
Investigation of Gas Diffusion Electrode Systems for the Electrochemical CO2 Conversion

 ,
,  , , ,
, , , 
Abstract
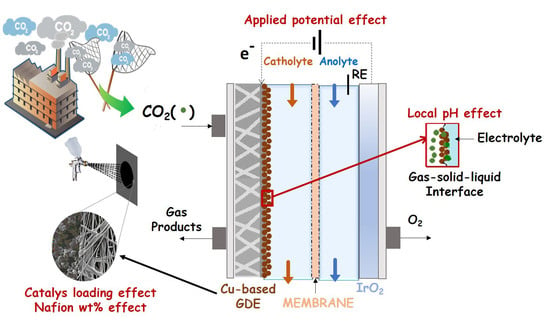
1. Introduction
2. Results and Discussion
2.1. Physico-Chemical Characterisation of Synthesised Catalysts
2.2. Electrochemical Behaviour of the Employed Catalysts
2.3. Electrochemical CO2 Reduction
2.3.1. Effect of Applied Potential
2.3.2. Comparison between the Different Studied Catalysts
2.3.3. Effect of Catalyst Loading
2.3.4. Effect of Nafion and Electrolyte Concentration
2.3.5. Considerations about the Obtained Results
2.4. Effect of Local pH
3. Materials and Methods
3.1. Materials
3.2. Synthesis of CuZnAl-Oxide Based Catalysts
3.3. Electrochemical Cell and Experimental Conditions
3.4. Modelling of GDE Local pH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guzmán, H.; Salomone, F.; Batuecas, E.; Tommasi, T.; Russo, N.; Bensaid, S.; Hernández, S. How to make sustainable CO2 conversion to Methanol: Thermocatalytic versus electrocatalytic technology. Chem. Eng. J. 2020, 127973. [Google Scholar] [CrossRef]
- Jouny, M.; Luc, W.W.; Jiao, F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57, 2165–2177. [Google Scholar] [CrossRef]
- Vennekoetter, J.-B.; Sengpiel, R.; Wessling, M. Beyond the catalyst: How electrode and reactor design determine the product spectrum during electrochemical CO2 reduction. Chem. Eng. J. 2019, 364, 89–101. [Google Scholar] [CrossRef]
- Endrődi, B.; Bencsik, G.; Darvas, F.; Jones, R.; Rajeshwar, K.; Janáky, C. Continuous-flow electroreduction of carbon dioxide. Prog. Energy Combust. Sci. 2017, 62, 133–154. [Google Scholar] [CrossRef]
- Burdyny, T.; Smith, W.A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 2019, 12, 1442–1453. [Google Scholar] [CrossRef]
- Sacco, A.; Zeng, J.; Bejtka, K.; Chiodoni, A. Modeling of gas bubble-induced mass transport in the electrochemical reduction of carbon dioxide on nanostructured electrodes. J. Catal. 2019, 372, 39–48. [Google Scholar] [CrossRef]
- Raciti, D.; Mao, M.; Wang, C. Mass transport modelling for the electroreduction of CO2 on Cu nanowires. Nanotechnology 2017, 29, 044001. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.R.; Clark, E.L.; Bell, A.T. Effects of electrolyte, catalyst, and membrane composition and operating conditions on the performance of solar-driven electrochemical reduction of carbon dioxide. Phys. Chem. Chem. Phys. 2015, 17, 18924–18936. [Google Scholar] [CrossRef] [PubMed]
- Schouten, K.J.P.; Gallent, E.P.; Koper, M.T. The influence of pH on the reduction of CO and CO2 to hydrocarbons on copper electrodes. J. Electroanal. Chem. 2014, 716, 53–57. [Google Scholar] [CrossRef]
- Ooka, H.; Figueiredo, M.C.; Koper, M.T.M. Competition between Hydrogen Evolution and Carbon Dioxide Reduction on Copper Electrodes in Mildly Acidic Media. Langmuir 2017, 33, 9307–9313. [Google Scholar] [CrossRef]
- Guzmán, H.; Russo, N.; Hernández, S. CO2 valorisation towards alcohols by Cu-based electrocatalysts: Challenges and perspectives. Green Chem. 2021, 23, 1896–1920. [Google Scholar] [CrossRef]
- Albo, J.; Perfecto-Irigaray, M.; Beobide, G.; Irabien, A. Cu/Bi metal-organic framework-based systems for an enhanced electrochemical transformation of CO2 to alcohols. J. CO2 Util. 2019, 33, 157–165. [Google Scholar] [CrossRef]
- Li, Y.C.; Wang, Z.; Yuan, T.; Nam, D.-H.; Luo, M.; Wicks, J.; Chen, B.; Li, J.; Li, F.; De Arquer, F.P.G.; et al. Binding Site Diversity Promotes CO2 Electroreduction to Ethanol. J. Am. Chem. Soc. 2019, 141, 8584–8591. [Google Scholar] [CrossRef]
