
Ferrocenylimine Palladium (II) Complexes: Synthesis, Characterization and Application in Mizoroki-Heck and Suzuki-Miyaura Cross-Coupling Reactions

Abstract
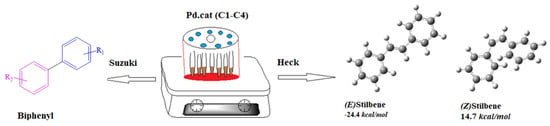
1. Introduction
2. Results and Discussion
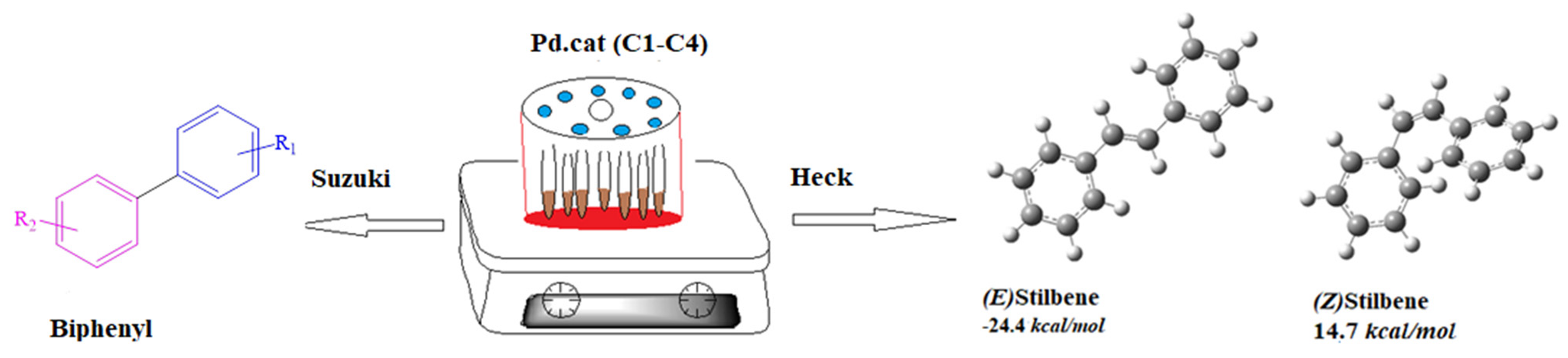
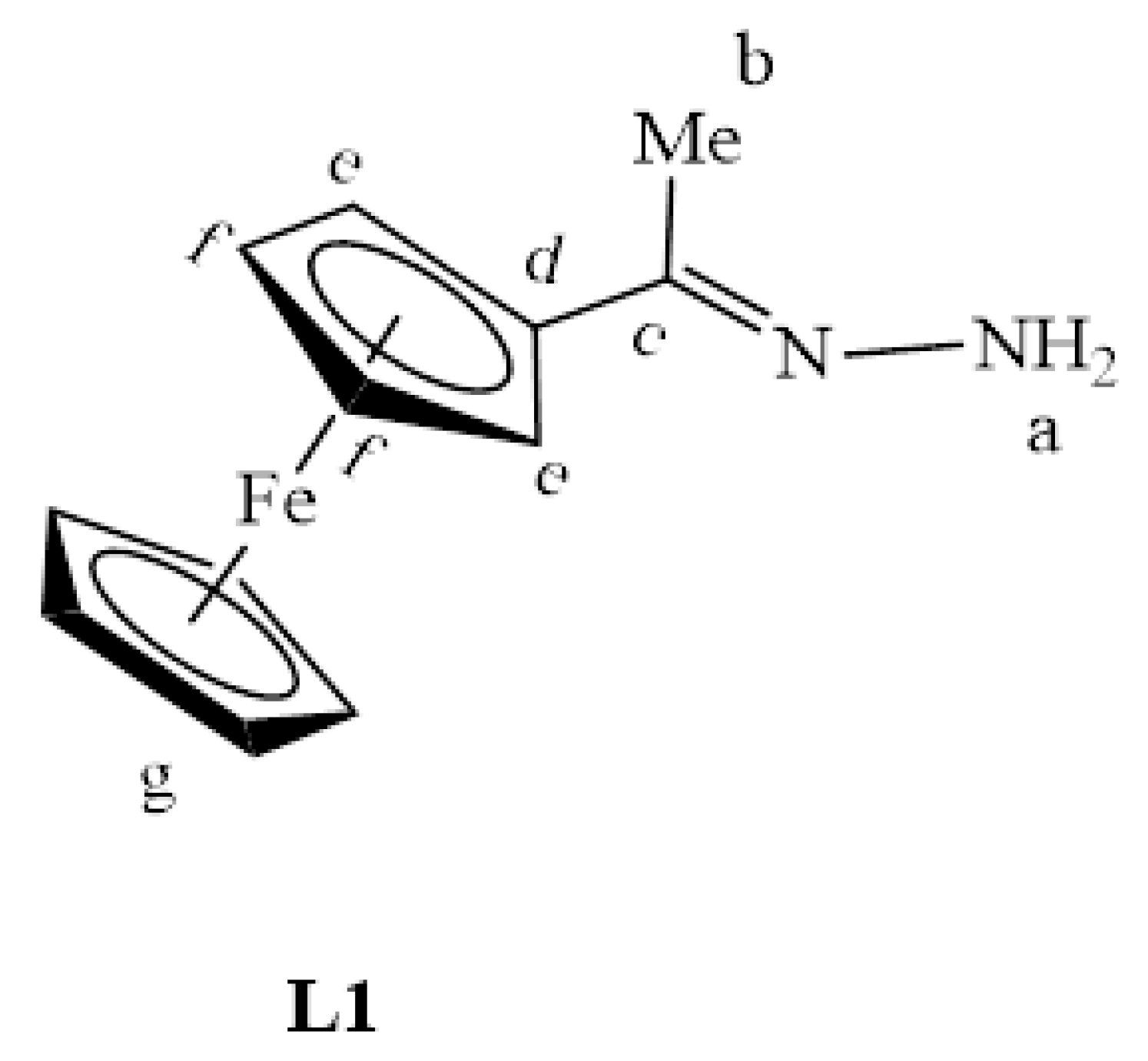
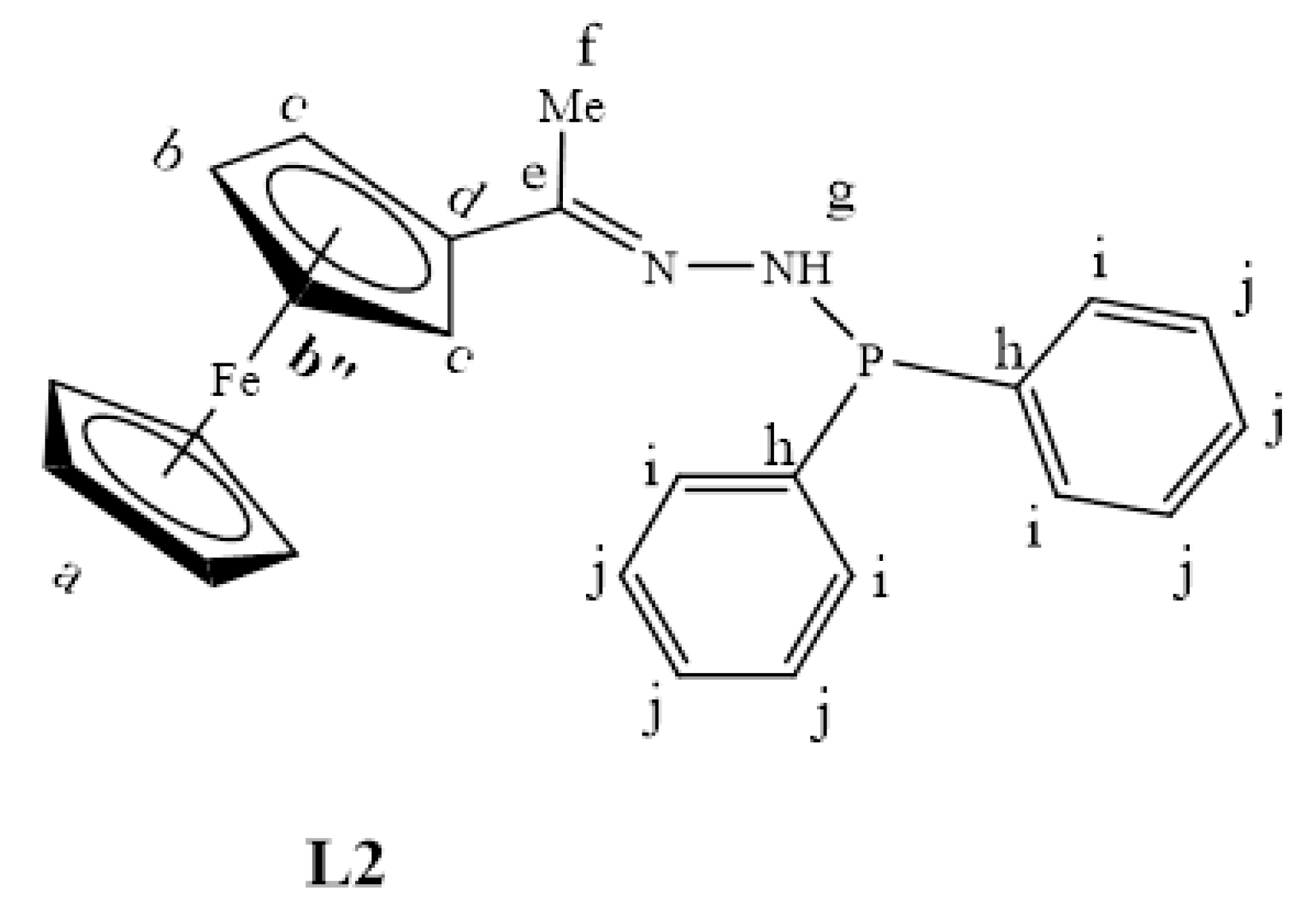
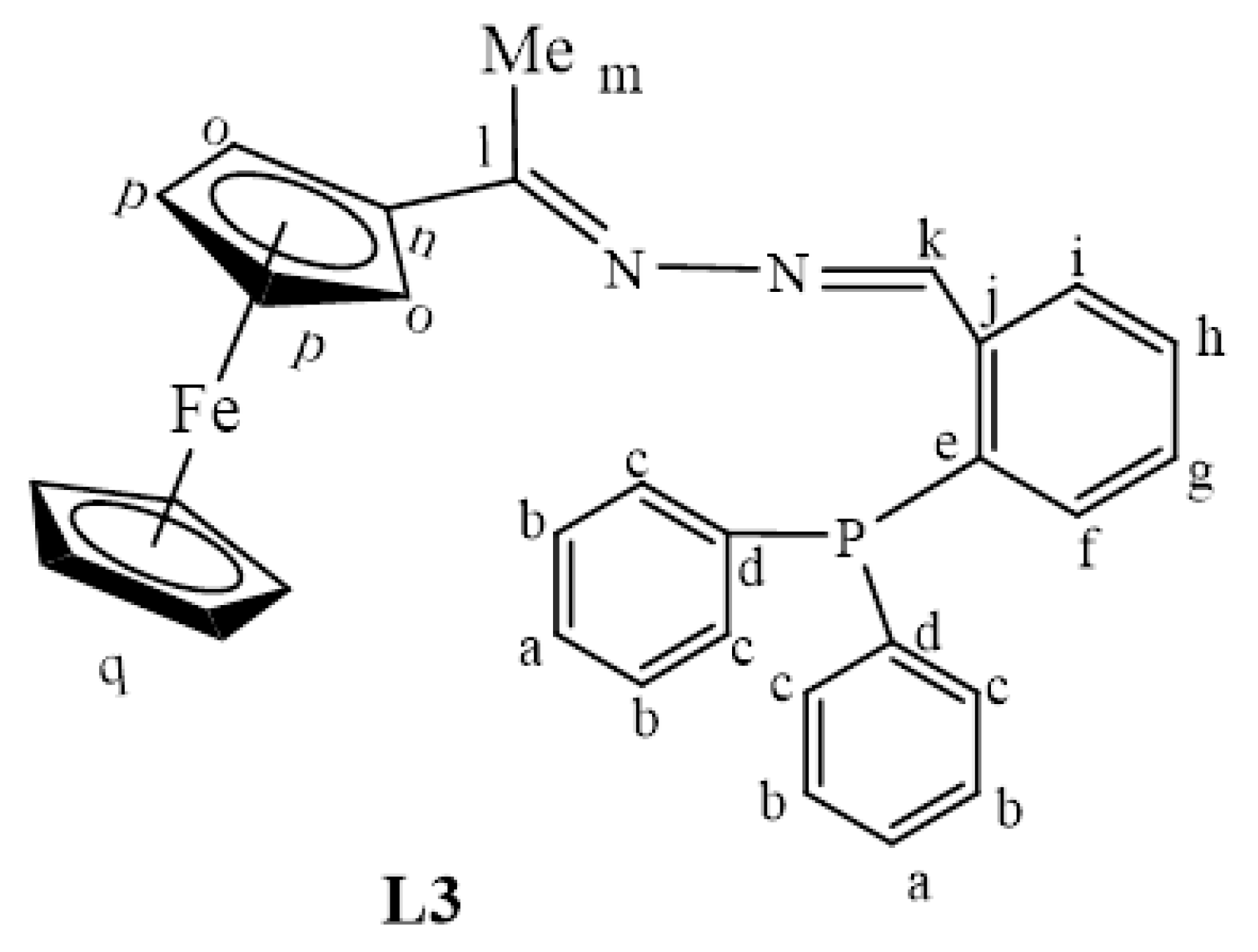
2.1. Synthesis and Characterization of Ligands, L1–L4
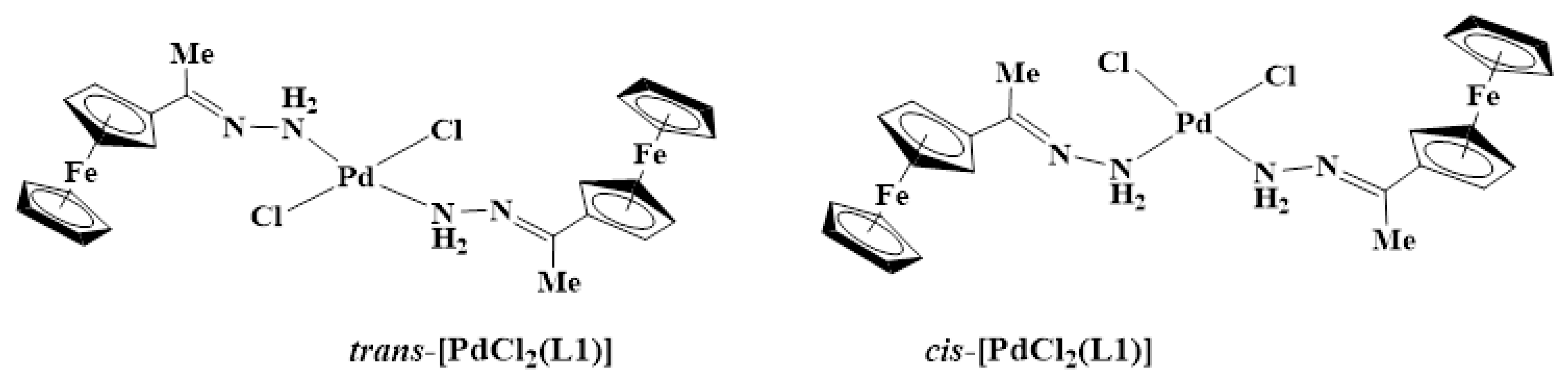
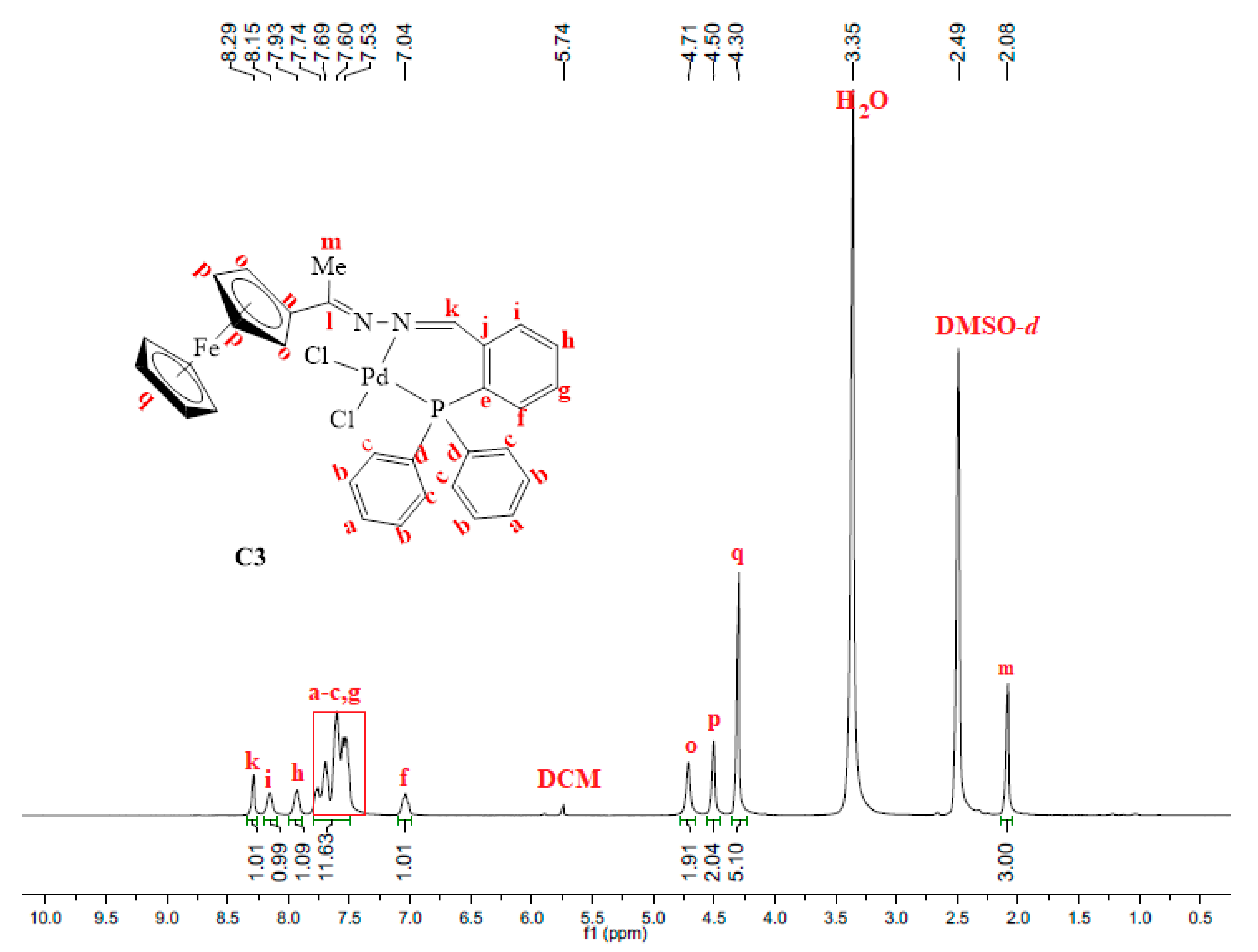
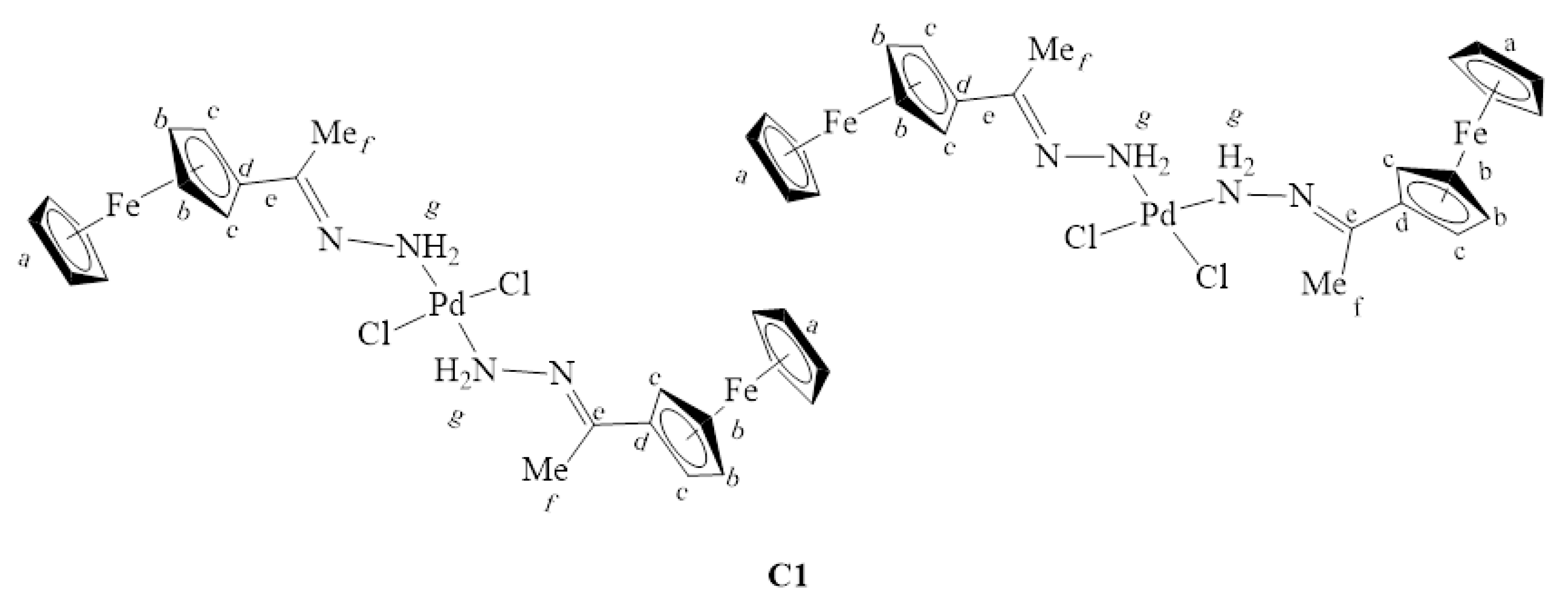
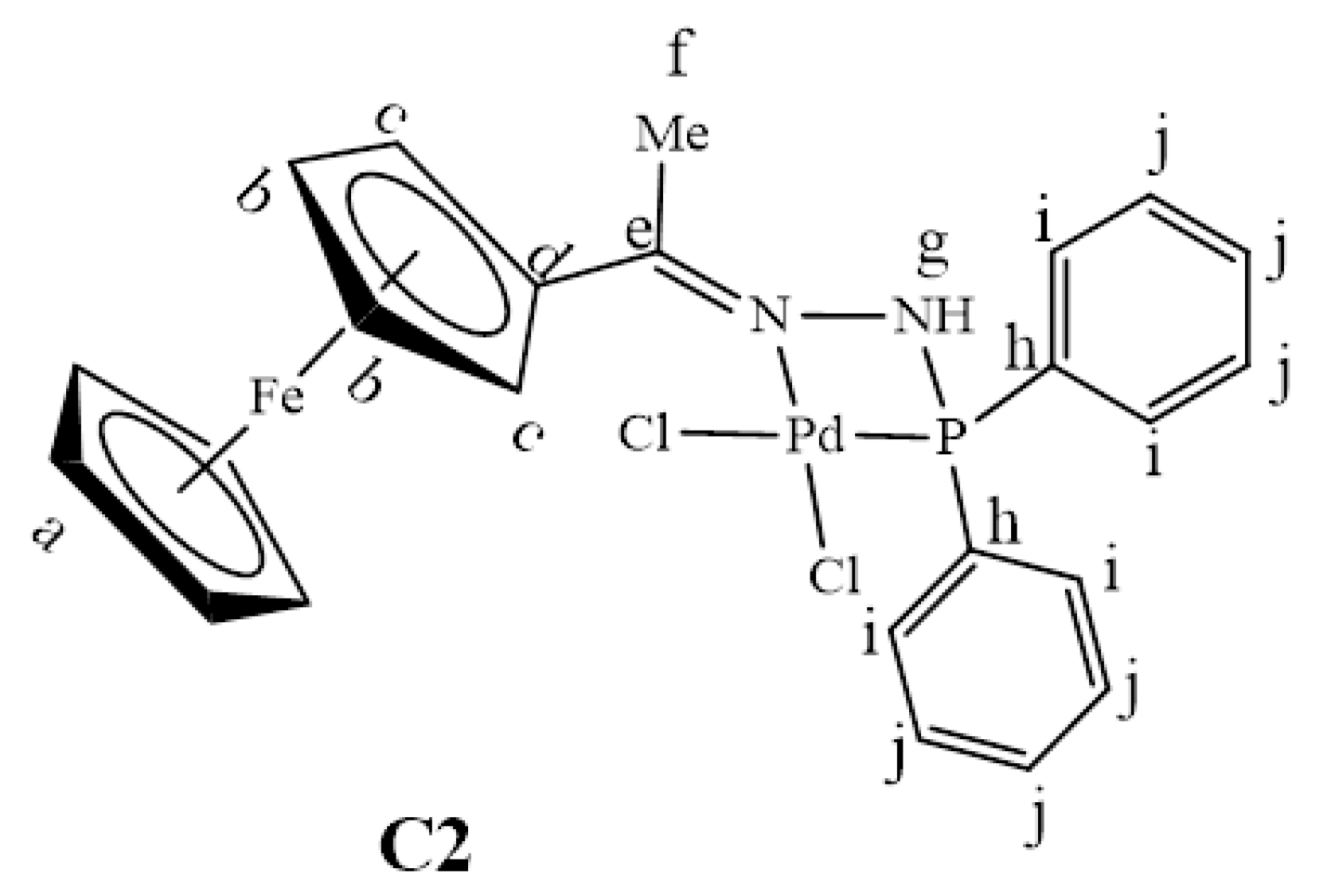
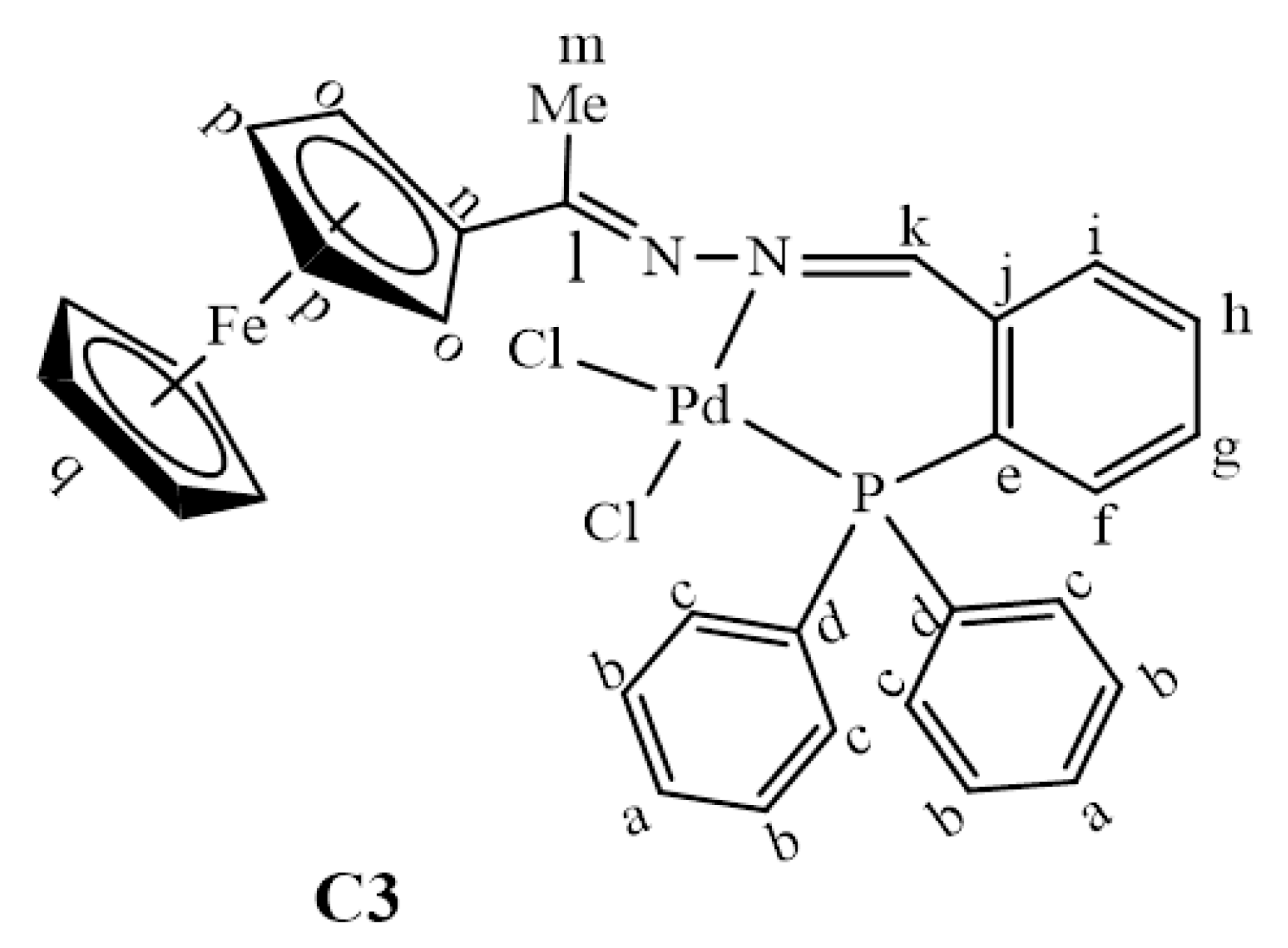
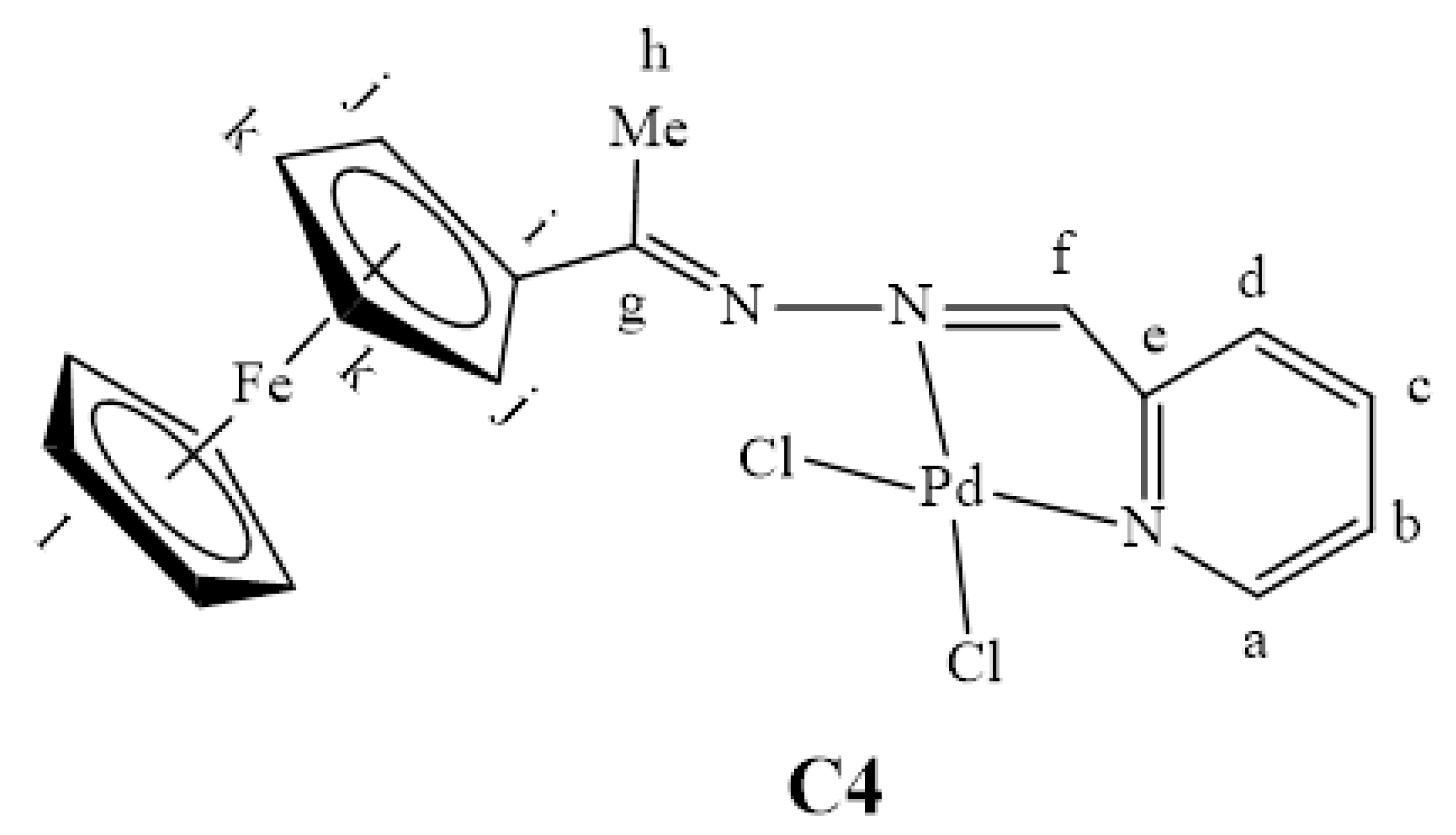
2.2. Synthesis and Characterization of Complexes, C1–C4
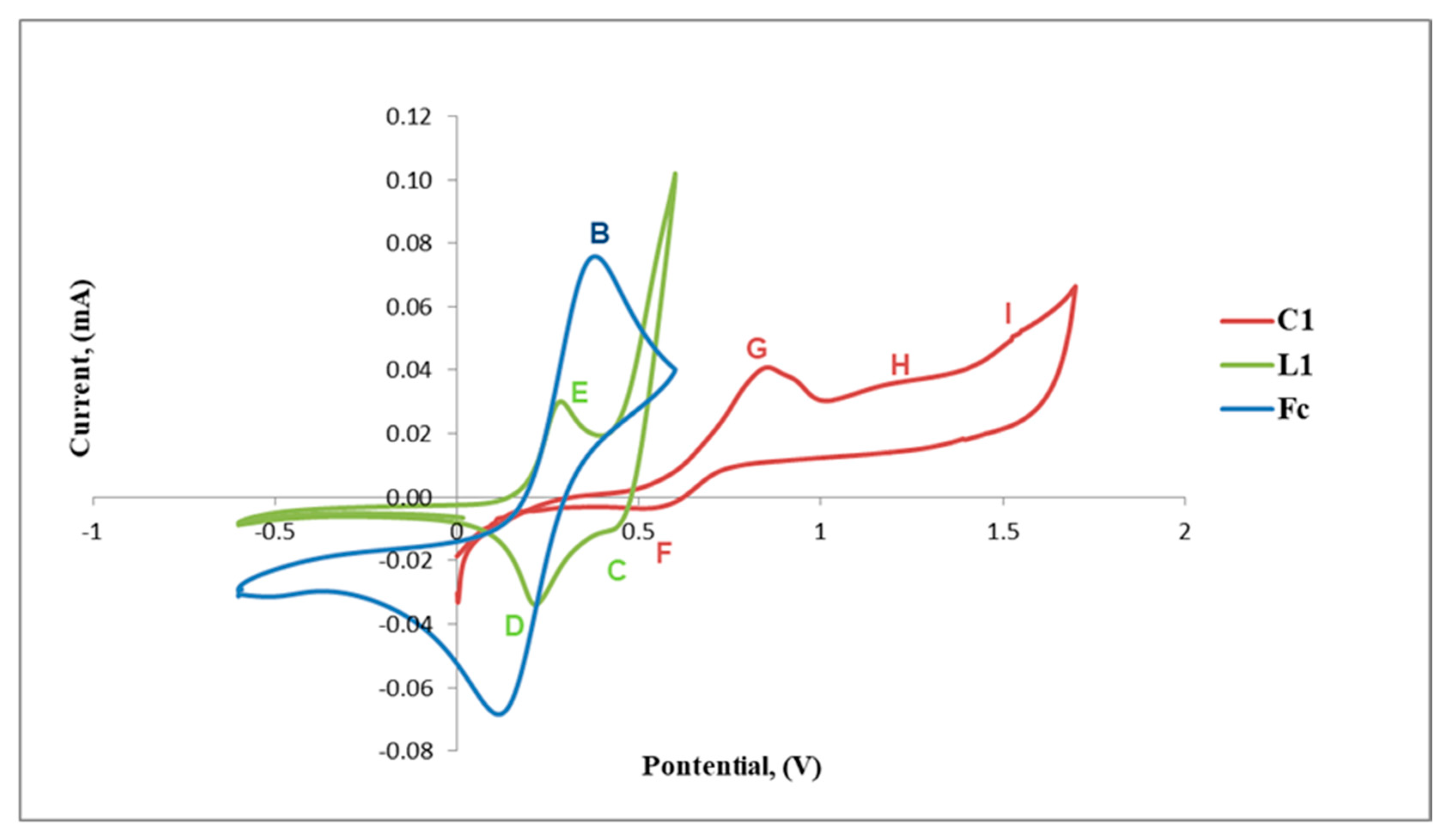
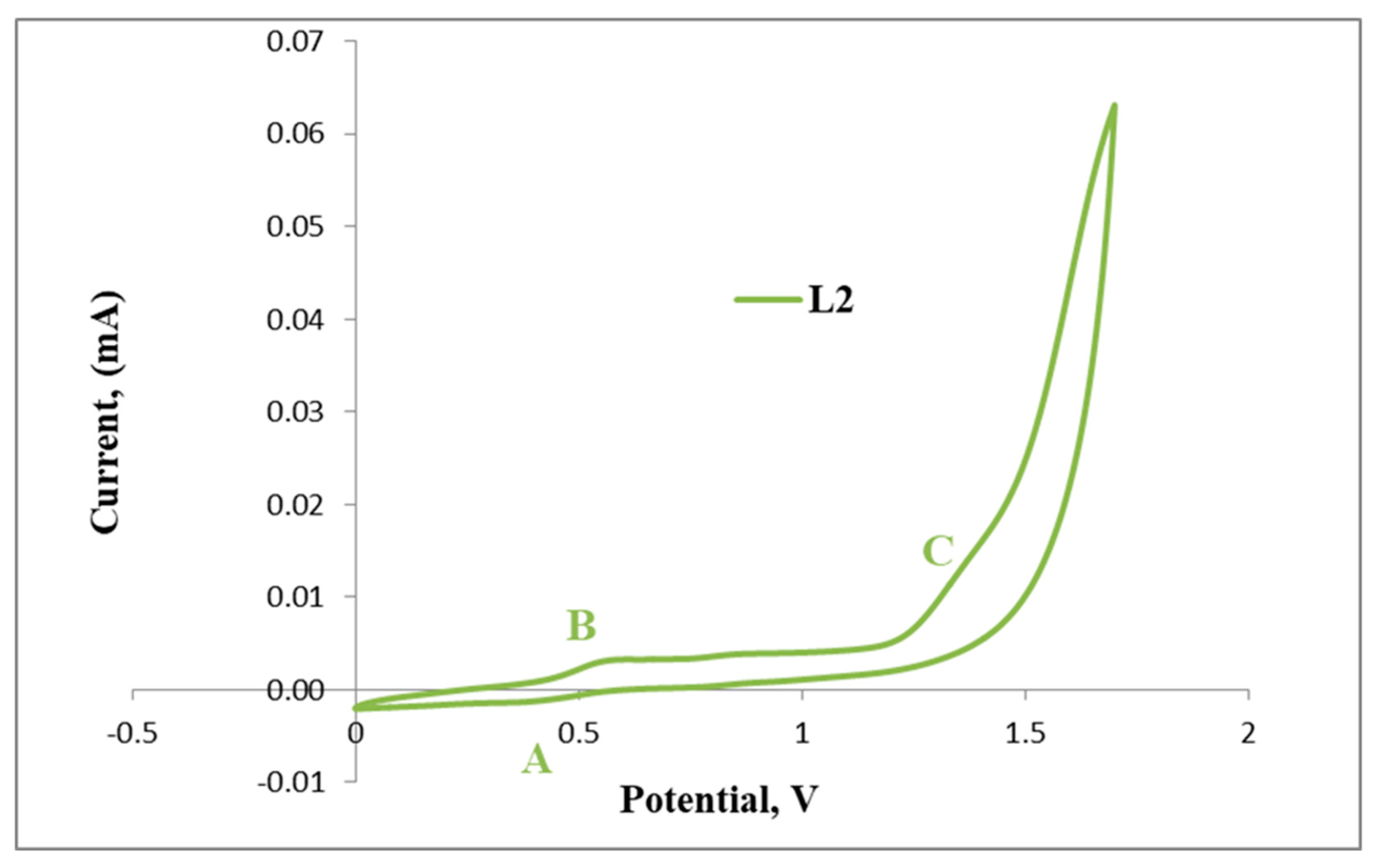
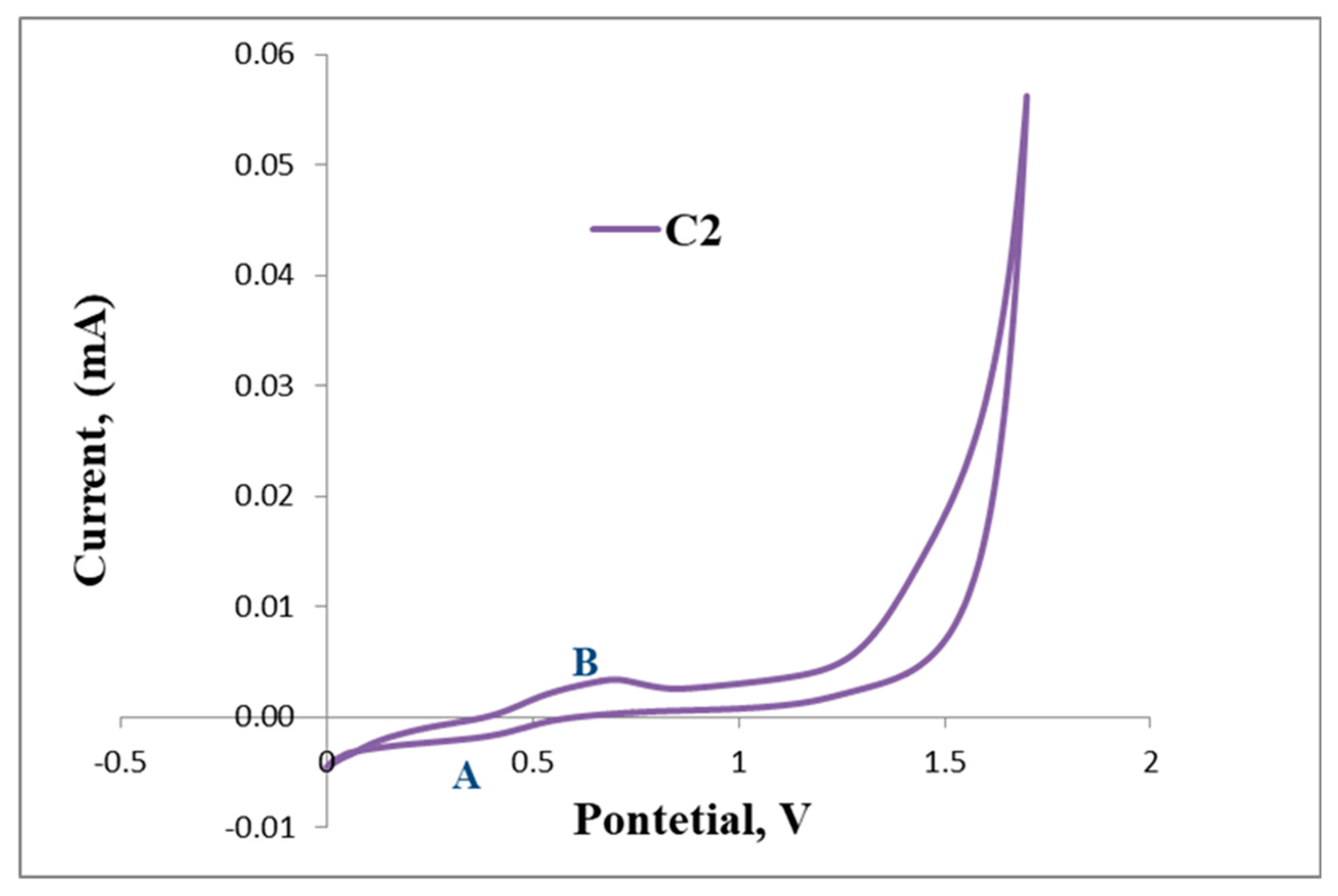
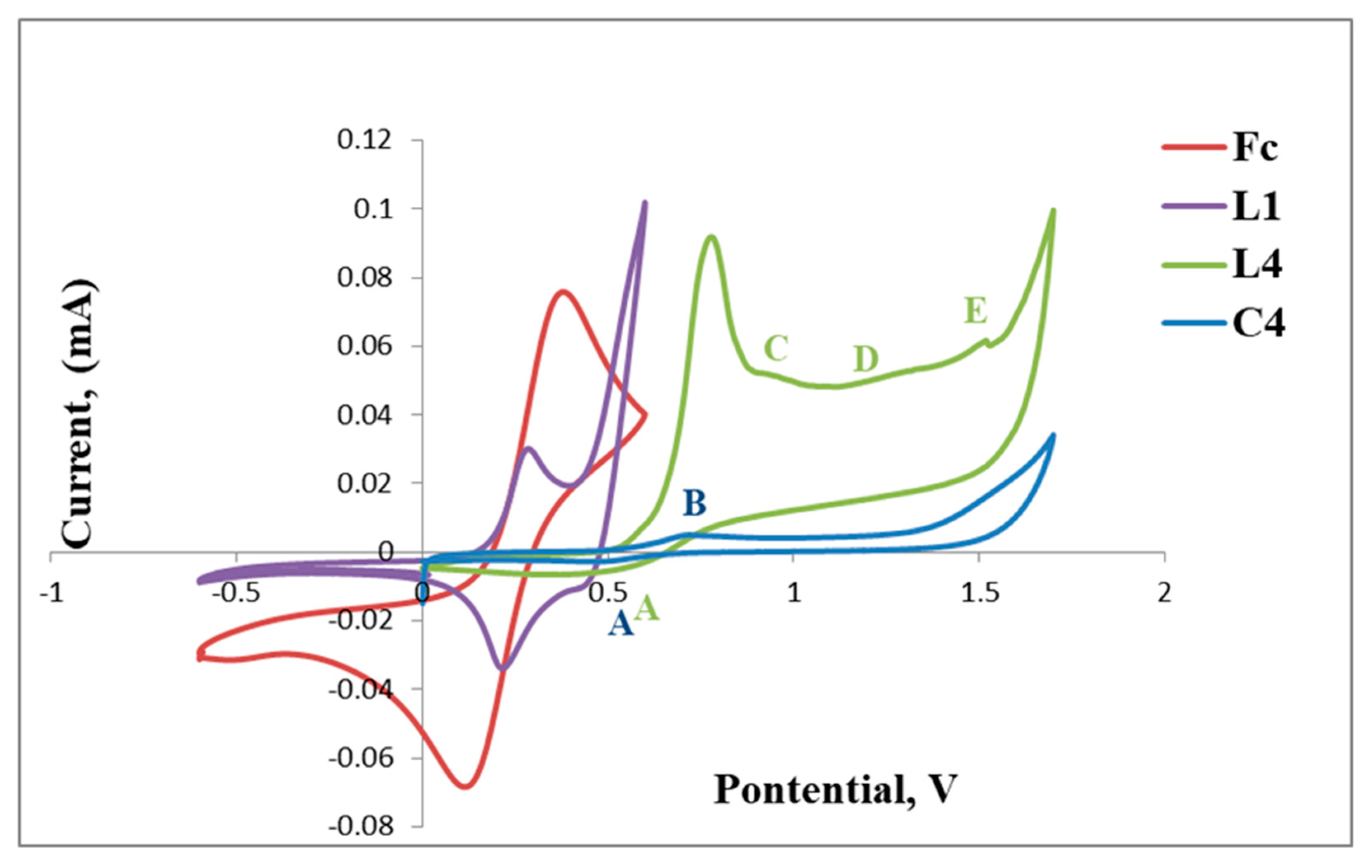
2.3. Electrochemical Studies
Cyclic Voltammetry Results of L1 and L2 with C1, C2 and C4
2.4. Mizoroki-Heck Catalytic Studies
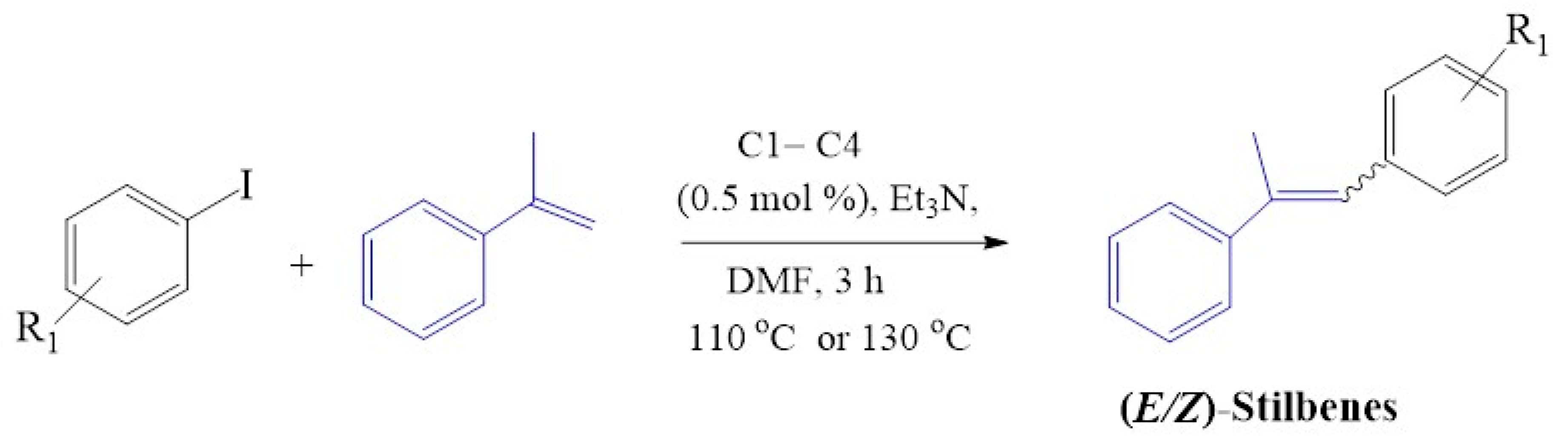
2.4.1. Mizoroki-Heck Cross-Coupling Reactions
2.4.2. Optimization of Reaction Conditions for Mizoroki-Heck Cross-Coupling Reactions
2.4.3. Mercury Poisoning Test
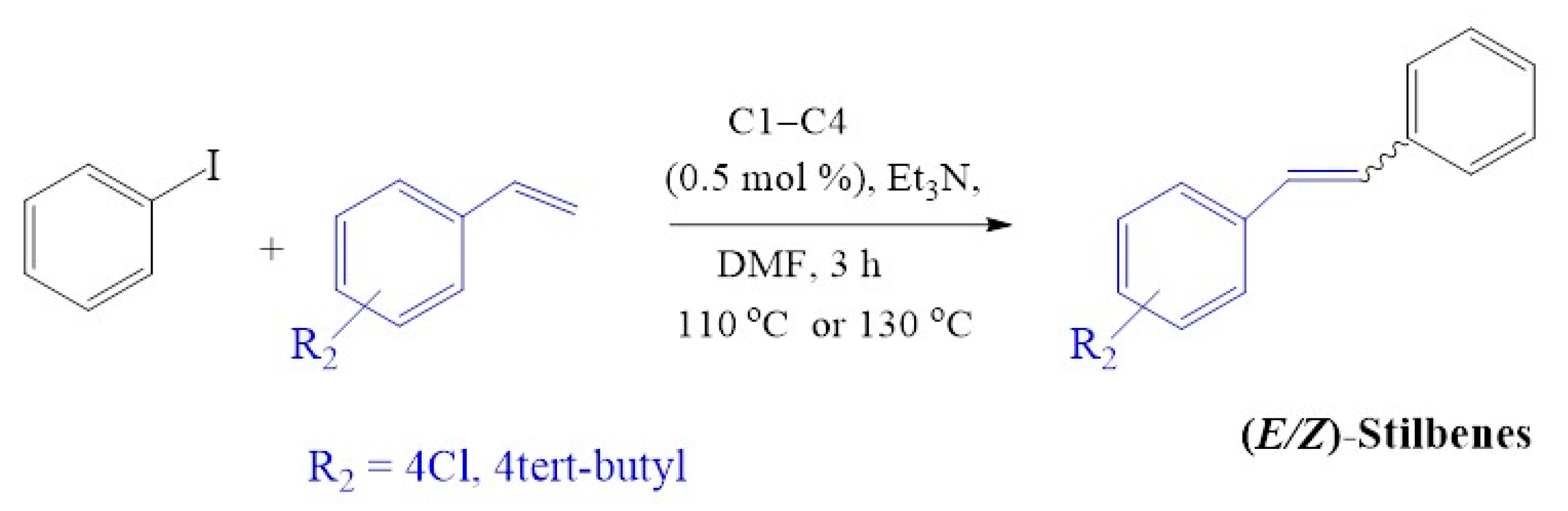
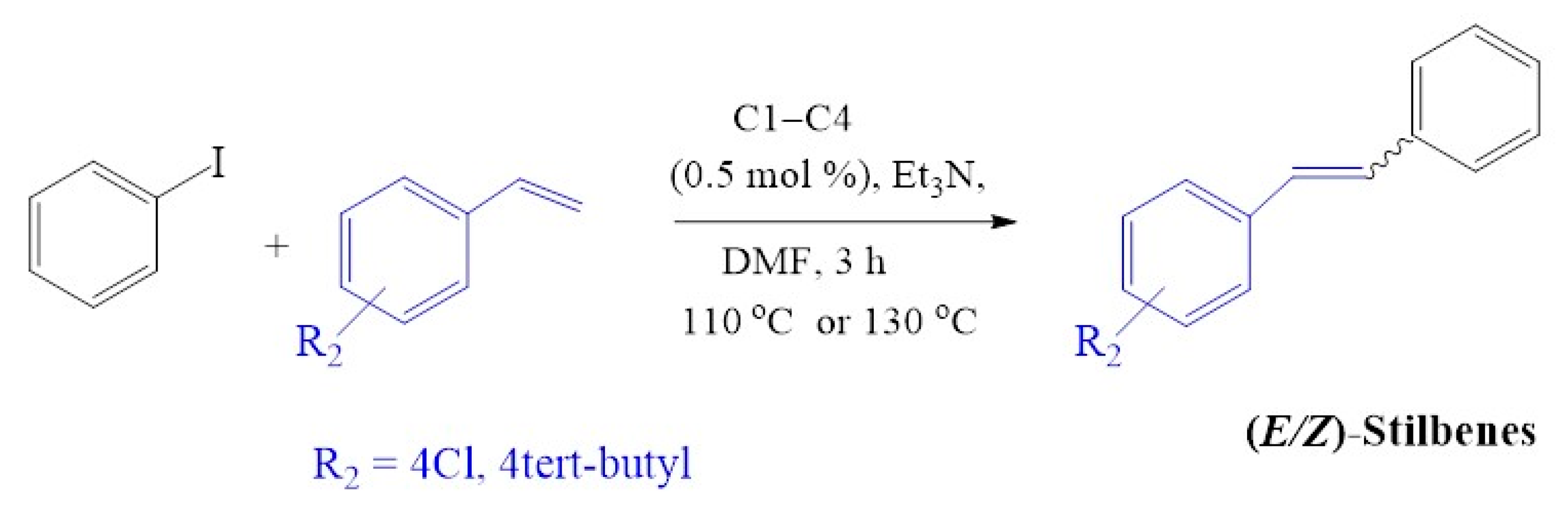
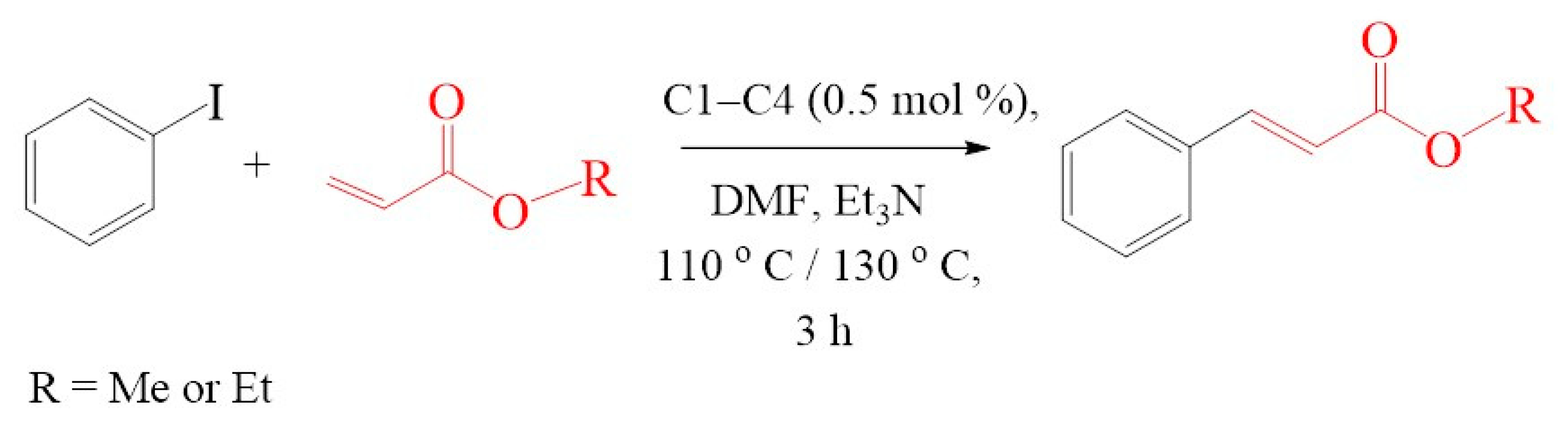
2.4.4. Evaluation of Substrate Variation
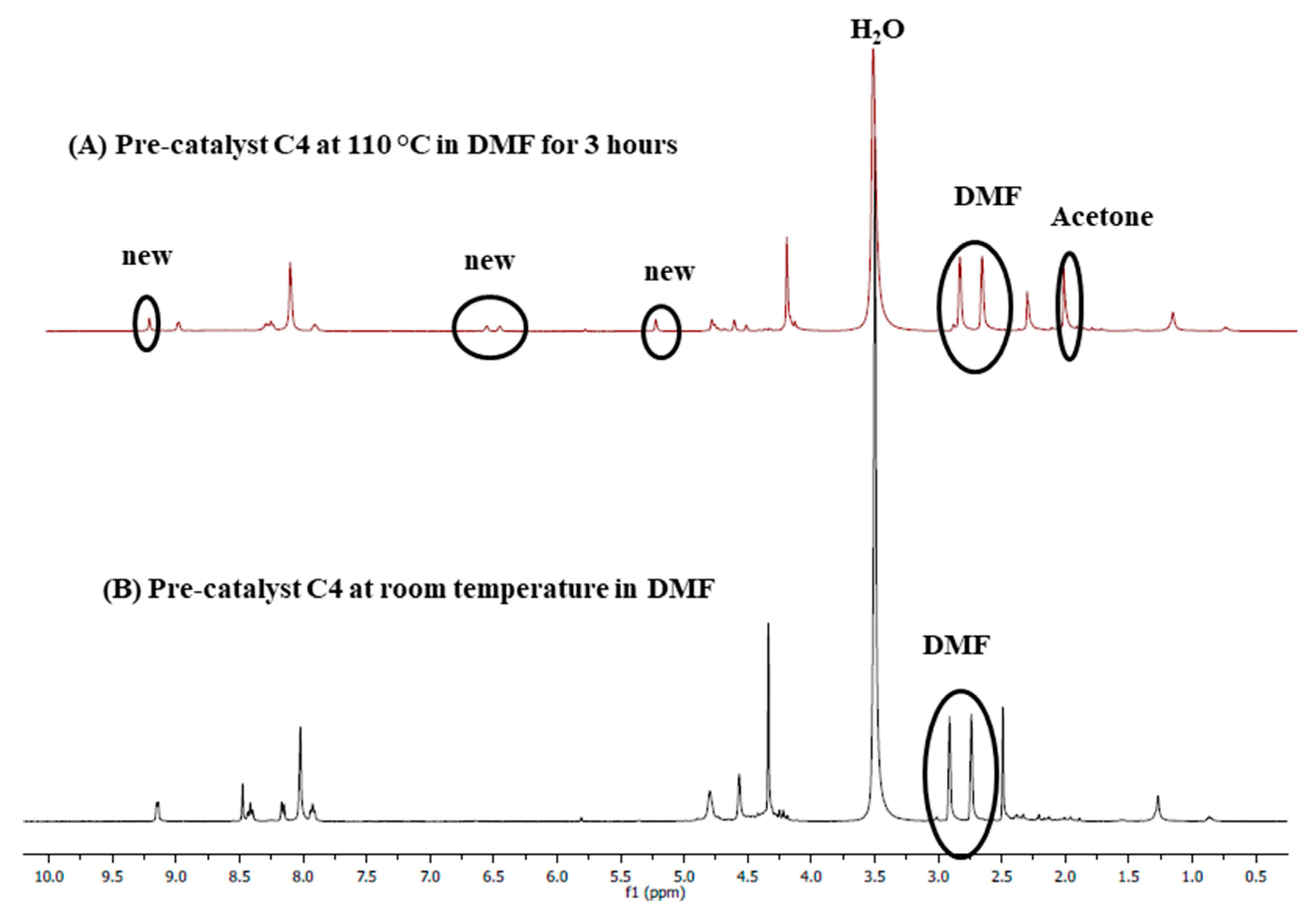
2.4.5. Pre-Catalyst Stability Studies
2.5. Computational Studies
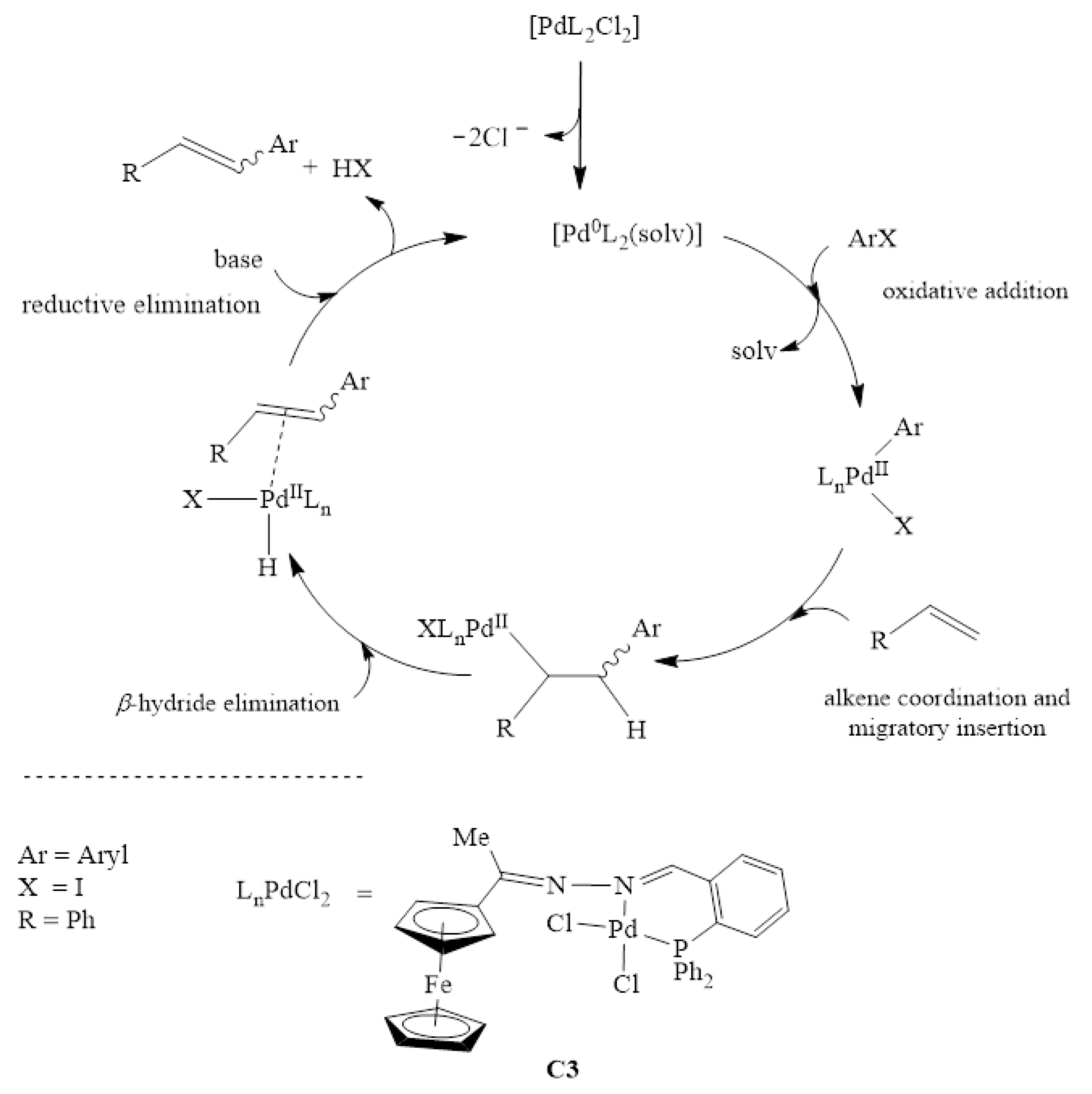
2.5.1. General Computational Approach to the Mizoroki-Heck Cross-Coupling Reaction
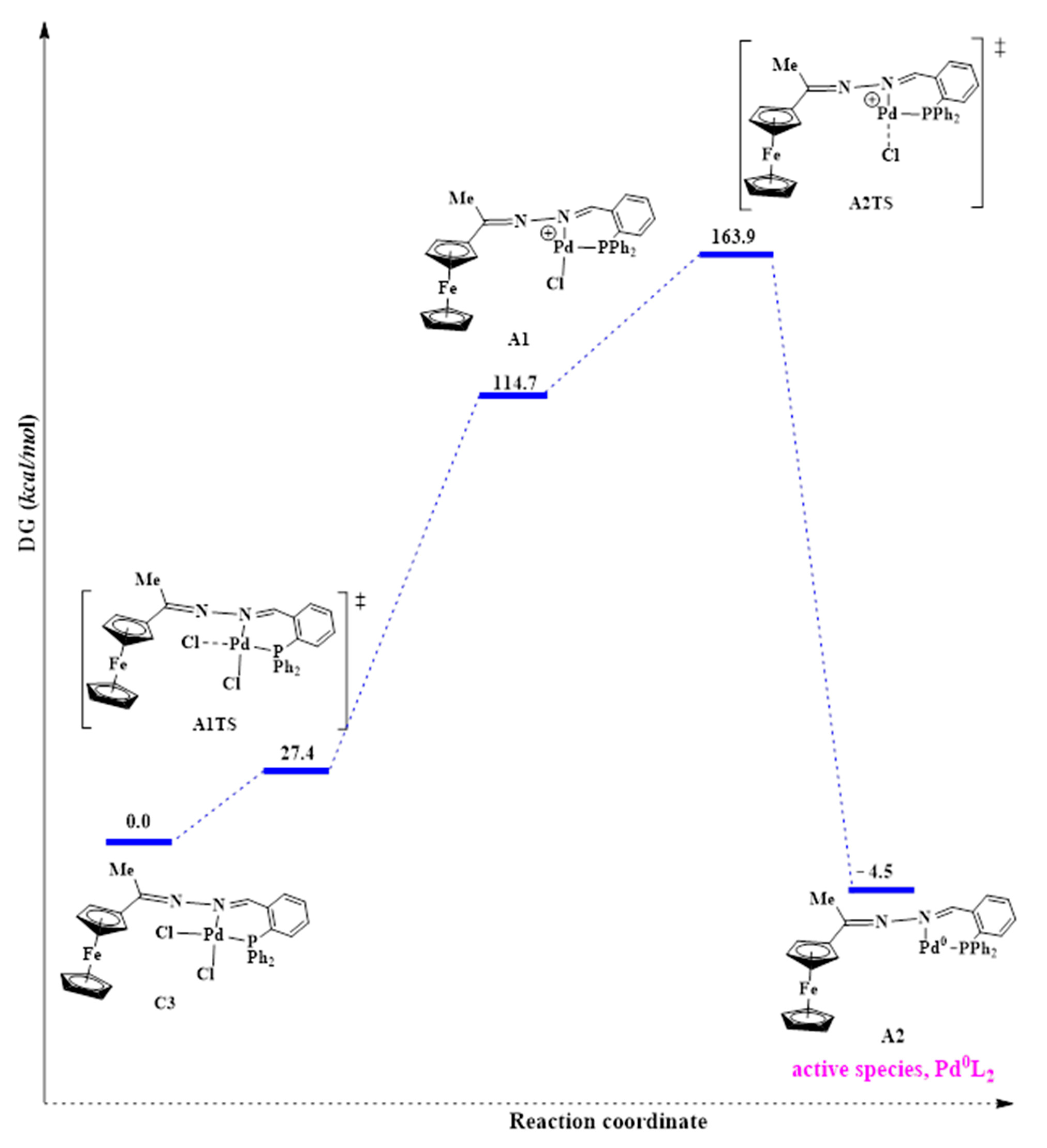
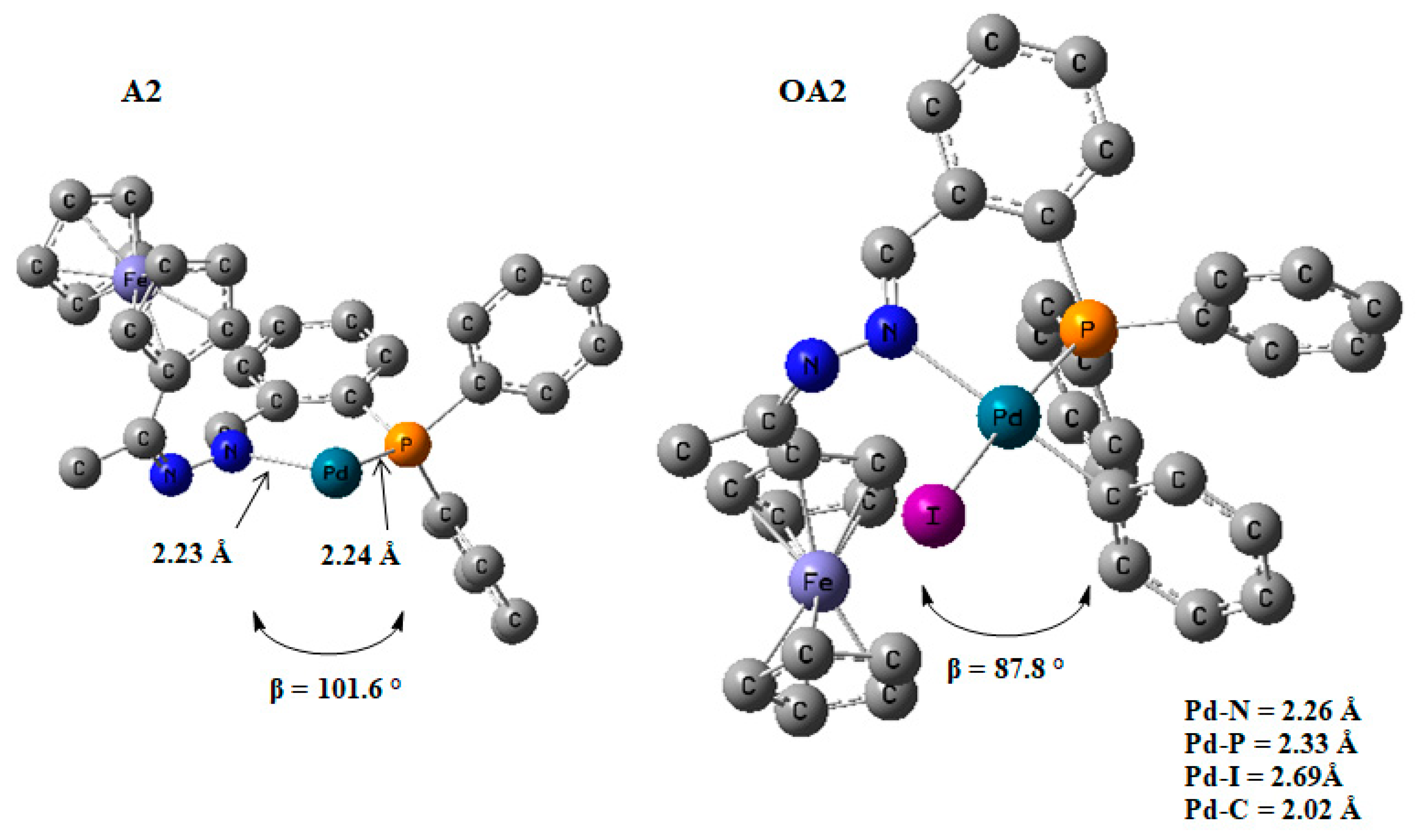
2.5.2. The Formation of the Active Species, Pd0L2
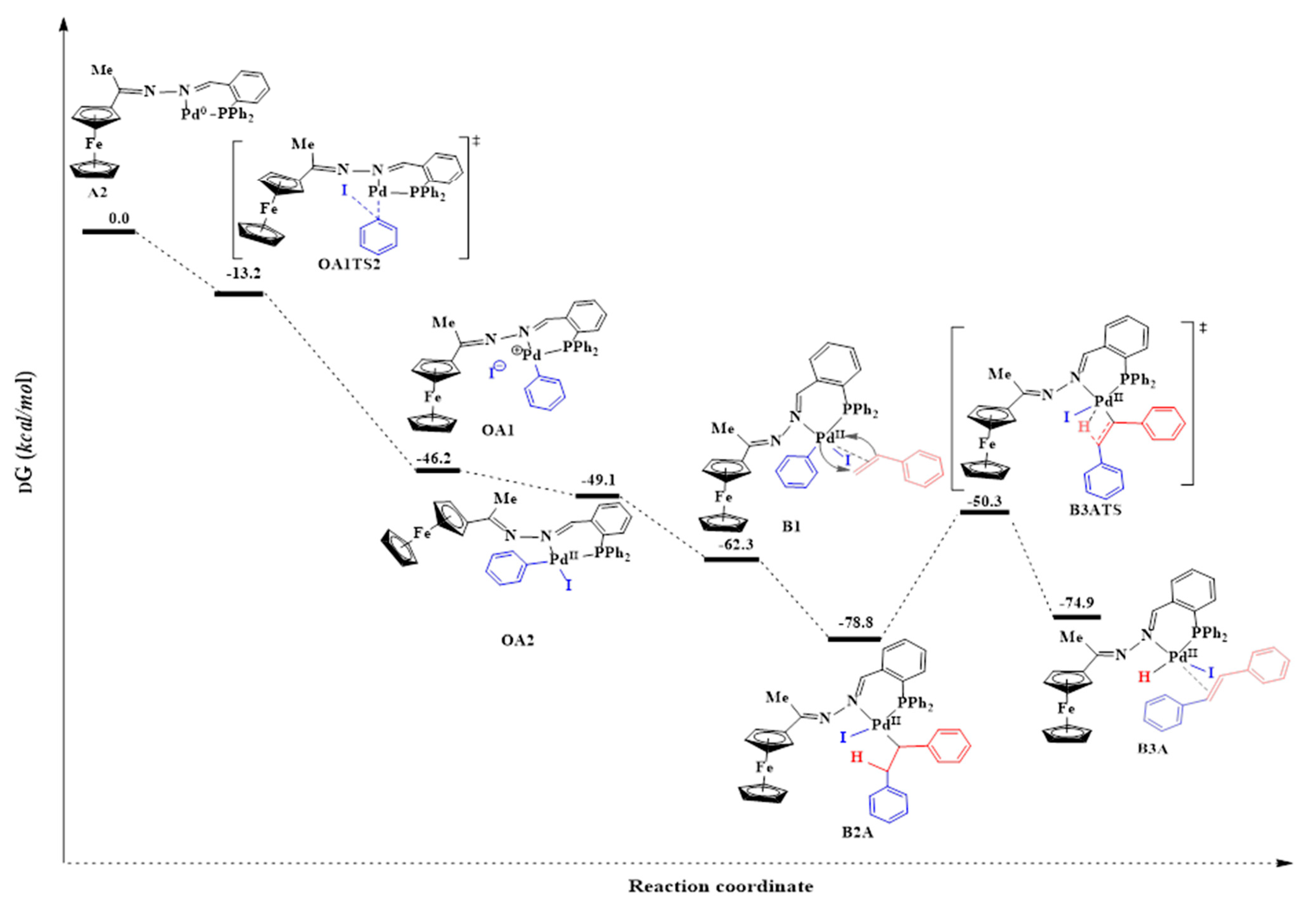
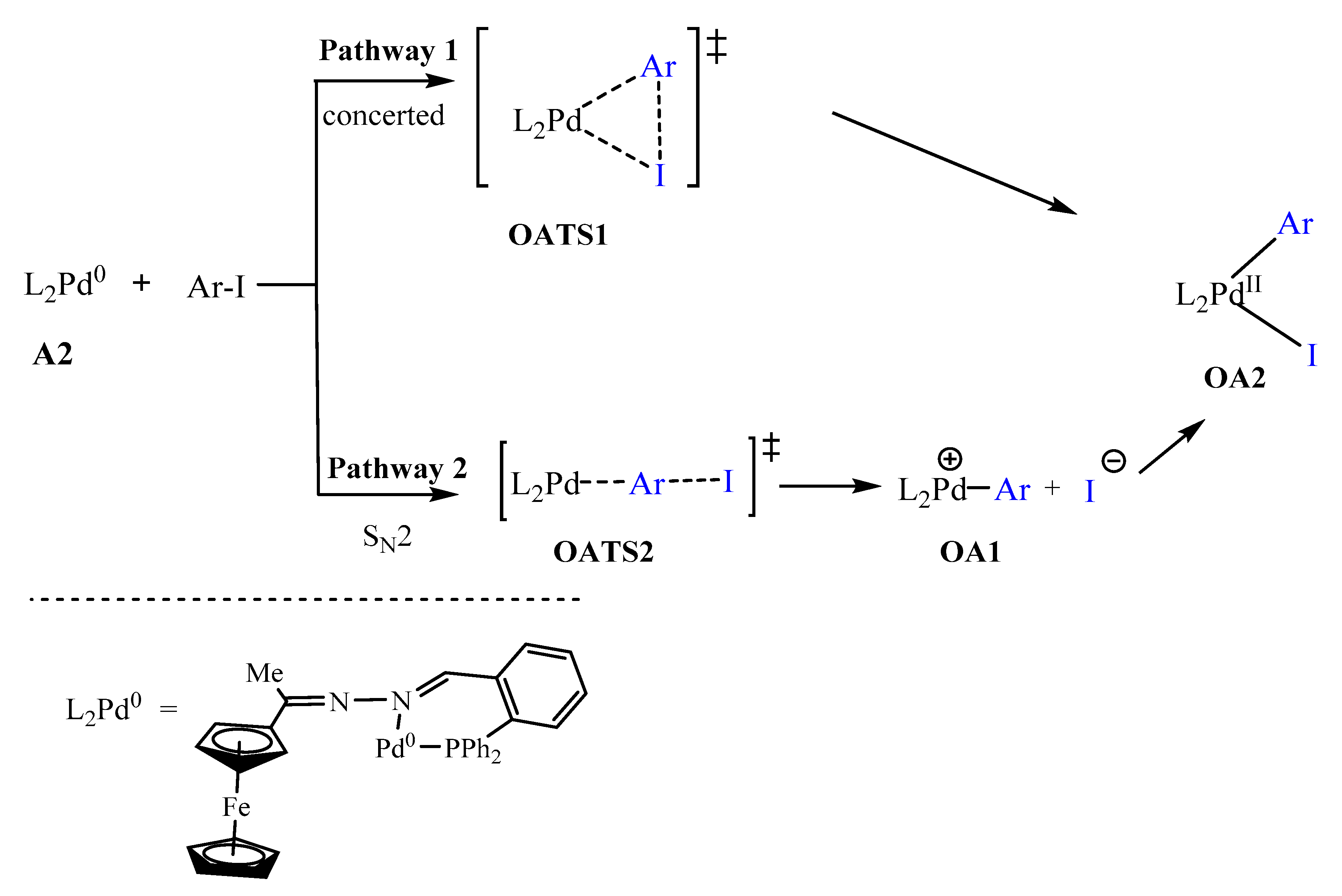
2.5.3. Oxidative Addition
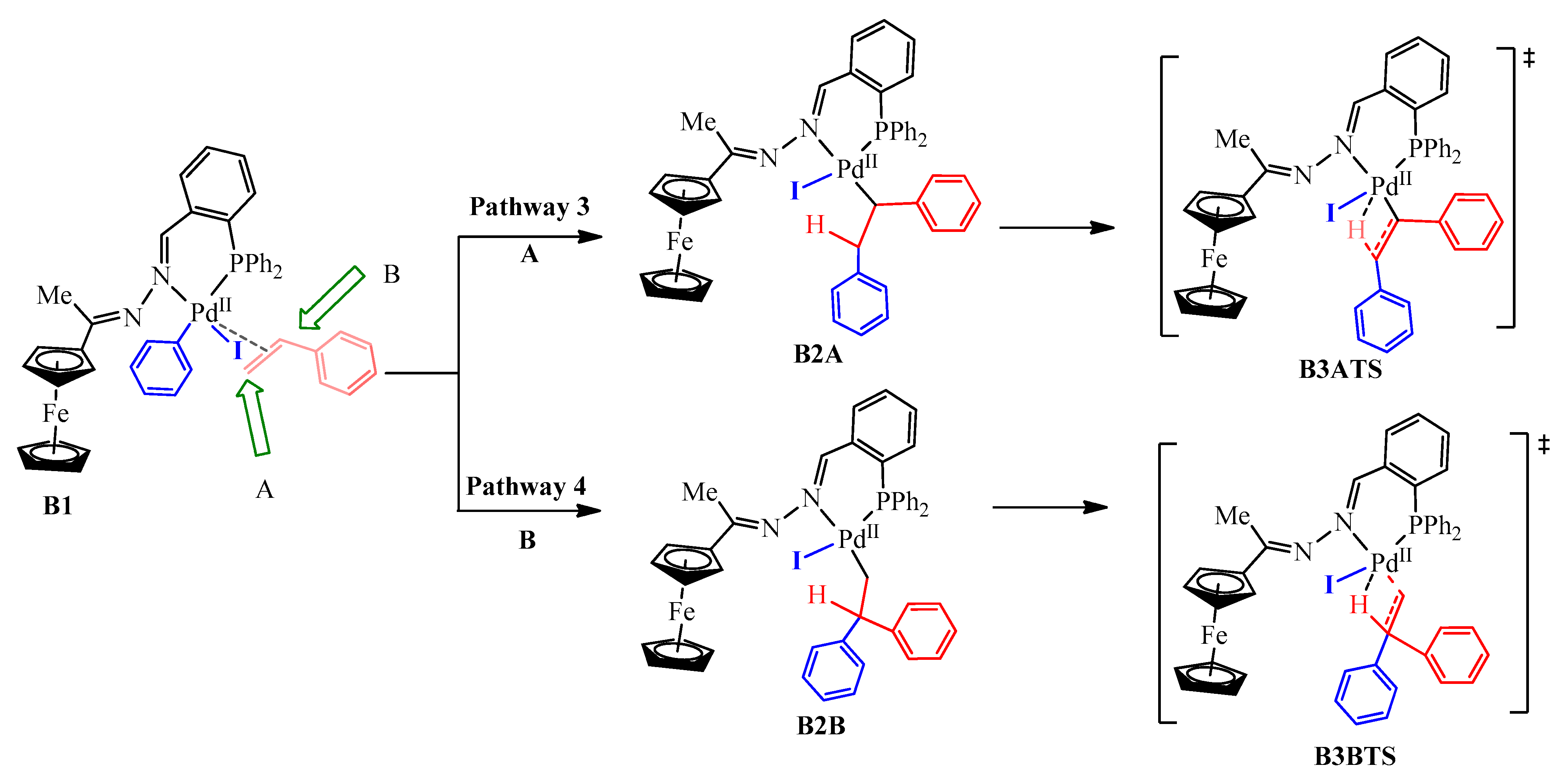
2.5.4. Alkene Coordination and Insertion → β-Hydride Elimination
2.5.5. Reductive Elimination
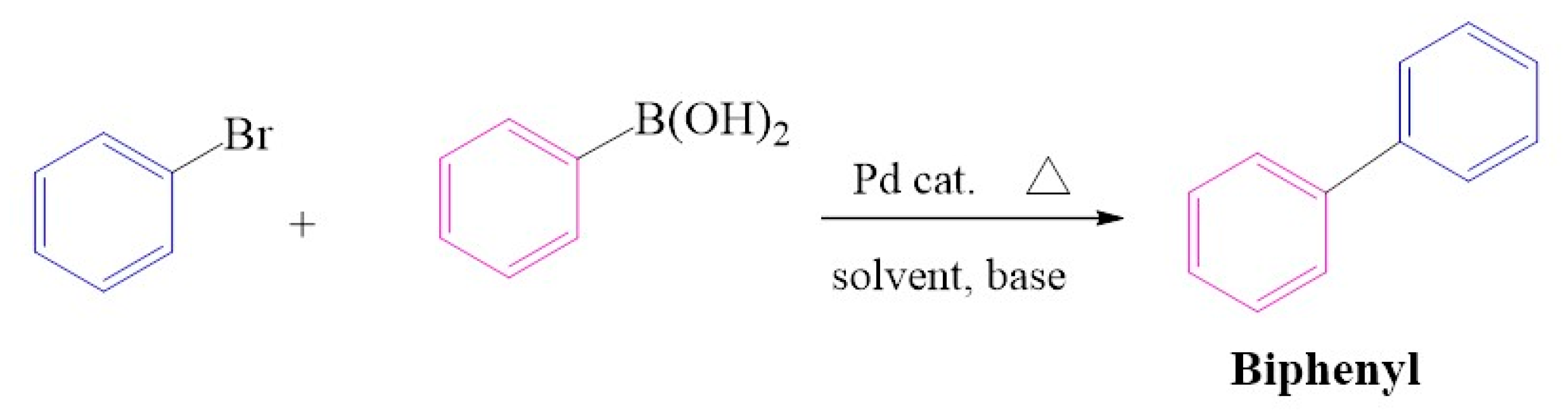
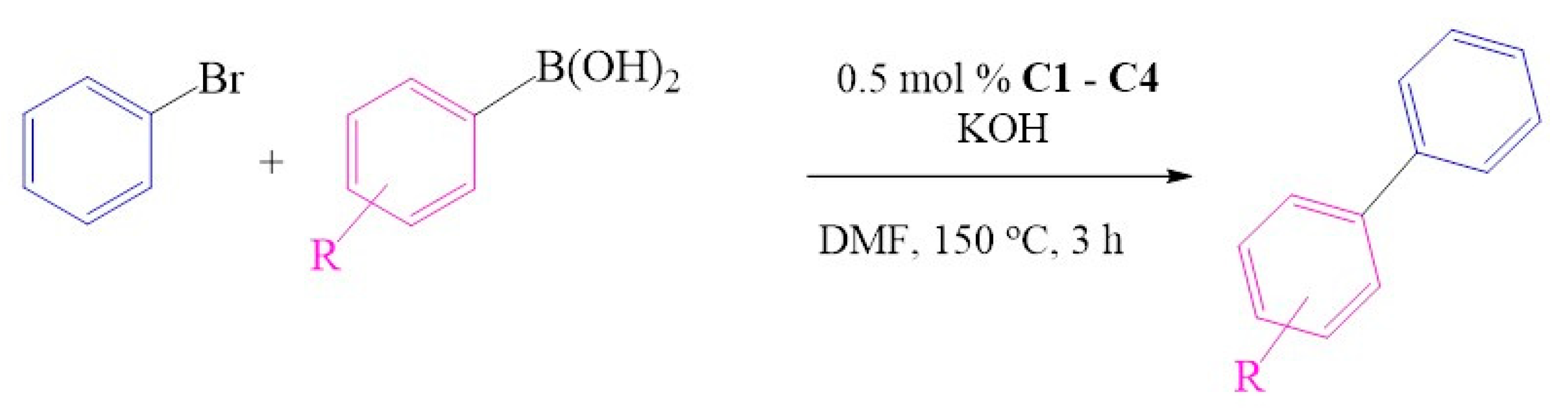
2.6. Suzuki-Miyaura C-C Cross-Coupling Reactions
2.6.1. Substrate Variation
2.6.2. Mercury Poisoning Test
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Characterization of Ligands, L1–L4
3.2.1. Preparation of Ferrocenyl Methyl Hydrazone Ligand, L1
3.2.2. Preparation of Ferrocenyl Aminophosphine Ligand, L2
