
Advancements on Basic Working Principles of Photo-Driven Oxidative Degradation of Organic Substrates over Pristine and Noble Metal-Modified TiO2. Model Case of Phenol Photo Oxidation
.jpg)


 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
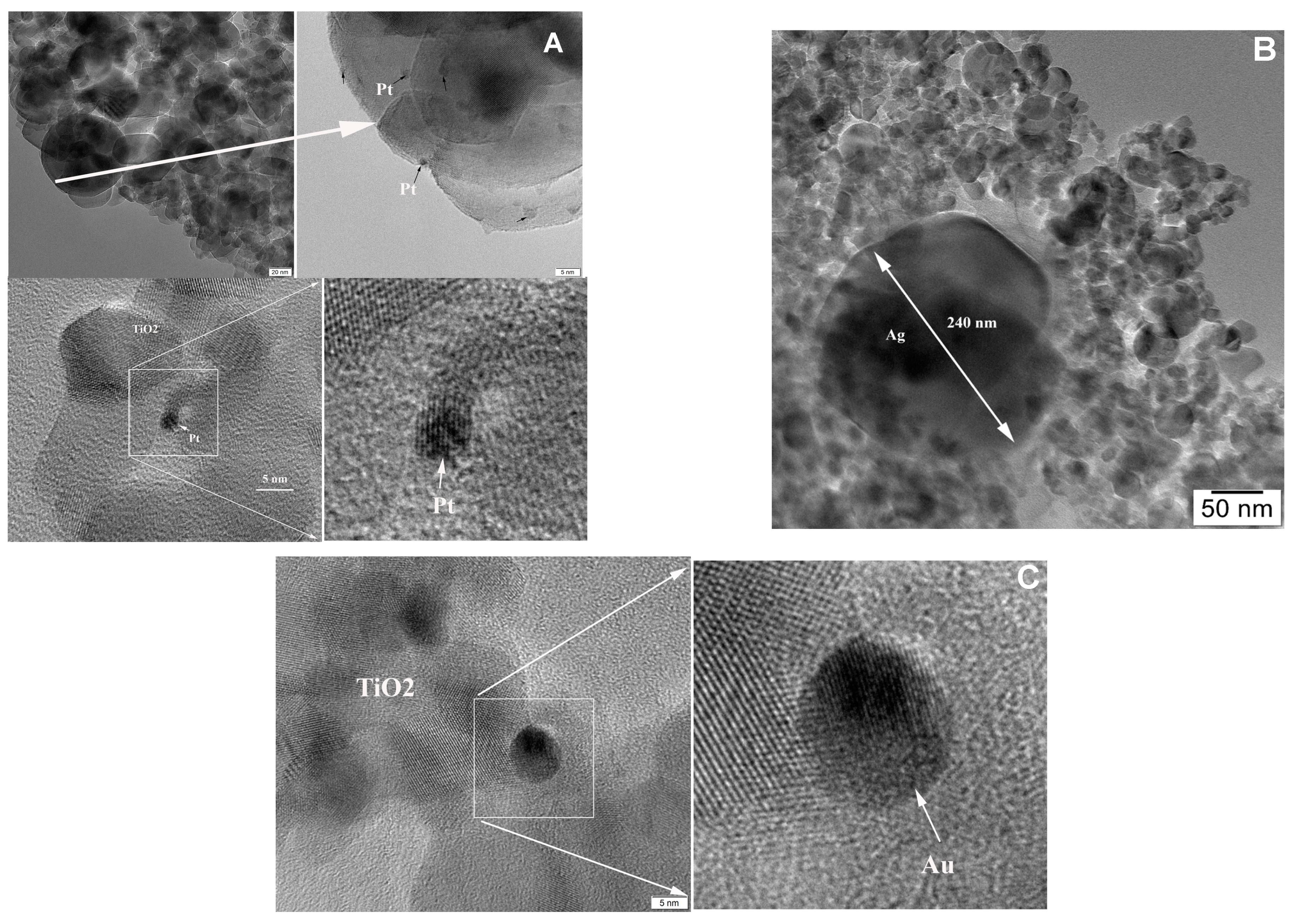
2.1. Characterization Data
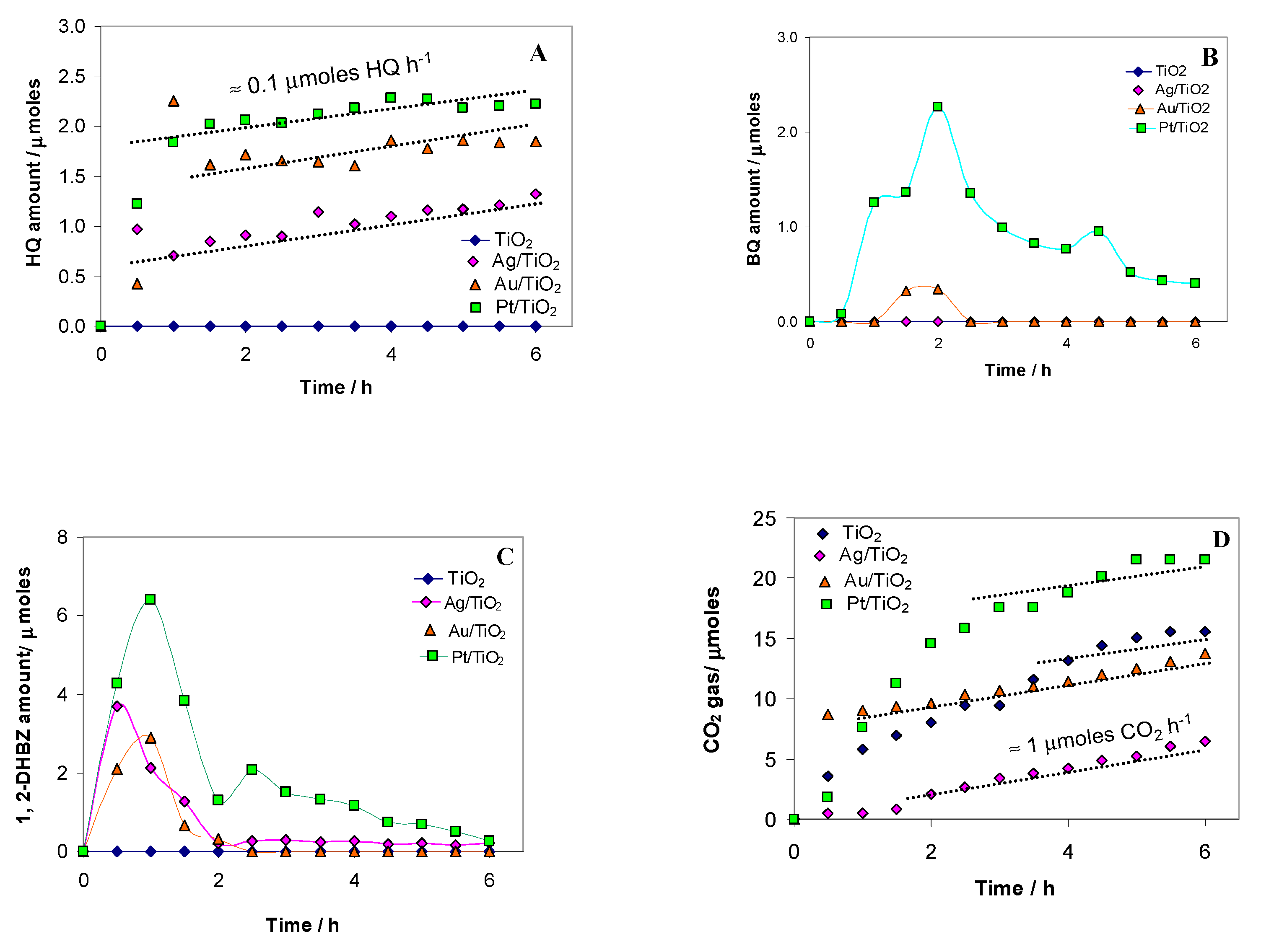
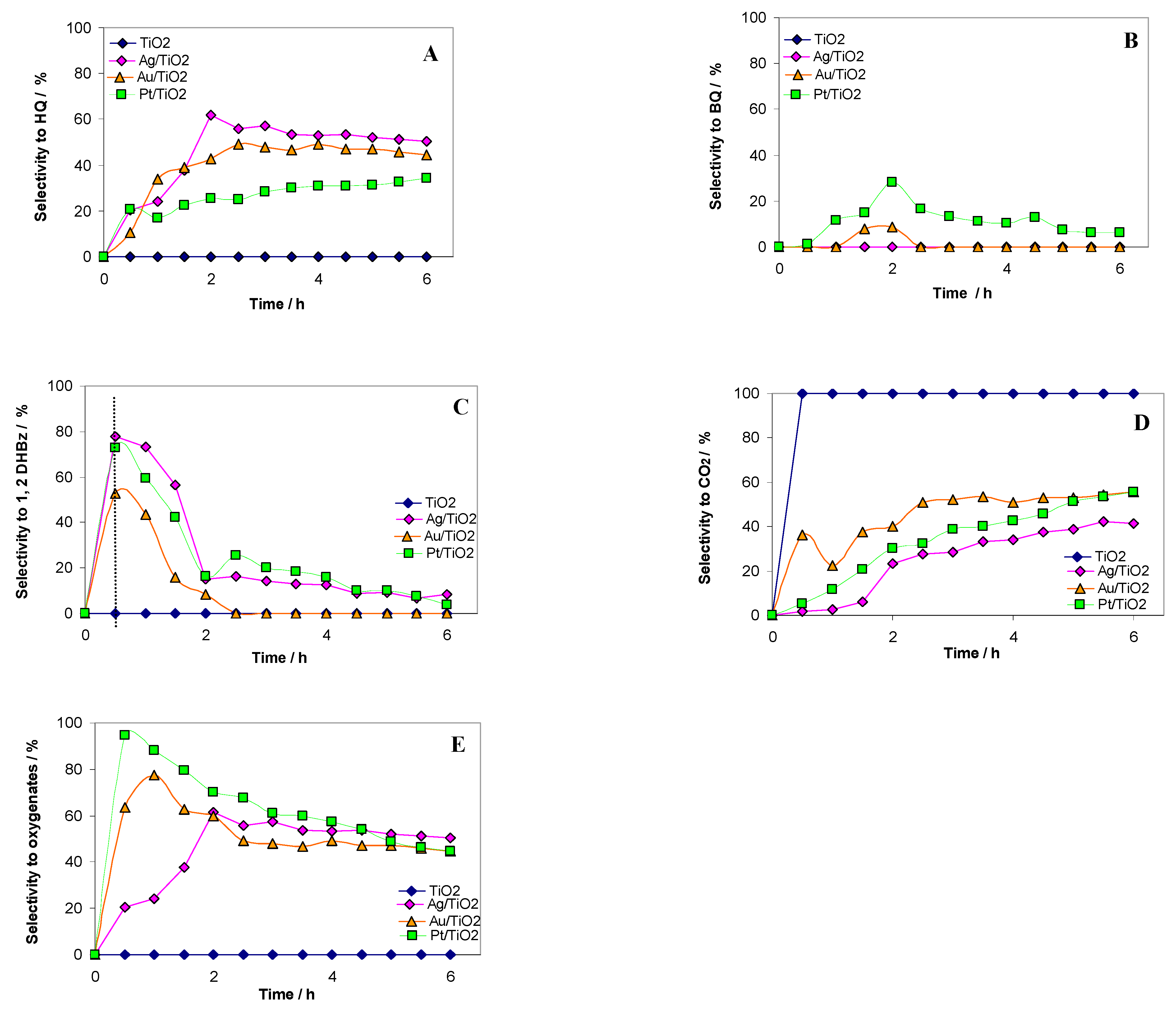
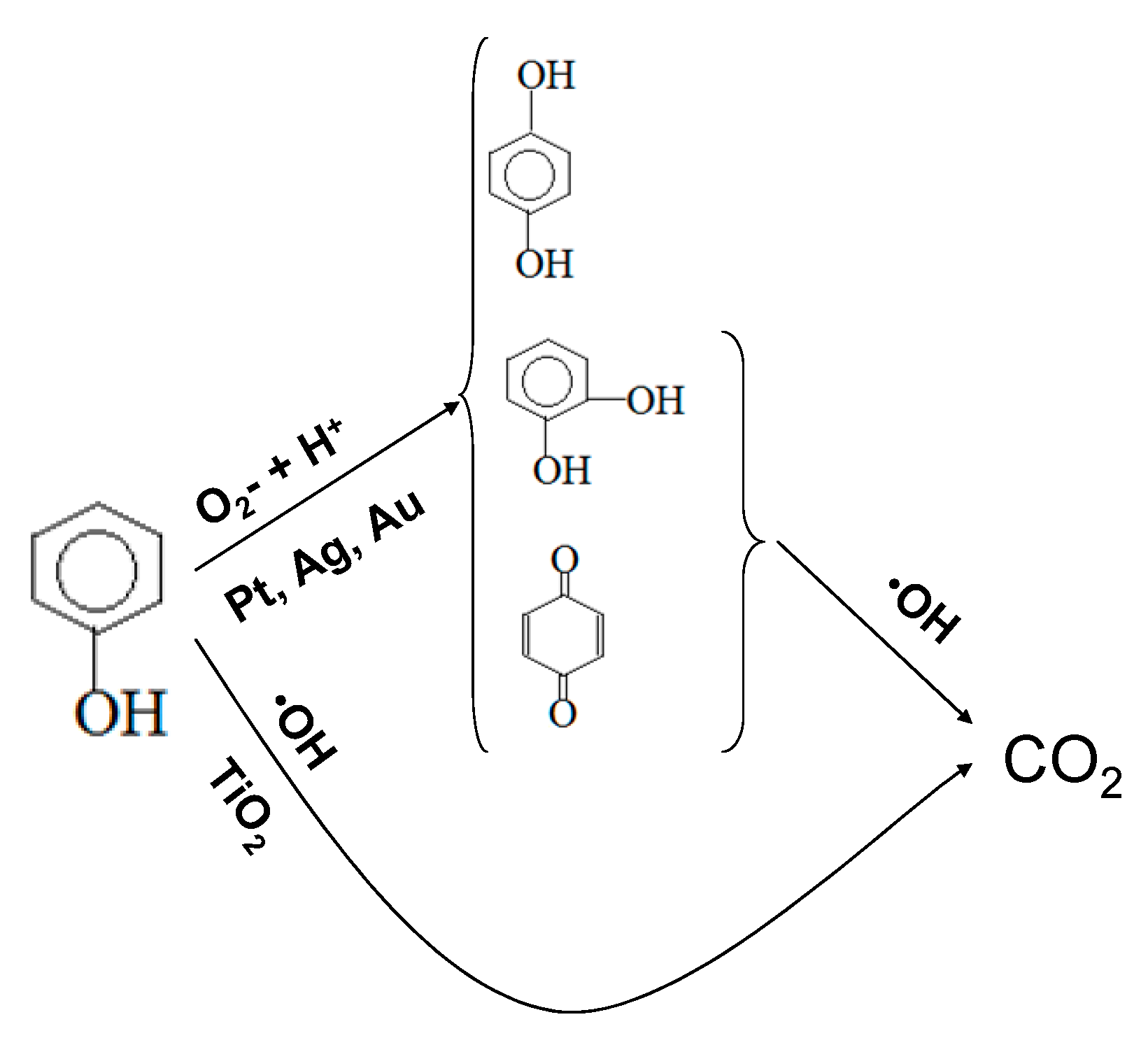
2.2. Photocatalytic Test Results
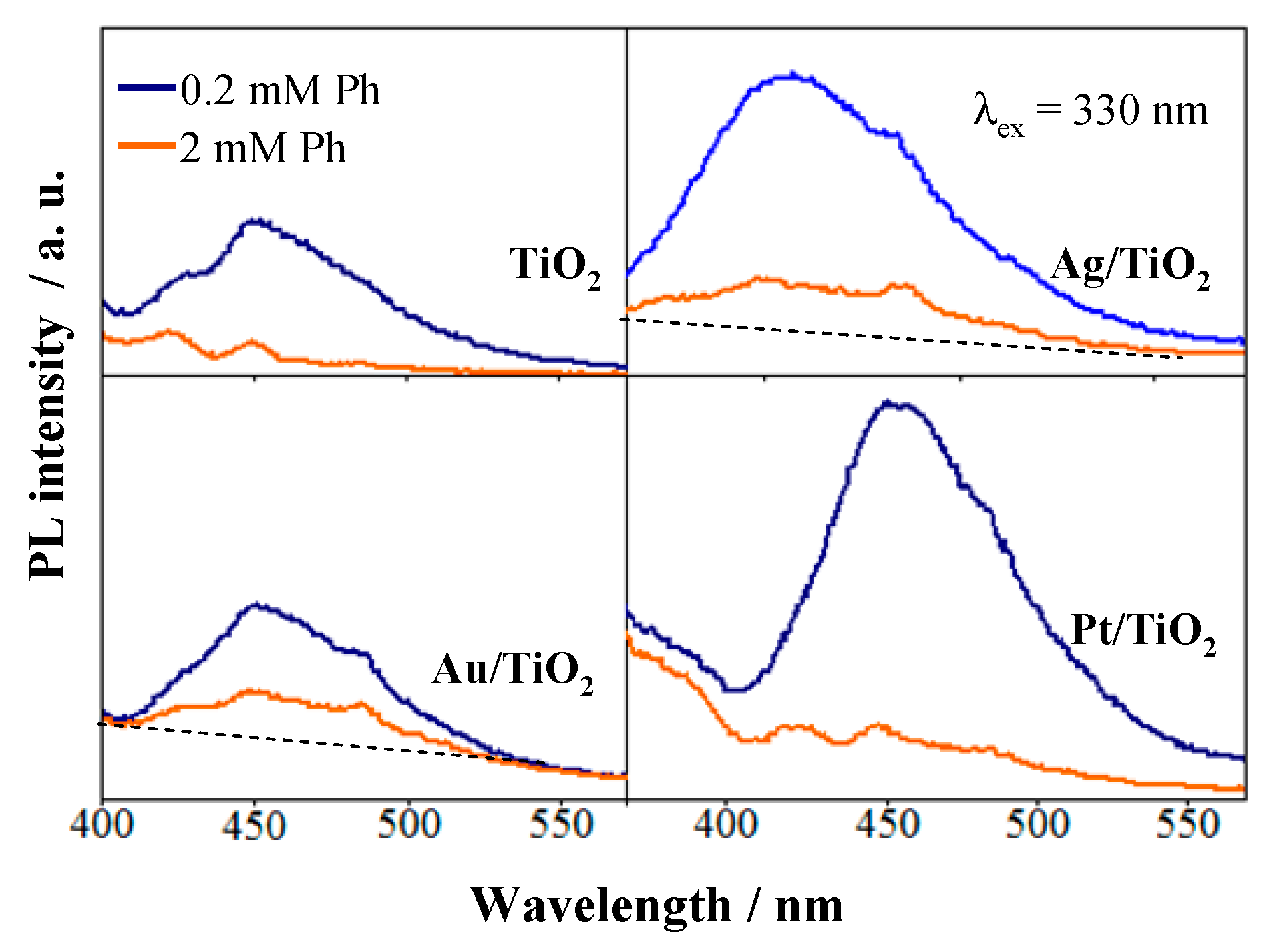
2.3. Noble Metals Role in Charge Separation and ROS Generation
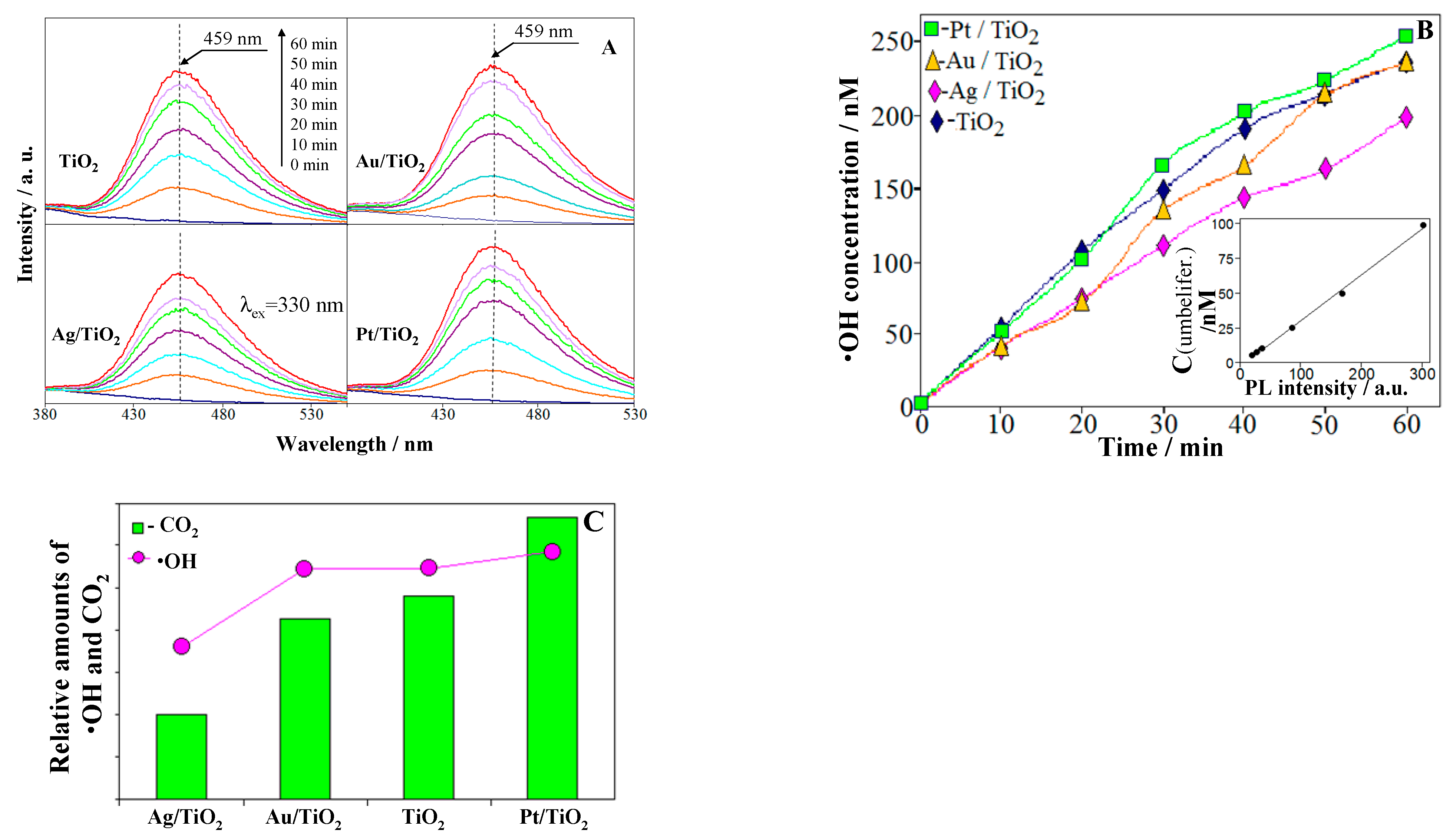
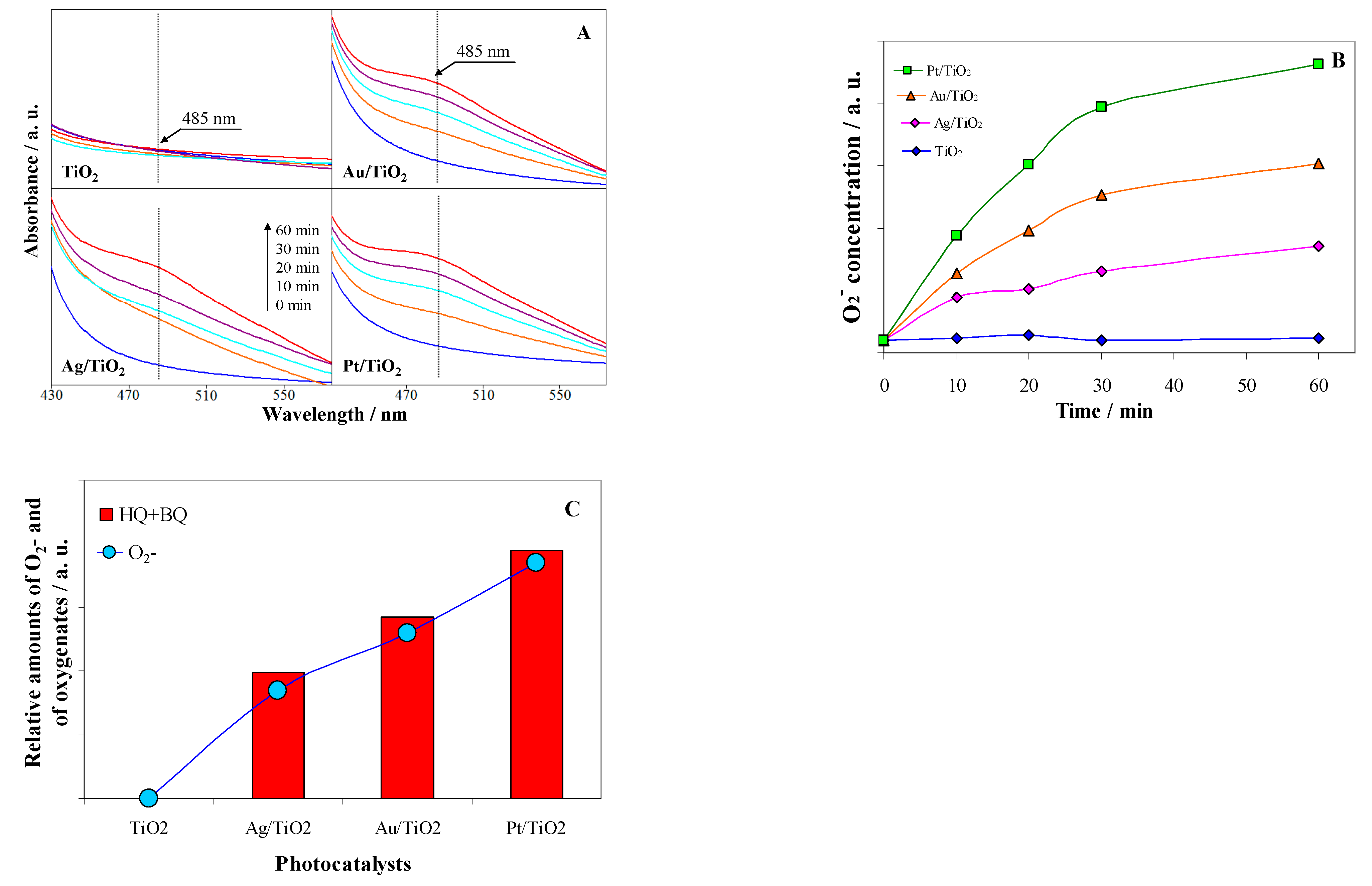
2.4. ROS Formation on Bare and Metal-Loaded TiO2
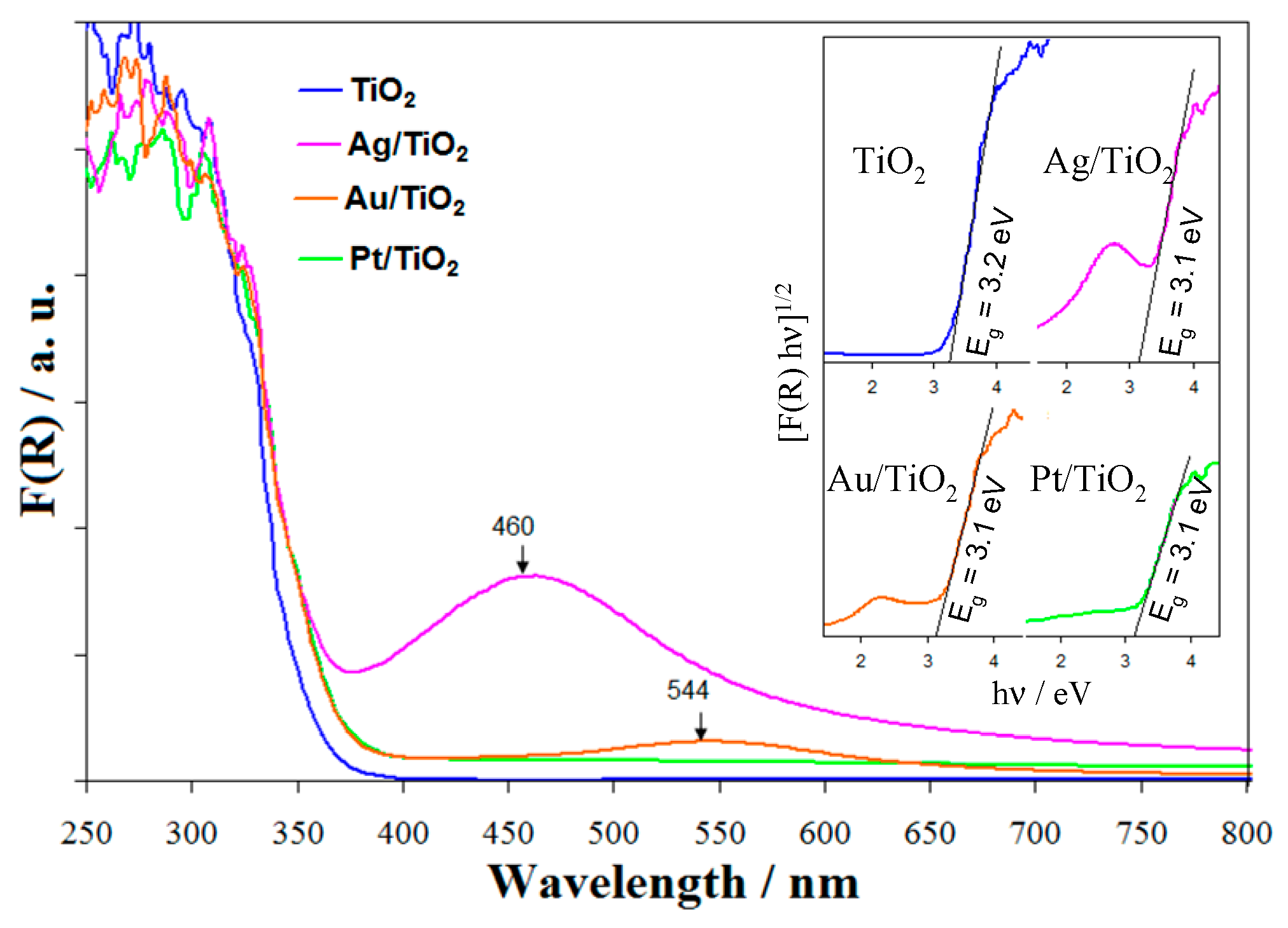
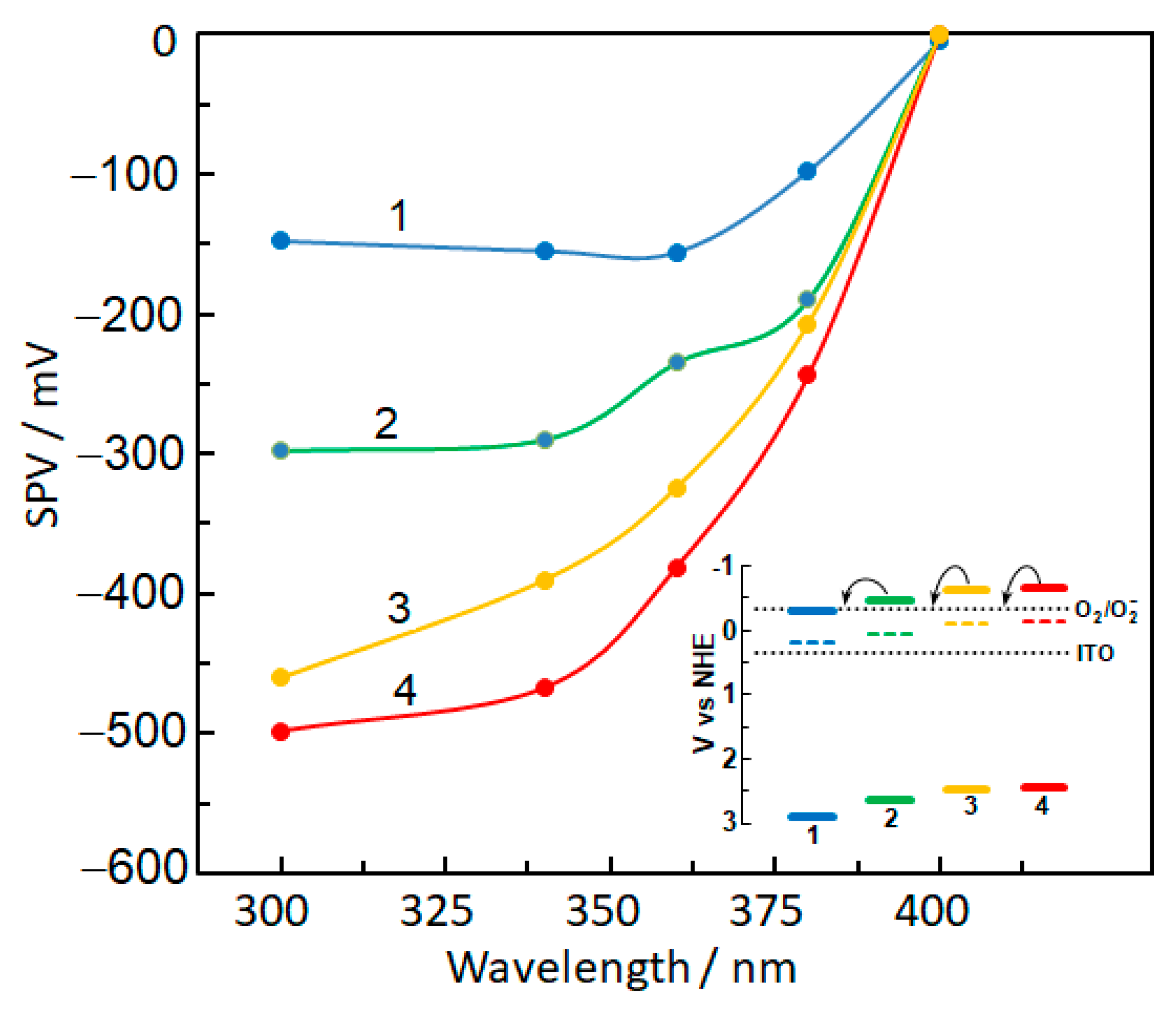
2.5. Photoelectric Properties of Bare and Metal-Modified TiO2
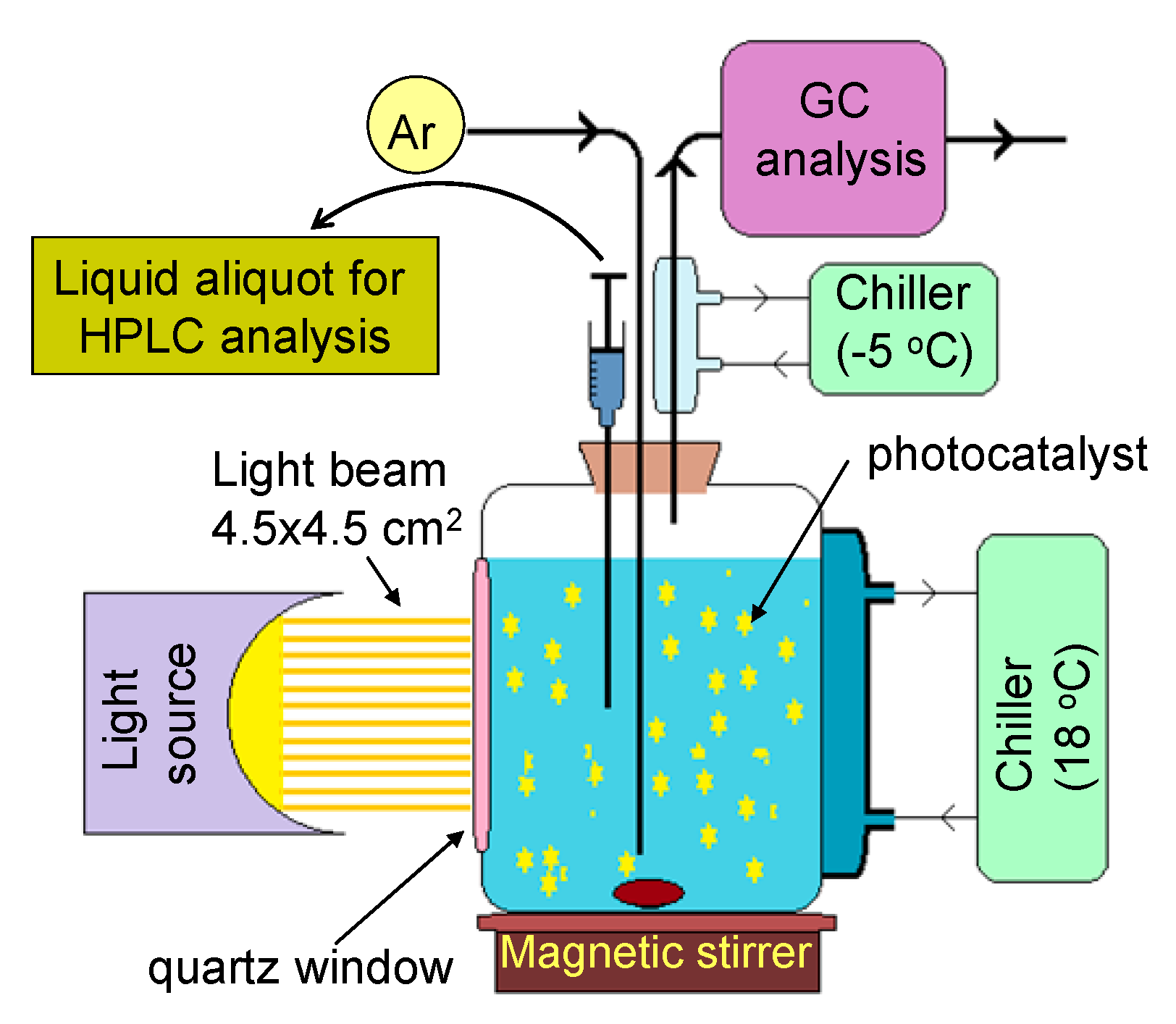
3. Experimental
3.1. Preparation of Catalytic Materials
3.2. Detection of ·OH Radicals
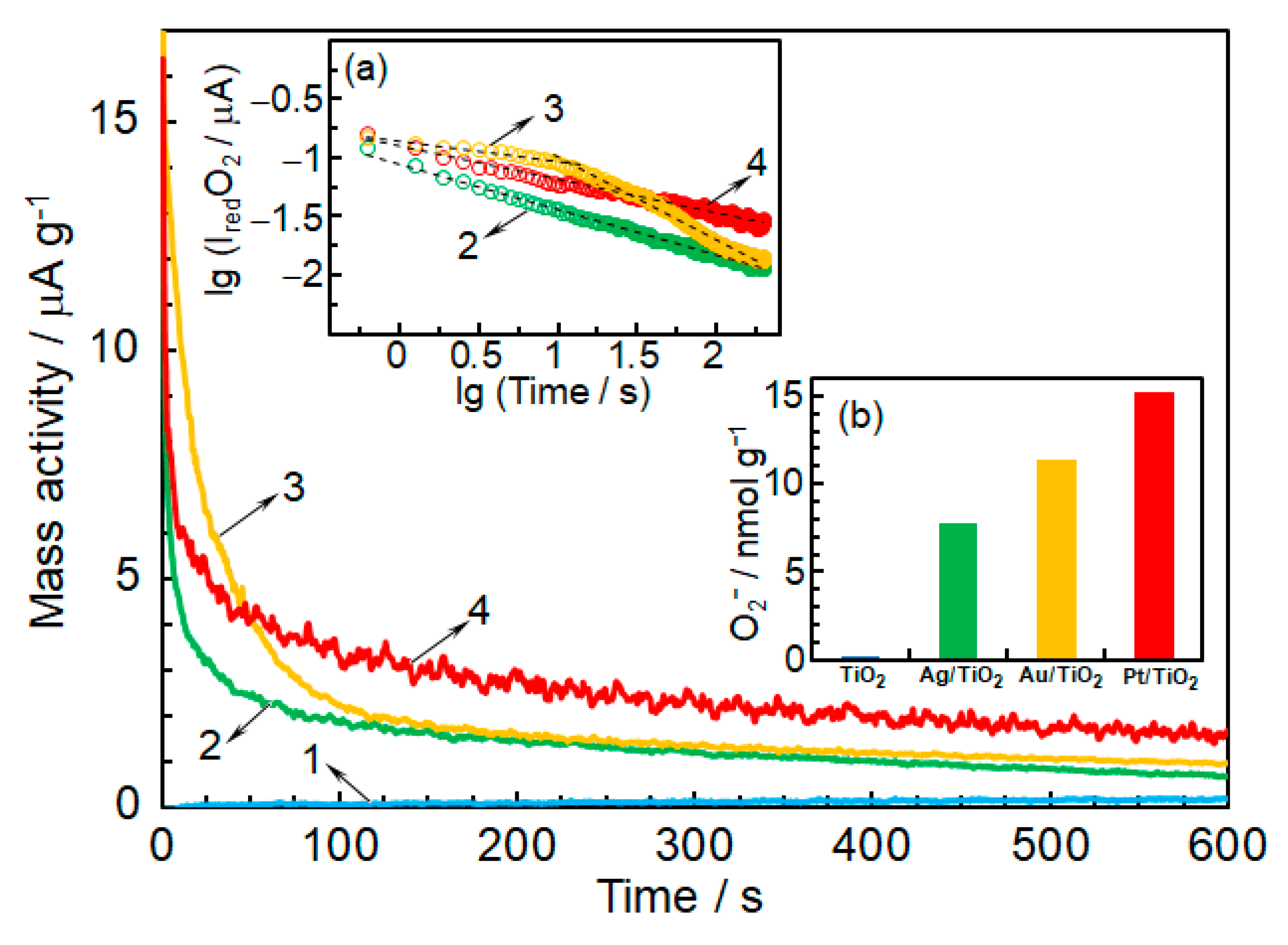
3.3. Detection of O2−
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, X.; Xie, J.; Jiang, C.; Yu, J.; Zhang, P. Review on design and evaluation of environmental photocatalysts. Front. Environ. Sci. Eng. 2018, 12, 14. [Google Scholar] [CrossRef]
- Paz, Y. Application of TiO2 photocatalysis for air treatment: Patents’ overview. Appl. Catal. B 2010, 99, 448–460. [Google Scholar] [CrossRef]
- Shaham-Waldmann, N.; Paz, Y. Away from TiO2: A critical minireview on the developing of new photocatalysts for degradation of contaminants in water. Mat. Sci. Semicond. Process. 2016, 42, 72–80. [Google Scholar] [CrossRef]
