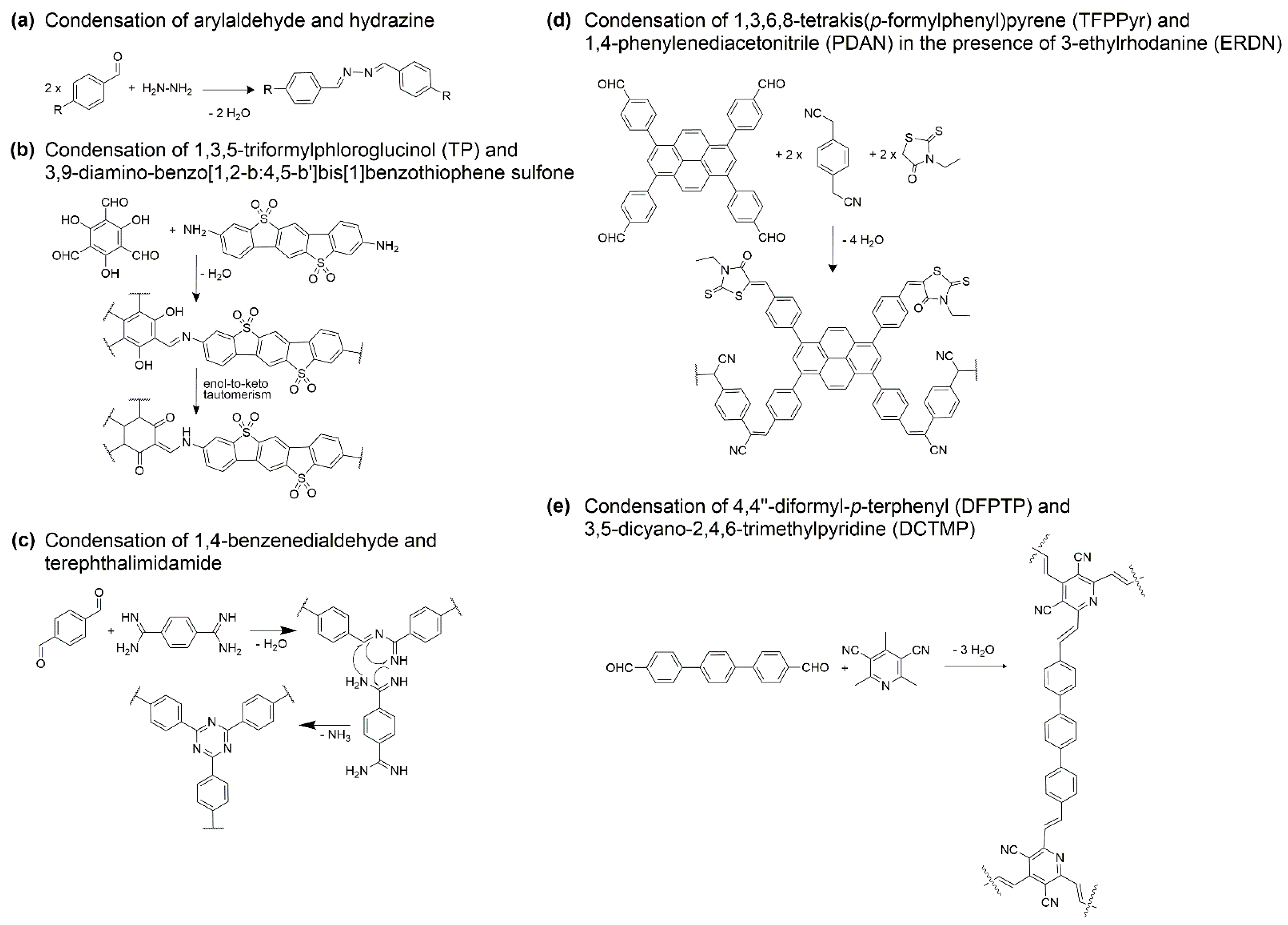
2.1. Common Synthetic Pathways to Build Efficient and Robust COFs
2.2. Secondary Interactions
2.3. Electronic Effects
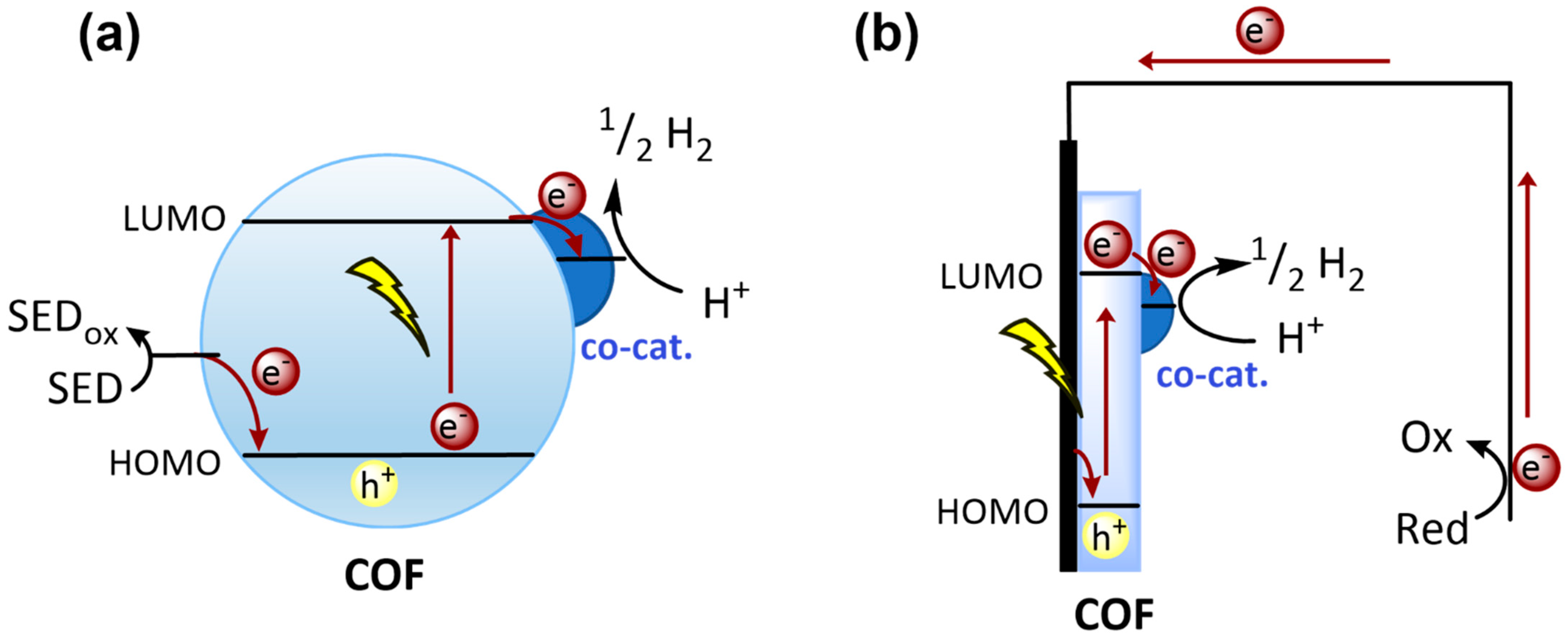
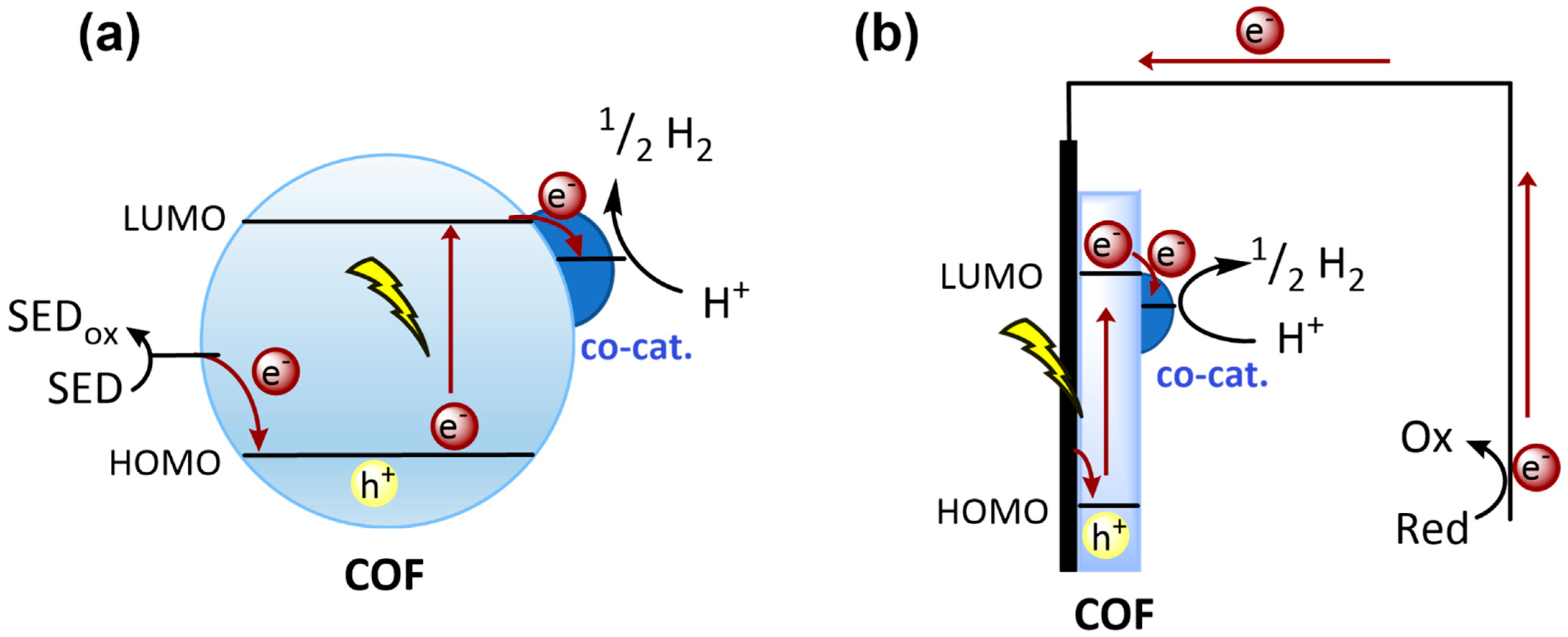
2.3.1. Charge Separation and Transfer
2.3.2. Charge Generation
2.3.3. Stabilization of Key Intermediates
2.4. Heterojunctions and Encapsulation of COFs with Other Materials
2.5. Choice of Metal Co-Catalyst
3. Final Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A | acceptor |
| AA | ascorbic acid |
| Ant | anthracene |
| AQE | apparent quantum efficiency |
| APTES | 3-aminopropyltriethoxysilane |
| BDBA | benzenediboronic acid |
| BDDA | 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline |
| BET | Brunauer–Emmett–Teller |
| Bpy | 2,2′-bipyridine |
| BPDC | 2,2′-bipyridine-5,5′-dicarbohydrazine |
| BT | benzothiadiazole |
| CB | conduction band |
| COF | covalent organic framework |
| CCP-Th | bithiophene-bridged C-linked conjugated polymer |
| CTF | covalent triazine framework |
| D | donor |
| DCTMP | 3,5-dicyano-2,4,6-trimethylpyridine |
| DEA | diethylamine |
| DFP | 2,6-diformylphenol |
| DFPTP | 4,4″-diformyl-p-terphenyl |
| DTz | 2,5-diphenyl-[1,3]-thiazolo[5,4-d][1,3]thiazole |
| EDDA | 4,4′-(ethyne-1,2-diyl)dianiline |
| ERDN | 3-ethylrhodanine |
| FS | fused sulfone |
| GCNS | graphitic carbon nitride nanosheets |
| HATP | 2,3,6,7,10,11-hexaaminotriphenylene |
| HER | hydrogen evolution reaction |
| HOMO | highest occupied molecular orbital |
| HHTP | hexahydroxytriphenylene |
| HUST | Huazhong University of Science and Technology |
| LUMO | lowest unoccupied molecular orbital |
| M-COF | metal coordinated COF |
| MOF | metal organic framework |
| NP | nanoparticle |
| PDAN | 1,4-phenylenediacetonitrile |
| POP | porous organic polymer |
| PTP | phenyl tripyridyl |
| PVP | polyvinylpyrrolidone |
| Pa-1 | p-phenylenediamine |
| Pa-2 | 2,5-dimethyl-p-phenylenediamine |
| Pyr | pyrene |
| Pyr-CHO | 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl)benzaldehyde |
| rGO | reduced graphene oxide |
| RHE | reversible hydrogen electrode |
| SED | sacrificial electron donor |
| STH | solar-to-hydrogen conversion efficiency |
| TEA | triethylamine |
| TEOA | triethanolamine |
| TFPPyr | 1,3,6,8-tetrakis(p-formylphenyl)pyrene |
| TFPT | 1,3,5-tris-(4-formyl-phenyl)triazine |
