
From Enzyme Stability to Enzymatic Bioelectrode Stabilization Processes


Abstract
1. Introduction
2. Intrinsic (in)Stability of Enzymes
3. Strategies for All Enzyme Stabilization
3.1. Addition of Stabilizers in the Medium
3.1.1. Chemical Stabilizer Addition
3.1.2. Salt Effect
3.2. Chemical Modification of Enzymes, Directed Evolution
3.2.1. Chemical Modification of Enzymes
3.2.2. Enzyme Engineering
3.3. Screening of the Biodiversity and Outstanding Properties of Extremophiles
3.3.1. Thermophiles
3.3.2. Halophiles
3.3.3. Cross-Stabilities of Extremophiles
4. Strategies for Enzyme Stabilization in the Immobilized State
4.1. Some Fundamentals on Enzyme Immobilization on a Solid Support
4.1.1. Various Types of Interactions
4.1.2. Enzyme Orientation on the Surface
4.1.3. Conformation Changes upon Immobilization
4.1.4. Adsorption Versus Multipoint Attachment
4.2. Nanomaterials for Stabilizing Enzyme in the Immobilized State
4.3. Encapsulation in Porous Materials
4.3.1. Key Role of Pore Size
4.3.2. Metal-Based Porous Matrix
4.3.3. Other Polymeric Matrices
4.4. Cross-Linked Enzyme Aggregates (CLEAs)
4.5. Bio-Surface Display of Enzymes
5. Specificity of the Stabilization of Redox Enzymes on Electrodes for Bioelectrocatalysis
5.1. Bioelectrocatalysis: When Enzymes Meet a Foreign Conductive Surface
5.1.1. Electron Transfer in Metabolic Energy Chains
5.1.2. Electron Transfer Mechanisms Involving Redox Enzymes at Electrochemical Interfaces
5.2. Stabilization Strategies for Bioelectrocatalysis
5.2.1. How Far Can General Enzyme Stabilization Strategies Be Extended to Bioelectrocatalysis?
5.2.2. Engineering of Enzymes Seeking for Both Enhanced ET Efficiency and Stability
5.2.3. Conductive Porous Material as Redox Enzyme Host Matrices Favoring Fast ET
5.2.4. Redox Enzyme Encapsulation in Hydrogels for Bioelectrocatalysis
5.2.5. When Aggregates or Partially Unfolded Enzymes Operate in Bioelectrocatalysis
5.3. Parameters Specific to Bioelectrocatalysis Stabilization
5.3.1. Reorientation of the Immobilized Enzyme
5.3.2. Effect of the Electric Field
5.3.3. Protection against Reactive Oxygen Species (ROS) Production
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Woodley, J.M. New frontiers in biocatalysis for sustainable synthesis. Curr. Opin. Green Sustain. Chem. 2020, 21, 22–26. [Google Scholar] [CrossRef]
- Reetz, M.T. What are the Limitations of Enzymes in Synthetic Organic Chemistry? Chem. Rec. 2016, 16, 2449–2459. [Google Scholar] [CrossRef] [PubMed]
- Hauer, B. Embracing Nature’s Catalysts: A Viewpoint on the Future of Biocatalysis. ACS Catal. 2020, 10, 8418–8427. [Google Scholar] [CrossRef]
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565. [Google Scholar] [CrossRef] [PubMed]
- Mandpe, P.; Prabhakar, B.; Gupta, H.; Shende, P. Glucose oxidase-based biosensor for glucose detection from biological fluids. Sens. Rev. 2020, 40, 497–511. [Google Scholar] [CrossRef]
- Mano, N.; de Poulpiquet, A. O-2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [PubMed]
- Lojou, E. Hydrogenases as catalysts for fuel cells: Strategies for efficient immobilization at electrode interfaces. Electrochim. Acta 2011, 56, 10385–10397. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rudiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Alfano, M.; Cavazza, C. Structure, function, and biosynthesis of nickel-dependent enzymes. Protein Sci. 2020, 29, 1071–1089. [Google Scholar] [CrossRef]
- Najafpour, M.M.; Zaharieva, I.; Zand, Z.; Hosseini, S.M.; Kouzmanova, M.; Holynska, M.; Tranca, I.; Larkum, A.W.; Shen, J.R.; Allakhverdiev, S.I. Water-oxidizing complex in Photosystem II: Its structure and relation to Manganese-oxide based catalysts. Coord. Chem. Rev. 2020, 409. [Google Scholar] [CrossRef]
- Morello, G.; Megarity, C.F.; Armstrong, F.A. The power of electrified nanoconfinement for energising, controlling and observing long enzyme cascades. Nat. Commun. 2021, 12, 340. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, K.; Rogalski, J.; Bilewicz, R. NAD(P)-dependent glucose dehydrogenase: Applications for biosensors, bioelectrodes, and biofuel cells. Bioelectrochemistry 2020, 135, 107574. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Malmagro, J.; Garcia-Molina, G.; De Lacey, A.L. Electrochemical Biosensors Based on Membrane-Bound Enzymes in Biomimetic Configurations. Sensors 2020, 20, 3393. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Katz, E. Bioelectrocatalysis at carbon nanotubes. In Methods in Enzymology: Nanoarmoring of Enzymes with Carbon Nanotubes and Magnetic Nanoparticles; Kumar, C.V., Pyle, A.M., Christianson, D.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 630, pp. 215–247. [Google Scholar]
- Wu, R.R.; Ma, C.L.; Zhu, Z.G. Enzymatic electrosynthesis as an emerging electrochemical synthesis platform. Curr. Opin. Electrochem. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Yuan, M.W.; Kummer, M.J.; Minteer, S.D. Strategies for Bioelectrochemical CO2 Reduction. Chem. A Eur. J. 2019, 25, 14258–14266. [Google Scholar] [CrossRef]
- Mazurenko, I.; Wang, X.; de Poulpiquet, A.; Lojou, E. H-2/O-2 enzymatic fuel cells: From proof-of-concept to powerful devices. Sustain. Energy Fuels 2017, 1, 1475–1501. [Google Scholar] [CrossRef]
- Xiao, X.X.; Xia, H.Q.; Wu, R.R.; Bai, L.; Yan, L.; Magner, E.; Cosnier, S.; Lojou, E.; Zhu, Z.G.; Liu, A.H. Tackling the Challenges of Enzymatic (Bio)Fuel Cells. Chem. Rev. 2019, 119, 9509–9558. [Google Scholar] [CrossRef] [PubMed]
- Mazurenko, I.; de Poulpiquet, A.; Lojou, E. Recent developments in high surface area bioelectrodes for enzymatic fuel cells. Curr. Opin. Electrochem. 2017, 5, 74–84. [Google Scholar] [CrossRef]
- Mazurenko, I.; Hitaishi, V.P.; Lojou, E. Recent advances in surface chemistry of electrodes to promote direct enzymatic bioelectrocatalysis. Curr. Opin. Electrochem. 2020, 19, 113–121. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; de Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts 2018, 8, 192. [Google Scholar] [CrossRef]
- Iyer, P.V.; Ananthanarayan, L. Enzyme stability and stabilization—Aqueous and non-aqueous environment. Process Biochemistry 2008, 43, 1019–1032. [Google Scholar] [CrossRef]
- Balcao, V.M.; Vila, M. Structural and functional stabilization of protein entities: State-of-the-art. Adv. Drug Deliv. Rev. 2015, 93, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Polizzi, K.M.; Bommarius, A.S.; Broering, J.M.; Chaparro-Riggers, J.F. Stability of biocatalysts. Curr. Opin. Chem. Biol. 2007, 11, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Gianfreda, L.; Scarfi, M.R. Enzyme Stabilization—State-Of-The-Art. Mol. Cell. Biochem. 1991, 100, 97–128. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, V.; Bidmanova, S.; Koudelakova, T.; Prokop, Z.; Chaloupkova, R.; Damborsky, J. Strategies for Stabilization of Enzymes in Organic Solvents. ACS Catal. 2013, 3, 2823–2836. [Google Scholar] [CrossRef]
- Lalande, M.; Schwob, L.; Vizcaino, V.; Chirot, F.; Dugourd, P.; Schlatholter, T.; Poully, J.C. Direct Radiation Effects on the Structure and Stability of Collagen and Other Proteins. Chembiochem 2019. [Google Scholar] [CrossRef]
- Doster, W.; Settles, M. Protein-water displacement distributions. Biochim. Biophys. Acta Proteins Proteom. 2005, 1749, 173–186. [Google Scholar] [CrossRef]
- Miyawaki, O. Hydration state change of proteins upon unfolding in sugar solutions. Biochim. Biophys. Acta Proteins Proteom. 2007, 1774, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Ganjalikhany, M.R.; Ranjbar, B.; Hosseinkhani, S.; Khalifeh, K.; Hassani, L. Roles of trehalose and magnesium sulfate on structural and functional stability of firefly luciferase. J. Mol. Catal. B Enzym. 2010, 62, 127–132. [Google Scholar] [CrossRef]
- Haque, I.; Singh, R.; Moosavi-Movahedi, A.A.; Ahmad, F. Effect of polyol osmolytes on Delta G(D), the Gibbs energy of stabilisation of proteins at different pH values. Biophys. Chem. 2005, 117, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Bhat, R. Stabilization of yeast hexokinase A by polyol osmolytes: Correlation with the physicochemical properties of aqueous solutions. Biophys. Chem. 2006, 124, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, S.R.; Costa-Silva, T.A.; Pessoa, A.; Madeira, P.; Monteiro, G. Effect of osmolytes on the activity of anti-cancer enzyme L-Asparaginase II from Erwinia chrysanthemi. Process Biochem. 2019, 81, 123–131. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Q.P.; Lu, Q.Y.; Feng, B. Protection effect of polyols on Rhizopus chinensis lipase counteracting the deactivation from high pressure and high temperature treatment. Int. J. Biol. Macromol. 2019, 127, 555–562. [Google Scholar] [CrossRef]
- Pazhang, M.; Mardi, N.; Mehrnejad, F.; Chaparzadeh, N. The combinatorial effects of osmolytes and alcohols on the stability of pyrazinamidase: Methanol affects the enzyme stability through hydrophobic interactions and hydrogen bonds. Int. J. Biol. Macromol. 2018, 108, 1339–1347. [Google Scholar] [CrossRef]
- Khan, M.V.; Ishtikhar, M.; Rabbani, G.; Zaman, M.; Abdelhameed, A.S.; Khan, R.H. Polyols (Glycerol and Ethylene glycol) mediated amorphous aggregate inhibition and secondary structure restoration of metalloproteinase-conalbumin (ovotransferrin). Int. J. Biol. Macromol. 2017, 94, 290–300. [Google Scholar] [CrossRef]
- Kaushik, J.K.; Bhat, R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J. Biol. Chem. 2003, 278, 26458–26465. [Google Scholar] [CrossRef]
- Silva, C.; Martins, M.; Jing, S.; Fu, J.J.; Cavaco-Paulo, A. Practical insights on enzyme stabilization. Crit. Rev. Biotechnol. 2018, 38, 335–350. [Google Scholar] [CrossRef]
- Scharnagl, C.; Reif, M.; Friedrich, J. Local compressibilities of proteins: Comparison of optical experiments and simulations for horse heart cytochrome-c. Biophys. J. 2005, 89, 64–75. [Google Scholar] [CrossRef]
- Mangiagalli, M.; Carvalho, H.; Natalello, A.; Ferrario, V.; Pennati, M.L.; Barbiroli, A.; Lotti, M.; Pleiss, J.; Brocca, S. Diverse effects of aqueous polar co-solvents on Candida antarctica lipase B. Int. J. Biol. Macromol. 2020, 150, 930–940. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sarkar, S.; Gupta, S.; Banerjee, S.; Senapati, S.; Chakrabarty, R.; Gachhui, R. DMSO strengthens chitin deacetylase-chitin interaction: Physicochemical, kinetic, structural and catalytic insights. Carbohydr. Polym. 2019, 223. [Google Scholar] [CrossRef] [PubMed]
- Woll, A.K.; Hubbuch, J. Investigation of the reversibility of freeze/thaw stress-induced protein instability using heat cycling as a function of different cryoprotectants. Bioprocess Biosyst. Eng. 2020, 43, 1309–1327. [Google Scholar] [CrossRef] [PubMed]
- Dal Magro, L.; Kornecki, J.F.; Klein, M.P.; Rodrigues, R.C.; Fernandez-Lafuente, R. Stability/activity features of the main enzyme components of rohapect 10L. Biotechnol. Prog. 2019, 35. [Google Scholar] [CrossRef] [PubMed]
- Nolan, V.; Collin, A.; Rodriguez, C.; Perillo, M.A. Effect of Polyethylene Glycol-Induced Molecular Crowding on the Enzymatic Activity and Thermal Stability of beta-Galactosidase from Kluyveromyces lactis. J. Agric. Food Chem. 2020, 68, 8875–8882. [Google Scholar] [CrossRef] [PubMed]
- Jain, E.; Flanagan, M.; Sheth, S.; Patel, S.; Gan, Q.; Patel, B.; Montano, A.M.; Zustiak, S.P. Biodegradable polyethylene glycol hydrogels for sustained release and enhanced stability of rhGALNS enzyme. Drug Deliv. Transl. Res. 2020, 10, 1341–1352. [Google Scholar] [CrossRef]