- Di Paola, A.; García-López, E.; Marcì, G.; Palmisano, L. A survey of photocatalytic materials for environmental remediation. J. Hazard. Mater. 2012, 211, 3–29. [Google Scholar] [CrossRef]
- Chen, L.; Tang, J.; Song, L.N.; Chen, P.; He, J.; Au, C.T.; Yin, S.F. Heterogeneous photocatalysis for selective oxidation of alcohols and hydrocarbons. Appl. Catal. B 2019, 242, 379–388. [Google Scholar] [CrossRef]
- Meng, S.; Ye, X.; Zhang, J.; Fu, X.; Chen, S.; Chen, S. Effective use of photogenerated electrons and holes in a system: Photocatalytic selective oxidation of aromatic alcohols to aldehydes and hydrogen production. J. Catal. 2018, 367, 159–170. [Google Scholar] [CrossRef]
- Kou, J.; Lu, C.; Wang, J.; Chen, Y.; Xu, Z.; Varma, R.S. Selectivity Enhancement in Heterogeneous Photocatalytic Transformations. Chem. Rev. 2017, 117, 1445–1514. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Cronin, S.B. A Review of Surface Plasmon Resonance-Enhanced Photocatalysis. Adv. Funct. Mater. 2013, 23, 1612–1619. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.L.; Reller, A. Photoinduced reactivity of titanium dioxide. Progress Solid State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Fujishima, A.; Rao, T.N.; Tryk, D.A. Titanium Dioxide Photocatalysis. J. Photochem. Photobiol. C Photochem. Rev. 2000, 1, 1–21. [Google Scholar] [CrossRef]
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 Surfaces: Principles, Mechanisms and Selected Results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Anastasescu, C.; Negrila, C.; Angelescu, D.G.; Atkinson, I.; Anastasescu, M.; Spataru, N.; Zaharescu, M.; Balint, I. Particularities of photocatalysis and formation of reactive oxygen species on insulators and semiconductors: Cases of SiO2, TiO2 and their composite SiO2-TiO2. Catal. Sci. Technol. 