3.2.3. Preparation of Ferrocenyl(Diphenylphosphine)Phenylimine Ligand, L3
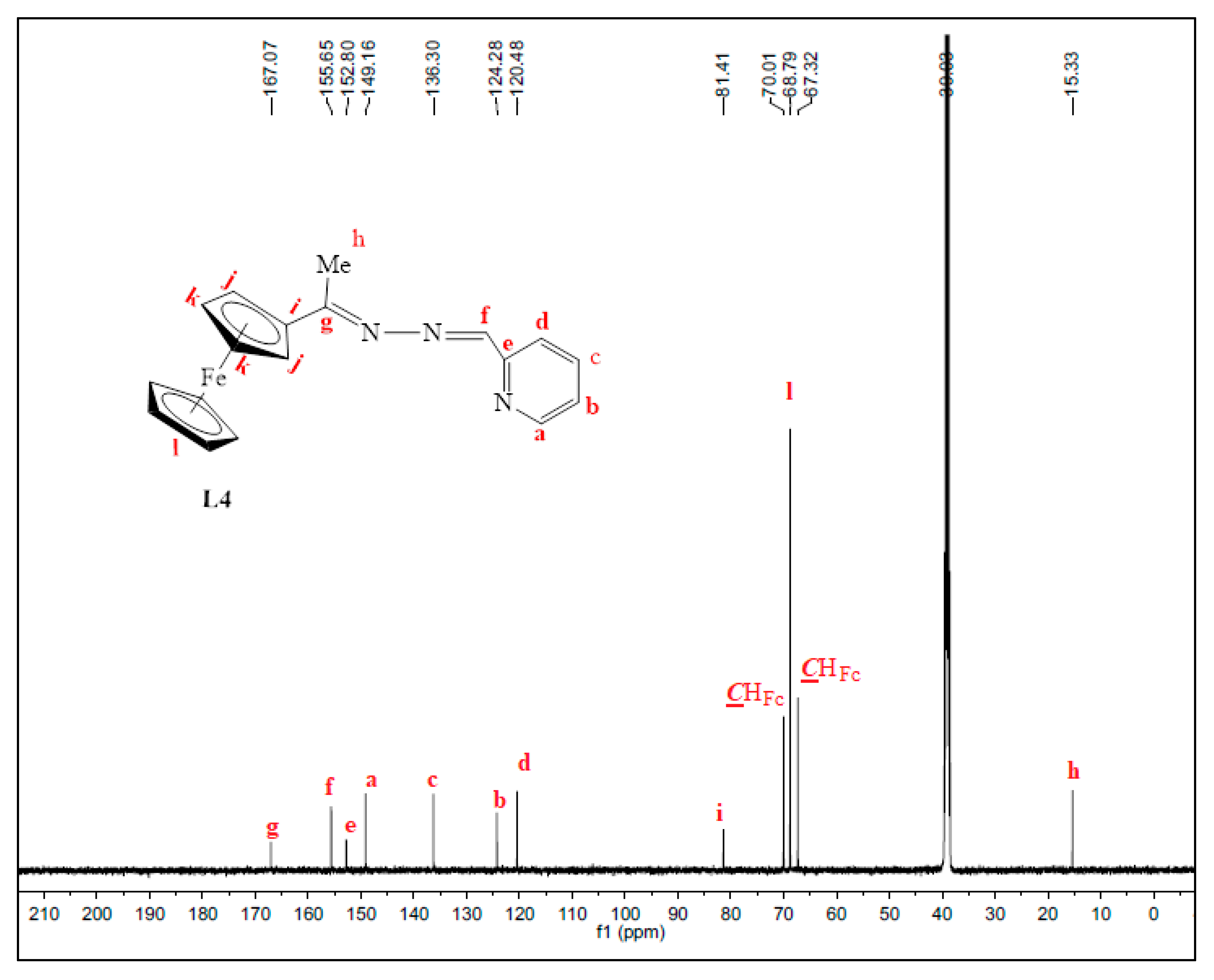
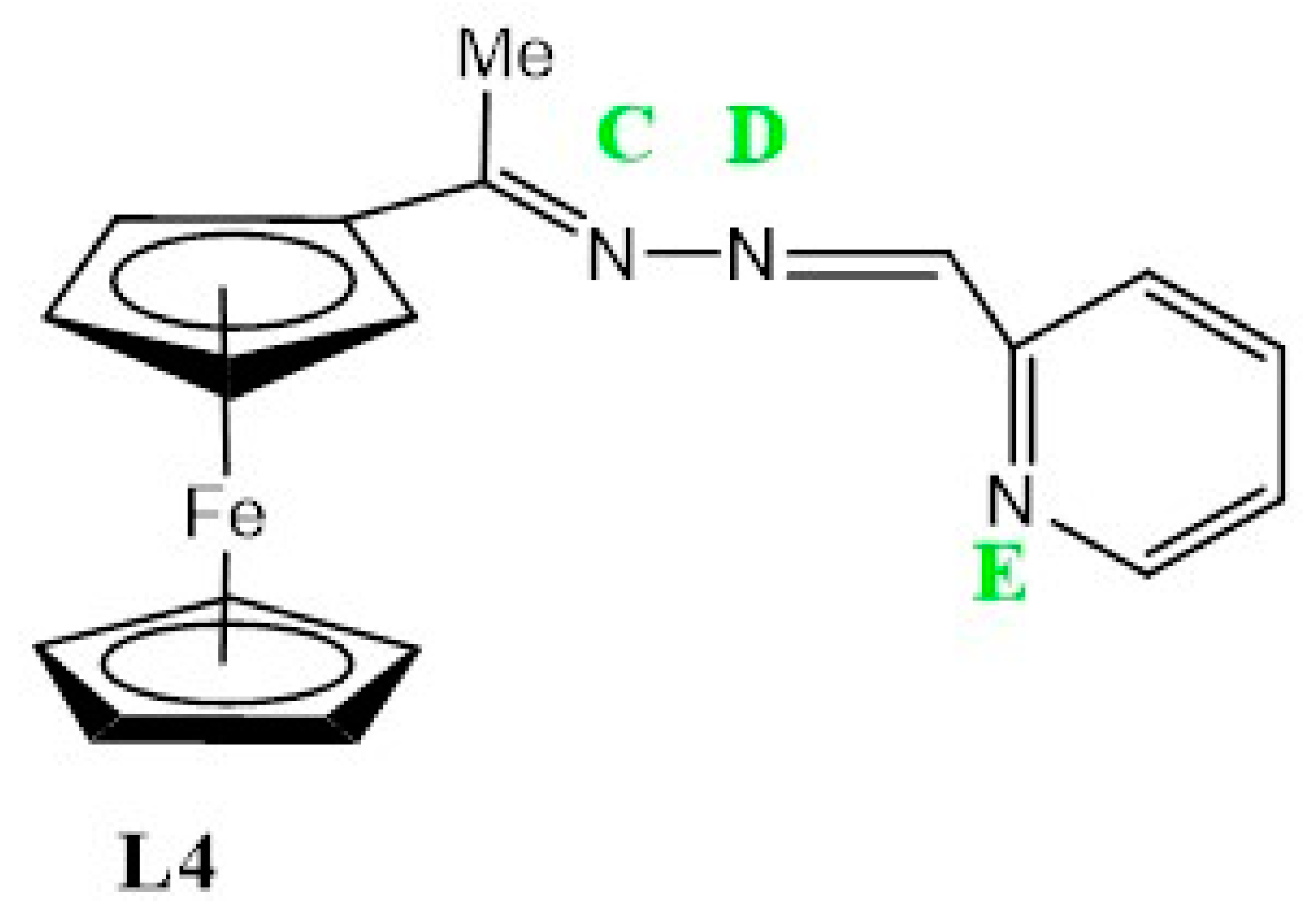
3.2.4. Preparation of Ferrocenyl(Pyridyl)imine Ligand, L4
3.3. Synthesis and Characterization of Complexes, C1–C4
3.3.1. Synthesis of [PdCl2(L1)2], C1
3.3.2. Synthesis of [PdCl2(L2)], C2
3.3.3. Synthesis of [PdCl2(L3)], C3
3.3.4. Synthesis of [PdCl2(L4)], C4
3.4. General Experimental Description of Mizoroki-Heck C-C Cross-Coupling Reactions
3.5. General Experimental Details for the Suzuki-Miyaura C-C Cross-Coupling Reactions
3.6. Computational Details on Mizoroki-Heck Cross-Coupling Reaction
3.7. General Procedure for the Isolation of Cross-Coupling Products
3.8. Electrochemical Studies Experimental Details
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tamao, K.N.M. Cross-Coupling Reactions. In Cross-Coupling Reaction, A Practical Guide; Springer: New York, NY, USA, 2002; pp. 1–20. [Google Scholar]
- Kantam, M.L.; Srinivas, P.; Yadav, J.; Likhar, P.R.; Bhargava, S. Trifunctional N,N,O-terdentate amido/pyridyl carboxylate ligated Pd(II) complexes for heck and suzuki reactions. J. Org. Chem. 2009, 74, 4882–4885. [Google Scholar] [CrossRef] [PubMed]
- Marion, N.; Nolan, S.P. Well-Defined N-Heterocyclic Carbenes-Palladium(II) Precatalysts for Cross-Coupling Reactions. Acc. Chem. Res. 2008, 41, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Oh-Tani, S.; Miyaura, N. Synthesis of Biaryls via a Nickel(0)-Catalyzed Cross-Coupling Reaction of Cholorarenes with Arylboronic Acids. J. Org. Chem. 1997, 62, 8024–8030. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chutia, A.; Brett, D.J.L.; He, G.; Parkin, I.P.; Shearing, P.R. Environmental Science Palladium alloys used as electrocatalysts for the oxygen reduction reaction. Energy Environ. Sci. 2021, 14, 2639–2669. [Google Scholar] [CrossRef]
- Indolese, A.F. Suzuki-type coupling of chloroarenes with arylboronic acids catalysed by nickel complexes. Tetrahedron Lett. 1997, 38, 3513–3516. [Google Scholar] [CrossRef]
- Farina, V. High-turnover palladium catalysts in cross-coupling and Heck chemistry: A critical overview. Adv. Synth. Catal. 2004, 346, 1553–1582. [Google Scholar] [CrossRef]
- Zhang, Y.; Lavigne, G.; Ce, V. Buchwald—Hartwig Amination of (Hetero)Aryl Tosylates Using a Well-Defined N-Heterocyclic Carbene/Palladium(II) Precatalyst. J. Org. Chem. 2015, 80, 7666–7673. [Google Scholar] [CrossRef]