- Sasahara, K.; McPhie, P.; Minton, A.P. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 2003, 326, 1227–1237. [Google Scholar] [CrossRef]
- Kuchler, A.; Yoshimoto, M.; Luginbuhl, S.; Mavelli, F.; Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409–420. [Google Scholar] [CrossRef]
- Li, J.H.; Zheng, H.Y.; Feng, C.J. Effect of Macromolecular Crowding on the FMN-Herne Intraprotein Electron Transfer in Inducible NO Synthase. Biochemistry 2019, 58, 3087–3096. [Google Scholar] [CrossRef]
- Shahid, S.; Hassan, M.I.; Islam, A.; Ahmad, F. Size-dependent studies of macromolecular crowding on the thermodynamic stability, structure and functional activity of proteins: In vitro and in silico approaches. Biochim. Et Biophys. Acta-Gen. Subj. 2017, 1861, 178–197. [Google Scholar] [CrossRef]
- Christiansen, A.; Wang, Q.; Samiotakis, A.; Cheung, M.S.; Wittung-Stafshede, P. Factors Defining Effects of Macromolecular Crowding on Protein Stability: An in Vitro/in Silico Case Study Using Cytochrome c. Biochemistry 2010, 49, 6519–6530. [Google Scholar] [CrossRef]
- Timasheff, S.N. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc. Natl. Acad. Sci. USA 2002, 99, 9721–9726. [Google Scholar] [CrossRef]
- Hasan, S.; Isar, M.; Naeem, A. Macromolecular crowding stabilises native structure of alpha-chymotrypsinogen-A against hexafluoropropanol-induced aggregates. Int. J. Biol. Macromol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Shahid, S.; Raina, N.; Ahmad, F.; Hassan, M.I.; Islam, A. Molecular and macromolecular crowding-induced stabilization of proteins: Effect of dextran and its building block alone and their mixtures on stability and structure of lysozyme. Int. J. Biol. Macromol. 2020, 150, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Baykal, E.; Vardar, G.; Attar, A.; Yapaoz, M.A. Complexes of glucose oxidase with chitosan and dextran possessing enhanced stability. Prep. Biochem. Biotechnol. 2020, 50, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Sen, P. Size-dependent macromolecular crowding effect on the thermodynamics of protein unfolding revealed at the single molecular level. Int. J. Biol. Macromol. 2019, 141, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Chowhan, R.K.; Singh, L.R. Macromolecular crowding: Macromolecules friend or foe. Biochim. Et Biophys. Acta-Gen. Subj. 2015, 1850, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, L.; Fresco, J.R. Stabilization of nucleic acid triplexes by high concentrations of sodium and ammonium salts follows the Hofmeister series. Biophys. Chem. 2003, 105, 681–699. [Google Scholar] [CrossRef]
- Okur, H.I.; Hladilkova, J.; Rembert, K.B.; Cho, Y.; Heyda, J.; Dzubiella, J.; Cremer, P.S.; Jungwirth, P. Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and Their Biological Functions. J. Phys. Chem. B 2017, 121, 1997–2014. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, F. Zur Lehre von der Wirkung der Salze. Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260. [Google Scholar] [CrossRef]
- Salis, A.; Ninham, B.W. Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited. Chem. Soc. Rev. 2014, 43, 7358–7377. [Google Scholar] [CrossRef]
- Ghobadi, R.; Divsalar, A. Enzymatic behavior of bovine liver catalase in aqueous medium of sugar based deep eutectic solvents. J. Mol. Liq. 2020, 310. [Google Scholar] [CrossRef]
- Usoltsev, D.; Sitnikova, V.; Kajava, A.; Uspenskaya, M. FTIR Spectroscopy Study of the Secondary Structure Changes in Human Serum Albumin and Trypsin under Neutral Salts. Biomolecules 2020, 10, 606. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, E.; Sedlakova, D.; Marek, J.; Hancar, J.; Garajova, K.; Zoldak, G. Ion-Specific Protein/Water Interface Determines the Hofmeister Effect on the Kinetic Stability of Glucose Oxidase. J. Phys. Chem. B 2019, 123, 7965–7973. [Google Scholar] [CrossRef] [PubMed]
- Garajova, K.; Balogova, A.; Dusekova, E.; Sedlakova, D.; Sedlak, E.; Varhac, R. Correlation of lysozyme activity and stability in the presence of Hofmeister series anions. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, M.; Sugihara, K.; Adachi, T.; Murata, K.; Shiraki, K.; Tsujimura, S. Effect of Electrolyte Ions on the Stability of Flavin Adenine Dinucleotide-Dependent Glucose Dehydrogenase. Chemelectrochem 2019, 6, 1028–1031. [Google Scholar] [CrossRef]
- Hani, F.M.; Cole, A.E.; Altman, E. The ability of salts to stabilize proteins in vivo or intracellularly correlates with the Hofmeister series of ions. Int. J. Biochem. Mol. Biol. 2019, 10, 23–31. [Google Scholar]
- Banerjee, S.; Arora, A.; Vijayaraghavan, R.; Patti, A.F. Extraction and crosslinking of bromelain aggregates for improved stability and reusability from pineapple processing waste. Int. J. Biol. Macromol. 2020, 158, 318–326. [Google Scholar] [CrossRef]
- Kulkarni, N.H.; Muley, A.B.; Bedade, D.K.; Singhal, R.S. Cross-linked enzyme aggregates of arylamidase from Cupriavidus oxalaticus ICTDB921: Process optimization, characterization, and application for mitigation of acrylamide in industrial wastewater. Bioprocess Biosyst. Eng. 2020, 43, 457–471. [Google Scholar] [CrossRef]
- Mayolo-Deloisa, K.; Gonzalez-Gonzalez, M.; Simental-Martinez, J.; Rito-Palomares, M. Aldehyde PEGylation of laccase from Trametes versicolor in route to increase its stability: Effect on enzymatic activity. J. Mol. Recognit. 2015, 28, 173–179. [Google Scholar] [CrossRef]
- El-Sayed, A.S.A.; Shindia, A.A.; Abou Zeid, A.A.; Yassin, A.M.; Sitohy, M.Z.; Sitohy, B. Aspergillus nidulans thermostable arginine deiminase-Dextran conjugates with enhanced molecular stability, proteolytic resistance, pharmacokinetic properties and anticancer activity. Enzym. Microb. Technol. 2019, 131. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Huang, H.F.; Fang, L.; Xue, L.; Zhang, T.; Kim, K.B.; Hou, S.R.; Zheng, F.; Zhan, C.G. PEGylation but Not Fc-Fusion Improves in Vivo Residence Time of a Thermostable Mutant of Bacterial Cocaine Esterase. Bioconjugate Chem. 2019, 30, 3021–3027. [Google Scholar] [CrossRef]
- Cummings, C.; Murata, H.; Koepsel, R.; Russell, A.J. Tailoring enzyme activity and stability using polymer-based protein engineering. Biomaterials 2013, 34, 7437–7443. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.F.; Kim, C.F.; Kwok, S.Y.; Tam, S.Y.; Chen, Y.W.; Chong, H.C.; Leung, S.L.; So, P.K.; Wong, K.Y.; Leung, Y.C.; et al. Mono-PEGylation of a Thermostable Arginine-Depleting Enzyme for the Treatment of Lung Cancer. Int. J. Mol. Sci. 2020, 21, 4234. [Google Scholar] [CrossRef] [PubMed]
- Kovaliov, M.; Zhang, B.; Konkolewicz, D.; Szczesniak, K.; Jurga, S.; Averick, S. Polymer grafting from a metallo-centered enzyme improves activity in non-native environments. Poly. Int. 2020. [Google Scholar] [CrossRef]
- Ritter, D.W.; Newton, J.M.; McShane, M.J. Modification of PEGylated enzyme with glutaraldehyde can enhance stability while avoiding intermolecular crosslinking. RSC Adv. 2014, 4, 28036–28040. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, G.; Ahmad, T.; Mansoor, S.; Kaur, B. Enzyme Engineering: Current Trends and Future Perspectives. Food Rev. Int. 2021. [Google Scholar] [CrossRef]
- Korendovych, I.V. Rational and Semirational Protein Design. Methods Mol. Biol. 2018, 1685, 15–23. [Google Scholar] [CrossRef]
- de Morais, M.A.B.; Polo, C.C.; Domingues, M.N.; Persinoti, G.F.; Pirolla, R.A.S.; de Souza, F.H.M.; Correa, J.B.D.; dos Santos, C.R.; Murakami, M.T. Exploring the Molecular Basis for Substrate Affinity and Structural Stability in Bacterial GH39 beta-Xylosidases. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Ito, Y.; Ikeuchi, A.; Imamura, C. Advanced evolutionary molecular engineering to produce thermostable cellulase by using a small but efficient library. Protein Eng. Des. Sel. 2013, 26, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Minges, H.; Schnepel, C.; Bottcher, D.; Weiss, M.S.; Spross, J.; Bornscheuer, U.T.; Sewald, N. Targeted Enzyme Engineering Unveiled Unexpected Patterns of Halogenase Stabilization. Chemcatchem 2020, 12, 818–831. [Google Scholar] [CrossRef]
- Bulut, H.; Yuksel, B.; Gul, M.; Eren, M.; Karatas, E.; Kara, N.; Yilmazer, B.; Kocyigit, A.; Labrou, N.E.; Binay, B. Conserved Amino Acid Residues that Affect Structural Stability ofCandida boidiniiFormate Dehydrogenase. Appl. Biochem. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Alvarez, C.E.; Bovdilova, A.; Hoppner, A.; Wolff, C.C.; Saigo, M.; Trajtenberg, F.; Zhang, T.; Buschiazzo, A.; Nagel-Steger, L.; Drincovich, M.F.; et al. Molecular adaptations of NADP-malic enzyme for its function in C-4 photosynthesis in grasses. Nat. Plants 2019, 5, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Yu, X.; Hu, X.; Zhang, H. Engineering Leuconostoc mesenteroides dextransucrase by inserting disulfide bridges for enhanced thermotolerance. Enzym. Microb. Technol. 2020, 139. [Google Scholar] [CrossRef]
- Gomez-Fernandez, B.J.; Risso, V.A.; Sanchez-Ruiz, J.M.; Alcalde, M. Consensus Design of an Evolved High-Redox Potential Laccase. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.X.; Lan, D.M.; Tan, X.Y.; Hollmann, F.; Bornscheuer, U.T.; Yang, B.; Wang, Y.H. How To Break the Janus Effect of H2O2 in Biocatalysis? Understanding Inactivation Mechanisms To Generate more Robust Enzymes. ACS Catal. 2019, 9, 2916–2921. [Google Scholar] [CrossRef]
- Liu, Q.; Xun, G.H.; Feng, Y. The state-of-the-art strategies of protein engineering for enzyme stabilization. Biotechnol. Adv. 2019, 37, 530–537. [Google Scholar] [CrossRef]
- Roger, M.; Castelle, C.; Guiral, M.; Infossi, P.; Lojou, E.; Giudici-Orticoni, M.T.; Ilbert, M. Mineral respiration under extreme acidic conditions: From a supramolecular organization to a molecular adaptation in Acidithiobacillus ferrooxidans. Biochem. Soc. Trans. 2012, 40, 1324–1329. [Google Scholar] [CrossRef]
- Hassan, N.; Rafiq, M.; Rehman, M.; Sajjad, W.; Hasan, F.; Abdullah, S. Fungi in acidic fire: A potential source of industrially important enzymes. Fungal Biol. Rev. 2019, 33, 58–71. [Google Scholar] [CrossRef]
- Jin, M.; Gai, Y.B.; Guo, X.; Hou, Y.P.; Zeng, R.Y. Properties and Applications of Extremozymes from Deep-Sea Extremophilic Microorganisms: A Mini Review. Mar. Drugs 2019, 17, 656. [Google Scholar] [CrossRef]
- Guiral, M.; Prunetti, L.; Aussignargues, C.; Ciaccafava, A.; Infossi, P.; Ilbert, M.; Lojou, E.; Giudici-Orticoni, M.T. The hyperthermophilic bacterium Aquifex aeolicus: From respiratory pathways to extremely resistant enzymes and biotechnological applications. Adv. Microb. Physiol. 2012, 61, 125–194. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, R.; Toma, W.; Yamazaki, K.; Akanuma, S. Ancestral sequence reconstruction produces thermally stable enzymes with mesophilic enzyme-like catalytic properties. Sci. Rep. 2020, 10, 15493. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, P.; Ghane, M.; Babaeekhou, L. Isolation and Characterization of Thermophilic Bacteria from Gavmesh Goli Hot Spring in Sabalan Geothermal Field, Iran: Thermomonas hydrothermalis and Bacillus altitudinis Isolates as a Potential Source of Thermostable Protease. Geomicrobiol. J. 2020. [Google Scholar] [CrossRef]
- Hilden, K.; Hakala, T.K.; Lundell, T. Thermotolerant and thermostable laccases. Biotechnol. Lett. 2009, 31, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.; Liebl, W. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 39–106. [Google Scholar] [CrossRef]