2018, 8, 5657–5668. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D.A. Heterogeneous photocatalysis: From water photolysis to applications in environmental cleanup. Int. J. Hydrog. Energy 2007, 32, 2664–2672. [Google Scholar] [CrossRef]
- Bumajdad, A.; Madkour, M. Understanding the superior photocatalytic activity of noble metals modified titania under UV and visible light irradiation. Phys. Chem. Chem. Phys. 2014, 16, 7146–7158. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Lee, S.W. Photocatalytic activity of Ag, Au-Deposited TiO2 nanoparticles prepared by sonochemical reduction method. Surface Rev. Lett. 2010, 17, 21–26. [Google Scholar] [CrossRef]
- Tian, Y.; Tatsuma, T. Mechanisms and Applications of Plasmon-Induced Charge Separation at TiO2 Films Loaded with Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127, 7632–7637. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Preparation of Au/TiO2 with Metal Cocatalysts Exhibiting Strong Surface Plasmon Resonance Effective for Photoinduced Hydrogen Formation under Irradiation of Visible Light. ACS Catal. 2013, 3, 79–85. [Google Scholar] [CrossRef]
- Zhai, W.; Xue, S.; Zhu, A.; Luo, Y.; Tian, Y. Plasmon-Driven Selective Oxidation of Aromatic Alcohols to Aldehydes in Water with Recyclable Pt/TiO2 Nanocomposites. Chem. Cat. Chem. 2011, 3, 127–130. [Google Scholar] [CrossRef]
- Osterloh, F.E. Nanoscale Effects in Water Splitting Photocatalysis. Top. Curr. Chem. 2016, 371, 105–142. [Google Scholar] [CrossRef]