| TP | 1,3,5-triformylphloroglucinol |
| TPh | terphenyl |
| TTA | 4,4′,4″-(1,3,5-triazine-2,4,6-triyl)trianiline |
| Tz | tetrazine |
| TZ | thiazole |
| VB | valence band |
References
- Côté, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, H.; Wang, Z.; Tang, L.; Zeng, G.; Xu, P.; Chen, M.; Xiong, T.; Zhou, C.; Li, X.; et al. Covalent Organic Framework Photocatalysts: Structures and Applications. Chem. Soc. Rev. 2020, 49, 4135–4165. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; He, T.; Gong, Y.; Jiang, D. Covalent Organic Frameworks: Pore Design and Interface Engineering. Acc. Chem. Res. 2020, 53, 1672–1685. [Google Scholar] [CrossRef]
- Peng, Y.; Li, L.; Zhu, C.; Chen, B.; Zhao, M.; Zhang, Z.; Lai, Z.; Zhang, X.; Tan, C.; Han, Y.; et al. Intramolecular Hydrogen Bonding-Based Topology Regulation of Two-Dimensional Covalent Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 13162–13169. [Google Scholar] [CrossRef]
- Zhao, X.; Pachfule, P.; Li, S.; Langenhahn, T.; Ye, M.; Schlesiger, C.; Praetz, S.; Schmidt, J.; Thomas, A. Macro/Microporous Covalent Organic Frameworks for Efficient Electrocatalysis. J. Am. Chem. Soc. 2019, 141, 6623–6630. [Google Scholar] [CrossRef]
- Low, J.; Dai, B.; Tong, T.; Jiang, C.; Yu, J. In Situ Irradiated X-Ray Photoelectron Spectroscopy Investigation on a Direct Z-Scheme TiO 2 /CdS Composite Film Photocatalyst. Adv. Mater. 2019, 31, 1802981. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.J.; Liu, G.; Moniz, S.J.A.; Bi, Y.; Beale, A.M.; Ye, J.; Tang, J. Efficient Visible Driven Photocatalyst, Silver Phosphate: Performance, Understanding and Perspective. Chem. Soc. Rev. 2015, 44, 7808–7828. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Zhang, Y.; Pan, Q.; Qiu, J. A Fantastic Graphitic Carbon Nitride (g-C3N4) Material: Electronic Structure, Photocatalytic and Photoelectronic Properties. J. Photochem. Photobiol. C Photochem. Rev. 2014, 20, 33–50. [Google Scholar] [CrossRef]
- Stegbauer, L.; Schwinghammer, K.; Lotsch, B.V. A Hydrazone-Based Covalent Organic Framework for Photocatalytic Hydrogen Production. Chem. Sci. 2014, 5, 2789–2793. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Xing, G.; Chen, W.; Chen, L. Porous Organic Polymers: A Promising Platform for Efficient Photocatalysis. Mater. Chem. Front. 2020, 4, 332–353. [Google Scholar] [CrossRef]