- Zhang, Y.; Lavigne, G. Efficient and Versatile Buchwald–Hartwig Amination of (Hetero) aryl Chlorides Using the Pd-PEPPSI-IPr (NMe2) 2 Precatalyst in the Presence of Carbonate. J. Org. Chem. 2015, 2042–2050. [Google Scholar]
- Tolman, C.A. Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Mom, S.; Beaupérin, M.; Roy, D.; Royer, S.; Amardeil, R.; Cattey, H.; Doucet, H.; Hierso, J.C. Congested ferrocenyl polyphosphanes bearing electron-donating or electron-withdrawing phosphanyl groups: Assessment of metallocene conformation from NMR spin couplings and use in palladium-catalyzed chloroarenes activation. Inorg. Chem. 2011, 50, 11592–11603. [Google Scholar] [CrossRef]
- Xue, L.; Lin, Z. Theoretical aspects of palladium-catalysed carbon–carbon cross-coupling reactions. Chem. Soc. Rev. 2010, 39, 1692–1705. [Google Scholar] [CrossRef]
- Anderson, K.W.; Ikawa, T.; Tundel, R.E.; Buchwald, S.L. The selective reaction of aryl halides with KOH: Synthesis of phenols, aromatic ethers, and benzofurans. J. Am. Chem. Soc. 2006, 128, 10694–10695. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, A.; Roche, M.; Alami, M.; Messaoudi, S. 2-Aminobiphenyl palladacycles: The “most powerful” precatalysts in C-C and C-heteroatom cross-couplings. ACS Catal. 2015, 5, 1386–1396. [Google Scholar] [CrossRef]
- Kataoka, N.; Shelby, Q.; Stambuli, J.P.; Hartwig, J.F. Air stable, sterically hindered ferrocenyl dialkylphosphines for palladium-catalyzed C-C, C-N, and C-O bond-forming cross-couplings. J. Org. Chem. 2002, 67, 5553–5566. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Buchwald, S.L. Use of precatalysts greatly facilitate palladium-catalyzed alkynylations in batch and continuous-flow conditions †. Chem. Sci. 2011, 2, 2321–2325. [Google Scholar] [CrossRef]
- Fihri, A.; Meunier, P.; Hierso, J.C. Performances of symmetrical achiral ferrocenylphosphine ligands in palladium-catalyzed cross-coupling reactions: A review of syntheses, catalytic applications and structural properties. Coord. Chem. Rev. 2007, 251, 2017–2055. [Google Scholar] [CrossRef]
- Feyrer, A.; Breher, F. Palladium complexes of ferrocene-based phosphine ligands as redox-switchable catalysts in Buchwald–Hartwig cross-coupling reactions. Inorg. Chem. Front. 2017, 4, 1125–1134. [Google Scholar] [CrossRef]