- Yoffe, A.D. Semiconductor quantum dots and related systems: Electronic, optical, luminescence and related properties of low dimensional systems. Adv. Phys. 2001, 50, 1–208. [Google Scholar] [CrossRef]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef]
- Suh, M.; Bagus, P.S.; Pak, S.; Rosynek, M.P.; Lunsford, J.H. Reactions of Hydroxyl Radicals on Titania, Silica, Alumina and Gold Surfaces. J. Phys. Chem. B 2000, 104, 2736–2742. [Google Scholar] [CrossRef]
- Goto, H.; Hanada, Y.; Ohno, T.; Matsumura, M. Quantitative Analysis of Superoxide Ion and Hydrogen Peroxide Produced from Molecular Oxygen on Photoirradiated TiO2 Particles. J. Catal. 2004, 225, 223–229. [Google Scholar] [CrossRef]
- Zhu, S.; Liang, S.; Gu, Q.; Xie, L.; Wang, J.; Ding, Z.; Liu, P. Effect of Au supported TiO2 with dominant exposed facets on the visible-light photocatalytic activity. Appl. Catal. B 2012, 119, 146–155. [Google Scholar] [CrossRef]
- Watanabe, K.; Menzel, D.; Nilius, N.; Freund, H.J. Photochemistry on Metal Nanoparticles. Chem. Rev. 2006, 106, 4301–4320. [Google Scholar] [CrossRef]
- Ueno, K.; Misawa, H. Surface plasmon-enhanced photochemical reactions. J. Photochem. Photobiol. C Photochem. Rev. 2013, 15, 31–52. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Tung, R.T. Recent Advances in Schottky Barrier Concepts. Mat. Sci. Eng. R 2001, 35, 1–138. [Google Scholar] [CrossRef]