- Jayakumar, J.; Chou, H.H. Recent Advances in Visible-Light-Driven Hydrogen Evolution from Water Using Polymer Photocatalysts. ChemCatChem 2020, 12, 689–704. [Google Scholar] [CrossRef]
- Rahman, M.Z.; Kibria, M.G.; Mullins, C.B. Metal-Free Photocatalysts for Hydrogen Evolution. Chem. Soc. Rev. 2020, 49, 1887–1931. [Google Scholar] [CrossRef]
- Guo, L.; Jin, S. Stable Covalent Organic Frameworks for Photochemical Applications. ChemPhotoChem 2019, 3, 973–983. [Google Scholar] [CrossRef]
- Banerjee, T.; Gottschling, K.; Savasci, G.; Ochsenfeld, C.; Lotsch, B.V. H2 Evolution with Covalent Organic Framework Photocatalysts. ACS Energy Lett. 2018, 3, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Podjaski, F.; Kröger, J.; Biswal, B.P.; Lotsch, B.V. Polymer Photocatalysts for Solar-to-Chemical Energy Conversion. Nat. Rev. Mater. 2020, 6. [Google Scholar] [CrossRef]
- You, J.; Zhao, Y.; Wang, L.; Bao, W. Recent Developments in the Photocatalytic Applications of Covalent Organic Frameworks: A Review. J. Clean. Prod. 2021, 291, 125822. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Y.-B. Covalent Organic Frameworks for Sunlight-Driven Hydrogen Evolution. Chem. Lett. 2021, 50, 676–686. [Google Scholar] [CrossRef]
- Chen, W.; Wang, L.; Mo, D.; He, F.; Wen, Z.; Wu, X.; Xu, H.; Chen, L. Modulating Benzothiadiazole-Based Covalent Organic Frameworks via Halogenation for Enhanced Photocatalytic Water Splitting. Angew. Chem. Int. Ed. 2020, 59, 16902–16909. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.S.; Haase, F.; Stegbauer, L.; Savasci, G.; Podjaski, F.; Ochsenfeld, C.; Lotsch, B.V. A Tunable Azine Covalent Organic Framework Platform for Visible Light-Induced Hydrogen Generation. Nat. Commun. 2015, 6, 8508. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Huang, Q.; Wang, S.; Li, Z.; Li, B.; Jin, S.; Tan, B. Crystalline Covalent Triazine Frameworks by In Situ Oxidation of Alcohols to Aldehyde Monomers. Angew. Chem. Int. Ed. 2018, 57, 11968–11972. [Google Scholar] [CrossRef]
- Wang, K.; Yang, L.-M.; Wang, X.; Guo, L.; Cheng, G.; Zhang, C.; Jin, S.; Tan, B.; Cooper, A. Covalent Triazine Frameworks via a Low-Temperature Polycondensation Approach. Angew. Chem. Int. Ed. 2017, 56, 14149–14153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Cheng, G.; Guo, L.; Wang, N.; Tan, B.; Jin, S. Strong-Base-Assisted Synthesis of a Crystalline Covalent Triazine Framework with High Hydrophilicity via Benzylamine Monomer for Photocatalytic Water Splitting. Angew. Chem. 2020, 132, 6063–6070. [Google Scholar] [CrossRef]