- Yuan, J.; Yang, M.-P.; Hu, Q.-L.; Li, S.-M.; Wang, H.; Lu, J.-X. Cu/TiO2 nanoparticles modified nitrogen-doped graphene as a highly efficient catalyst for the selective electroreduction of CO2 to different alcohols. J. CO2 Util. 2018, 24, 334–340. [Google Scholar] [CrossRef]
- Albo, J.; Irabien, A. Cu2O-loaded gas diffusion electrodes for the continuous electrochemical reduction of CO2 to methanol. J. Catal. 2016, 343, 232–239. [Google Scholar] [CrossRef]
- Fan, L.; Xia, C.; Yang, F.; Wang, J.; Wang, H.; Lu, Y. Strategies in catalysts and electrolyzer design for electrochemical CO2 reduction toward C2+ products. Sci. Adv. 2020, 6, eaay3111. [Google Scholar] [CrossRef]
- Liu, G.; Tran-Phu, T.; Chen, H.; Tricoli, A. A Review of Metal- and Metal-Oxide-Based Heterogeneous Catalysts for Electroreduction of Carbon Dioxide. Adv. Sustain. Syst. 2018, 2, 1800028. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef]
- Marcos-Madrazo, A.; Casado-Coterillo, C.; Irabien, Á. Sustainable Membrane-Coated Electrodes for CO2 Electroreduction to Methanol in Alkaline Media. ChemElectroChem 2019, 6, 5273–5282. [Google Scholar] [CrossRef]
- Gabrielli, C.; Grand, P.P.; Lasia, A.; Perrot, H. Investigation of Hydrogen Adsorption and Absorption in Palladium Thin Films. J. Electrochem. Soc. 2004, 151, A1937–A1942. [Google Scholar] [CrossRef]
- Rodríguez, J.M.D.; Alberto, J.; Melián, H.; Peña, J.P.B.C.G. In the laboratory. Lancet 1928, 211, 408–409. [Google Scholar]
- Albo, J.; Sáez, A.; Solla-Gullón, J.; Montiel, V.; Irabien, A. Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl. Catal. B: Environ. 2015, 176–177, 709–717. [Google Scholar] [CrossRef]
- Hori, Y.; Koga, O.; Yamazaki, H.; Matsuo, T. Infrared spectroscopy of adsorbed CO and intermediate species in electrochemical reduction of CO2 to hydrocarbons on a Cu electrode. Electrochim. Acta 1995, 40, 2617–2622. [Google Scholar] [CrossRef]
- Khurana, R.; Mohanty, J.; Padma, N.; Barooah, N.; Bhasikuttan, A.C. Redox-mediated Negative Differential Resistance (NDR) Behavior in Perylenediimide Derivative: A Supramolecular Approach. Chem. Eur. J. 2012, 25, 13939–13944. [Google Scholar] [CrossRef]
- Lv, J.-J.; Jouny, M.; Luc, W.; Zhu, W.; Zhu, J.-J.; Jiao, F. A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction. Adv. Mater. 2018, 30, e1803111. [Google Scholar] [CrossRef] [PubMed]
- Dinh, C.-T.; Burdyny, T.; Kibria, G.; Seifitokaldani, A.; Gabardo, C.M.; De Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed]
- De Mot, B.; Hereijgers, J.; Duarte, M.; Breugelmans, T. Influence of flow and pressure distribution inside a gas diffusion electrode on the performance of a flow-by CO2 electrolyzer. Chem. Eng. J. 2019, 378, 122224. [Google Scholar] [CrossRef]
- Duarte, M.; De Mot, B.; Hereijgers, J.; Breugelmans, T. Electrochemical Reduction of CO2: Effect of Convective CO2 Supply in Gas Diffusion Electrodes. ChemElectroChem 2019, 6, 5596–5602. [Google Scholar] [CrossRef]
- Gutiérrez-Guerra, N.; González, J.; Serrano-Ruiz, J.; López-Fernández, E.; Valverde, J.; De Lucas-Consuegra, A. Gas-phase electrocatalytic conversion of CO2 to chemicals on sputtered Cu and Cu–C catalysts electrodes. J. Energy Chem. 2019, 31, 46–53. [Google Scholar] [CrossRef]
- Pander, J.E.; Ren, D.; Huang, Y.; Loo, N.W.X.; Hong, S.H.L.; Yeo, B.S. Understanding the Heterogeneous Electrocatalytic Reduction of Carbon Dioxide on Oxide-Derived Catalysts. ChemElectroChem 2018, 5, 219–237. [Google Scholar] [CrossRef]