- Henderson, M.A. A surface science perspective on TiO2 photocatalysis. Surface Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Fan, H.; Jiang, T.; Li, H.; Wang, D.; Wang, L.; Zhai, J.; He, D.; Wang, P.; Xie, T. Effect of BiVO4 Crystalline Phases on the Photoinduced Carriers Behavior and Photocatalytic Activity. J. Phys. Chem. C 2012, 116, 2425–2430. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, T.; Jiang, T.; Wei, X.; Pang, S.; Wang, X.; Wang, D. Surface photovoltage characterization of a ZnO nanowire array/CdS quantum dot heterogeneous film and its application for photovoltaic devices. Nanotechnology 2009, 20, 155707. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wang, J.; Zhao, J.; Guo, L.; Osterloh, F.E. Structure defects in g-C3N4 limit visible light driven hydrogen evolution and photovoltage. J. Mat. Chem. A 2014, 2, 20338–20344. [Google Scholar] [CrossRef]
- Duzhko, V.V.; Timoshenko, V.; Koch, F.; Dittrich, T. Photovoltage in nanocrystalline porous TiO2. Phys. Rev. B 2001, 64, 075204. [Google Scholar] [CrossRef]
- Osterloh, F.E.; Holmes, M.A.; Zhao, J.; Chang, L.; Kawula, S.; Roehling, J.D.; Moule, A.J. P3HT:PCBM Bulk-Heterojunctions: Observing Interfacial and Charge Transfer States with Surface Photovoltage Spectroscopy. J. Phys. Chem. C 2014, 118, 14723–14731. [Google Scholar] [CrossRef]
- Kronik, L.; Shapira, Y. Surface photovoltage spectroscopy of semiconductor structures: At the crossroads of physics, chemistry and electrical engineering. Surf. Interface Anal. 2001, 31, 954–965. [Google Scholar] [CrossRef]
- Frame, F.A.; Townsend, T.K.; Chamousis, R.L.; Sabio, E.M.; Dittrich, T.; Browning, N.D.; Osterloh, F.E. Photocatalytic Water Oxidation with Nonsensitizied IrO2 Nanocrystals under Visible and UV Light. J. Am. Chem. Soc. 2011, 133, 7264–7267. [Google Scholar] [CrossRef]