- Pachfule, P.; Acharjya, A.; Roeser, J.; Langenhahn, T.; Schwarze, M.; Schomäcker, R.; Thomas, A.; Schmidt, J. Diacetylene Functionalized Covalent Organic Framework (COF) for Photocatalytic Hydrogen Generation. J. Am. Chem. Soc. 2018, 140, 1423–1427. [Google Scholar] [CrossRef]
- Wang, X.; Chen, L.; Chong, S.Y.; Little, M.A.; Wu, Y.; Zhu, W.H.; Clowes, R.; Yan, Y.; Zwijnenburg, M.A.; Sprick, R.S.; et al. Sulfone-Containing Covalent Organic Frameworks for Photocatalytic Hydrogen Evolution from Water. Nat. Chem. 2018, 10, 1180–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswal, B.P.; Vignolo-González, H.A.; Banerjee, T.; Grunenberg, L.; Savasci, G.; Gottschling, K.; Nuss, J.; Ochsenfeld, C.; Lotsch, B.V. Sustained Solar H2 Evolution from a Thiazolo[5,4-d]Thiazole-Bridged Covalent Organic Framework and Nickel-Thiolate Cluster in Water. J. Am. Chem. Soc. 2019, 141, 11082–11092. [Google Scholar] [CrossRef] [Green Version]
- Jin, E.; Lan, Z.; Jiang, Q.; Geng, K.; Li, G.; Wang, X.; Jiang, D. 2D Sp2 Carbon-Conjugated Covalent Organic Frameworks for Photocatalytic Hydrogen Production from Water. Chem 2019, 5, 1632–1647. [Google Scholar] [CrossRef]
- Xu, S.; Sun, H.; Addicoat, M.; Biswal, B.P.; He, F.; Park, S.W.; Paasch, S.; Zhang, T.; Sheng, W.; Brunner, E.; et al. Thiophene-Bridged Donor–Acceptor Sp2-Carbon-Linked 2D Conjugated Polymers as Photocathodes for Water Reduction. Adv. Mater. 2021, 33, 2006274. [Google Scholar] [CrossRef]
- Bi, S.; Yang, C.; Zhang, W.; Xu, J.; Liu, L.; Wu, D.; Wang, X.; Han, Y.; Liang, Q.; Zhang, F. Two-Dimensional Semiconducting Covalent Organic Frameworks via Condensation at Arylmethyl Carbon Atoms. Nat. Commun. 2019, 10, 2467. [Google Scholar] [CrossRef]
- Huang, W.; Luo, W.; Li, Y. Two-Dimensional Semiconducting Covalent Organic Frameworks for Photocatalytic Solar Fuel Production. Mater. Today 2020, 40, 160–172. [Google Scholar] [CrossRef]
- He, T.; Geng, K.; Jiang, D. Engineering Covalent Organic Frameworks for Light-Driven Hydrogen Production from Water. Acs Mater. Lett. 2019, 1, 203–208. [Google Scholar] [CrossRef]