- Maiello, F.; Gallo, G.; Coelho, C.; Sucharski, F.; Hardy, L.; Wurtele, M. Crystal structure of Thermus thermophilus methylenetetrahydrofolate dehydrogenase and determinants of thermostability. PLoS ONE 2020, 15. [Google Scholar] [CrossRef]
- McManus, T.J.; Wells, S.A.; Walker, A.B. Salt bridge impact on global rigidity and thermostability in thermophilic citrate synthase. Phys. Biol. 2020, 17. [Google Scholar] [CrossRef]
- Xie, Z.H.; Zhai, L.X.; Meng, D.; Tian, Q.P.; Guan, Z.B.; Cai, Y.J.; Liao, X.R. Improving the catalytic thermostability ofBacillus altitudinisW3 omega-transaminase by proline substitutions. 3 Biotech 2020, 10. [Google Scholar] [CrossRef]
- Kargar, F.; Mortezavi, M.; Torkzadeh-Mahani, M.; Lotfi, S.; Shakeri, S. Evaluation of Luciferase Thermal Stability by Arginine Saturation in the Flexible Loops. Curr. Proteom. 2020, 17, 30–39. [Google Scholar] [CrossRef]
- Enguita, F.J.; Martins, L.O.; Henriques, A.O.; Carrondo, M.A. Crystal structure of a bacterial endospore coat component—A laccase with enhanced thermostability properties. J. Biol. Chem. 2003, 278, 19416–19425. [Google Scholar] [CrossRef]
- Brininger, C.; Spradlin, S.; Cobani, L.; Evilia, C. The more adaptive to change, the more likely you are to survive: Protein adaptation in extremophiles. Semin. Cell Dev. Biol. 2018, 84, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Cea, P.A.; Araya, G.; Vallejos, G.; Recabarren, R.; Alzate-Morales, J.; Babul, J.; Guixe, V.; Castro-Fernandez, V. Characterization of hydroxymethylpyrimidine phosphate kinase from mesophilic and thermophilic bacteria and structural insights into their differential thermal stability. Arch. Biochem. Biophys. 2020, 688. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, I.; Laage, D.; Stirnemann, G.; Sterpone, F. Differences in thermal structural changes and melting between mesophilic and thermophilic dihydrofolate reductase enzymes. Phys. Chem. Chem. Phys. 2020, 22, 18361–18373. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yang, X.Y.; Xu, Q.; Cui, H.L. Characterization of a novel Cu-containing dissimilatory nitrite reductase from the haloarchaeon Halorussus sp. YCN54. Extremophiles 2020, 24, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G.; Merlino, A. Molecular bases of protein halotolerance. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.M.; Gao, D.K.; Zhu, M.L.; Li, C.; Zhu, Z.L.; Wang, H.B.; Liu, W.D.; Tanokura, M.; Lu, F.P. Biochemical characterization and structural analysis of ulvan lyase from marine Alteromonas sp. reveals the basis for its salt tolerance. Int. J. Biol. Macromol. 2020, 147, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, Z.; Yang, H.; Zhang, T.; Niu, H.; Liu, D.; Wang, J.; Ying, H. Computation-aided rational design of a halophilic choline kinase for cytidine diphosphate choline production in high-salt condition. J. Biotechnol. 2019, 290, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yuan, M.; Zhang, X.; Liang, Q.; Yang, M.; Mou, H.; Zhu, C. A thermostable glucose oxidase from Aspergillus heteromophus CBS 117.55 with broad pH stability and digestive enzyme resistance. Protein Expr. Purif. 2020, 176. [Google Scholar] [CrossRef]
- Yi, Z.; Cai, Z.; Zeng, B.; Zeng, R.; Zhang, G. Identification and Characterization of a Novel Thermostable and Salt-Tolerant beta-1,3 Xylanase from Flammeovirga pacifica Strain WPAGA1. Biomolecules 2020, 10, 1287. [Google Scholar] [CrossRef]
- Zhu, T.; Li, R.; Sun, J.; Cui, Y.; Wu, B. Characterization and efficient production of a thermostable, halostable and organic solvent-stable cellulase from an oil reservoir. Int. J. Biol. Macromol. 2020, 159, 622–629. [Google Scholar] [CrossRef]
- Monsalve, K.; Mazurenko, I.; Gutierrez-Sanchez, C.; Ilbert, M.; Infossi, P.; Frielingsdorf, S.; Giudici-Orticoni, M.T.; Lenz, O.; Lojou, E. Impact of Carbon Nanotube Surface Chemistry on Hydrogen Oxidation by Membrane-Bound Oxygen-Tolerant Hydrogenases. Chemelectrochem 2016, 3, 2179–2188. [Google Scholar] [CrossRef]
- Oteri, F.; Baaden, M.; Lojou, E.; Sacquin-Mora, S. Multiscale Simulations Give Insight into the Hydrogen In and Out Pathways of NiFe -Hydrogenases from Aquifex aeolicus and Desulfovibrio fructosovorans. J. Phys. Chem. B 2014, 118, 13800–13811. [Google Scholar] [CrossRef]
- Coglitore, D.; Janot, J.M.; Balme, S. Protein at liquid solid interfaces: Toward a new paradigm to change the approach to design hybrid protein/solid-state materials. Adv. Colloid Interface Sci. 2019, 270, 278–292. [Google Scholar] [CrossRef]
- Arai, T.; Norde, W. The behavior of some model proteins at solid liquid interfaces 1. adsorption from single protein solutions. Adv. Colloid Interface Sci. 1990, 51, 1–15. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Hitaishi, V.P.; Mazurenko, I.; Harb, M.; Clement, R.; Taris, M.; Castano, S.; Duche, D.; Lecomte, S.; Ilbert, M.; de Poulpiquet, A.; et al. Electrostatic-Driven Activity, Loading, Dynamics, and Stability of a Redox Enzyme on Functionalized-Gold Electrodes for Bioelectrocatalysis. ACS Catal. 2018, 8, 12004–12014. [Google Scholar] [CrossRef]
- Bourassin, N.; Baaden, M.; Lojou, E.; Sacquin-Mora, S. Implicit Modeling of the Impact of Adsorption on Solid Surfaces for Protein Mechanics and Activity with a Coarse-Grained Representation. J. Phys. Chem. B 2020, 124, 8516–8523. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, D.; Sotres, J.; Barrantes, A.; Arnebrant, T.; Shleev, S. Interfacial Behavior and Activity of Laccase and Bilirubin Oxidase on Bare Gold Surfaces. Langmuir 2014, 30, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Kienle, D.F.; Falatach, R.M.; Kaar, J.L.; Schwartz, D.K. Correlating Structural and Functional Heterogeneity of Immobilized Enzymes. ACS Nano 2018, 12, 8091–8103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Ge, J.; Liu, Z. Enhanced Activity of Immobilized or Chemically Modified Enzymes. ACS Catal. 2015, 5, 4503–4513. [Google Scholar] [CrossRef]
- Garcia-Garcia, P.; Guisan, J.M.; Fernandez-Lorente, G. A mild intensity of the enzyme-support multi-point attachment promotes the optimal stabilization of mesophilic multimeric enzymes: Amine oxidase from Pisum sativum. J. Biotechnol. 2020, 318, 39–44. [Google Scholar] [CrossRef]
- Weltz, J.S.; Kienle, D.F.; Schwartz, D.K.; Kaar, J.L. Reduced Enzyme Dynamics upon Multipoint Covalent Immobilization Leads to Stability-Activity Trade-off. J. Am. Chem. Soc. 2020, 142, 3463–3471. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Mazurenko, I.; Vengasseril Murali, A.; de Poulpiquet, A.; Coustillier, G.; Delaporte, P.; Lojou, E. Nanosecond Laser-Fabricated Monolayer of Gold Nanoparticles on ITO for Bioelectrocatalysis. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Sigurdardottir, S.B.; Lehmann, J.; Grivel, J.C.; Zhang, W.J.; Kaiser, A.; Pinelo, M. Alcohol dehydrogenase on inorganic powders: Zeta potential and particle agglomeration as main factors determining activity during immobilization. Colloids Surf. B-Biointerfaces 2019, 175, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.J.; Algar, W.R.; Malanoski, A.P.; Ancona, M.G.; Medintz, I.L. Understanding enzymatic acceleration at nanoparticle interfaces: Approaches and challenges. Nano Today 2014, 9, 102–131. [Google Scholar] [CrossRef]
- Sharifi, M.; Sohrabi, M.J.; Hosseinali, S.H.; Hasan, A.; Kani, P.H.; Talaei, A.J.; Karim, A.Y.; Nanakali, N.M.Q.; Salihi, A.; Aziz, F.M.; et al. Enzyme immobilization onto the nanomaterials: Application in enzyme stability and prodrug-activated cancer therapy. Int. J. Biol. Macromol. 2020, 143, 665–676. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Z.; Wang, J.; Wu, H.; Lin, C.-T.; Lin, Y. Apoferritin nanoparticle: A novel and biocompatible carrier for enzyme immobilization with enhanced activity and stability. J. Mater. Chem. 2011, 21, 17468–17475. [Google Scholar] [CrossRef]
- Yang, Y.L.; Zhu, G.X.; Wang, G.C.; Li, Y.L.; Tang, R.K. Robust glucose oxidase with a Fe3O4@C-silica nanohybrid structure. J. Mater. Chem. B 2016, 4, 4726–4731. [Google Scholar] [CrossRef] [PubMed]
- Suo, H.B.; Gao, Z.; Xu, L.L.; Xu, C.; Yu, D.H.; Xiang, X.R.; Huang, H.; Hu, Y. Synthesis of functional ionic liquid modified magnetic chitosan nanoparticles for porcine pancreatic lipase immobilization. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 96, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Husain, Q.; Qayyum, S.; Azam, A. Designing and surface modification of zinc oxide nanoparticles for biomedical applications. Food Chem. Toxicol. 2011, 49, 2107–2115. [Google Scholar] [CrossRef]
- Grewal, J.; Ahmad, R.; Khare, S.K. Development of cellulase-nanoconjugates with enhanced ionic liquid and thermal stability for in situ lignocellulose saccharification. Bioresour. Technol. 2017, 242, 236–243. [Google Scholar] [CrossRef]
- Shang, W.; Nuffer, J.H.; Dordick, J.S.; Siegel, R.W. Unfolding of ribonuclease A on silica nanoparticle surfaces. Nano Lett. 2007, 7, 1991–1995. [Google Scholar] [CrossRef]
- Song, Y.; Zhong, D.; Luo, D.; Huang, M.; Huang, Z.; Tan, H.; Sun, L.; Wang, L. Effect of particle size on conformation and enzymatic activity of EcoRI adsorbed on CdS nanoparticles. Colloids Surf. B-Biointerfaces 2014, 114, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Asuri, P.; Karajanagi, S.S.; Yang, H.C.; Yim, T.J.; Kane, R.S.; Dordick, J.S. Increasing protein stability through control of the nanoscale environment. Langmuir 2006, 22, 5833–5836. [Google Scholar] [CrossRef] [PubMed]
- Vertegel, A.A.; Siegel, R.W.; Dordick, J.S. Silica nanoparticle size influences the structure and enzymatic activity of adsorbed lysozyme. Langmuir 2004, 20, 6800–6807. [Google Scholar] [CrossRef] [PubMed]
- Gagner, J.E.; Lopez, M.D.; Dordick, J.S.; Siegel, R.W. Effect of gold nanoparticle morphology on adsorbed protein structure and function. Biomaterials 2011, 32, 7241–7252. [Google Scholar] [CrossRef]
- Bilal, M.; Asgher, M.; Cheng, H.R.; Yan, Y.J.; Iqbal, H.M.N. Multi-point enzyme immobilization, surface chemistry, and novel platforms: A paradigm shift in biocatalyst design. Crit. Rev. Biotechnol. 2019, 39, 202–219. [Google Scholar] [CrossRef]
- Vila, N.; Andre, E.; Ciganda, R.; Ruiz, J.; Astruc, D.; Walcarius, A. Molecular Sieving with Vertically Aligned Mesoporous Silica Films and Electronic Wiring through Isolating Nanochannels. Chem. Mater. 2016, 28, 2511–2514. [Google Scholar] [CrossRef]
- Abdelbar, M.F.; Shams, R.S.; Morsy, O.M.; Hady, M.A.; Shoueir, K.; Abdelmonem, R. Highly ordered functionalized mesoporous silicate nanoparticles reinforced poly (lactic acid) gatekeeper surface for infection treatment. Int. J. Biol. Macromol. 2020, 156, 858–868. [Google Scholar] [CrossRef]
- Rodriguez-Abetxuko, A.; Sanchez-deAlcazar, D.; Munumer, P.; Beloqui, A. Tunable Polymeric Scaffolds for Enzyme Immobilization. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Inagaki, M.; Toyoda, M.; Soneda, Y.; Tsujimura, S.; Morishita, T. Templated mesoporous carbons: Synthesis and applications. Carbon 2016, 107, 448–473. [Google Scholar] [CrossRef]
- Ruff, A.; Szczesny, J.; Markovic, N.; Conzuelo, F.; Zacarias, S.; Pereira, I.A.C.; Lubitz, W.; Schuhmann, W. A fully protected hydrogenase/polymer-based bioanode for high-performance hydrogen/glucose biofuel cells. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Stine, K.J. Enzyme Immobilization on Nanoporous Gold: A Review. Biochem. Insights 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Baruch-Shpigler, Y.; Avnir, D. Enzymes in a golden cage. Chem. Sci. 2020, 11, 3965–3977. [Google Scholar] [CrossRef]