- Shi, F.; Baker, L.R.; Hervier, A.; Somorjai, G.A.; Komvopoulos, K. Tuning the electronic structure of titanium oxide support to enhance the electrochemical activity of platinum nanoparticles. Nano Lett. 2013, 13, 4469–4474. [Google Scholar] [CrossRef]
- Mirceski, V.; Guziejewski, D.; Bozem, M.; Bogeski, I. Characterizing electrode reactions by multisampling the current in square-wave voltammetry. Electrochim. Acta 2016, 213, 520–528. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Valentine, J.S. How super is superoxide? Acc. Chem. Res. 1981, 14, 393–400. [Google Scholar] [CrossRef]
- TBui, D.; Kimura, A.; Ikeda, S.; Matsumura, M. Determination of Oxygen Sources for Oxidation of Benzene on TiO2 Photocatalysts in Aqueous Solutions Containing Molecular Oxygen. J. Am. Chem. Soc. 2010, 132, 8453–8458. [Google Scholar] [CrossRef]
- Ilie, A.G.; Scarisoreanu, M.; Dutu, E.; Dumitrache, F.; Banici, A.M.; Fleaca, C.T.; Vasile, E.; Mihailescu, I. Study of phase development and thermal stability in as synthesized TiO2 nanoparticles by laser pyrolysis: Ethylene uptake and oxygen enrichment. Appl. Surf. Sci. 2018, 427, 798–806. [Google Scholar] [CrossRef]
- Gupta, N.; Bansal, P.; Pal, B. Metal ion-TiO2 nanocomposites for the selective photooxidation of benzene to phenol and cycloalkanol to cycloalkanone. J. Experim. Nanosci. 2015, 10, 148–160. [Google Scholar] [CrossRef]
- State, R.; Scurtu, M.; Miyazaki, A.; Papa, F.; Atkinson, I.; Munteanu, C.; Balint, I. Influence of metal-support interaction on nitrate hydrogenation over Rh and Rh-Cu nanoparticles dispersed on Al2O3 and TiO2 supports. Arab. J. Chem. 2017, 10, 975–984. [Google Scholar] [CrossRef]