- Sollot, G.; Snead, J.; Portnoy, S.; Peterson, W., Jr.; Mertwoy, H. Aluminum chloride-catalyzed reactions of ferrocene with phosphorus(III) amides. Novel coordination of the PN system. J. Organomet. Chem. 1969, 19, 143–159. [Google Scholar] [CrossRef]
- Colacot, T.J. Ferrocenyl Phosphme Complexes of the Platinum Metals in Non-Chral Catalysis. Platin. Met. Rev. 2001, 45, 22–30. [Google Scholar]
- Ishiyama, T.; Itoh, Y.; Kitano, T.; Miyaura, N. Synthesis of arylboronates via the palladium(0)-catalyzed cross-coupling reaction of tetra(alkoxo)diborons with aryl triflates. Tetrahedron Lett. 1997, 38, 3447–3450. [Google Scholar] [CrossRef]
- Colacot, T.J.; Qian, H.; Cea-Olivares, R.; Hernandez-Ortega, S. Synthesis, X-ray, spectroscopic and a preliminary Suzuki coupling screening studies of a complete series of dppfMX 2 (M = Pt, Pd; X = Cl, Br, I). J. Organomet. Chem. 2001, 637–639, 691–697. [Google Scholar] [CrossRef]
- Vedejs, E.; Barda, D.A. Progress toward synthesis of diazonamide A. Preparation of a 3-(oxazol-5-yl)-4-trifluoromethyl-sulfonyloxyindole and its use in biaryl coupling reactions. Org. Lett. 2000, 2, 1033–1035. [Google Scholar] [CrossRef]
- Cousaert, N.; Toto, P.; Willand, N.; Deprez, B. Efficient, protection-free Suzuki-Miyaura synthesis of ortho-biphenyltetrazoles. Tetrahedron Lett. 2005, 46, 6529–6532. [Google Scholar] [CrossRef]
- Olofsson, K.; Larhed, M.; Hallberg, A. Highly Regioselective Palladium-Catalyzed Internal Arylation of Allyltrimethylsilane with Aryl Triflates synthesis, and their utility is mainly attributed to the. J. Org. Chem. 1998, 3263, 5076–5079. [Google Scholar] [CrossRef]
- Kang, S.; Choi, S.; Ryu, H. Palladium-Catalyzed Coupling of Organolead Compounds with Olefins. J. Org. Chem. 1998, 3263, 5748–5749. [Google Scholar] [CrossRef]
- Qadir, M.; Möchel, T.; Hii, K.K. Examination of ligand effects in the heck arylation reaction. Tetrahedron 2000, 56, 7975–7979. [Google Scholar] [CrossRef]
- Shelby, Q.; Kataoka, N.; Mann, G.; Hartwig, J. Unusual in situ ligand modification to generate a catalyst for room temperature aromatic C-O bond formation. J. Am. Chem. Soc. 2000, 122, 10718–10719. [Google Scholar] [CrossRef]
- Stambuli, J.P.; Stauffer, S.R.; Shaughnessy, K.H.; Hartwig, J.F. Screening of homogeneous catalysts by fluorescence resonance energy transfer. Identification of catalysts for room-temperature heck reactions. J. Am. Chem. Soc. 2001, 123, 2677–2678. [Google Scholar] [CrossRef]