- Ge, J.; Lei, J.D.; Zare, R.N. Protein-inorganic hybrid nanoflowers. Nat. Nanotechnol. 2012, 7, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Wied, P.; Carraro, F.; Sumby, C.J.; Nidetzky, B.; Tsung, C.-K.; Falcaro, P.; Doonan, C.J. Metal-Organic Framework-Based Enzyme Biocomposites. Chem. Rev. 2021. [Google Scholar] [CrossRef]
- Lian, X.Z.; Fang, Y.; Joseph, E.; Wang, Q.; Li, J.L.; Banerjee, S.; Lollar, C.; Wang, X.; Zhou, H.C. Enzyme-MOF (metal-organic framework) composites. Chem. Soc. Rev. 2017, 46, 3386–3401. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Moon, S.Y.; Guelta, M.A.; Harvey, S.P.; Hupp, J.T.; Farha, O.K. Encapsulation of a Nerve Agent Detoxifying Enzyme by a Mesoporous Zirconium Metal-Organic Framework Engenders Thermal and Long-Term Stability. J. Am. Chem. Soc. 2016, 138, 8052–8055. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Zhong, L.; Hou, Y.; Jia, S.R.; Cui, J.D. Acid-resistant enzyme@MOF nanocomposites with mesoporous silica shells for enzymatic applications in acidic environments. J. Biotechnol. 2019, 306, 54–61. [Google Scholar] [CrossRef]
- He, H.M.; Han, H.B.; Shi, H.; Tian, Y.Y.; Sun, F.X.; Song, Y.; Li, Q.S.; Zhu, G.S. Construction of Thermophilic Lipase-Embedded Metal Organic Frameworks via Biomimetic Mineralization: A Biocatalyst for Ester Hydrolysis and Kinetic Resolution. ACS Appl. Mater. Interfaces 2016, 8, 24517–24524. [Google Scholar] [CrossRef]
- Wu, X.L.; Yang, C.; Ge, J.; Liu, Z. Polydopamine tethered enzyme/metal-organic framework composites with high stability and reusability. Nanoscale 2015, 7, 18883–18886. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Yang, C.; Ge, J. Green synthesis of enzyme/metal-organic framework composites with high stability in protein denaturing solvents. Bioresour. Bioprocess. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Knedel, T.O.; Ricklefs, E.; Schlusener, C.; Urlacher, V.B.; Janiak, A.C. Laccase Encapsulation in ZIF-8 Metal-Organic Framework Shows Stability Enhancement and Substrate Selectivity. Chemistryopen 2019, 8, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; He, W.T.; Shen, H.; Zhou, Z.X.; Li, M.Q.; Su, P.; Yang, Y. Construction of multiple enzyme metal-organic frameworks biocatalyst via DNA scaffold: A promising strategy for enzyme encapsulation. Chem. Eng. J. 2019, 363, 174–182. [Google Scholar] [CrossRef]
- Gao, Y.; Doherty, C.M.; Mulet, X. A Systematic Study of the Stability of Enzyme/Zeolitic Imidazolate Framework-8 Composites in Various Biologically Relevant Solutions. Chemistryselect 2020, 5, 13766–13774. [Google Scholar] [CrossRef]
- Nadar, S.S.; Rathod, V.K. Magnetic-metal organic framework (magnetic-MOF): A novel platform for enzyme immobilization and nanozyme applications. Int. J. Biol. Macromol. 2018, 120, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Bagheri, A.R.; Aramesh, N.; Gul, I.; Franco, M.; Almulaiky, Y.Q.; Bilal, M. Covalent organic frameworks as emerging host platforms for enzyme immobilization and robust biocatalysis—A review. Int. J. Biol. Macromol. 2021, 167, 502–515. [Google Scholar] [CrossRef]
- Chapman, R.; Stenzel, M.H. All Wrapped up: Stabilization of Enzymes within Single Enzyme Nanoparticles. J. Am. Chem. Soc. 2019, 141, 2754–2769. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Luo, Z.G.; Lu, X.X. Construction of Novel Enzyme-Graphene Oxide Catalytic Interface with Improved Enzymatic Performance and Its Assembly Mechanism. ACS Appl. Mater. Interfaces 2019, 11, 11349–11359. [Google Scholar] [CrossRef]
- Wu, L.; Lu, X.; Niu, K.; Ma, D.; Chen, J. Tyrosinase nanocapsule based nano-biosensor for ultrasensitive and rapid detection of bisphenol A with excellent stability in different application scenarios. Biosens. Bioelectron. 2020, 165. [Google Scholar] [CrossRef]
- Shen, X.T.; Yang, M.; Cui, C.X.; Cao, H. In situ immobilization of glucose oxidase and catalase in a hybrid interpenetrating polymer network by 3D bioprinting and its application. Colloids Surf. A Physicochem. Eng. Asp. 2019, 568, 411–418. [Google Scholar] [CrossRef]
- Zhang, S.T.; Wu, Z.F.; Chen, G.; Wang, Z. An Improved Method to Encapsulate Laccase from Trametes versicolor with Enhanced Stability and Catalytic Activity. Catalysts 2018, 8, 286. [Google Scholar] [CrossRef]
- Wan, L.; Chen, Q.S.; Liu, J.B.; Yang, X.H.; Huang, J.; Li, L.; Guo, X.; Zhang, J.; Wang, K.M. Programmable Self-Assembly of DNA-Protein Hybrid Hydrogel for Enzyme Encapsulation with Enhanced Biological Stability. Biomacromolecules 2016, 17, 1543–1550. [Google Scholar] [CrossRef]
- Liang, H.; Jiang, S.H.; Yuan, Q.P.; Li, G.F.; Wang, F.; Zhang, Z.J.; Liu, J.W. Co-immobilization of multiple enzymes by metal coordinated nucleotide hydrogel nanofibers: Improved stability and an enzyme cascade for glucose detection. Nanoscale 2016, 8, 6071–6078. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kiyota, Y.; Miyazaki, M. Techniques for Preparation of Cross-Linked Enzyme Aggregates and Their Applications in Bioconversions. Catalysts 2018, 8, 174. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soci. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; van Pelt, S.; Kanbak-Aksu, S.; Rasmussen, J.A.; Janssen, M.H.A. Cross-Linked Enzyme Aggregates (CLEAs) in Organic Synthesis. Aldrichimica Acta 2013, 46, 81–93. [Google Scholar]
- Qian, J.; Zhao, C.; Ding, J.; Chen, Y.; Guo, H. Preparation of nano-enzyme aggregates by crosslinking lipase with sodium tripolyphosphate. Process Biochem. 2020, 97, 19–26. [Google Scholar] [CrossRef]
- Nadar, S.S.; Muley, A.B.; Ladole, M.R.; Joshi, P.U. Macromolecular cross-linked enzyme aggregates (M-CLEAs) of alpha-amylase. Int. J. Biol. Macromol. 2016, 84, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, D.; Yin, L.; Wang, F. Preparation, activity and structure of cross-linked enzyme aggregates (CLEAs) with nanoparticle. Enzym. Microb. Technol. 2017, 107, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.Z.; Li, C.H.; Jiao, X.B.; Jia, S.R.; Jiang, Y.J.; Bilal, M.; Cui, J.D. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Monteiro, R.R.C.; dos Santos, J.C.S.; Alcantara, A.R.; Fernandez-Lafuente, R. Enzyme-Coated Micro-Crystals: An Almost Forgotten but Very Simple and Elegant Immobilization Strategy. Catalysts 2020, 10, 891. [Google Scholar] [CrossRef]
- Zerva, A.; Pentari, C.; Topakas, E. Crosslinked Enzyme Aggregates (CLEAs) of Laccases from Pleurotus citrinopileatus Induced in Olive Oil Mill Wastewater (OOMW). Molecules 2020, 25, 2221. [Google Scholar] [CrossRef]
- Kopp, W.; da Costa, T.P.; Pereira, S.C.; Jafelicci, M., Jr.; Giordano, R.C.; Marques, R.F.C.; Araujo-Moreira, F.M.; Giordano, R.L.C. Easily handling penicillin G acylase magnetic cross-linked enzymes aggregates: Catalytic and morphological studies. Process Biochem. 2014, 49, 38–46. [Google Scholar] [CrossRef]
- Cui, J.D.; Zhang, S.; Sun, L.M. Cross-Linked Enzyme Aggregates of Phenylalanine Ammonia Lyase: Novel Biocatalysts for Synthesis of L-Phenylalanine. Appl. Biochem. Biotechnol. 2012, 167, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.D.; Jia, S.R. Optimization protocols and improved strategies of cross-linked enzyme aggregates technology: Current development and future challenges. Crit. Rev. Biotechnol. 2015, 35, 15–28. [Google Scholar] [CrossRef]
- Tandjaoui, N.; Tassist, A.; Abouseoud, M.; Couvert, A.; Amrane, A. Preparation and characterization of cross-linked enzyme aggregates (CLEAs) of Brassica rapa peroxidase. Biocatal. Agric. Biotechnol. 2015, 4, 208–213. [Google Scholar] [CrossRef]
- Dinh, T.H.; Jang, N.Y.; McDonald, K.A.; Won, K. Cross-linked aggregation of glutamate decarboxylase to extend its activity range toward alkaline pH. J. Chem. Technol. Biotechnol. 2015, 90, 2100–2105. [Google Scholar] [CrossRef]
- Gupta, K.; Jana, A.K.; Kumar, S.; Jana, M.M. Solid state fermentation with recovery of Amyloglucosidase from extract by direct immobilization in cross linked enzyme aggregate for starch hydrolysis. Agric. Biotechnol. 2015, 4, 486–492. [Google Scholar] [CrossRef]
- Lucena, G.N.; dos Santos, C.C.; Pinto, G.C.; Piazza, R.D.; Guedes, W.N.; Jafelicci Junior, M.; de Paula, A.V.; Marques, R.F.C. Synthesis and characterization of magnetic cross-linked enzyme aggregate and its evaluation of the alternating magnetic field (AMF) effects in the catalytic activity. J. Magn. Magn. Mater. 2020, 516. [Google Scholar] [CrossRef]
- Primozic, M.; Kravanja, G.; Knez, Z.; Crnjac, A.; Leitgeb, M. Immobilized laccase in the form of (magnetic) cross-linked enzyme aggregates for sustainable diclofenac (bio) degradation. J. Clean. Prod. 2020, 275. [Google Scholar] [CrossRef]
- Xu, M.Q.; Wang, S.S.; Li, L.N.; Gao, J.; Zhang, Y.W. Combined Cross-Linked Enzyme Aggregates as Biocatalysts. Catalysts 2018, 8, 460. [Google Scholar] [CrossRef]
- Xu, M.Q.; Li, F.L.; Yu, W.Q.; Li, R.F.; Zhang, Y.W. Combined cross-linked enzyme aggregates of glycerol dehydrogenase and NADH oxidase for high efficiency in situ NAD(+) regeneration. Int. J. Biol. Macromol. 2020, 144, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Sellami, K.; Couvert, A.; Nasrallah, N.; Maachi, R.; Tandjaoui, N.; Abouseoud, M.; Amrane, A. Bio-based and cost effective method for phenolic compounds removal using cross-linked enzyme aggregates. J. Hazard. Mater. 2021, 403. [Google Scholar] [CrossRef] [PubMed]
- Mai-Lan, P.; Polakovic, M. Microbial cell surface display of oxidoreductases: Concepts and applications. Int. J. Biol. Macromol. 2020, 165, 835–841. [Google Scholar] [CrossRef]
- Ugwuodo, C.J.; Nwagu, T.N. Stabilizing enzymes by immobilization on bacterial spores: A review of literature. Int. J. Biol. Macromol. 2021, 166, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-Y.; Lin, C.-H.; Hsu, S.-Y.; Stewart, G.C. A Bacillus Spore-Based Display System for Bioremediation of Atrazine. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Tang, X.J.; Liang, B.; Yi, T.Y.; Manco, G.; Palchetti, I.; Liu, A.H. Cell surface display of organophosphorus hydrolase for sensitive spectrophotometric detection of p-nitrophenol substituted organophosphates. Enzym. Microb. Technol. 2014, 55, 107–112. [Google Scholar] [CrossRef]
- Padkina, M.V.; Sambuk, E.V. Prospects for the Application of Yeast Display in Biotechnology and Cell Biology (Review). Appl. Biochem. Microbiol. 2018, 54, 337–351. [Google Scholar] [CrossRef]
- Chen, H.; Chen, Z.; Ni, Z.; Tian, R.; Zhang, T.; Jia, J.; Chen, K.; Yang, S. Display of Thermotoga maritima MSB8 nitrilase on the spore surface of Bacillus subtilis using out coat protein CotG as the fusion partner. J. Mol. Catal. B Enzym. 2016, 123, 73–80. [Google Scholar] [CrossRef]