- Preda, S.; Anastasescu, C.; Balint, I.; Umek, P.; Sluban, M.; Negrila, C.C.; Angelescu, D.G.; Bratan, V.; Rusu, A.; Zaharescu, M. Charge separation and ROS generation on tubular sodium titanates exposed to simulated solar light. Appl. Surf. Sci. 2019, 470, 1053–1063. [Google Scholar] [CrossRef]
- Anastasescu, C.; Zaharescu, M.; Angelescu, D.; Munteanu, C.; Bratan, V.; Spataru, T.; Negrila, C.; Spataru, N.; Balint, I. Defect-related light absorption, photoluminescence and photocatalytic activity of SiO2 with tubular morphology. Solar Energ. Mater. Solar Cells 2017, 159, 325–335. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Cryst. Size /nm a | Metal Content /at.% (wt%) | Metal Particle Size /nm | Ti3+/ at. % f | Oxid. State g | ||
|---|---|---|---|---|---|---|---|
| EDAX b | XPS c | TEM d | Chem. e | ||||
| TiO2 | 18.8 | - | - | - | - | 0 | - |
| Ag/TiO2 | 19.3 | 0.04 (2.7) | 0.04 (2.7) | >50 | - | 22.9 | Ag0 |
| Au/TiO2 | 19.8 | 0.01 (2.5) | 0.02 (5.0) | 6 | - | 10.9 | Au0 |
| Pt/TiO2 | 20.6 | 0.01 (2.4) | 0.002 (4.8) | 2 | 1 | 8.8 | Pt0 |
| Photocatalysts | C Inlet a | C Outlet a | ||||||
|---|---|---|---|---|---|---|---|---|
| Ph/μmoles | Ph/μmoles | CO2/μmoles | HQ/μmoles | BQ/μmoles | 1, 2 DHBz/μmoles | Carbon Balance/ % | Phenol Conversion/% b | |
| TiO2 | 362.9 | 331.5 | 15.6 | 0 | 0 | 0 | 95.6 | 8.7 |
| Ag/TiO2 | 369.8 | 324.5 | 6.5 | 7.9 | 0 | 1.3 | 92.0 | 12.2 |
| Au/TiO2 | 356.2 | 324.6 | 13.8 | 11.1 | 0 | 0 | 98.1 | 8.9 |