- Teo, S.; Weng, Z.; Hor, T.S.A. 1,1′-P/O-ferrocenyl ligands in palladium-catalyzed Suzuki coupling of aryl chlorides. Organometallics 2006, 25, 1199–1205. [Google Scholar] [CrossRef]
- Itoh, T.; Sato, K.; Mase, T. A novel practical synthesis of C-2-arylpurines. Adv. Synth. Catal. 2004, 346, 1859–1867. [Google Scholar] [CrossRef]
- Itoh, T.; Mase, T. Direct synthesis of hetero-biaryl compounds containing an unprotected NH2 group via Suzuki-Miyaura reaction. Tetrahedron Lett. 2005, 46, 3573–3577. [Google Scholar] [CrossRef]
- Shu-Sheng, Z.; Bo, Y.; Hui-Xiang, L.; Kui, J. Facile Synthesis and Characterization of Bioactive N-[(1-Ferrocenylethylidene) amino]-N’-β-d-glycopyranosylthiourea. Chin. J. Chem. 2005, 23, 1253–1256. [Google Scholar]
- Vera-oyarce, C.; Aguirre, P.A.; Lagos, C.A.; Moya, S.A.; Sola, E.; Peris, G.; Bay, J.C. Methoxycarbonylation of olefins catalyzed by palladium complexes bearing P, N-donor ligands. Dalton Trans. 2007, 5419–5426. [Google Scholar]
- Wang, P.; Liu, H.; Li, Y.; Zhao, X.; Lu, Y.; Liu, Y. Catalysis Science & Technology bifunctional ligands for sequential catalysis of. Catal. Sci. Technol. 2016, 6, 3854–3861. [Google Scholar]
- Garrou, P.E. Ar Ring Contributions to 31P NMR Parameters of Transition-Metal-Phosphorus Chelate Complexes. Chem. Rev. 1981, 81, 229–266. [Google Scholar] [CrossRef]
- Van Delden, R.A.; Ter Wiel, M.K.J.; Pollard, M.M.; Vicario, J.; Koumura, N.; Feringa, B.L. Unidirectional molecular motor on a gold surface. Nature 2005, 437, 1337–1340. [Google Scholar] [CrossRef]
- Ozawa, F.; Ito, T.; Nakamura, Y.; Yamamoto, A. Mechanism of Thermal Decomposition of trans- and cis-Dialkylbis-(tertiary phosphine)palladium(II). Bull. Chem. Soc. Jpn. 1981, 54, 1868–1880. [Google Scholar] [CrossRef]
- Oroshnik, B.Y.W.; Brown, P.K.; Habburd, R.; George, W. Rainn. Proc. Natl. Acad. Sci. USA 1956, 42, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.M.; Meetsma, A.; Feringa, B.L. A redesign of light-driven rotary molecular motors. Org. Biomol. Chem. 2008, 6, 507–512. [Google Scholar] [CrossRef]
- Makhubela, B.C.E.; Jardine, A.; Smith, G.S. Pd nanosized particles supported on chitosan and 6-deoxy-6-amino chitosan as recyclable catalysts for Suzuki-Miyaura and Heck cross-coupling reactions. Appl. Catal. A Gen. 2011, 393, 231–241. [Google Scholar] [CrossRef]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2018, 95, 197–206. [Google Scholar] [CrossRef]