- Lozancic, M.; Hossain, A.S.; Mrsa, V.; Teparic, R. Surface Display-An Alternative to Classic Enzyme Immobilization. Catalysts 2019, 9, 728. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. 1. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Richardson, D.J.; Butt, J.N.; Fredrickson, J.K.; Zachara, J.M.; Shi, L.; Edwards, M.J.; White, G.; Baiden, N.; Gates, A.J.; Marritt, S.J.; et al. The porin-cytochrome’ model for microbe-to-mineral electron transfer. Mol. Microbiol. 2012, 85, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Hitaishi, V.P.; Clement, R.; Quattrocchi, L.; Parent, P.; Duche, D.; Zuily, L.; Ilbert, M.; Lojou, E.; Mazurenko, I. Interplay between Orientation at Electrodes and Copper Activation of Thermus thermophilus Laccase for O-2 Reduction. J. Am. Chem. Soc. 2020, 142, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Mazurenko, I.; Monsalve, K.; Rouhana, J.; Parent, P.; Laffon, C.; Le Goff, A.; Szunerits, S.; Boukherroub, R.; Giudici-Orticoni, M.T.; Mano, N.; et al. How the Intricate Interactions between Carbon Nanotubes and Two Bilirubin Oxidases Control Direct and Mediated O-2 Reduction. ACS Appl. Mater. Interfaces 2016, 8, 23074–23085. [Google Scholar] [CrossRef] [PubMed]
- Oteri, F.; Ciaccafava, A.; de Poulpiquet, A.; Baaden, M.; Lojou, E.; Sacquin-Mora, S. The weak, fluctuating, dipole moment of membrane-bound hydrogenase from Aquifex aeolicus accounts for its adaptability to charged electrodes. Phys. Chem. Chem. Phys. 2014, 16, 11318–11322. [Google Scholar] [CrossRef]
- Mazurenko, I.; Monsalve, K.; Infossi, P.; Giudici-Orticoni, M.T.; Topin, F.; Mano, N.; Lojou, E. Impact of substrate diffusion and enzyme distribution in 3D-porous electrodes: A combined electrochemical and modelling study of a thermostable H-2/O-2 enzymatic fuel cell. Energy Environ. Sci. 2017, 10, 1966–1982. [Google Scholar] [CrossRef]
- Atalah, J.; Zhou, Y.; Espina, G.; Blamey, J.M.; Ramasamy, R.P. Improved stability of multicopper oxidase-carbon nanotube conjugates using a thermophilic laccase. Catal. Sci. Technol. 2018, 8, 1272–1276. [Google Scholar] [CrossRef]
- Zafar, M.N.; Aslam, I.; Ludwig, R.; Xu, G.B.; Gorton, L. An efficient and versatile membraneless bioanode for biofuel cells based on Corynascus thermophilus cellobiose dehydrogenase. Electrochim. Acta 2019, 295, 316–324. [Google Scholar] [CrossRef]
- Malinowski, S.; Wardak, C.; Jaroszynska-Wolinska, J.; Herbert, P.A.F.; Panek, R. Cold Plasma as an Innovative Construction Method of Voltammetric Biosensor Based on Laccase. Sensors 2018, 18, 4086. [Google Scholar] [CrossRef] [PubMed]
- Bathinapatla, A.; Kanchi, S.; Sabela, M.I.; Ling, Y.C.; Bisetty, K.; Inamuddin. Experimental and Computational Studies of a Laccase Immobilized ZnONPs/GO-Based Electrochemical Enzymatic Biosensor for the Detection of Sucralose in Food Samples. Food Anal. Methods 2020, 13, 2014–2027. [Google Scholar] [CrossRef]
- Aleksejeva, O.; Mateljak, I.; Ludwig, R.; Alcalde, M.; Shleev, S. Electrochemistry of a high redox potential laccase obtained by computer-guided mutagenesis combined with directed evolution. Electrochem. Commun. 2019, 106. [Google Scholar] [CrossRef]
- Mohtar, L.G.; Aranda, P.; Messina, G.A.; Nazaren, M.A.; Pereira, S.V.; Raba, J.; Bertolino, F.A. Amperometric biosensor based on laccase immobilized onto a nanostructured screen-printed electrode for determination of polyphenols in propolis. Microchem. J. 2019, 144, 13–18. [Google Scholar] [CrossRef]
- Chen, T.; Xu, Y.H.; Peng, Z.; Li, A.H.; Liu, J.Q. Simultaneous Enhancement of Bioactivity and Stability of Laccase by Cu2+/PAA/PPEGA Matrix for Efficient Biosensing and Recyclable Decontamination of Pyrocatechol. Anal. Chem. 2017, 89, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, S.; Ramaraj, S.K.; Chen, S.M.; Yang, T.C.K.; Yi-Fan, P.; Chen, T.W.; Velusamy, V.; Selvam, S. A novel Laccase Biosensor based on Laccase immobilized Graphene-Cellulose Microfiber Composite modified Screen-Printed Carbon Electrode for Sensitive Determination of Catechol. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Lee, J.Y.Y.; Elouarzaki, K.; Sabharwal, H.S.; Fisher, A.C.; Lee, J.-M. A hydrogen/oxygen hybrid biofuel cell comprising an electrocatalytically active nanoflower/laccase-based biocathode. Catal. Sci. Technol. 2020, 10, 6235–6243. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Li, X.; Li, D.W.; Wei, Q.F. A laccase based biosensor on AuNPs-MoS2 modified glassy carbon electrode for catechol detection. Colloids Surf. B Biointerfaces 2020, 186. [Google Scholar] [CrossRef]
- El Ichi-Ribault, S.; Zebda, A.; Tingry, S.; Petit, M.; Suherman, A.L.; Boualam, A.; Cinquin, P.; Martin, D.K. Performance and stability of chitosan-MWCNTs-laccase biocathode: Effect of MWCNTs surface charges and ionic strength. J. Electroanal. Chem. 2017, 799, 26–33. [Google Scholar] [CrossRef]
- Yang, Y.; Zeng, H.; Huo, W.S.; Zhang, Y.H. Direct Electrochemistry and Catalytic Function on Oxygen Reduction Reaction of Electrodes Based on Two Kinds of Magnetic Nano-particles with Immobilized Laccase Molecules. J. Inorg. Organomet. Polym. Mater. 2017, 27, 201–214. [Google Scholar] [CrossRef]
- Zappi, D.; Masci, G.; Sadun, C.; Tortolini, C.; Antonelli, M.L.; Bollella, P. Evaluation of new cholinium-amino acids based room temperature ionic liquids (RTILs) as immobilization matrix for electrochemical biosensor development: Proof-of-concept with Trametes Versicolor laccase. Microchem. J. 2018, 141, 346–352. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, Z.Y.; Zhou, J.; Fang, Y.; Wu, H.; Xin, F.X.; Zhang, W.M.; Ma, J.F.; Xu, N.; He, A.Y.; et al. Amperometric Biosensors Based on Recombinant Bacterial Laccase CotA for Hydroquinone Determination. Electroanalysis 2020, 32, 142–148. [Google Scholar] [CrossRef]
- Xu, R.; Cui, J.Y.; Tang, R.Z.; Li, F.T.; Zhang, B.R. Removal of 2,4,6-trichlorophenol by laccase immobilized on nano-copper incorporated electrospun fibrous membrane-high efficiency, stability and reusability. Chem. Eng. J. 2017, 326, 647–655. [Google Scholar] [CrossRef]
- Wang, A.Q.; Ding, Y.P.; Li, L.; Duan, D.D.; Mei, Q.W.; Zhuang, Q.; Cui, S.Q.; He, X.Y. A novel electrochemical enzyme biosensor for detection of 17 beta-estradiol by mediated electron-transfer system. Talanta 2019, 192, 478–485. [Google Scholar] [CrossRef]
- Yashas, S.R.; Sandeep, S.; Shivakumar, B.P.; Swamy, N.K. A matrix of perovskite micro-seeds and polypyrrole nanotubes tethered laccase/graphite biosensor for sensitive quantification of 2,4-dichlorophenol in wastewater. Anal. Methods 2019, 11, 4511–4519. [Google Scholar] [CrossRef]
- Castrovilli, M.C.; Bolognesi, P.; Chiarinelli, J.; Avaldi, L.; Cartoni, A.; Calandra, P.; Tempesta, E.; Giardi, M.T.; Antonacci, A.; Arduini, F.; et al. Electrospray deposition as a smart technique for laccase immobilisation on carbon black-nanomodified screen-printed electrodes. Biosens. Bioelectron. 2020, 163. [Google Scholar] [CrossRef]
- Lee, Y.G.; Liao, B.X.; Weng, Y.C. Ascorbic acid sensor using a PVA/laccase-Au-NPs/Pt electrode. RSC Adv. 2018, 8, 37872–37879. [Google Scholar] [CrossRef]
- Lou, C.Q.; Jing, T.; Zhou, J.Y.; Tian, J.Z.; Zheng, Y.J.; Wang, C.; Zhao, Z.Y.; Lin, J.; Liu, H.; Zhao, C.Q.; et al. Laccase immobilized polyaniline/magnetic graphene composite electrode for detecting hydroquinone. Int. J. Biol. Macromol. 2020, 149, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xu, Y.H.; Wei, S.; Li, A.H.; Huang, L.; Liu, J.Q. A signal amplification system constructed by bi-enzymes and bi-nanospheres for sensitive detection of norepinephrine and miRNA. Biosens. Bioelectron. 2019, 124, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Mazlan, S.Z.; Lee, Y.H.; Abu Hanifah, S. A New Laccase Based Biosensor for Tartrazine. Sensors 2017, 17, 2859. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.T.; Salamanca-Neto, C.A.R.; Svorc, L.; Schirmann, J.G.; Barbosa-Dekker, A.M.; Dekker, R.F.H.; Sartori, E.R. Laccase from Botryosphaeria rhodina MAMB-05 as a biological component in electrochemical biosensing devices. Anal. Methods 2019, 11, 717–720. [Google Scholar] [CrossRef]
- Sedenho, G.C.; Hassan, A.; Macedo, L.J.A.; Crespilho, F.N. Stabilization of bilirubin oxidase in a biogel matrix for high-performance gas diffusion electrodes. J. Power Sources 2021, 482. [Google Scholar] [CrossRef]
- Gentil, S.; Carriere, M.; Cosnier, S.; Gounel, S.; Mano, N.; Le Goff, A. Direct Electrochemistry of Bilirubin Oxidase from Magnaporthe orizae on Covalently-Functionalized MWCNT for the Design of High-Performance Oxygen-Reducing Biocathodes. Chem. A Eur. J. 2018, 24, 8404–8408. [Google Scholar] [CrossRef]
- Szczesny, J.; Markovic, N.; Conzuelo, F.; Zacarias, S.; Pereira, I.A.C.; Lubitz, W.; Plumere, N.; Schuhmann, W.; Ruff, A. A gas breathing hydrogen/air biofuel cell comprising a redox polymer/hydrogenase-based bioanode. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Markovic, N.; Conzuelo, F.; Szczesny, J.; Garcia, M.B.G.; Santos, D.H.; Ruff, A.; Schuhmann, W. An Air-breathing Carbon Cloth-based Screen-printed Electrode for Applications in Enzymatic Biofuel Cells. Electroanalysis 2019, 31, 217–221. [Google Scholar] [CrossRef]
- Trifonov, A.; Stemmer, A.; Tel-Vered, R. Carbon-coated magnetic nanoparticles as a removable protection layer extending the operation lifetime of bilirubin oxidase-based bioelectrode. Bioelectrochemistry 2021, 137, 107640. [Google Scholar] [CrossRef] [PubMed]
- Al-Lolage, F.A.; Bartlett, P.N.; Gounel, S.; Staigre, P.; Mano, N. Site-Directed Immobilization of Bilirubin Oxidase for Electrocatalytic Oxygen Reduction. ACS Catal. 2019, 9, 2068–2078. [Google Scholar] [CrossRef]
- Takimoto, D.; Tsujimura, S. Oxygen Reduction Reaction Activity and Stability of Electrochemically Deposited Bilirubin Oxidase. Chem. Lett. 2018, 47, 1269–1271. [Google Scholar] [CrossRef]
- Tsujimura, S.; Oyama, M.; Funabashi, H.; Ishii, S. Effects of pore size and surface properties of MgO-templated carbon on the performance of bilirubin oxidase-modified oxygen reduction reaction cathode. Electrochim. Acta 2019, 322. [Google Scholar] [CrossRef]
- Tang, J.; Yan, X.M.; Huang, W.; Engelbrekt, C.; Duus, J.O.; Ulstrup, J.; Xiao, X.X.; Zhang, J.D. Bilirubin oxidase oriented on novel type three-dimensional biocathodes with reduced graphene aggregation for biocathode. Biosens. Bioelectron. 2020, 167. [Google Scholar] [CrossRef]
- Walgama, C.; Pathiranage, A.; Akinwale, M.; Montealegre, R.; Niroula, J.; Echeverria, E.; McIlroy, D.N.; Harriman, T.A.; Lucca, D.A.; Krishnan, S. Buckypaper–Bilirubin Oxidase Biointerface for Electrocatalytic Applications: Buckypaper Thickness. ACS Appl. Bio Mater. 2019, 2, 2229–2236. [Google Scholar] [CrossRef]
- Zhang, L.; Carucci, C.; Reculusa, S.; Goudeau, B.; Lefrancois, P.; Gounel, S.; Mano, N.; Kuhn, A. Rational Design of Enzyme-Modified Electrodes for Optimized Bioelectrocatalytic Activity. Chemelectrochem 2019, 6, 4980–4984. [Google Scholar] [CrossRef]
- Mukha, D.; Cohen, Y.; Yehezkeli, O. Bismuth Vanadate/Bilirubin Oxidase Photo(bio)electrochemical Cells for Unbiased, Light-Triggered Electrical Power Generation. Chemsuschem 2020, 13, 2684–2692. [Google Scholar] [CrossRef]
- Wang, Y.; Song, Y.; Ma, C.; Kang, Z.; Zhu, Z. A heterologously-expressed thermostable Pyrococcus furiosus cytoplasmic [NiFe]-hydrogenase I used as the catalyst of H2/air biofuel cells. Int. J. Hydrogen Energy 2021, 46, 3035–3044. [Google Scholar] [CrossRef]
- Gentil, S.; Mansor, S.M.C.; Jamet, H.; Cosnier, S.; Cavazza, C.; Le Goff, A. Oriented Immobilization of NiFeSe Hydrogenases on Covalently and Noncovalently Functionalized Carbon Nanotubes for H-2/Air Enzymatic Fuel Cells. ACS Catal. 2018, 8, 3957–3964. [Google Scholar] [CrossRef]
- Szczesny, J.; Birrell, J.A.; Conzuelo, F.; Lubitz, W.; Ruff, A.; Schuhmann, W. Redox-Polymer-Based High-Current-Density Gas-Diffusion H-2-Oxidation Bioanode Using FeFe Hydrogenase fromDesulfovibrio desulfuricansin a Membrane-free Biofuel Cell. Angew. Chem. Int. Ed. 2020, 59, 16506–16510. [Google Scholar] [CrossRef]