- Qian, Z.; Wang, Z.J.; Zhang, K.A.I. Covalent Triazine Frameworks as Emerging Heterogeneous Photocatalysts. Chem. Mater. 2021, 33, 1909–1926. [Google Scholar] [CrossRef]
- Ghosh, S.; Nakada, A.; Springer, M.A.; Kawaguchi, T.; Suzuki, K.; Kaji, H.; Baburin, I.; Kuc, A.; Heine, T.; Suzuki, H.; et al. Identification of Prime Factors to Maximize the Photocatalytic Hydrogen Evolution of Covalent Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 9752–9762. [Google Scholar] [CrossRef]
- Zang, Y.; Wang, R.; Shao, P.P.; Feng, X.; Wang, S.; Zang, S.Q.; Mak, T.C.W. Prefabricated Covalent Organic Framework Nanosheets with Double Vacancies: Anchoring Cu for Highly Efficient Photocatalytic H2evolution. J. Mater. Chem. A 2020, 8, 25094–25100. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Peh, S.B.; Liu, G.; Kundu, T.; Dong, J.; Ying, Y.; Qian, Y.; Zhao, D. Cobalt-Containing Covalent Organic Frameworks for Visible Light-Driven Hydrogen Evolution. Sci. China Chem. 2020, 63, 192–197. [Google Scholar] [CrossRef]
- Mi, Z.; Zhou, T.; Weng, W.; Unruangsri, J.; Hu, K.; Yang, W.; Wang, C.; Zhang, K.A.I.; Guo, J. Covalent Organic Frameworks Enabling Site-Isolation of Viologen-Derived Electron Transfer Mediators for Stable Photocatalytic Hydrogen Evolution. Angew. Chem. Int. Ed. 2021, 60, 9642–9649. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, M.F.; Guo, T.; Zheng, L.L.; Wang, D.; Mu, Y.; Xing, Q.J.; Zou, J.P. Chlorine-Mediated Photocatalytic Hydrogen Production Based on Triazine Covalent Organic Framework. Appl. Catal. B Environ. 2020, 272, 118989. [Google Scholar] [CrossRef]
- Haase, F.; Banerjee, T.; Savasci, G.; Ochsenfeld, C.; Lotsch, B.V. Structure-Property-Activity Relationships in a Pyridine Containing Azine-Linked Covalent Organic Framework for Photocatalytic Hydrogen Evolution. Faraday Discuss. 2017, 201, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Jia, Z.; Bai, Y.; Wang, X.; Hodgkiss, S.E.; Chen, L.; Chong, S.Y.; Wang, X.; Yang, H.; Xu, Y.; et al. Synthesis of Stable Thiazole-Linked Covalent Organic Frameworks via a Multicomponent Reaction. J. Am. Chem. Soc. 2020, 142, 11131–11138. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.Z.; Wang, Z.K.; Wei, L.; Yin, G.; Tian, J.; Liu, C.Z.; Wang, H.; Zhang, D.W.; Zhang, Y.B.; Li, X.; et al. Water-Soluble 3D Covalent Organic Framework That Displays an Enhanced Enrichment Effect of Photosensitizers and Catalysts for the Reduction of Protons to H2. ACS Appl. Mater. Interfaces 2020, 12, 1404–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, C.; Yang, M.; Sun, F.; Jian, J.; Zhong, L.; Fang, Z.; Feng, J.; Yu, D. Alkene-Linked Covalent Organic Frameworks Boosting Photocatalytic Hydrogen Evolution by Efficient Charge Separation and Transfer in the Presence of Sacrificial Electron Donors. Adv. Sci. 2020, 7, 1902988. [Google Scholar] [CrossRef]
- Chen, R.; Wang, Y.; Ma, Y.; Mal, A.; Gao, X.Y.; Gao, L.; Qiao, L.; Li, X.B.; Wu, L.Z.; Wang, C. Rational Design of Isostructural 2D Porphyrin-Based Covalent Organic Frameworks for Tunable Photocatalytic Hydrogen Evolution. Nat. Commun. 2021, 12, 1354. [Google Scholar] [CrossRef]
- Li, L.; Zhu, Y.; Gong, N.; Zhang, W.; Peng, W.; Li, Y.; Zhang, F.; Fan, X. Band-Gap Engineering of Layered Covalent Organic Frameworks via Controllable Exfoliation for Enhanced Visible-Light-Driven Hydrogen Evolution. Int. J. Hydrog. Energy 2020, 45, 2689–2698. [Google Scholar] [CrossRef]
- Wang, H.; Qian, C.; Liu, J.; Zeng, Y.; Wang, D.; Zhou, W.; Gu, L.; Wu, H.; Liu, G.; Zhao, Y. Integrating Suitable Linkage of Covalent Organic Frameworks into Covalently Bridged Inorganic/Organic Hybrids toward Efficient Photocatalysis. J. Am. Chem. Soc. 2020, 142, 4862–4871. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Han, C.; Gong, L.; Chen, X.; Deng, J.; Qi, D.; Bian, Y.; Wang, K.; Jiang, J. Donor–Acceptor Covalent Organic Framework/g-C 3 N 4 Hybrids for Efficient Visible Light Photocatalytic H 2 Production. Catal. Sci. Technol. 2021, 11, 2616–2621. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Huang, X.; Tao, L.; Bi, Y. Direct Observation of Dynamic Interfacial Bonding and Charge Transfer in Metal-Free Photocatalysts for Efficient Hydrogen Evolution. Appl. Catal. B Environ. 2021, 283, 119633. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, D.; Shi, B.; Dai, W.; Ren, H.; An, K.; Zhou, Z.; Zhao, Z.; Wang, W.; Jiang, Z. In Situ Construction of Hydrazone-Linked COF-Based Core-Shell Hetero-Frameworks for Enhanced Photocatalytic Hydrogen Evolution. J. Mater. Chem. A 2020, 8, 7724–7732. [Google Scholar] [CrossRef]