- Mabbott, G.A. An introduction to cyclic voltammetry. J. Chem. Educ. 1983, 60, 697–702. [Google Scholar] [CrossRef]
- Powers, M.J.; Meyer, T.J. Medium and Distance Effects in Optical and Thermal Electron Transfer. J. Am. Chem. Soc. 1980, 102, 1289–1297. [Google Scholar] [CrossRef]
- Dagdevren, M.; Yilmaz, I.; Yucel, B.; Emirik, M. A Novel Ferrocenyl Naphthoquinone Fused Crown Ether as a Multisensor for Water Determination in Acetonitrile and Selective Cation Binding. J. Phys. Chem. B 2015, 119, 12464–12479. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, H.R.; Fereidoonnezhad, M.; Niazi, M.; Mosavi, S.T.; Habib Kazemi, S.; Kia, R.; Shirkhan, S.; Abdollahi Aghdam, S.; Raithby, P.R. Cyclometalated platinum(ii) complexes of 2,2′-bipyridine N-oxide containing a 1,1′-bis(diphenylphosphino)ferrocene ligand: Structural, computational and electrochemical studies. Dalton Trans. 2017, 46, 2013–2022. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A. σ-Donor and π-Acceptor Properties of Phosphorus Ligands: An Insight from the Natural Orbitals for Chemical Valence. Inorg. Chem. 2010, 49, 578–582. [Google Scholar] [CrossRef]
- Gagne, R.R.; Koval, C.A.; Lisensky, G.C. Ferrocene as an Internal Standard for Electrochemical Measurements. Inorg. Chem. 1980, 19, 2854–2855. [Google Scholar] [CrossRef]
- Rapakousiou, A.; Deraedt, C.; Irigoyen, J.; Wang, Y.; Pinaud, N.; Salmon, L.; Ruiz, J.; Moya, S.; Astruc, D. Synthesis and redox activity of “clicked” triazolylbiferrocenyl polymers, network encapsulation of gold and silver nanoparticles and anion sensing. Inorg. Chem. 2015, 54, 2284–2299. [Google Scholar] [CrossRef] [PubMed]
- Berben, L.A.; Faia, M.C.; Crawford, N.R.M.; Long, J.R. Angle-dependent electronic effects in 4,4′-bipyridine-bridged Ru 3 triangle and Ru4 square complexes. Inorg. Chem. 2006, 45, 6378–6386. [Google Scholar] [CrossRef] [PubMed]
- Toma, S.H.; Uemi, M.; Nikolaou, S.; Tomazela, D.M.; Eberlin, M.N.; Toma, H.E. {trans-1,4=bis[(4-pyridyl)ethenyl]benzene}(2,2′-bipyridine) rutherium(II) complexes and their supramolecular assemblies with β-cyclodextrin. Inorg. Chem. 2004, 43, 3521–3527. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Keene, F.R. Intervalence charge transfer (IVCT) in trinuclear and tetranuclear complexes of iron, ruthenium, and osmium. Chem. Rev. 2006, 106, 2270–2298. [Google Scholar] [CrossRef] [PubMed]