- Ruff, A.; Szczesny, J.; Vega, M.; Zacarias, S.; Matias, P.M.; Gounel, S.; Mano, N.; Pereira, I.A.C.; Schuhmann, W. Redox-Polymer-Wired NiFeSe Hydrogenase Variants with Enhanced O-2 Stability for Triple-Protected High-Current-Density H-2-Oxidation Bioanodes. Chemsuschem 2020, 13, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. [Google Scholar] [CrossRef]
- Pereira, A.R.; Luz, R.A.S.; Lima, F.; Crespilho, F.N. Protein Oligomerization Based on Bronsted Acid Reaction. ACS Catal. 2017, 7, 3082–3088. [Google Scholar] [CrossRef]
- Bulutoglu, B.; Macazo, F.C.; Bale, J.; King, N.; Baker, D.; Minteer, S.D.; Banta, S. Multimerization of an Alcohol Dehydrogenase by Fusion to a Designed Self-Assembling Protein Results in Enhanced Bioelectrocatalytic Operational Stability. ACS Appl. Mater. Interfaces 2019, 11, 20022–20028. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Silveira, C.M.; Silva, D.; Brissos, V.; Hildebrandt, P.; Martins, L.O.; Todorovic, S. Immobilized dye-decolorizing peroxidase (DyP) and directed evolution variants for hydrogen peroxide biosensing. Biosens. Bioelectron. 2020, 153. [Google Scholar] [CrossRef] [PubMed]
- Mate, D.M.; Alcalde, M. Laccase engineering: From rational design to directed evolution. Biotechnol. Adv. 2015, 33, 25–40. [Google Scholar] [CrossRef]
- Mateljak, I.; Monza, E.; Lucas, M.F.; Guallar, V.; Aleksejeva, O.; Ludwig, R.; Leech, D.; Shleev, S.; Alcalde, M. Increasing Redox Potential, Redox Mediator Activity, and Stability in a Fungal Laccase by Computer-Guided Mutagenesis and Directed Evolution. ACS Catal. 2019, 9, 4561–4572. [Google Scholar] [CrossRef]
- Lopes, P.; Koschorreck, K.; Pedersen, J.N.; Ferapontov, A.; Lorcher, S.; Pedersen, J.S.; Urlacher, V.B.; Ferapontova, E.E. Bacillus Licheniformis CotA Laccase Mutant: ElectrocatalyticReduction of O-2 from 0.6 V (SHE) at pH 8 and in Seawater. Chemelectrochem 2019, 6, 2043–2049. [Google Scholar] [CrossRef]
- Al-Lolage, F.A.; Meneghello, M.; Ma, S.; Ludwig, R.; Bartlett, P.N. A Flexible Method for the Stable, Covalent Immobilization of Enzymes at Electrode Surfaces. Chemelectrochem 2017, 4, 1528–1534. [Google Scholar] [CrossRef]
- Meneghello, M.; Al-Lolage, F.A.; Ma, S.; Ludwig, R.; Bartlett, P.N. Studying Direct Electron Transfer by Site-Directed Immobilization of Cellobiose Dehydrogenase. Chemelectrochem 2019, 6, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, D.W.; Zhang, Y.N.; Lv, P.F.; Feng, Q.; Wei, Q.F. Encapsulation of enzyme by metal-organic framework for single-enzymatic biofuel cell-based self-powered biosensor. Nano Energy 2020, 68. [Google Scholar] [CrossRef]
- Itoh, T.; Shibuya, Y.; Yamaguchi, A.; Hoshikawa, Y.; Tanaike, O.; Tsunoda, T.; Hanaoka, T.A.; Hamakawa, S.; Mizukami, F.; Hayashi, A.; et al. High-performance bioelectrocatalysts created by immobilization of an enzyme into carbon-coated composite membranes with nano-tailored structures. J. Mater. Chem. A 2017, 5, 20244–20251. [Google Scholar] [CrossRef]
- Funabashi, H.; Murata, K.; Tsujimura, S. Effect of Pore Size of MgO-templated Carbon on the Direct Electrochemistry of D-fructose Dehydrogenase. Electrochemistry 2015, 83, 372–375. [Google Scholar] [CrossRef]
- Mazurenko, I.; Clement, R.; Byrne-Kodjabachian, D.; de Poulpiquet, A.; Tsujimura, S.; Lojou, E. Pore size effect of MgO-templated carbon on enzymatic H-2 oxidation by the hyperthermophilic hydrogenase from Aquifex aeolicus. J. Electroanal. Chem. 2018, 812, 221–226. [Google Scholar] [CrossRef]
- Wanibuchi, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Enhancement of the Direct Electron Transfer-type Bioelectrocatalysis of Bilirubin Oxidase at the Interface between Carbon Particles. Electrochemistry 2021, 89, 43–48. [Google Scholar] [CrossRef]
- Takahashi, Y.; Wanibuchi, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Improved direct electron transfer-type bioelectrocatalysis of bilirubin oxidase using porous gold electrodes. J. Electroanal. Chem. 2019, 843, 47–53. [Google Scholar] [CrossRef]
- Wu, F.; Su, L.; Yu, P.; Mao, L.Q. Role of Organic Solvents in Immobilizing Fungus Laccase on Single Walled Carbon Nanotubes for Improved Current Response in Direct Bioelectrocatalysis. J. Am. Chem. Soc. 2017, 139, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Hickey, D.P.; Lim, K.; Cai, R.; Patterson, A.R.; Yuan, M.W.; Sahin, S.; Abdellaoui, S.; Minteer, S.D. Pyrene hydrogel for promoting direct bioelectrochemistry: ATP-independent electroenzymatic reduction of N-2. Chem. Sci. 2018, 9, 5172–5177. [Google Scholar] [CrossRef] [PubMed]
- Kuk, S.K.; Gopinath, K.; Singh, R.K.; Kim, T.D.; Lee, Y.; Choi, W.S.; Lee, J.K.; Park, C.B. NADH-Free Electroenzymatic Reduction of CO2 by Conductive Hydrogel-Conjugated Formate Dehydrogenase. ACS Catal. 2019, 9, 5584–5589. [Google Scholar] [CrossRef]
- El Ichi, S.; Zebda, A.; Laaroussi, A.; Reverdy-Bruas, N.; Chaussy, D.; Belgacem, M.N.; Cinquin, P.; Martin, D.K. Chitosan improves stability of carbon nanotube biocathodes for glucose biofuel cells. Chem. Commun. 2014, 50, 14535–14538. [Google Scholar] [CrossRef]
- El Ichi, S.; Zebda, A.; Alcaraz, J.P.; Laaroussi, A.; Boucher, F.; Boutonnat, J.; Reverdy-Bruas, N.; Chaussy, D.; Belgacem, M.N.; Cinquin, P.; et al. Bioelectrodes modified with chitosan for long-term energy supply from the body. Energy Environ. Sci. 2015, 8, 1017–1026. [Google Scholar] [CrossRef]
- Matsumoto, T.; Isogawa, Y.; Tanaka, T.; Kondo, A. Streptavidin-hydrogel prepared by sortase A-assisted click chemistry for enzyme immobilization on an electrode. Biosens. Bioelectron. 2018, 99, 56–61. [Google Scholar] [CrossRef]
- Ghimire, A.; Pattamrnattel, A.; Maher, C.E.; Kasi, R.M.; Kumar, C.V. Three-Dimensional, Enzyme Biohydrogel Electrode for Improved Bioelectrocatalysis. ACS Appl. Mater. Interfaces 2017, 9, 42556–42565. [Google Scholar] [CrossRef]
- Heller, A. Electrical connection of enzyme redox centers to electrodes. J. Phys. Chem. 1992, 96, 3579–3587. [Google Scholar] [CrossRef]
- Cadoux, C.; Milton, R.D. Recent Enzymatic Electrochemistry for Reductive Reactions. Chemelectrochem 2020, 7, 1974–1986. [Google Scholar] [CrossRef]
- Ruth, J.C.; Milton, R.D.; Gu, W.Y.; Spormann, A.M. Enhanced Electrosynthetic Hydrogen Evolution by Hydrogenases Embedded in a Redox-Active Hydrogel. Chem. A Eur. J. 2020, 26, 7323–7329. [Google Scholar] [CrossRef]
- Xiao, X.X.; Conghaile, P.O.; Leech, D.; Magner, E. Use of Polymer Coatings to Enhance the Response of Redox-Polymer-Mediated Electrodes. Chemelectrochem 2019, 6, 1344–1349. [Google Scholar] [CrossRef]
- Diaz-Gonzalez, J.C.M.; Escalona-Villalpando, R.A.; Arriaga, L.G.; Minteer, S.D.; Casanova-Moreno, J.R. Effects of the cross-linker on the performance and stability of enzymatic electrocatalytic films of glucose oxidase and dimethylferrocene-modified linear poly(ethyleneimine). Electrochim. Acta 2020, 337. [Google Scholar] [CrossRef]
- Tsujimura, S.; Takeuchi, S. Toward an ideal platform structure based on MgO-templated carbon for flavin adenine dinucleotide-dependent glucose dehydrogenase-Os polymer-hydrogel electrodes. Electrochim. Acta 2020, 343. [Google Scholar] [CrossRef]
- Huang, X.C.; Zhang, L.L.; Zhang, Z.; Guo, S.; Shang, H.; Li, Y.B.; Liu, J. Wearable biofuel cells based on the classification of enzyme for high power outputs and lifetimes. Biosens. Bioelectron. 2019, 124, 40–52. [Google Scholar] [CrossRef]
- Kim, J.H.; Hong, S.G.; Wee, Y.; Hu, S.; Kwon, Y.; Ha, S.; Kim, J. Enzyme precipitate coating of pyranose oxidase on carbon nanotubes and their electrochemical applications. Biosens. Bioelectron. 2017, 87, 365–372. [Google Scholar] [CrossRef]
- Garcia, K.E.; Babanova, S.; Scheffler, W.; Hans, M.; Baker, D.; Atanassov, P.; Banta, S. Designed Protein Aggregates Entrapping Carbon Nanotubes for Bioelectrochemical Oxygen Reduction. Biotechnol. Bioeng. 2016, 113, 2321–2327. [Google Scholar] [CrossRef]
- Caserta, G.; Lorent, C.; Ciaccafava, A.; Keck, M.; Breglia, R.; Greco, C.; Limberg, C.; Hildebrandt, P.; Cramer, S.P.; Zebger, I.; et al. The large subunit of the regulatory NiFe -hydrogenase from Ralstonia eutropha—A minimal hydrogenase? Chem. Sci. 2020, 11, 5453–5465. [Google Scholar] [CrossRef]
- Rodriguez-Padron, D.; Puente-Santiago, A.R.; Caballero, A.; Balu, A.M.; Romero, A.A.; Luque, R. Highly efficient direct oxygen electro-reduction by partially unfolded laccases immobilized on waste-derived magnetically separable nanoparticles. Nanoscale 2018, 10, 3961–3968. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sanchez, C.; Ciaccafava, A.; Blanchard, P.Y.; Monsalve, K.; Giudici-Orticoni, M.T.; Lecomte, S.; Lojou, E. Efficiency of Enzymatic O-2 Reduction by Myrothecium verrucaria Bilirubin Oxidase Probed by Surface Plasmon Resonance, PMIRRAS, and Electrochemistry. ACS Catal. 2016, 6, 5482–5492. [Google Scholar] [CrossRef]
- Singh, K.; McArdle, T.; Sullivan, P.R.; Blanford, C.F. Sources of activity loss in the fuel cell enzyme bilirubin oxidase. Energy Environ. Sci. 2013, 6, 2460–2464. [Google Scholar] [CrossRef]
- Zelechowska, K.; Stolarczyk, K.; Lyp, D.; Rogalski, J.; Roberts, K.P.; Bilewicz, R.; Biernat, J.F. Aryl and N-arylamide carbon nanotubes for electrical coupling of laccase to electrodes in biofuel cells and biobatteries. Biocybern. Biomed. Eng. 2013, 33, 235–245. [Google Scholar] [CrossRef]
- Yahata, N.; Saitoh, T.; Takayama, Y.; Ozawa, K.; Ogata, H.; Higuchi, Y.; Akutsu, H. Redox interaction of cytochrome c(3) with NiFe hydrogenase from Desulfovibrio vulgaris Miyazaki F. Biochemistry 2006, 45, 1653–1662. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Nishikawa, K.; Higuchi, Y.; Yamamoto, M.; Kano, K. Electrostatic roles in electron transfer from NiFe hydrogenase to cytochrome c(3) from Desulfovibrio vulgaris Miyazaki F. Biochim. Et Biophys. Acta Proteins Proteom. 2017, 1865, 481–487. [Google Scholar] [CrossRef]
- McArdle, T.; McNamara, T.P.; Fei, F.; Singh, K.; Blanford, C.F. Optimizing the Mass-Specific Activity of Bilirubin Oxidase Adlayers through Combined Electrochemical Quartz Crystal Microbalance and Dual Polarization Interferometry Analyses. ACS Appl. Mater. Interfaces 2015, 7, 25270–25280. [Google Scholar] [CrossRef] [PubMed]
- Kitazumi, Y.; Shirai, O.; Yamamoto, M.; Kano, K. Numerical simulation of diffuse double layer around microporous electrodes based on the Poisson-Boltzmann equation. Electrochim. Acta 2013, 112, 171–175. [Google Scholar] [CrossRef]
- Olloqui-Sariego, J.L.; Calvente, J.J.; Andreu, R. Immobilizing redox enzymes at mesoporous and nanostructured electrodes. Curr. Opin. Electrochem. 2021, 26, 100658. [Google Scholar] [CrossRef]
- Sakai, K.; Xia, H.-Q.; Kitazumi, Y.; Shirai, O.; Kano, K. Assembly of direct-electron-transfer-type bioelectrodes with high performance. Electrochim. Acta 2018, 271, 305–311. [Google Scholar] [CrossRef]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Kano, K. Nanostructured Porous Electrodes by the Anodization of Gold for an Application as Scaffolds in Direct-electron-transfer-type Bioelectrocatalysis. Electrochim. Acta 2018, 34, 1317–1322. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Bond, A.M.; Hill, H.A.O.; Oliver, B.N.; Psalti, I.S.M. Electrochemistry of cytochrome-c, plastocyanin, and ferredoxin at edge-plane and basal-plane graphite-electrodes interpreted via a model based on electron-transfer at electroactive sites of microscopic dimensions in size. J. Am. Chem. Soc. 1989, 111, 9185–9189. [Google Scholar] [CrossRef]