- Zhang, F.M.; Sheng, J.L.; Yang, Z.D.; Sun, X.J.; Tang, H.L.; Lu, M.; Dong, H.; Shen, F.C.; Liu, J.; Lan, Y.Q. Rational Design of MOF/COF Hybrid Materials for Photocatalytic H2 Evolution in the Presence of Sacrificial Electron Donors. Angew. Chem. Int. Ed. 2018, 57, 12106–12110. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Cui, Y.; Guo, B.; Li, H.Y.; Li, H.X. Boosting Visible-Light-Driven H2 Evolution of Covalent Triazine Framework from Water by Modifying Ni(II) Pyrimidine-2-Thiolate Cocatalyst. ChemCatChem 2021, 13, 958–965. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Wang, R.; Wang, S.; Xi, X.J.; Ma, L.F.; Zang, S.Q. Encapsulating [Mo3S13]2- Clusters in Cationic Covalent Organic Frameworks: Enhancing Stability and Recyclability by Converting a Homogeneous Photocatalyst to a Heterogeneous Photocatalyst. Chem. Commun. 2018, 54, 13563–13566. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Yang, Q.; Yang, W.; Wang, J.; He, F.; Liu, K.; Cao, H.; Yan, H. Defects Engineering Leads to Enhanced Photocatalytic H2 Evolution on Graphitic Carbon Nitride–Covalent Organic Framework Nanosheet Composite. Small 2020, 16, 1–8. [Google Scholar] [CrossRef]
- Yao, Y.H.; Li, J.; Zhang, H.; Tang, H.L.; Fang, L.; Niu, G.D.; Sun, X.J.; Zhang, F.M. Facile Synthesis of a Covalently Connected RGO-COF Hybrid Material by: In Situ Reaction for Enhanced Visible-Light Induced Photocatalytic H2 Evolution. J. Mater. Chem. A 2020, 8, 8949–8956. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Tang, H.L.; Dong, H.; Gao, M.Y.; Li, C.C.; Sun, X.J.; Wei, J.Z.; Qu, Y.; Li, Z.J.; Zhang, F.M. Covalent-Organic Framework Based Z-Scheme Heterostructured Noble-Metal-Free Photocatalysts for Visible-Light-Driven Hydrogen Evolution. J. Mater. Chem. A 2020, 8, 4334–4340. [Google Scholar] [CrossRef]
- Gao, M.Y.; Li, C.C.; Tang, H.L.; Sun, X.J.; Dong, H.; Zhang, F.M. Boosting Visible-Light-Driven Hydrogen Evolution of Covalent Organic Frameworks through Compositing with MoS2: A Promising Candidate for Noble-Metal-Free Photocatalysts. J. Mater. Chem. A 2019, 7, 20193–20200. [Google Scholar] [CrossRef]
- Thote, J.; Aiyappa, H.B.; Deshpande, A.; Díaz Díaz, D.; Kurungot, S.; Banerjee, R. A Covalent Organic Framework-Cadmium Sulfide Hybrid as a Prototype Photocatalyst for Visible-Light-Driven Hydrogen Production. Chemistry 2014, 20, 15961–15965. [Google Scholar] [CrossRef]