- Argazzi, R.; Bertolasi, E.; Chiorboli, C.; Bignozzi, C.A.; Itokazu, M.K.; Murakami Iha, N.Y. Intramolecular energy transfer processes in binuclear Re-Os complexes. Inorg. Chem. 2001, 40, 6885–6891. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Colby, D.A.; Graham, S.R.; Ferrence, G.M.; Szczepura, L.F. Organometallic Chemistry of Azuliporphyrins: Synthesis, Spectroscopy, Electrochemistry, and Structural Characterization of Nickel(II), Palladium(II), and Platinum(II) Complexes of Azuliporphyrins. Inorg. Chem. 2003, 42, 7326–7338. [Google Scholar] [CrossRef]
- van Asselt, R.; Elsevier, C.J.; Amatore, C.; Jutand, A. Divalent Palladium and Platinum Complexes Containing Rigid Bidentate Nitrogen Ligands and Electrochemistry of the Palladium Complexes 1. Organometallics 1997, 16, 317–328. [Google Scholar] [CrossRef]
- Leonhardt, S.E.S.; Stolle, A.; Ondruschka, B.; Cravotto, G.; De Leo, C.; Jandt, K.D.; Keller, T.F. Chitosan as a support for heterogeneous Pd catalysts in liquid phase catalysis. Appl. Catal. A Gen. 2010, 379, 30–37. [Google Scholar] [CrossRef]
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef]
- Hierso, J.C.; Fihri, A.; Amardeil, R.; Meunier, P.; Doucet, H.; Santelli, M. Use of a bulky phosphine of weak σ-donicity with palladium as a versatile and highly-active catalytic system: Allylation and arylation coupling reactions at 10-1-10-4mol% catalyst loadings of ferrocenyl bis(difurylphosphine)/Pd. Tetrahedron 2005, 61, 9759–9766. [Google Scholar] [CrossRef]
- Wu, X.F.; Anbarasan, P.; Neumann, H.; Beller, M. From noble metal to Nobel Prize: Palladium-catalyzed coupling reactions as key methods in organic synthesis. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef]
- Littke, A.F.; Fu, G.C. Palladium-catalyzed coupling reactions of aryl chlorides. Angew. Chem. Int. Ed. Engl. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Hierso, J.C.; Fihri, A.; Amardeil, R.; Meunier, P.; Doucet, H.; Santelli, M.; Donnadieu, B. A palladium-ferrocenyl tetraphosphine system as catalyst for suzuki cross-coupling and heck vinylation of aryl halides: Dynamic behavior of the palladium/phosphine species. Organometallics 2003, 22, 4490–4499. [Google Scholar] [CrossRef]
- Collman, J.; Hedegus, S.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry; University Science Books: Sausalito, CA, USA, 1987; p. 103. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.198809841 (accessed on 10 June 2021).