- Komori, K.; Huang, J.; Mizushima, N.; Ko, S.; Tatsuma, T.; Sakai, Y. Controlled direct electron transfer kinetics of fructose dehydrogenase at cup-stacked carbon nanofibers. Phys. Chem. Chem. Phys. 2017, 19, 27795–27800. [Google Scholar] [CrossRef] [PubMed]
- Kavetskyy, T.; Smutok, O.; Demkiv, O.; Mat’ko, I.; Svajdlenkova, H.; Sausa, O.; Novak, I.; Berek, D.; Cechova, K.; Pecz, M.; et al. Microporous carbon fibers as electroconductive immobilization matrixes: Effect of their structure on operational parameters of laccase-based amperometric biosensor. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 109. [Google Scholar] [CrossRef] [PubMed]
- de Poulpiquet, A.; Marques-Knopf, H.; Wernert, V.; Giudici-Orticoni, M.T.; Gadiou, R.; Lojou, E. Carbon nanofiber mesoporous films: Efficient platforms for bio-hydrogen oxidation in biofuel cells. Phys. Chem. Chem. Phys. 2014, 16, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.E.; Fourmond, V.; Tron-Infossi, P.; Lojou, E.; Bertrand, P.; Leger, C.; Giudici-Orticoni, M.T.; Lubitz, W. Membrane-Bound Hydrogenase I from the Hyperthermophilic Bacterium Aquifex aeolicus: Enzyme Activation, Redox Intermediates and Oxygen Tolerance. J. Am. Chem. Soc. 2010, 132, 6991–7004. [Google Scholar] [CrossRef]
- Ciaccafava, A.; De Poulpiquet, A.; Techer, V.; Giudici-Orticoni, M.T.; Tingry, S.; Innocent, C.; Lojou, E. An innovative powerful and mediatorless H-2/O-2 biofuel cell based on an outstanding bioanode. Electrochem. Commun. 2012, 23, 25–28. [Google Scholar] [CrossRef]
- Oughli, A.A.; Velez, M.; Birrell, J.A.; Schuhmann, W.; Lubitz, W.; Plumere, N.; Rudiger, O. Viologen-modified electrodes for protection of hydrogenases from high potential inactivation while performing H-2 oxidation at low overpotential. Dalton Trans. 2018, 47, 10685–10691. [Google Scholar] [CrossRef]
- Plumere, N.; Rudiger, O.; Oughli, A.A.; Williams, R.; Vivekananthan, J.; Poller, S.; Schuhmann, W.; Lubitz, W. A redox hydrogel protects hydrogenase from high-potential deactivation and oxygen damage. Nat. Chem. 2014, 6, 822–827. [Google Scholar] [CrossRef]
- Ruff, A.; Szczesny, J.; Zacarias, S.; Pereira, I.A.C.; Plumere, N.; Schuhmann, W. Protection and Reactivation of the NiFeSe Hydrogenase from Desulfovibrio vulgaris Hildenborough under Oxidative Conditions. ACS Energy Lett. 2017, 2, 964–968. [Google Scholar] [CrossRef]
- Mano, N. Engineering glucose oxidase for bioelectrochemical applications. Bioelectrochemistry 2019, 128, 218–240. [Google Scholar] [CrossRef]
- Elouarzaki, K.; Bourourou, M.; Holzinger, M.; Le Goff, A.; Marks, R.S.; Cosnier, S. Freestanding HRP-GOx redox buckypaper as an oxygen-reducing biocathode for biofuel cell applications. Energy Environ. Sci. 2015, 8, 2069–2074. [Google Scholar] [CrossRef]
- Lopez, F.; Zerria, S.; Ruff, A.; Schuhmann, W. An O-2 Tolerant Polymer/Glucose Oxidase Based Bioanode as Basis for a Self-powered Glucose Sensor. Electroanalysis 2018, 30, 1311–1318. [Google Scholar] [CrossRef]
- Sahin, S.; Wongnate, T.; Chuaboon, L.; Chaiyen, P.; Yu, E.H. Enzymatic fuel cells with an oxygen resistant variant of pyranose-2-oxidase as anode biocatalyst. Biosens. Bioelectron. 2018, 107, 17–25. [Google Scholar] [CrossRef]
- Tremey, E.; Stines-Chaumeil, C.; Gounel, S.; Mano, N. Designing an O-2-Insensitive Glucose Oxidase for Improved Electrochemical Applications. Chemelectrochem 2017, 4, 2520–2526. [Google Scholar] [CrossRef]
- Li, H.; Münchberg, U.; Oughli, A.A.; Buesen, D.; Lubitz, W.; Freier, E.; Plumeré, N. Suppressing hydrogen peroxide generation to achieve oxygen-insensitivity of a [NiFe] hydrogenase in redox active films. Nat. Commun. 2020, 11, 920. [Google Scholar] [CrossRef]
- Zebda, A.; Alcaraz, J.P.; Vadgama, P.; Shleev, S.; Minteer, S.D.; Boucher, F.; Cinquin, P.; Martin, D.K. Challenges for successful implantation of biofuel cells. Bioelectrochemistry 2018, 124, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.H.; Liu, Y.C.; Williams, I.; Li, Y.; Qian, F.Y.; Wang, L.; Lei, Y.; Li, B.K. Flat enzyme-based lactate biofuel cell integrated with power management system: Towards long term in situ power supply for wearable sensors. Appl. Energy 2017, 194, 71–80. [Google Scholar] [CrossRef]
- Lv, J.; Jeerapan, I.; Tehrani, F.; Yin, L.; Silva-Lopez, C.A.; Jang, J.H.; Joshuia, D.; Shah, R.; Liang, Y.Y.; Xie, L.Y.; et al. Sweat-based wearable energy harvesting-storage hybrid textile devices. Energy Environ. Sci. 2018, 11, 3431–3442. [Google Scholar] [CrossRef]
- Ruff, A.; Conzuelo, F.; Schuhmann, W. Bioelectrocatalysis as the basis for the design of enzyme-based biofuel cells and semi-artificial biophotoelectrodes. Nat. Catal. 2020, 3, 214–224. [Google Scholar] [CrossRef]
- Clement, R.; Wang, X.; Biaso, F.; Ilbert, M.; Mazurenko, I.; Lojou, E. Mutations in the coordination spheres of T1 Cu affect Cu2+-activation of the laccase from Thermus thermophilus. Biochimie 2021. [Google Scholar] [CrossRef]
- Zhang, L.L.; Cui, H.Y.; Zou, Z.; Garakani, T.M.; Novoa-Henriquez, C.; Jooyeh, B.; Schwaneberg, U. Directed Evolution of a Bacterial Laccase (CueO) for Enzymatic Biofuel Cells. Angew. Chem. Int. Ed. 2019, 58, 4562–4565. [Google Scholar] [CrossRef]
- Herkendell, K.; Stemmer, A.; Tel-Vered, R. Extending the operational lifetimes of all-direct electron transfer enzymatic biofuel cells by magnetically assembling and exchanging the active biocatalyst layers on stationary electrodes. Nano Res. 2019, 12, 767–775. [Google Scholar] [CrossRef]
- Hammond, J.L.; Gross, A.J.; Giroud, F.; Travelet, C.; Borsali, R.; Cosnier, S. Solubilized Enzymatic Fuel Cell (SEFC) for Quasi-Continuous Operation Exploiting Carbohydrate Block Copolymer Glyconanoparticle Mediators. ACS Energy Lett. 2019, 4, 142–148. [Google Scholar] [CrossRef]
- Wang, D.D.; Jana, D.L.; Zhao, Y.L. Metal-Organic Framework Derived Nanozymes in Biomedicine. Acc. Chem. Res. 2020, 53, 1389–1400. [Google Scholar] [CrossRef]
- Navyatha, B.; Singh, S.; Nara, S. AuPeroxidase nanozymes: Promises and applications in biosensing. Biosens. Bioelectron. 2021, 175, 112882. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.E.; Dey, A. Recent developments in bioinspired modelling of NiFe- and FeFe-hydrogenases. Curr. Opin. Electrochem. 2019, 15, 155–164. [Google Scholar] [CrossRef]
- Laureanti, J.A.; O’Hagan, M.; Shaw, W.J. Chicken fat for catalysis: A scaffold is as important for molecular complexes for energy transformations as it is for enzymes in catalytic function. Sustain. Energy Fuels 2019, 3, 3260–3278. [Google Scholar] [CrossRef]
- Yang, Y.; Xiong, Y.; Zeng, R.; Lu, X.; Krumov, M.; Huang, X.; Xu, W.; Wang, H.; DiSalvo, F.J.; Brock, J.D.; et al. Operando Methods in Electrocatalysis. ACS Catal. 2021, 11, 1136–1178. [Google Scholar] [CrossRef]
- de Souza, J.C.P.; Macedo, L.J.A.; Hassan, A.; Sedenho, G.C.; Modenez, I.A.; Crespilho, F.N. In Situ and Operando Techniques for Investigating Electron Transfer in Biological Systems. Chemelectrochem 2020. [Google Scholar] [CrossRef]
- Kornienko, N.; Ly, K.H.; Robinson, W.E.; Heidary, N.; Zhang, J.Z.; Reisner, E. Advancing Techniques for Investigating the Enzyme-Electrode Interface. Acc. Chem. Res. 2019, 52, 1439–1448. [Google Scholar] [CrossRef]
- Singh, K.; Blanford, C.F. Electrochemical Quartz Crystal Microbalance with Dissipation Monitoring: A Technique to Optimize Enzyme Use in Bioelectrocatalysis. Chemcatchem 2014, 6, 921–929. [Google Scholar] [CrossRef]
- Sezer, M.; Kielb, P.; Kuhlmann, U.; Mohrmann, H.; Schulz, C.; Heinrich, D.; Schlesinger, R.; Heberle, J.; Weidinger, I.M. Surface Enhanced Resonance Raman Spectroscopy Reveals Potential Induced Redox and Conformational Changes of Cytochrome c Oxidase on Electrodes. J. Phys. Chem. B 2015, 119, 9586–9591. [Google Scholar] [CrossRef] [PubMed]
- Dagys, M.; Laurynenas, A.; Ratautas, D.; Kulys, J.; Vidziunaite, R.; Talaikis, M.; Niaura, G.; Marcinkeviciene, L.; Meskys, R.; Shleev, S. Oxygen electroreduction catalysed by laccase wired to gold nanoparticles via the trinuclear copper cluster. Energy Environ. Sci. 2017, 10, 498–502. [Google Scholar] [CrossRef]
- Olejnik, P.; Pawlowska, A.; Palys, B. Application of Polarization Modulated Infrared Reflection Absorption Spectroscopy for electrocatalytic activity studies of laccase adsorbed on modified gold electrodes. Electrochim. Acta 2013, 110, 105–111. [Google Scholar] [CrossRef]
- Macedo, L.J.A.; Hassan, A.; Sedenho, G.C.; Crespilho, F.N. Assessing electron transfer reactions and catalysis in multicopper oxidases with operando X-ray absorption spectroscopy. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Abdiaziz, K.; Salvadori, E.; Sokol, K.P.; Reisner, E.; Roessler, M.M. Protein film electrochemical EPR spectroscopy as a technique to investigate redox reactions in biomolecules. Chem. Commun. 2019, 55, 8840–8843. [Google Scholar] [CrossRef]
- Harris, T.; Heidary, N.; Kozuch, J.; Frielingsdorf, S.; Lenz, O.; Mroginski, M.A.; Hildebrandt, P.; Zebger, I.; Fischer, A. In Situ Spectroelectrochemical Studies into the Formation and Stability of Robust Diazonium-Derived Interfaces on Gold Electrodes for the Immobilization of an Oxygen-Tolerant Hydrogenase. ACS Appl. Mater. Interfaces 2018, 10, 23380–23391. [Google Scholar] [CrossRef]
- Abad, J.M.; Tesio, A.Y.; Martinez-Perinan, E.; Pariente, F.; Lorenzo, E. Imaging resolution of biocatalytic activity using nanoscale scanning electrochemical microscopy. Nano Res. 2018, 11, 4232–4244. [Google Scholar] [CrossRef]
- Tassy, B.; Dauphin, A.L.; Man, H.M.; Le Guenno, H.; Lojou, E.; Bouffier, L.; de Poulpiquet, A. In Situ Fluorescence Tomography Enables a 3D Mapping of Enzymatic O-2 Reduction at the Electrochemical Interface. Anal. Chem. 2020, 92, 7249–7256. [Google Scholar] [CrossRef] [PubMed]
- Zigah, D.; Lojou, E.; de Poulpiquet, A. Micro- and Nanoscopic Imaging of Enzymatic Electrodes: A Review. Chemelectrochem 2019, 6, 5524–5546. [Google Scholar] [CrossRef]
- Barrozo, A.; Orio, M. Molecular Electrocatalysts for the Hydrogen Evolution Reaction: Input from Quantum Chemistry. Chemsuschem 2019, 12, 4905–4915. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.Y.; Li, Q.Y.; Xu, Y.J.; Shen, S.H.; Sun, C.H. Learning from nature: Understanding hydrogenase enzyme using computational approach. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10. [Google Scholar] [CrossRef]
- Yang, S.J.; Liu, J.; Quan, X.B.; Zhou, J. Bilirubin Oxidase Adsorption onto Charged Self-Assembled Monolayers: Insights from Multiscale Simulations. Langmuir 2018, 34, 9818–9828. [Google Scholar] [CrossRef] [PubMed]















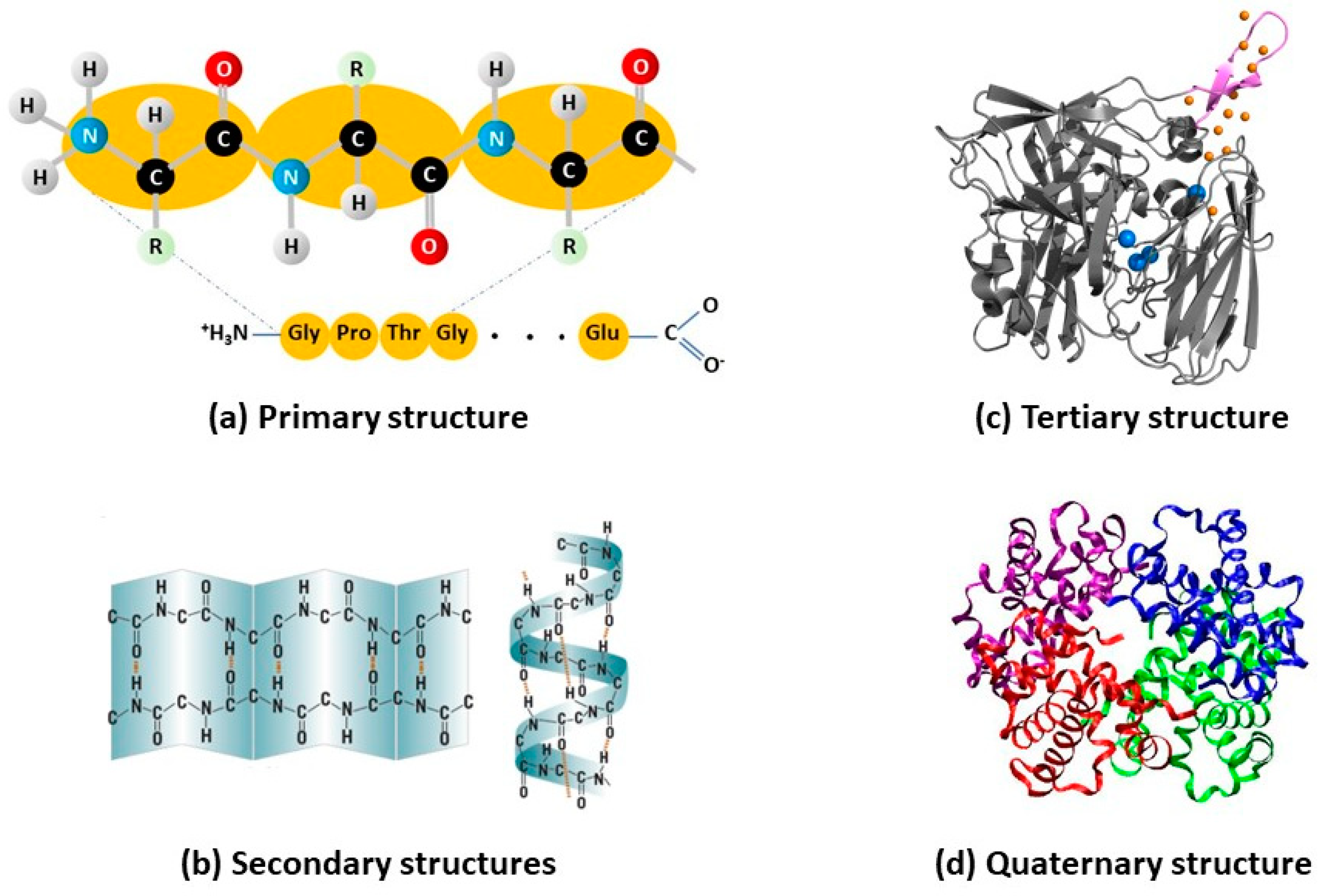{kind=link}
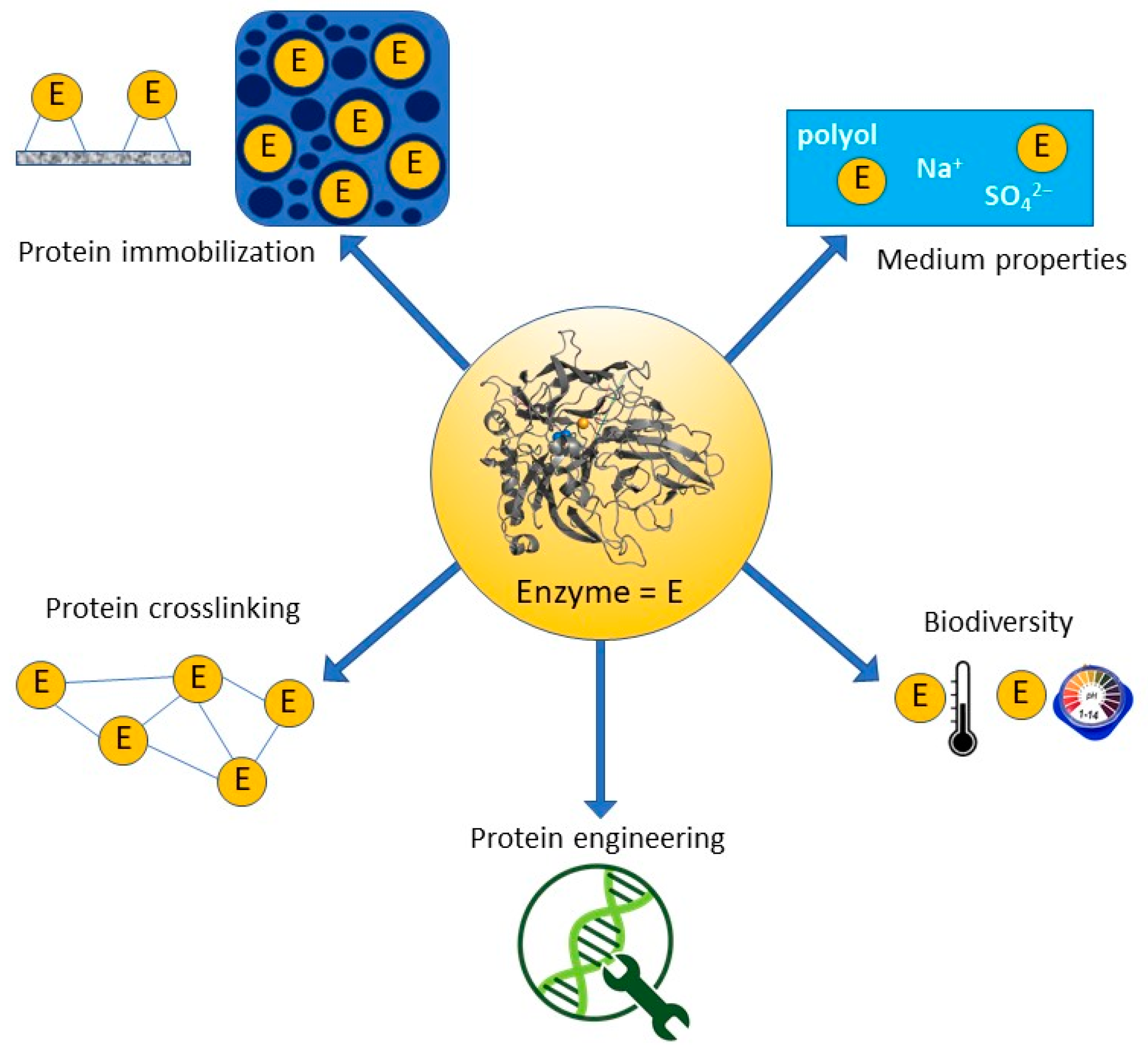{kind=link}
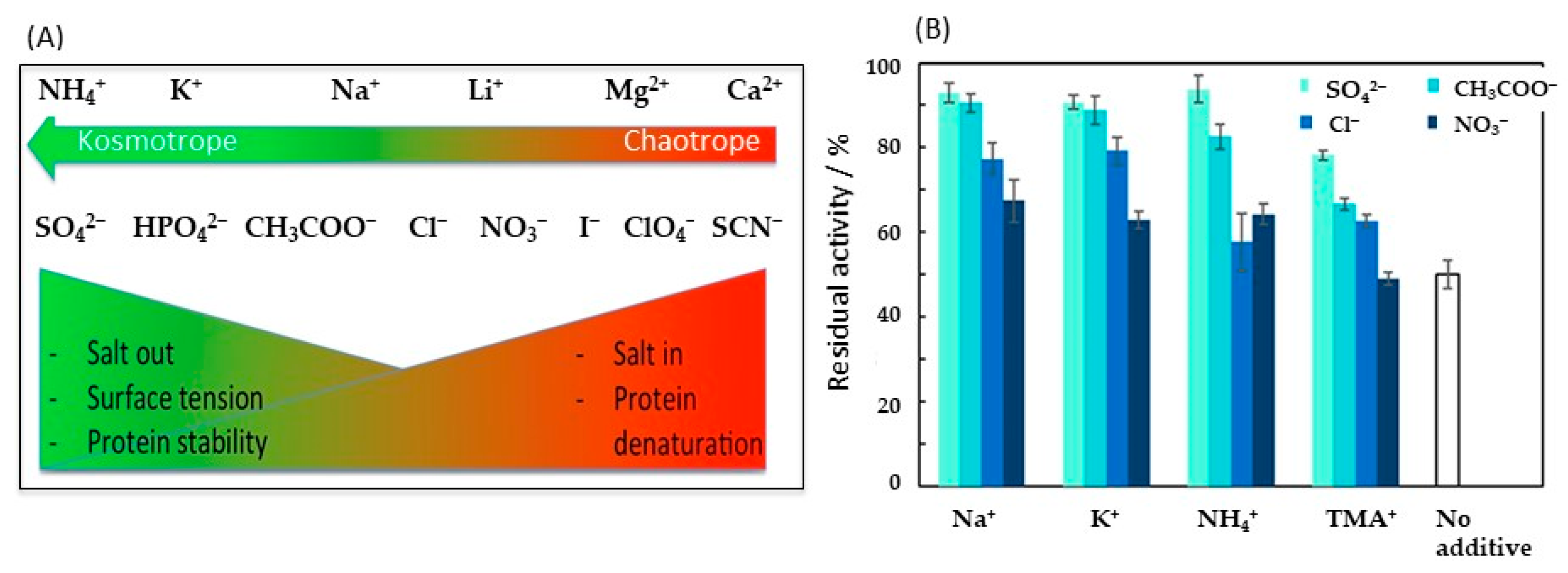{kind=link}
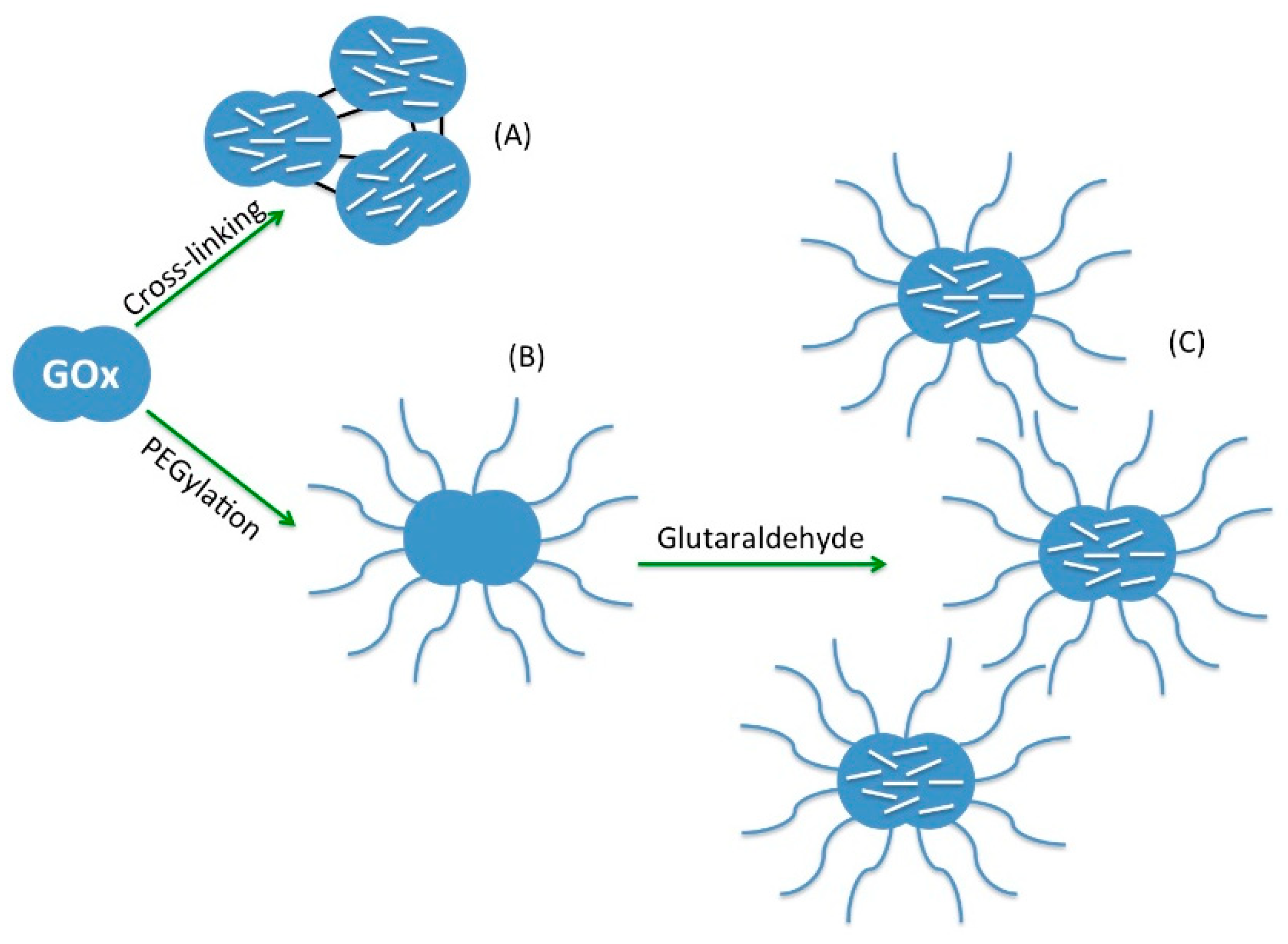{kind=link}
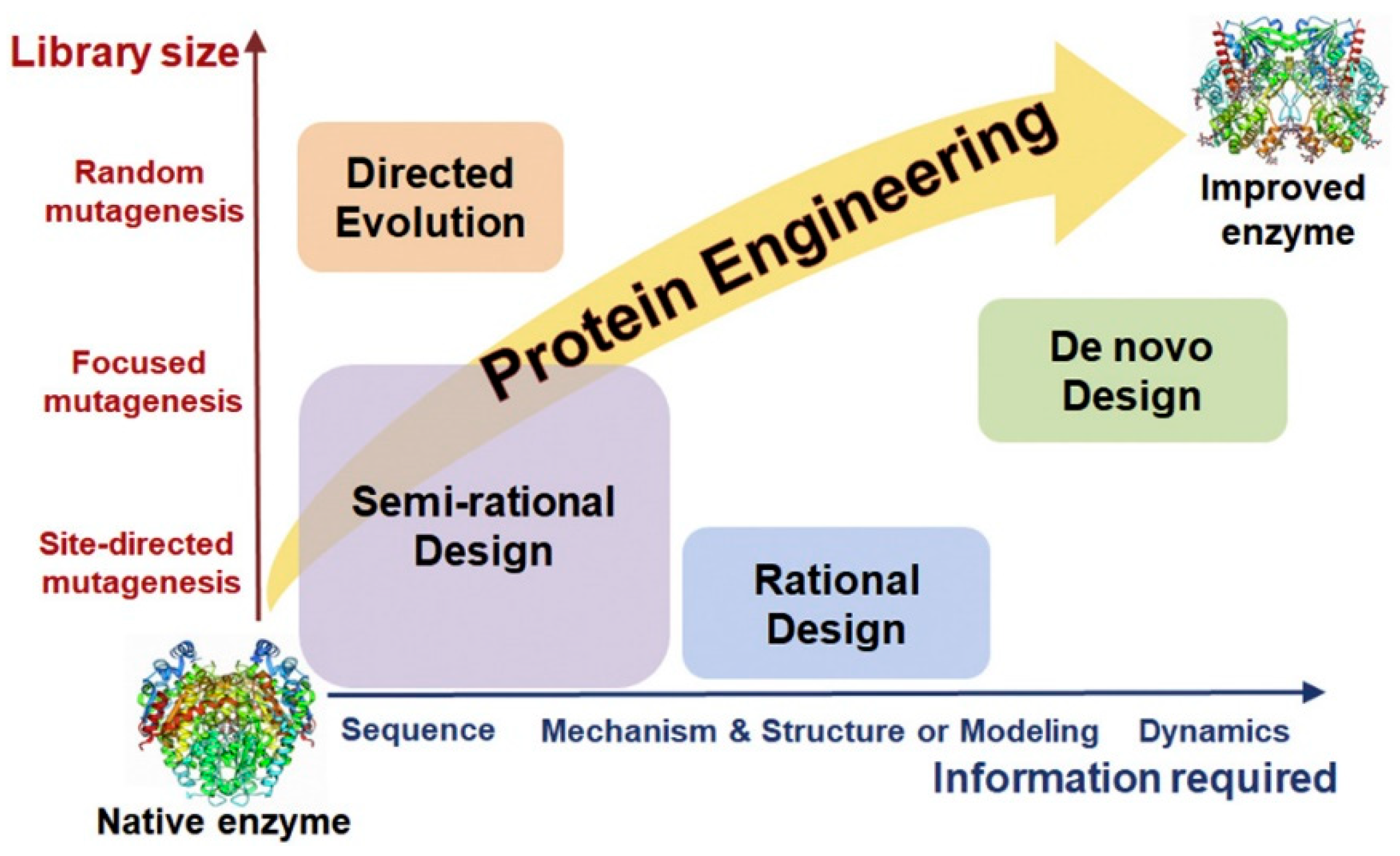{kind=link}
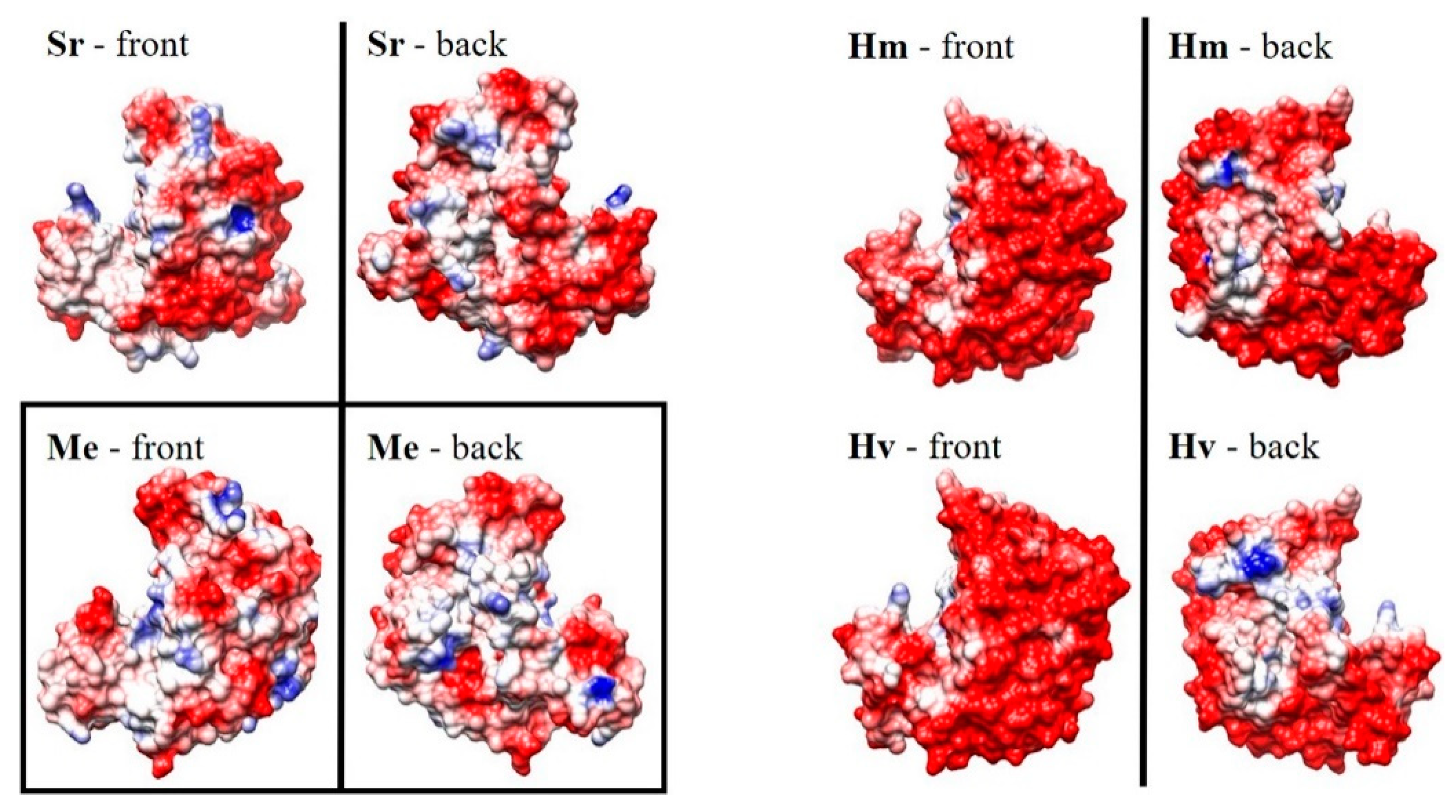{kind=link}
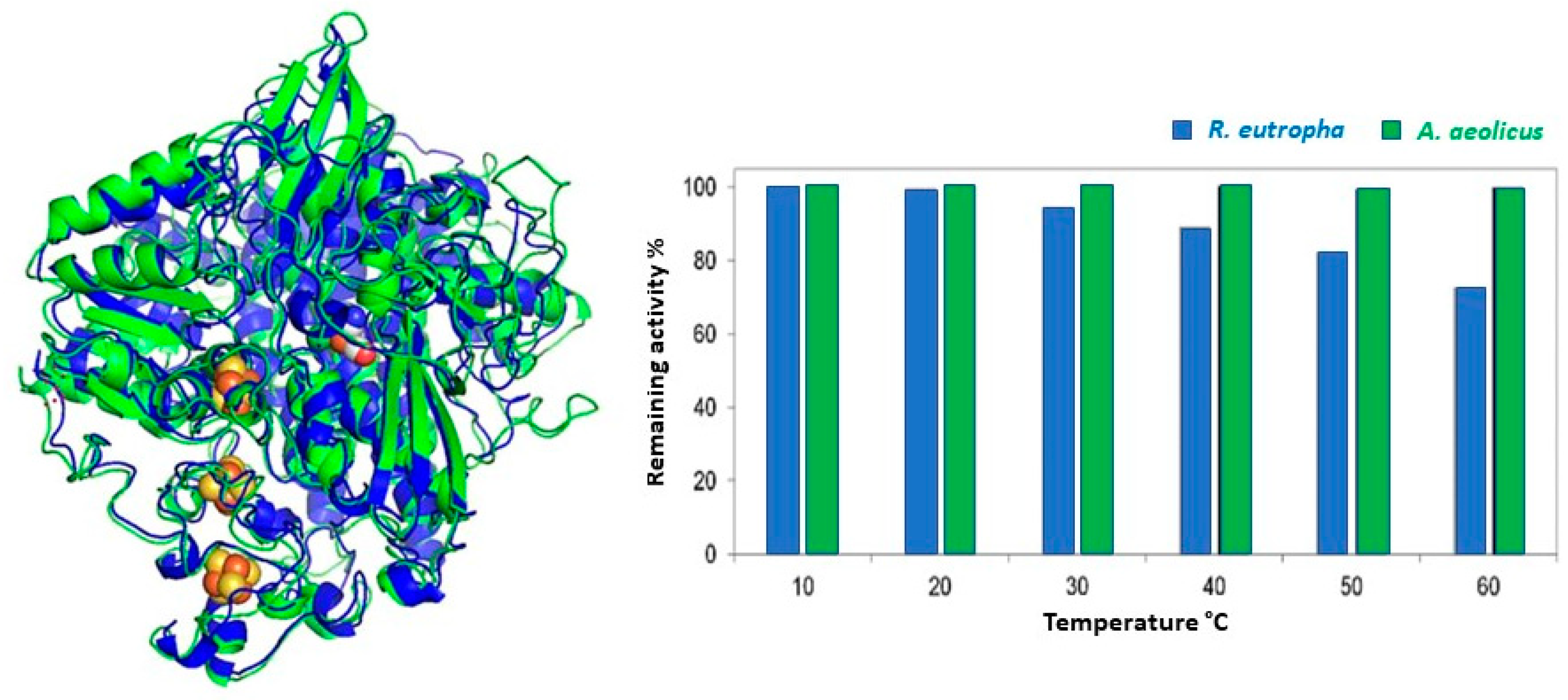{kind=link}
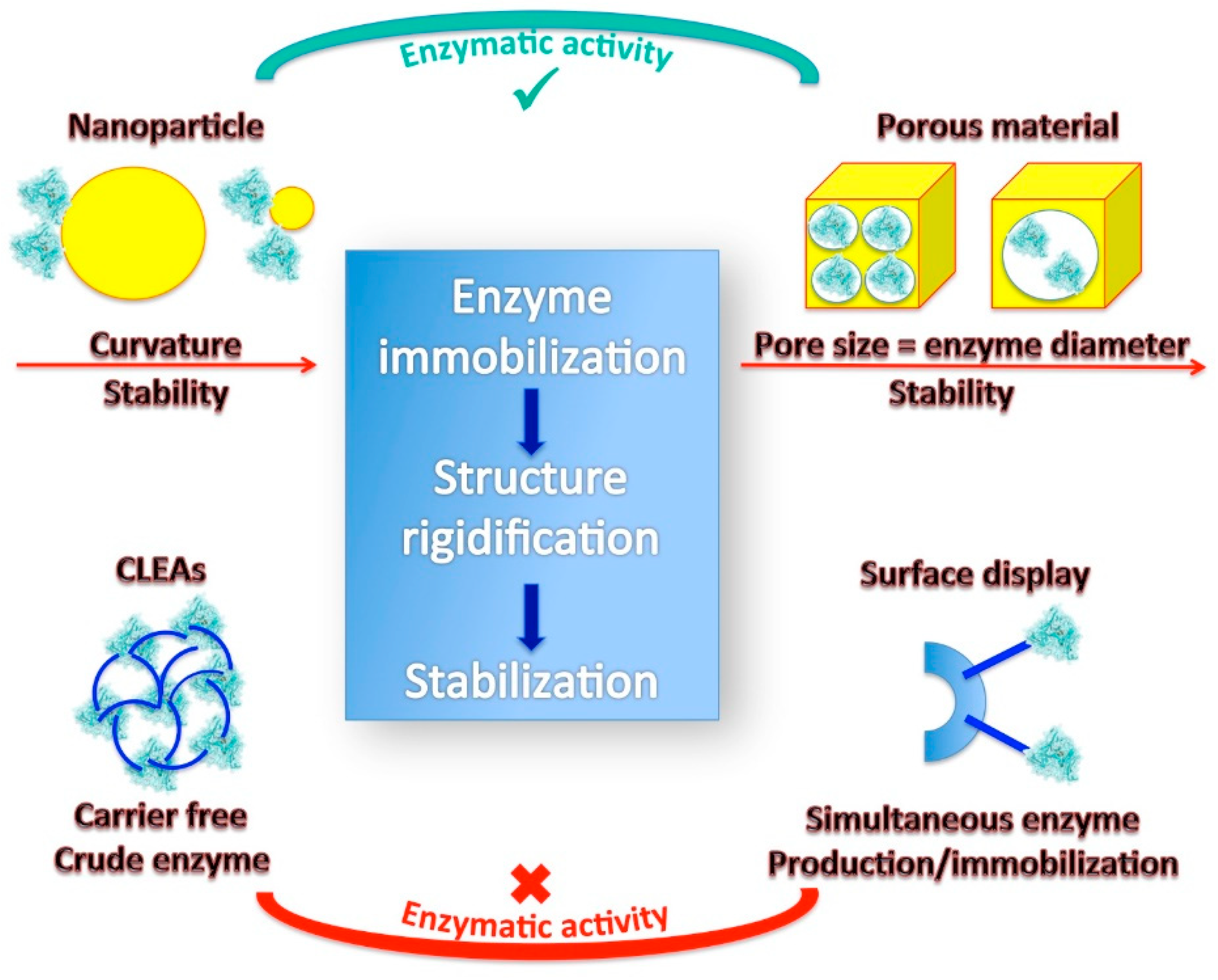{kind=link}
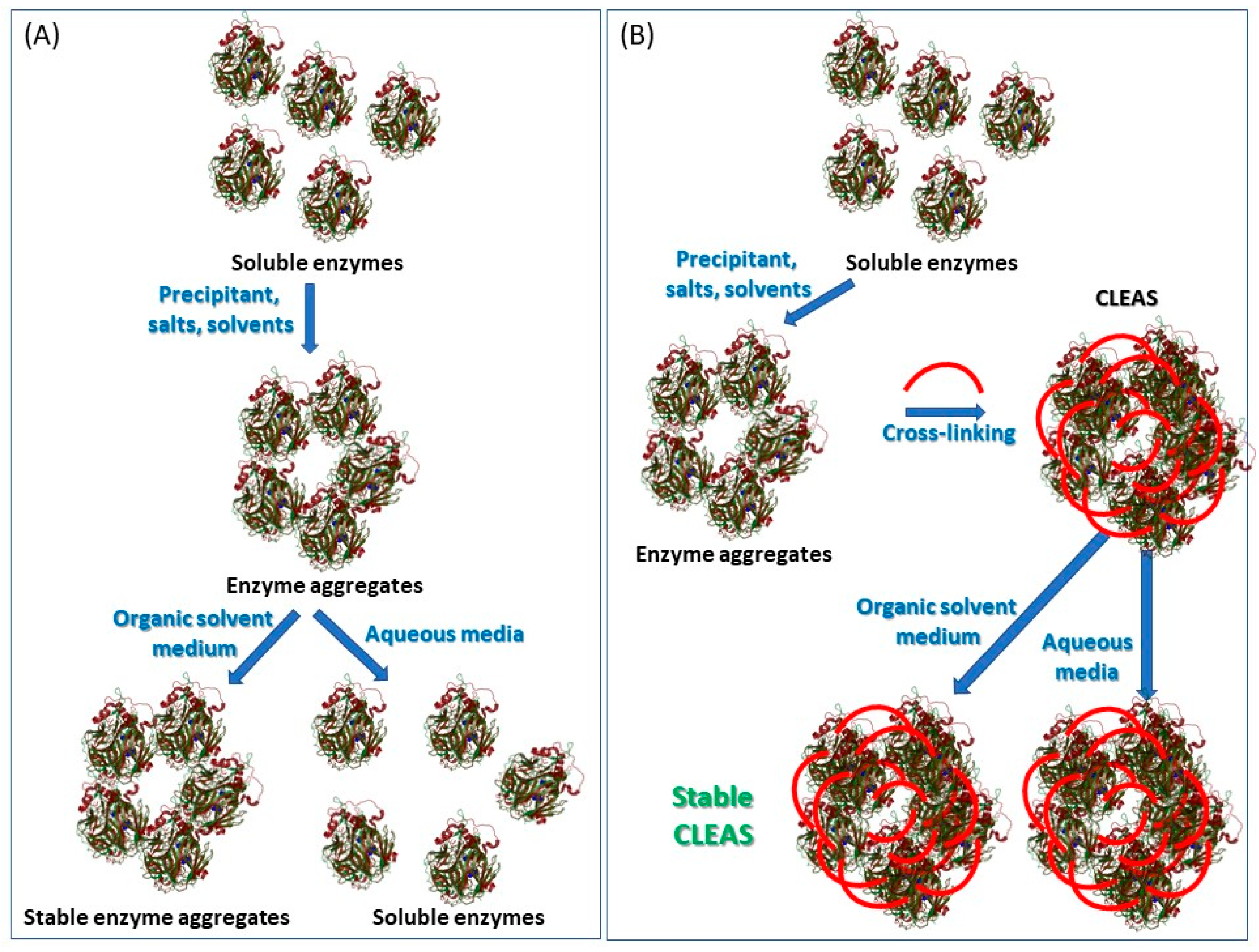{kind=link}
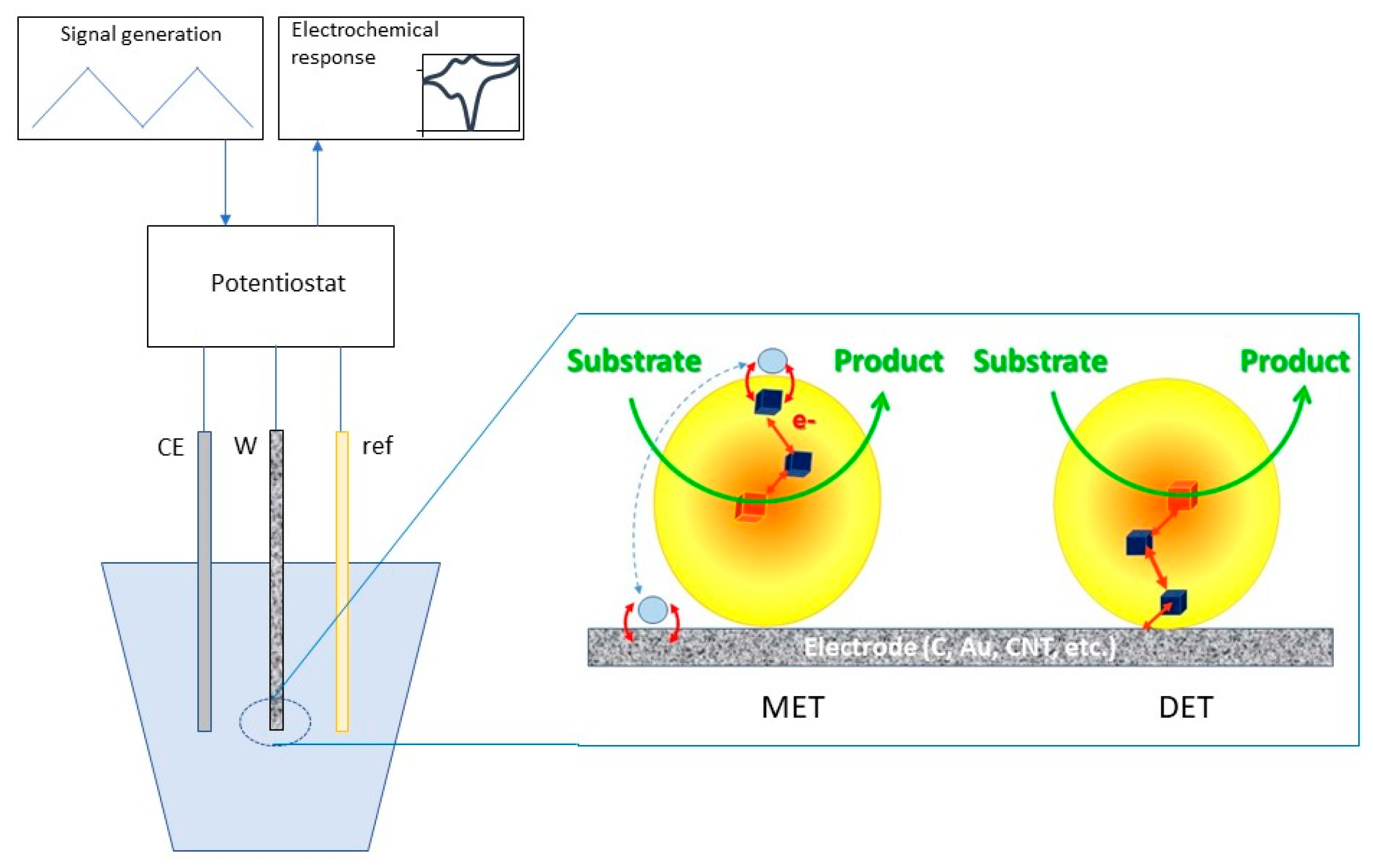{kind=link}
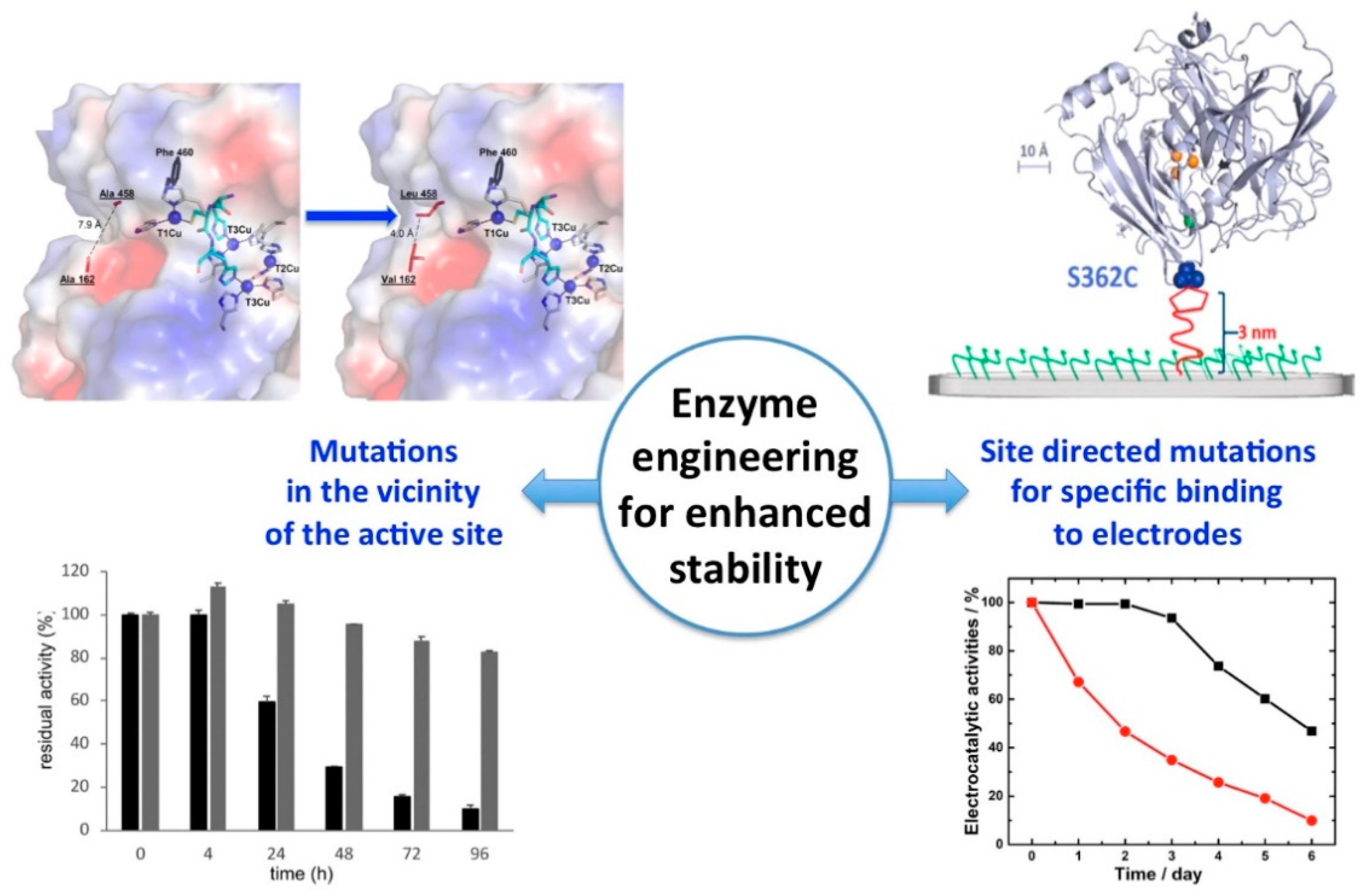{kind=link}
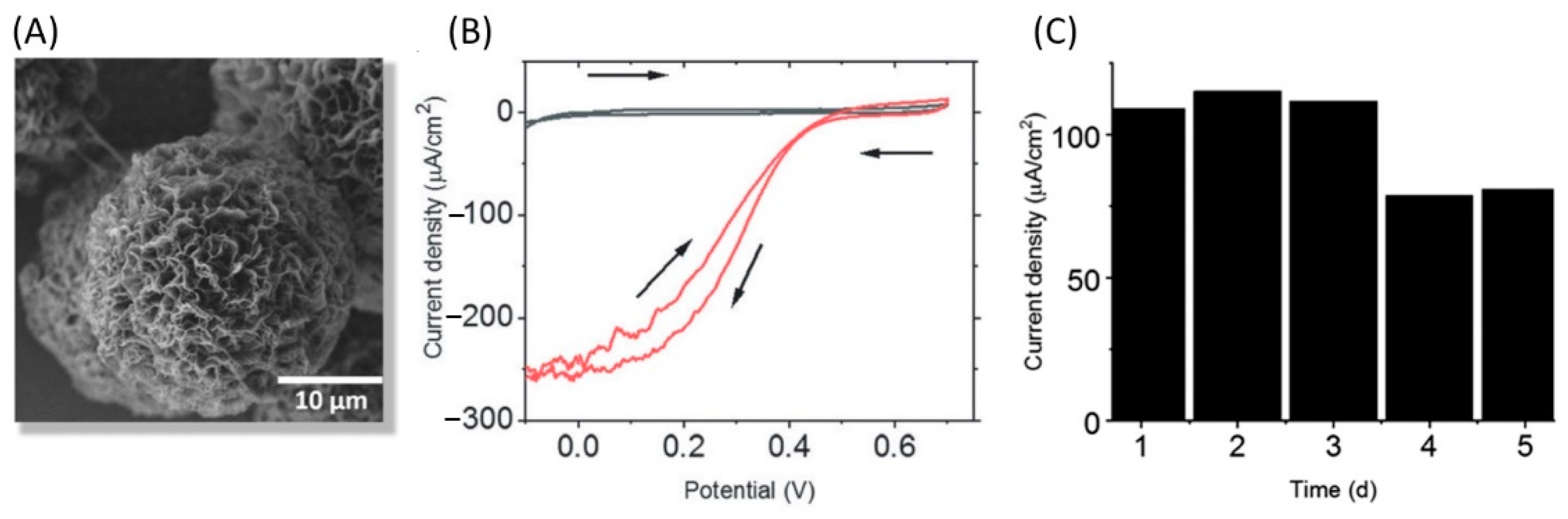{kind=link}
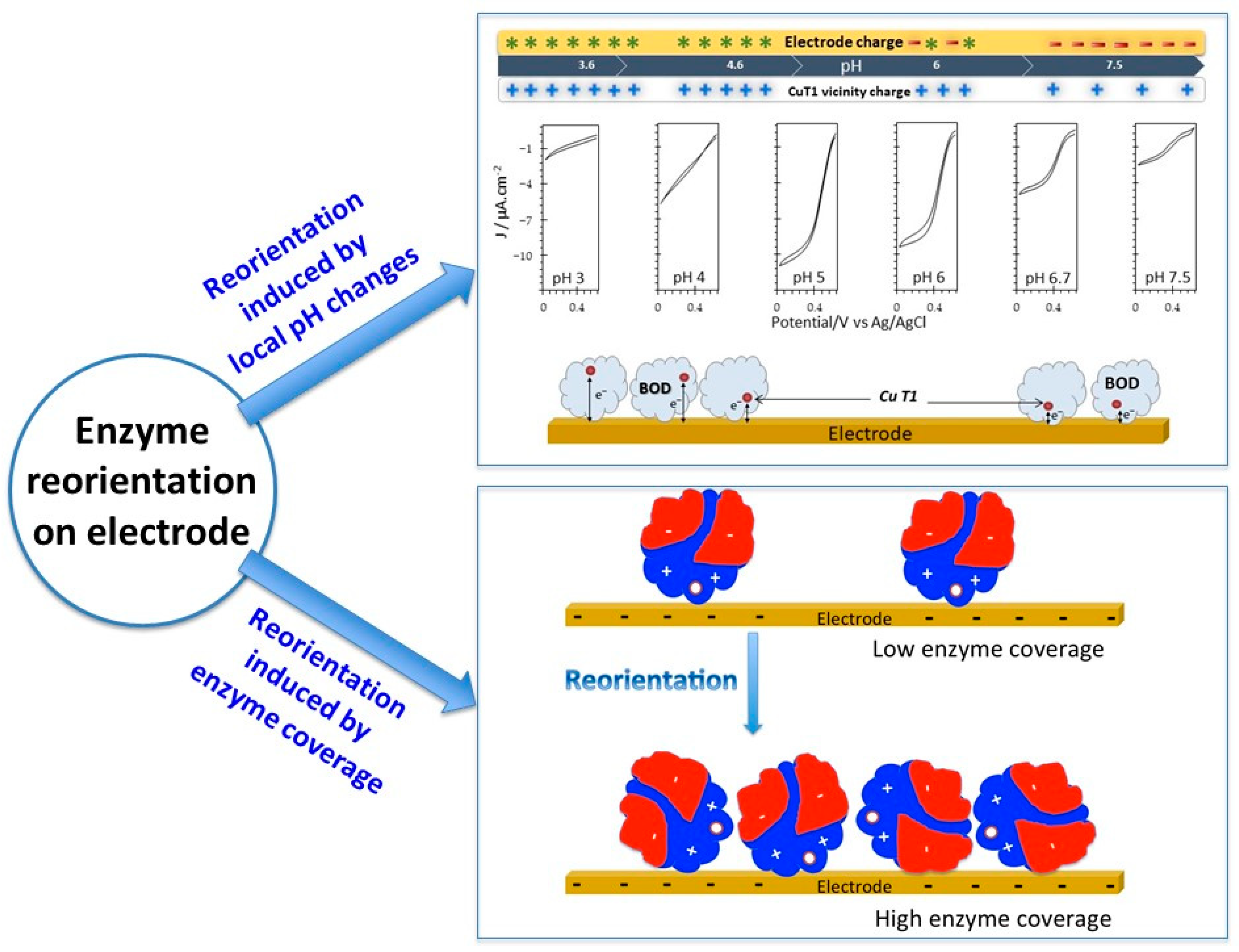{kind=link}
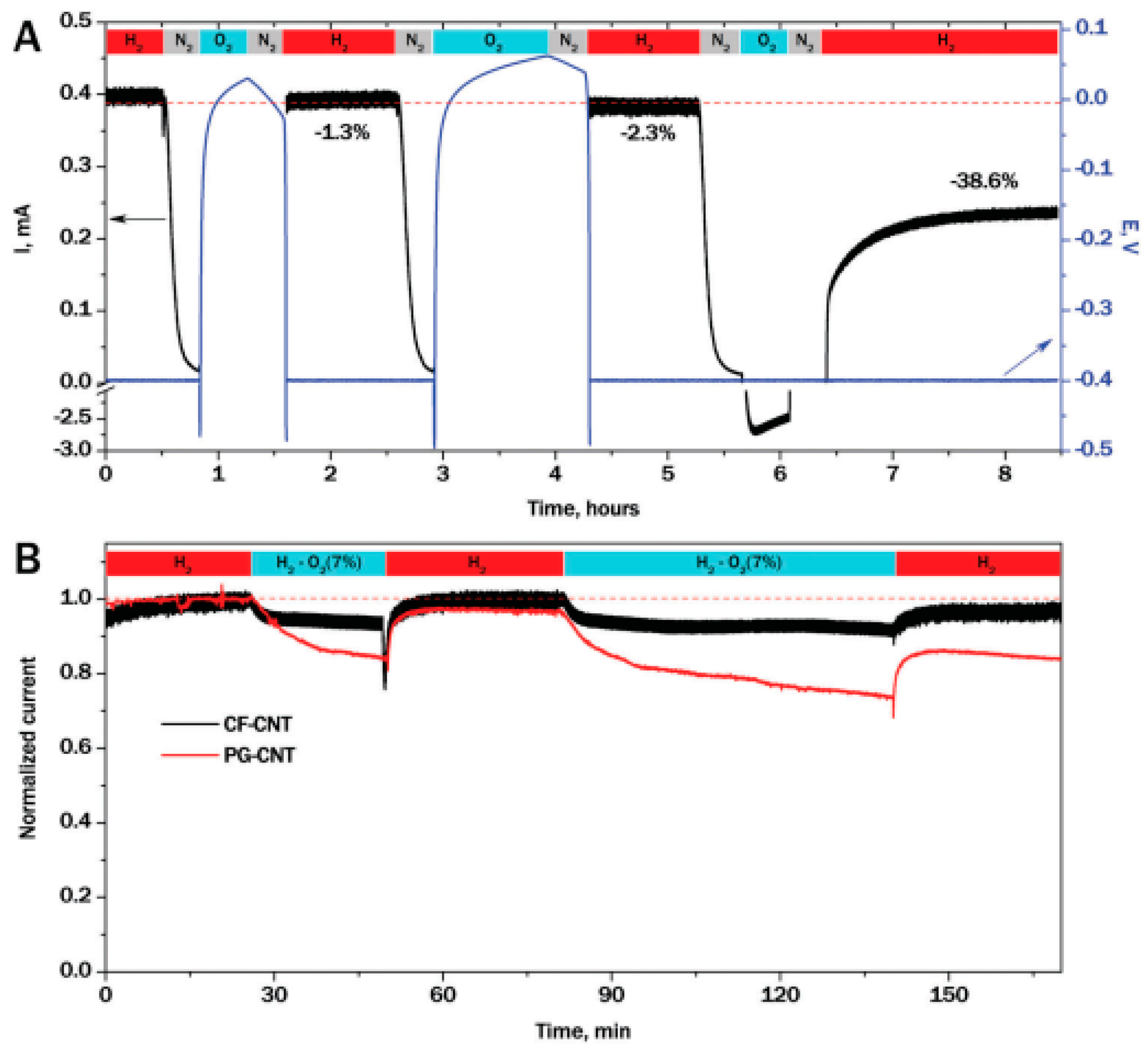{kind=link}
| Enzyme | Bioelectrode Design | Electrochemical Conditions | Reaction Followed/Purpose | Stability | |