- Albo, J.; Vallejo, D.; Beobide, G.; Castillo, O.; Castaño, P.; Irabien, A. Copper-Based Metal-Organic Porous Materials for CO2 Electrocatalytic Reduction to Alcohols. ChemSusChem 2017, 10, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Mandal, L.; Yang, K.R.; Motapothula, M.R.; Ren, D.; Lobaccaro, P.; Patra, A.; Sherburne, M.; Batista, V.S.; Yeo, B.S.; Ager, J.W.; et al. Investigating the Role of Copper Oxide in Electrochemical CO2 Reduction in Real Time. ACS Appl. Mater. Interfaces 2018, 10, 8574–8584. [Google Scholar] [CrossRef]
- Back, S.; Kim, H.; Jung, Y. Selective Heterogeneous CO2 Electroreduction to Methanol. ACS Catal. 2015, 5, 965–971. [Google Scholar] [CrossRef]
- Liu, X.; Schlexer, P.; Xiao, J.; Ji, Y.; Wang, L.; Sandberg, R.B.; Tang, M.; Brown, K.S.; Peng, H.; Ringe, S.; et al. pH effects on the electrochemical reduction of CO(2) towards C2 products on stepped copper. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Nie, X.; Luo, W.; Janik, M.J.; Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. J. Catal. 2014, 312, 108–122. [Google Scholar] [CrossRef]
- Velasco-Vélez, J.-J.; Jones, T.E.; Gao, D.; Carbonio, E.; Arrigo, R.; Hsu, C.-J.; Huang, Y.-C.; Dong, C.-L.; Chen, J.-M.; Lee, J.-F.; et al. The Role of the Copper Oxidation State in the Electrocatalytic Reduction of CO2 into Valuable Hydrocarbons. ACS Sustain. Chem. Eng. 2018, 7, 1485–1492. [Google Scholar] [CrossRef]
- Marepally, B.C.; Ampelli, C.; Genovese, C.; Tavella, F.; Veyre, L.; Quadrelli, E.A.; Perathoner, S.; Centi, G. Role of small Cu nanoparticles in the behaviour of nanocarbon-based electrodes for the electrocatalytic reduction of CO2. J. CO2 Util. 2017, 21, 534–542. [Google Scholar] [CrossRef]
- Zhong, H.; Fujii, K.; Nakano, Y.; Jin, F. Effect of CO2 Bubbling into Aqueous Solutions Used for Electrochemical Reduction of CO2 for Energy Conversion and Storage. J. Phys. Chem. C 2014, 119, 55–61. [Google Scholar] [CrossRef]
- Varela, A.S.; Kroschel, M.; Reier, T.; Strasser, P. Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH. Catal. Today 2016, 260, 8–13. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, H.; Yu, H.; Yu, H. Enhanced performance of gas diffusion electrode for electrochemical reduction of carbon dioxide to formate by adding polytetrafluoroethylene into catalyst layer. J. Power Sources 2015, 279, 1–5. [Google Scholar] [CrossRef]
- Garg, S.; Li, M.; Weber, A.Z.; Ge, L.; Li, L.; Rudolph, V.; Wang, G.; Rufford, T.E. Advances and challenges in electrochemical CO2 reduction processes: An engineering and design perspective looking beyond new catalyst materials. J. Mater. Chem. A 2020, 8, 1511–1544. [Google Scholar] [CrossRef]
- Wu, J.; Risalvato, F.G.; Ma, S.; Zhou, X.-D. Electrochemical reduction of carbon dioxide III. The role of oxide layer thickness on the performance of Sn electrode in a full electrochemical cell. J. Mater. Chem. A 2014, 2, 1647–1651. [Google Scholar] [CrossRef]
- Chen, C.; Sun, X.; Lu, L.; Yang, D.; Ma, J.; Zhu, Q.; Qian, Q.; Han, B. Efficient electroreduction of CO2 to C2 products over B-doped oxide-derived copper. Green Chem. 2018, 20, 4579–4583. [Google Scholar] [CrossRef]
- Zhou, Y.; Che, F.; Liu, M.; Zou, C.; Liang, Z.; De Luna, P.; Yuan, H.; Li, J.; Wang, Z.; Xie, H.; et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 2018, 10, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Woldu, A.R.; Akbar, M.B.; Louis, H.; Cong, H. A facile synthesis of Cu catalysts with multiple high-index facets for the suppression of competing H2 evolution during electrocatalytic CO2 reduction. Nanoscale 2021, 13, 3042–3048. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Luo, R.; Gold, J.I.; Yu, A.Z.; Kim, B.; Kenis, P.J.A. Carbon nanotube containing Ag catalyst layers for efficient and selective reduction of carbon dioxide. J. Mater. Chem. A 2016, 4, 8573–8578. [Google Scholar] [CrossRef]