- Shaughnessy, K.H. Beyond TPPTS: New approaches to the development of efficient palladium-catalyzed aqueous-phase cross-coupling reactions. Eur. J. Org. Chem. 2006, 1827–1835. [Google Scholar] [CrossRef]
- Matsinha, L.C.; Mao, J.; Mapolie, S.F.; Smith, G.S. Water-Soluble Palladium(II) Sulfonated Thiosemicarbazone Complexes: Facile Synthesis and Preliminary Catalytic Studies in the Suzuki-Miyaura Cross-Coupling Reaction in Water. Eur. J. Inorg. Chem. 2015, 2015, 4088–4094. [Google Scholar] [CrossRef]
- Siangwata, S.; Baartzes, N.; Makhubela, B.C.E.; Smith, G.S. Synthesis, characterisation and reactivity of water-soluble ferrocenylimine-Rh(I) complexes as aqueous-biphasic hydroformylation catalyst precursors. J. Organomet. Chem. 2015, 796, 26–32. [Google Scholar] [CrossRef]
- Widegren, J.A.; Bennett, M.A.; Finke, R.G. Is it homogeneous or heterogeneous catalysis? Identification of bulk ruthenium metal as the true catalyst in benzene hydrogenations starting with the monometallic precursor, Ru(II)(η6-C6Me 6)(OAc)2, plus kinetic characterization of the heterogeneous nucle. J. Am. Chem. Soc. 2003, 125, 10301–10310. [Google Scholar] [CrossRef]
- Atkins, P.; Overton, T.; Rourke, J.; Weller, M.; Amstrong, F. Shriver and Atkins. In Shriver and Atkins’ Inorganic Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2009; p. 514. [Google Scholar]
- Fitton, P.; Johnson, M.P.; McKeon, J.E. Oxidative additions to palladium(0). Chem. Commun. 1968, 6–7. [Google Scholar] [CrossRef]
- Senn, H.M.; Ziegler, T. Oxidative addition of aryl halides to palladium(0) complexes: A density-functional study including solvation. Organometallics 2004, 23, 2980–2988. [Google Scholar] [CrossRef]
- Vyskočil, Š.; Smrcina, M.; Hanuš, V.; Polášek, M.; Kočovský, P. Derivatives of 2-Amino-2′-diphenylphosphino-1,1′-binaphthyl (MAP) and Their Application in Asymmetric Palladium(0)-Catalyzed Allylic Substitution. J. Org. Chem. 1998, 63, 7738–7748. [Google Scholar] [CrossRef]
- García-Melchor, M.; Braga, A.A.C.; Lledós, A.; Ujaque, G.; Maseras, F. Computational perspective on Pd-catalyzed C-C cross-coupling reaction mechanisms. Acc. Chem. Res. 2013, 46, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. The SN1 and SN2 mechanisms for nucleophilic substitution. In Organic Chemistry; Oxford University Press Inc.: New York, NY, USA, 2001; pp. 411–414. [Google Scholar]
- van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Reek, J.N.H. The bite angle makes the catalyst. Pure Appl. Chem. 1999, 71, 1443–1452. [Google Scholar] [CrossRef]
- Chen, L.; Ren, P.; Carrow, B.P. Tri(1-adamantyl)phosphine: Expanding the Boundary of Electron- Releasing Character Available to Organophosphorus Compounds. J. Am. Chem. Soc. 2016, 138, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. Electrophilic aromatic substitution. In Organic Chemistry; Oxford University Press Inc.: New York, NY, USA, 2001; pp. 556–571. [Google Scholar]
- Kashin, A.S.; Ananikov, V.P. Catalytic C–C and C–Heteroatom Bond Formation Reactions: In Situ Generated or Preformed Catalysts? Complicated Mechanistic Picture Behind Well-Known Experimental Procedures. J. Org. Chem. 2013, 78, 11117–111125. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.K.; Lin, M.; Sen, A.; Gretz, E. Bis(Benzonitrile)Dichloro Complexes of Palladium and Platinum. In Inorganic Syntheses; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 60–63. [Google Scholar]



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Anodic Potentials Epa (V) | Cathodic Potentials, Epc (V) | |||
|---|---|---|---|---|---|
| Fc | 0.14 | 0.39 | |||
| L1 | 0.24 | 0.41 | 0.26 | ||
| C1 | 0.56 | 0.81 | 1.17 | 1.51 | |
| Compound | Anodic Potentials Epa (V) | Cathodic Potentials, Epc (V) | |
|---|---|---|---|
| Fc | 0.14 | 0.39 | |
| L2 | 0.39 | 0.49 | 1.33 |
| C2 | 0.39 | 0.62 | |
| Compound | Anodic Potentials Epa (V) | Cathodic Potentials, Epc (V) | |||
|---|---|---|---|---|---|
| Fc | 0.14 | 0.39 | |||
| L4 | 0.45 | 0.9 | 1.24 | 1.48 | 0.76 |
| C4 | 0.51 | 0.65 | |||
| Entry | Cat. | Conv. (%) | TON | TOF (hr−1)(3 h) | Selectivity (%) | Hg (mg) | |
|---|---|---|---|---|---|---|---|
| (E)-Stilbene | (Z)-Stilbene | ||||||
| 1 | C1 | 95 | 189 | 63 | 85 | 15 | 0 |
| 2 | C2 | 100 | 199 | 66 | 83 | 17 | 0 |
| 3 | C3 | 100 | 199 | 66 | 87 | 13 | 0 |
| 4 | C4 | 91 | 183 | 61 | 85 | 15 | 0 |
| 5 | C1 | 93 | 187 | 62 | 87 | 12 | 1 |
| 6 | C2 | 98 | 197 | 66 | 83 | 17 | 1 |
| 7 | C3 | 99 | 198 | 66 | 88 | 12 | 1 |
| 8 | C4 | 95 | 187 | 62 | 88 | 12 | 1 |
| Entry | Cat. | ArX | Conv. (%) | TON | TOF (h−1) (3 h) | Selectivity (%) | |
|---|---|---|---|---|---|---|---|
| (E)-Stilbene | (Z)-Stilbene | ||||||
| 1 | C1 |  | 99 | 199 | 66 | 88 | 12 |
| 2 | C2 |  | 95 | 179 | 63 | 88 | 12 |
| 3 | C3 |  | 96 | 190 | 64 | 88 | 12 |
| 4 | C4 |  | 73 | 148 | 49 | 88 | 12 |
| 5 | C3 |  | 2 | 4 | 1 | -- | 100 |
| 6 | C3 |  | -- | -- | -- | -- | -- |
| 7 | C3 |  | -- | -- | -- | -- | -- |
| 8 | C3 |  | 3 | 7 | 2 | -- | 100 |
| 9 | C3 |  | -- | -- | -- | -- | -- |
| Entry | Cat. | ArX | Conv. (%) | TON | TOF (h−1) (3 h) | Selectivity (%) | |
|---|---|---|---|---|---|---|---|
| (E)-Stilbene | (Z)-Stilbene | ||||||
| 1 | C1 |  | 19 | 38 | 13 | 70 | 30 |
| 2 | C2 |  | 48 | 93 | 31 | 82 | 18 |
| 3 | C3 |  | 41 | 84 | 28 | 85 | 15 |
| 4 | C4 |  | 19 | 38 | 13 | 70 | 30 |
| Entry | Cat. | ArX | Conv. (%) | TON | TOF (h−1) (3 h) | Selectivity (%) | |
|---|---|---|---|---|---|---|---|
| (E)-Stilbene | (Z)-Stilbene | ||||||
| 1 | C1 |  | 94 | 190 | 89 | 84 | 16 |
| 2 | C2 |  | 94 | 189 | 63 | 92 | 8 |
| 3 | C3 |  | 88 | 177 | 59 | 93 | 7 |
| 4 | C4 |  | 93 | 189 | 63 | 90 | 10 |
| 5 a | C1 |  | 97 | 196 | 65 | 96 | 4 |
| 6 a | C2 |  | 100 | 202 | 67 | 96 | 4 |
| 7 a | C3 |  | 95 | 192 | 64 | 99 | 1 |
| 8 a | C4 |  | 95 | 192 | 64 | 95 | 5 |
| Entry | Cat. | Alkene | Conv. (%) | TON | TOF (h−1) | Selectivity (%) | |
|---|---|---|---|---|---|---|---|
| (E)-Cinnamate | (Z)-Cinnamate | ||||||
| 1 | C1 |  | 99 | 202 | 67 | 98 | 2 |
| 2 | C2 |  | 99 | 200 | 67 | 98 | 2 |
| 3 | C3 |  | 99 | 201 | 67 | 99 | 1 |
| 4 | C4 |  | 99 | 201 | 67 | 99 | 1 |
| 5 | C1 |  | 100 | 201 | 67 | 98 | 2 |
| 6 | C2 |  | 100 | 202 | 67 | 98 | 2 |
| 7 | C3 |  | 98 | 197 | 66 | 98 | 2 |
| 8 | C4 |  | 99 | 199 | 66 | 98 | 2 |
| Entry | Cat. | Conv. (%) | Selectivity (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|
| 1 | [PdCl2(MeCN)2] | 37 | >99 | 74 | 25 |
| 2 | C1 | 93 | >99 | 188 | 62 |
| 3 | C2 | 96 | >99 | 196 | 63 |
| 4 | C3 | 87 | >99 | 175 | 58 |
| 5 | C4 | 95 | >99 | 189 | 64 |
| Entry | Cat. | Ph-B(OH)2 | Conv. (%) | Selectivity (%) | TON | TOF(h−1) |
|---|---|---|---|---|---|---|
| 1 | C1 |  | 56 | >99 | 113 | 38 |
| 2 | C2 |  | 61 | >99 | 123 | 41 |
| 3 | C3 |  | 52 | >99 | 105 | 35 |
| 4 | C4 |  | 56 | >99 | 114 | 38 |
| 5 | C1 |  | 52 | 98 | 106 | 35 |
| 6 | C2 |  | 51 | 99 | 104 | 34 |
| 7 | C3 |  | 51 | 100 | 103 | 34 |
| 9 | C4 |  | 54 | 100 | 108 | 36 |
| Entry | Cat. | Ph-B(OH)2 | Conv. (%) | Selectivity (%) | TON | TOF(h−1) |
|---|---|---|---|---|---|---|
| 1 | C1 |  | 69 | 100 | 139 | 46 |
| 2 | C2 |  | 70 | >99 | 140 | 47 |
| 3 | C3 |  | 53 | >99 | 113 | 38 |
| 4 | C4 |  | 65 | >99 | 131 | 43 |
| 5 | C1 |  | 28 | 100 | 57 | 19 |
| 6 | C2 |  | 30 | 100 | 62 | 20 |
| 7 | C3 |  | 66 | 100 | 134 | 44 |
| 8 | C4 |  | 19 | 100 | 38 | 12 |
| Entry | Cat. | Ph-B(OH)2 | Conv. (%) | Selectivity (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| 1 | C1 |  | 41 | >99 | 83 | 28 |
| 2 | C2 |  | 53 | >99 | 107 | 36 |
| 3 | C3 |  | 45 | >99 | 91 | 30 |
| 4 | C4 |  | 48 | >99 | 98 | 33 |
| Entry | Cat. | ArX | Conv. (%) | Selectivity (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| 1 | C3 |  | 68 | 93 | 137 | 46 |
| 2 | C3 |  | 100 | 77 | 202 | 67 |
| 3 | C3 |  | 32 | 100 | 65 | 22 |
| 4 | C3 |  | 81 | >99 | 163 | 54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsheku, A.C.; Tia, R.; Maumela, M.C.; Makhubela, B.C.E. Ferrocenylimine Palladium (II) Complexes: Synthesis, Characterization and Application in Mizoroki-Heck and Suzuki-Miyaura Cross-Coupling Reactions. Catalysts 2021, 11, 755. https://doi.org/10.3390/catal11070755
Matsheku AC, Tia R, Maumela MC, Makhubela BCE. Ferrocenylimine Palladium (II) Complexes: Synthesis, Characterization and Application in Mizoroki-Heck and Suzuki-Miyaura Cross-Coupling Reactions. Catalysts. 2021; 11(7):755. https://doi.org/10.3390/catal11070755
Chicago/Turabian StyleMatsheku, Asanda C., Richard Tia, Munaka C. Maumela, and Banothile C. E. Makhubela. 2021. "Ferrocenylimine Palladium (II) Complexes: Synthesis, Characterization and Application in Mizoroki-Heck and Suzuki-Miyaura Cross-Coupling Reactions" Catalysts 11, no. 7: 755. https://doi.org/10.3390/catal11070755
APA StyleMatsheku, A. C., Tia, R., Maumela, M. C., & Makhubela, B. C. E. (2021). Ferrocenylimine Palladium (II) Complexes: Synthesis, Characterization and Application in Mizoroki-Heck and Suzuki-Miyaura Cross-Coupling Reactions. Catalysts, 11(7), 755. https://doi.org/10.3390/catal11070755