- Wang, D.; Zeng, H.; Xiong, X.; Wu, M.F.; Xia, M.; Xie, M.; Zou, J.P.; Luo, S.L. Highly Efficient Charge Transfer in CdS-Covalent Organic Framework Nanocomposites for Stable Photocatalytic Hydrogen Evolution under Visible Light. Sci. Bull. 2020, 65, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Li, C.C.; Gao, M.Y.; Sun, X.J.; Tang, H.L.; Dong, H.; Zhang, F.M. Rational Combination of Covalent-Organic Framework and Nano TiO2 by Covalent Bonds to Realize Dramatically Enhanced Photocatalytic Activity. Appl. Catal. B Environ. 2020, 266, 118586. [Google Scholar] [CrossRef]
- Ming, J.; Liu, A.; Zhao, J.; Zhang, P.; Huang, H.; Lin, H.; Xu, Z.; Zhang, X.; Wang, X.; Hofkens, J.; et al. Hot π-Electron Tunneling of Metal–Insulator–COF Nanostructures for Efficient Hydrogen Production. Angew. Chem. Int. Ed. 2019, 58, 18290–18294. [Google Scholar] [CrossRef]
- Banerjee, T.; Haase, F.; Savasci, G.; Gottschling, K.; Ochsenfeld, C.; Lotsch, B.V. Single-Site Photocatalytic H2 Evolution from Covalent Organic Frameworks with Molecular Cobaloxime Co-Catalysts. J. Am. Chem. Soc. 2017, 139, 16228–16234. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Liu, J.; Wang, K.; Jin, S.; Tan, B. Palladium as a Superior Cocatalyst to Platinum for Hydrogen Evolution Using Covalent Triazine Frameworks as a Support. ACS Appl. Mater. Interfaces 2020, 12, 12774–12782. [Google Scholar] [CrossRef]
- Chen, Y.; Li, W.; Wang, X.H.; Gao, R.Z.; Tang, A.N.; Kong, D.M. Green Synthesis of Covalent Organic Frameworks Based on Reaction Media. Mater. Chem. Front. 2021, 5, 1253–1267. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, N.; Bofill, R.; Francàs, L.; García-Antón, J.; Sala, X. Light-Driven Hydrogen Evolution Assisted by Covalent Organic Frameworks. Catalysts 2021, 11, 754. https://doi.org/10.3390/catal11060754
Romero N, Bofill R, Francàs L, García-Antón J, Sala X. Light-Driven Hydrogen Evolution Assisted by Covalent Organic Frameworks. Catalysts. 2021; 11(6):754. https://doi.org/10.3390/catal11060754
Chicago/Turabian StyleRomero, Nuria, Roger Bofill, Laia Francàs, Jordi García-Antón, and Xavier Sala. 2021. "Light-Driven Hydrogen Evolution Assisted by Covalent Organic Frameworks" Catalysts 11, no. 6: 754. https://doi.org/10.3390/catal11060754
APA StyleRomero, N., Bofill, R., Francàs, L., García-Antón, J., & Sala, X. (2021). Light-Driven Hydrogen Evolution Assisted by Covalent Organic Frameworks. Catalysts, 11(6), 754. https://doi.org/10.3390/catal11060754