- Komatsu, S.; Tanaka, M.; Okumura, A.; Kungi, A. Preparation of cu-solid polymer electrolyte composite electrodes and application to gas-phase electrochemical reduction of CO2. Electrochim. Acta 1995, 40, 745–753. [Google Scholar] [CrossRef]
- Gabardo, C.M.; O’Brien, C.P.; Edwards, J.P.; McCallum, C.; Xu, Y.; Dinh, C.-T.; Li, J.; Sargent, E.H.; Sinton, D. Continuous Carbon Dioxide Electroreduction to Concentrated Multi-carbon Products Using a Membrane Electrode Assembly. Joule 2019, 3, 2777–2791. [Google Scholar] [CrossRef]
- Gao, D.; Arán-Ais, R.M.; Jeon, H.S.; Cuenya, B.R. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2019, 2, 198–210. [Google Scholar] [CrossRef]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Mul, G.; Baltrusaitis, J. Electrochemical CO2 reduction on Cu2O-derived copper nanoparticles: Controlling the catalytic selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Bejtka, K.; Di Martino, G.; Sacco, A.; Castellino, M.; Fiorentin, M.R.; Risplendi, F.; Farkhondehfal, M.A.; Hernández, S.; Cicero, G.; et al. Microwave-Assisted Synthesis of Copper-Based Electrocatalysts for Converting Carbon Dioxide to Tunable Syngas. ChemElectroChem 2020, 7, 229–238. [Google Scholar] [CrossRef]
- Ramdin, M.; Morrison, A.R.T.; De Groen, M.; Van Haperen, R.; De Kler, R.; Van Den Broeke, L.J.P.; Trusler, J.P.M.; De Jong, W.; Vlugt, T.J.H. High Pressure Electrochemical Reduction of CO2 to Formic Acid/Formate: A Comparison between Bipolar Membranes and Cation Exchange Membranes. Ind. Eng. Chem. Res. 2019, 58, 1834–1847. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte | Catalyst Material | Real Applied Potential, V vs. Ag/AgCl | Current Density, mA cm−2 | Bulk pH, before/after the Co-Electrolysis | Calculated Surface pH (±0.05) |
|---|---|---|---|---|---|
| 1 M KHCO3 | Cu-06 | −1.5 | 15.6 | 8.38/8.8 | 9.37 |
| Cu-06 | 1.75 | 26.3 | 8.38/9.3 | 9.62 | |
| Cu-06 | −2 | 34.6 | 8.38/9.7 | 9.76 | |
| CuZ-06-03 | −1.5 | 11.5 | 8.38/8.9 | 9.25 | |
| CuZA-06-03-01 | −1.5 | 13.5 | 8.38/8.7 | 9.31 | |
| Cu-06 | −1.5 | 4.7 | 8.38/8.6 | 8.93 | |
| 0.1 M KHCO3 | Cu-06 (15%wt Nafion) | −1.5 | 3.5 | 8.36/7.6 | 9.28 |
| Cu-06 (30%wt Nafion) | −1.5 | 2.25 | 8.36/7.5 | 9.09 | |
| Cu-06 (45%wt Nafion) | −1.5 | 2.1 | 8.36/7.5 | 9.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzmán, H.; Zammillo, F.; Roldán, D.; Galletti, C.; Russo, N.; Hernández, S. Investigation of Gas Diffusion Electrode Systems for the Electrochemical CO2 Conversion. Catalysts 2021, 11, 482. https://doi.org/10.3390/catal11040482
Guzmán H, Zammillo F, Roldán D, Galletti C, Russo N, Hernández S. Investigation of Gas Diffusion Electrode Systems for the Electrochemical CO2 Conversion. Catalysts. 2021; 11(4):482. https://doi.org/10.3390/catal11040482
Chicago/Turabian StyleGuzmán, Hilmar, Federica Zammillo, Daniela Roldán, Camilla Galletti, Nunzio Russo, and Simelys Hernández. 2021. "Investigation of Gas Diffusion Electrode Systems for the Electrochemical CO2 Conversion" Catalysts 11, no. 4: 482. https://doi.org/10.3390/catal11040482
APA StyleGuzmán, H., Zammillo, F., Roldán, D., Galletti, C., Russo, N., & Hernández, S. (2021). Investigation of Gas Diffusion Electrode Systems for the Electrochemical CO2 Conversion. Catalysts, 11(4), 482. https://doi.org/10.3390/catal11040482