|---|---|---|---|---|---|
| Cerrena unicolor C-139 LAC | Enzyme polymerized at a glassy carbon electrode through cold plasma | Acetate buffer pH 5, 25 °C | Rutin biosensing | 42% of current is recovered after 8 days at pH 5. | [201] |
| Streptomyces coelicor LAC | Enzyme adsorbed on zinc oxide NPs capped with p-amino thiophenol and attached to graphene oxide | 0.1 M phosphate buffer pH 5 | Sucralose sensing | 83% and 73% of the initial activity after 5 and 10 days, respectively. | [202] |
| Trametes hirsuta LAC GreeDo mutant obtained by combining computational design | Enzyme covalently attached to AuNPs grafted on graphite electrode | 0.1 M acetate buffer pH 4 | O2 reduction | 80% of initial activity after 96 h incubation at pH 4 60% of initial activity after 2 h of continuous operation | [203] |
| LAC | Enzyme adsorbed on AuNPs electrodeposited on a screen-printed carbon electrode (SPCE) modified with polypyrrole | 0.1 M acetate buffer pH 3.5 25 °C | Polyphenol sensing | 85% of the initial activity after 1 month (storage at 4 °C) | [204] |
| Trametes versicolor LAC | Enzyme and Cu2+co-adsorbed on pyrene-terminated block polymer on pyrolytic graphite surface and graphene papers | 0.1 M acetic acid buffer pH 5 | Pyrocatechol sensing | 70% of original activity upon 90 days of the freeze-dried powder. 80% of activity after 3 freeze-thaw cycles.31% activity at 70 °C. 96% of the initial activity after 30 days storage at 4 °C. | [205] |
| Trametes versicolor LAC | Enzyme entrapped within graphene-cellulose microfiber composite modified SPCE. | 0.1 M sodium phosphate buffer pH 5 | Catechol sensing | 97% of the initial activity after 130 h storage. | [206] |
| Trametes versicolor LAC | Enzyme with Cu-nanoflowers mixed with CNTs | 0.01 M phosphate buffer pH 7.4 | O2 reduction in a H2/O2 hybrid biofuel cell | 85% of biofuel cell initial power density for 15 days at room temperature. | [207] |
| LAC | Enzyme absorbed on AuNPs-MoS2 composite glued on a glassy carbon electrode (GCE) by Nafion | 0.2 M acetate buffer pH 4 | Catechol sensing | 95% initial activity after 15 days of storage stability. | [208] |
| Trametes versicolor LAC | Enzyme entrapped in chitosan-CNT matrix | 0.1 M phosphate buffer pH 7.4, 20 °C | O2 reduction | 82% of the initial activity within 10 days. | [209] |
| Trametes versicolor LAC | Enzyme entrapped with Fe3O4 magnetic nanoparticles in chitosan matrix | 0.2 M phosphate buffer | O2 reduction | 50% of the initial activity after 20 days. | [210] |
| Trametes versicolor LAC | Enzyme crosslinked with GA in a matrix made of room temperature IL-CNTs | 50 mM acetate buffer pH 5 | Polyphenol biosensing | 33% of the initial activity after 20 days. | [211] |
| Bacillus subtilis LAC | Enzyme adsorbed on the electrode modified with thiol graphene-AuNP nanocomposite film | 0.1 M HAc-NaAc buffer solution pH 4.6, 25 °C | Hydroquinone sensing | 87% of the initial current response after 100 days | [212] |
| Trametes versicolor LAC | Enzyme and nanocopper incorporated in polyacrylonitrile/polyvinylidene electrospun fibrous membranes | Trichlorophenol removal | 68% of the initial activity after 30 days. | [213] | |
| Bacillus subtilis LAC | Enzyme covalently bound on functionalized CNTs | O2 reduction | 70% of initial activity after two weeks of storage at room temperature. | [199] | |
| LAC | Enzyme cross-linked on electropolymerized L-lysine molecules on a graphene modified GCE | 0.1 M sodium phosphate buffer pH 6 | 17β-estradiol sensing | 68% of initial activity after 30 days storage at 4 °C. | [214] |
| Aspergillus sp. LAC | Enzyme adsorbed on polypyrrole-modified nanotubes and SrCuO2 microseeds composite on graphite electrode | 0.1 M phosphate buffer pH 7 | 2,4-dichlorophenol sensing | 80% of initial activity after 14 days of storage at 4 °C. | [215] |
| Trametes versicolor LAC | Enzyme deposited by electrospray on carbon black modified screen-printed electrodes | 0.1 M citric acid/sodium citrate buffer pH 4.5 | Catechol detection | Valuable working and storage stability of the biosensor that remains 100% of its performances for up to 25 measurements and can be preserved at room temperature for at least 90 days. | [216] |
| LAC | Enzyme mixed with polyvinylacetate and AuNPs electrospun on platinum electrode | Phosphate buffer | Ascorbic acid sensing | Stability for 76 days. | [217] |
| LAC | Enzyme adsorbed on polyaniline/magnetic graphene composite | pH 5 | Hydroquinone sensing | 90% initial response after 30 days. | [218] |
| Trametes versicolor LAC | Enzyme embedded in polydopamine NPs with anchored AuNPs | 0.1 M ABS pH 5 | Norepinephrine sensing | 91% initial response after 30 days. | [219] |
| LAC | Enzyme covalently bound to a composite made of acrylate microspheres and AuNPs coated on a carbon paste screen-printed electrode | 0.1 M phosphate buffer pH 5 | Tartrazine sensing | 51% initial response after 90 days. | [220] |
| Botryosphaeria rhodina LAC | Enzyme entrapment into nanostructured carbon black paste electrode | 0.1 M phosphate buffer pH 6 | Epinephrine sensing | 93% initial response after 7 days. | [221] |
| Myrothecium. Sp BOD | Enzyme crosslinked into a biogel matrix made of Nafion deposited on carbon NPs | 0.1 M phosphate buffer pH 7.2, 25 °C | O2 reduction | Electrode potential decrease from 0.37 to 0.31 V after 24 h of operation at 1 mA·cm−2 | [222] |
| Magnaporthe orizae BOD | Enzyme entrapped in osmium-based hydrogels deposited on CNTs modified by electrografting of aryldiazonium salts | 0.1 M phosphate buffer pH 5 | O2 reduction | After 1 day loss of 27% of the initial activity then stable for several days. | [223] |
| Myrothecium verrucaria BOD | Enzyme adsorbed on carbon cloth modified with aminobenzoic acid. Gas diffusion electrode. | 0.1 M phosphate buffer pH 7.4 | O2 reduction | 80% of initial activity after 1.3 h of operation of the H2/O2 biofuel cell. | [224] |
| Myrothecium verrucaria BOD | Enzyme covalently linked to carbon cloth modified by aminobenzoic acid and coated with a microporous hydrophobic Nafion layer. Gas diffusion electrode. | 0.1 M phosphate buffer pH 7, 25 °C | O2 reduction | 80% of initial current after 6 h of continuous operation at 0 V vs. Ag/AgCl. | [225] |
| Myrothecium verrucaria BOD | Enzyme adsorbed on CNTs and protected by a carbon-coated magnetic nanoparticle layer | 0.1 M HEPES buffer pH 7, | O2 reduction | 80% of initial activity after 48 h of continuous operation at 0 V vs. Ag/AgCl in the presence of 1 nM proteinase. 70% initial activity after 30 days of storage at room temperature. | [226] |
| Magnaporthe orizae BOD | Wild type enzyme adsorbed and mutants covalently bound to a maleimide CNT modified electrode | 200 mM phosphate buffer 100 mM citrate buffer pH 7 | O2 reduction | 40% and 10% initial activity after 3 days and 6 days, respectively, in the adsorbed mode. 95% and 52% initial activity after 3 days and 6 days, respectively, in the covalent attachment mode | [227] |
| Myrothecium verrucaria BOD | Enzyme adsorbed or electrochemically deposited on Au polycrystalline electrode | 0.1 M acetate buffer pH 5, 25 °C | O2 reduction | Adsorption mode: 5% initial activity after 100 potential cycles between 0 and 0.8 V vs. Ag/AgCl at 50 mV/s. Electrochemical deposition mode: 80% initial activity after 200 cycles | [228] |
| Myrothecium verrucaria BOD | Enzyme adsorbed on MgOC modified with 6 amino 2-naphtoïque acid | 0.1 M acetate buffer pH 5, 24 °C | O2 reduction | 50% of the initial activity after 5 days. | [229] |
| Myrothecium verrucaria BOD | Enzyme covalently bound to reduced graphene oxide modified by 4-aminobenzoic acid to reduce its aggregation | 0.1 M phosphate buffer pH 7 | O2 reduction | 55 h half-life. | [230] |
| Myrothecium verrucaria BOD | Enzyme adsorbed on buckypaper CNTs | Phosphate buffer pH 6.5, 25 °C | O2 reduction | 10 h half-life | [231] |
| Magnaporthe orizae BOD | Molecular engineering for covalent immobilization in macroporous gold electrode | 0.1 M phosphate buffer pH 7.2, 25 °C | O2 reduction | 75% of the initial activity after incubation for 5 days. | [232] |
| Myrothecium verrucaria BOD | Mv BOD entrapped with ABTS in polydopamine layer on CNTs | 0.1 M phosphate buffer pH 7, 25 °C | O2 reduction | Mv BOD stable for 22 h of operation at 0.1 V vs. Ag/AgCl. | [233] |
| Bacillus pumilus BOD | Bp BOD: entrapped in pyrenebetaine on CNTs | 50 °C | O2 reduction | Rapid drop in activity, almost 0 after 3 h of operation at 0.1 V vs. Ag/AgCl | [233] |
| Bacillus pumilus BOD | Enzyme entrapment in a carbon felt modified by pyrene-NH2 modified CNTs | 0.2 M phosphate buffer pH 6, | O2 reduction | 7 days half-life at 25 °C upon continuous operation of a H2/O2 biofuel cell. | [198] |
| Pyrococcus furiosus HASE | Enzyme adsorbed onto a CNT modified carbon felt electrode | 50 mM phosphate buffer pH 7, 50 °C | H2 oxidation | Voltage retains 90% of the initial value 33 h operation in a hybrid fuel cell. | [234] |
| Desulfovibrio vulgaris HASE | Enzyme embedded in viologen-based redox hydrogel modified porous carbon cloth. | 0.1 M phosphate buffer pH 7.4 | H2 oxidation | 60% of initial activity after 20 h at 0.16 V vs. SHE. | [224] |
| Desulfomicrobium baculatum HASE | Oriented covalent immobilization of the enzyme on modified CNTs | 50 mM phosphate buffer pH 7.6, 25 °C | H2 oxidation | 80% of initial activity after 1 h in the biofuel cell. | [235] |
| Desulfovibrio desulfuricans HASE | Enzyme embedded in viologen-based redox hydrogel on carbon cloth-based electrode | 0.1 M phosphate buffer pH 7.4 | H2 oxidation | 50% of initial activity after 11 h at 0.16 V vs. SHE. | [236] |
| Desulfovibrio vulgaris HASE | Enzyme embedded in viologen-based redox hydrogel and protected by catalase-GOx layer. | 0.1 M phosphate buffer pH 7.4 | H2 oxidation | 60% initial activity after 8 h in the presence of 5% O2 at 0.016 V vs. SHE. | [143] |
| Aquifex aeolicus HASE | Enzyme adsorption on CNT modified by π–π stacking with PyrNH2 | 0.2 M phosphate buffer pH 7, 25 °C | H2 oxidation | 1 month half-life at 25 °C upon continuous operation of a H2/O2 biofuel cell. | [198] |
| [NiFeSe] Desulfovibrio vulgaris Hildenborough HASE | Enzyme entrapped in methylviologen-based redox polymer films deposited on carbon cloths | 0.1 M phosphate buffer pH 7.4, room temperature | H2 oxidation | 25% and 50% of initial activity depending on the variant after 7 h in chronoamperometry experiments. | [237] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaufils, C.; Man, H.-M.; de Poulpiquet, A.; Mazurenko, I.; Lojou, E. From Enzyme Stability to Enzymatic Bioelectrode Stabilization Processes. Catalysts 2021, 11, 497. https://doi.org/10.3390/catal11040497
Beaufils C, Man H-M, de Poulpiquet A, Mazurenko I, Lojou E. From Enzyme Stability to Enzymatic Bioelectrode Stabilization Processes. Catalysts. 2021; 11(4):497. https://doi.org/10.3390/catal11040497
Chicago/Turabian StyleBeaufils, Charlène, Hiu-Mun Man, Anne de Poulpiquet, Ievgen Mazurenko, and Elisabeth Lojou. 2021. "From Enzyme Stability to Enzymatic Bioelectrode Stabilization Processes" Catalysts 11, no. 4: 497. https://doi.org/10.3390/catal11040497
APA StyleBeaufils, C., Man, H.-M., de Poulpiquet, A., Mazurenko, I., & Lojou, E. (2021). From Enzyme Stability to Enzymatic Bioelectrode Stabilization Processes. Catalysts, 11(4), 497. https://doi.org/10.3390/catal11040497