| Pt/TiO2 | 367.9 | 309.7 | 21.6 | 13.3 | 0.4 | 1.5 | 94.2 | 15.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandulescu, A.; Anastasescu, C.; Papa, F.; Raciulete, M.; Vasile, A.; Spataru, T.; Scarisoreanu, M.; Fleaca, C.; Mihailescu, C.N.; Teodorescu, V.S.; et al. Advancements on Basic Working Principles of Photo-Driven Oxidative Degradation of Organic Substrates over Pristine and Noble Metal-Modified TiO2. Model Case of Phenol Photo Oxidation. Catalysts 2021, 11, 487. https://doi.org/10.3390/catal11040487
Sandulescu A, Anastasescu C, Papa F, Raciulete M, Vasile A, Spataru T, Scarisoreanu M, Fleaca C, Mihailescu CN, Teodorescu VS, et al. Advancements on Basic Working Principles of Photo-Driven Oxidative Degradation of Organic Substrates over Pristine and Noble Metal-Modified TiO2. Model Case of Phenol Photo Oxidation. Catalysts. 2021; 11(4):487. https://doi.org/10.3390/catal11040487
Chicago/Turabian StyleSandulescu, Alexandra, Crina Anastasescu, Florica Papa, Monica Raciulete, Anca Vasile, Tanta Spataru, Monica Scarisoreanu, Claudiu Fleaca, Cristian N. Mihailescu, Valentin S. Teodorescu, and et al. 2021. "Advancements on Basic Working Principles of Photo-Driven Oxidative Degradation of Organic Substrates over Pristine and Noble Metal-Modified TiO2. Model Case of Phenol Photo Oxidation" Catalysts 11, no. 4: 487. https://doi.org/10.3390/catal11040487
APA StyleSandulescu, A., Anastasescu, C., Papa, F., Raciulete, M., Vasile, A., Spataru, T., Scarisoreanu, M., Fleaca, C., Mihailescu, C. N., Teodorescu, V. S., Spataru, N., Zaharescu, M., & Balint, I. (2021). Advancements on Basic Working Principles of Photo-Driven Oxidative Degradation of Organic Substrates over Pristine and Noble Metal-Modified TiO2. Model Case of Phenol Photo Oxidation. Catalysts, 11(4), 487. https://doi.org/10.3390/catal11040487