
Total Oxidation of Methane on Oxide and Mixed Oxide Ceria-Containing Catalysts

 ,
, 
 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ceria Generalities
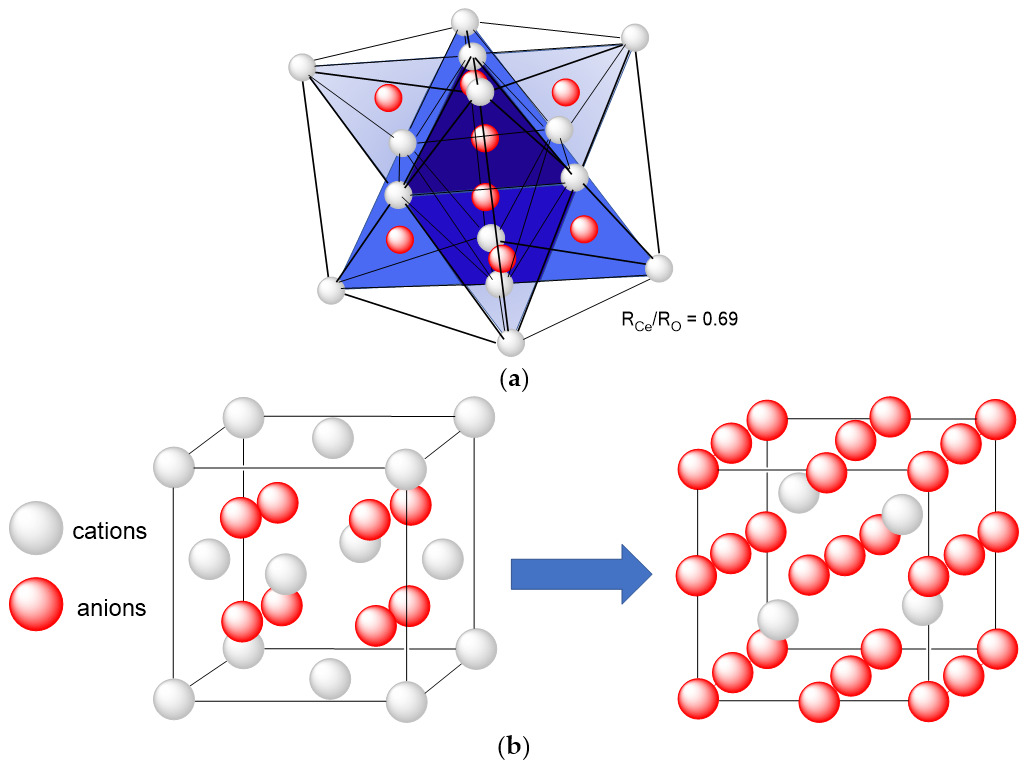
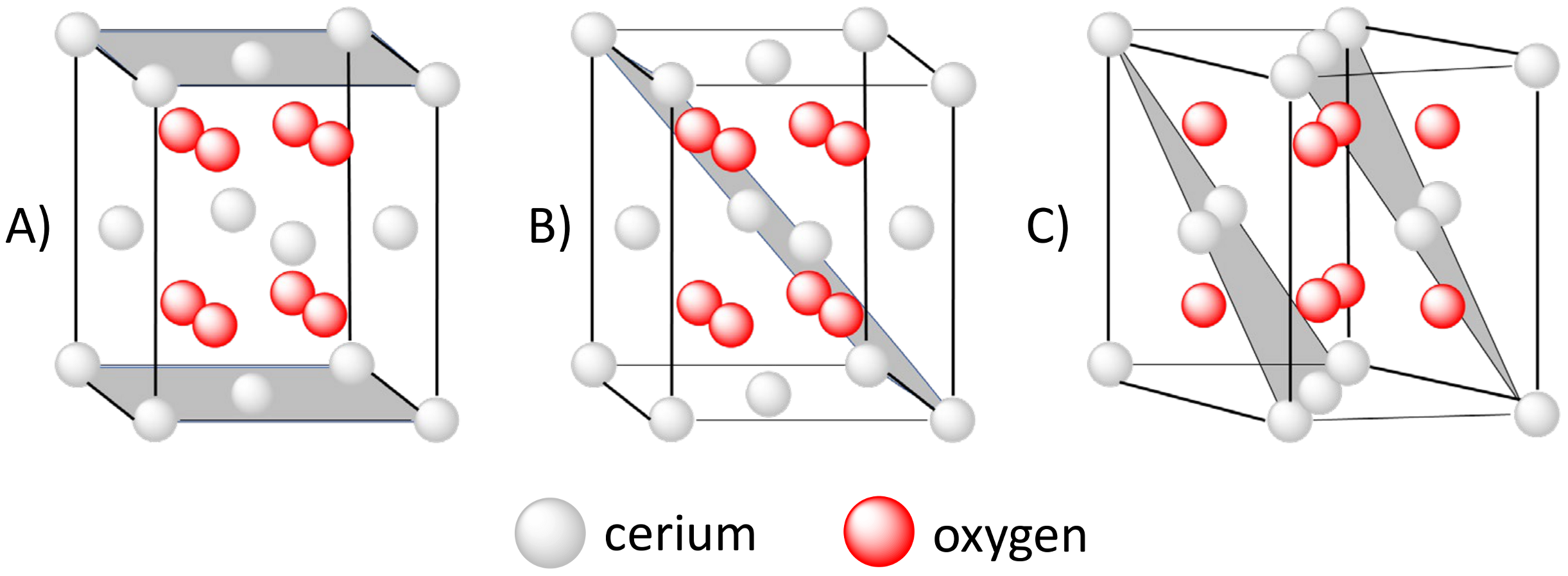
2.1. Structure
2.2. Oxygen Vacancy Defects
2.3. Morphology
3. Types of Mechanisms for Total Oxidation
4. Total Oxidation of Methane on Ceria-Containing Transition Metal Oxide-Based Catalysts
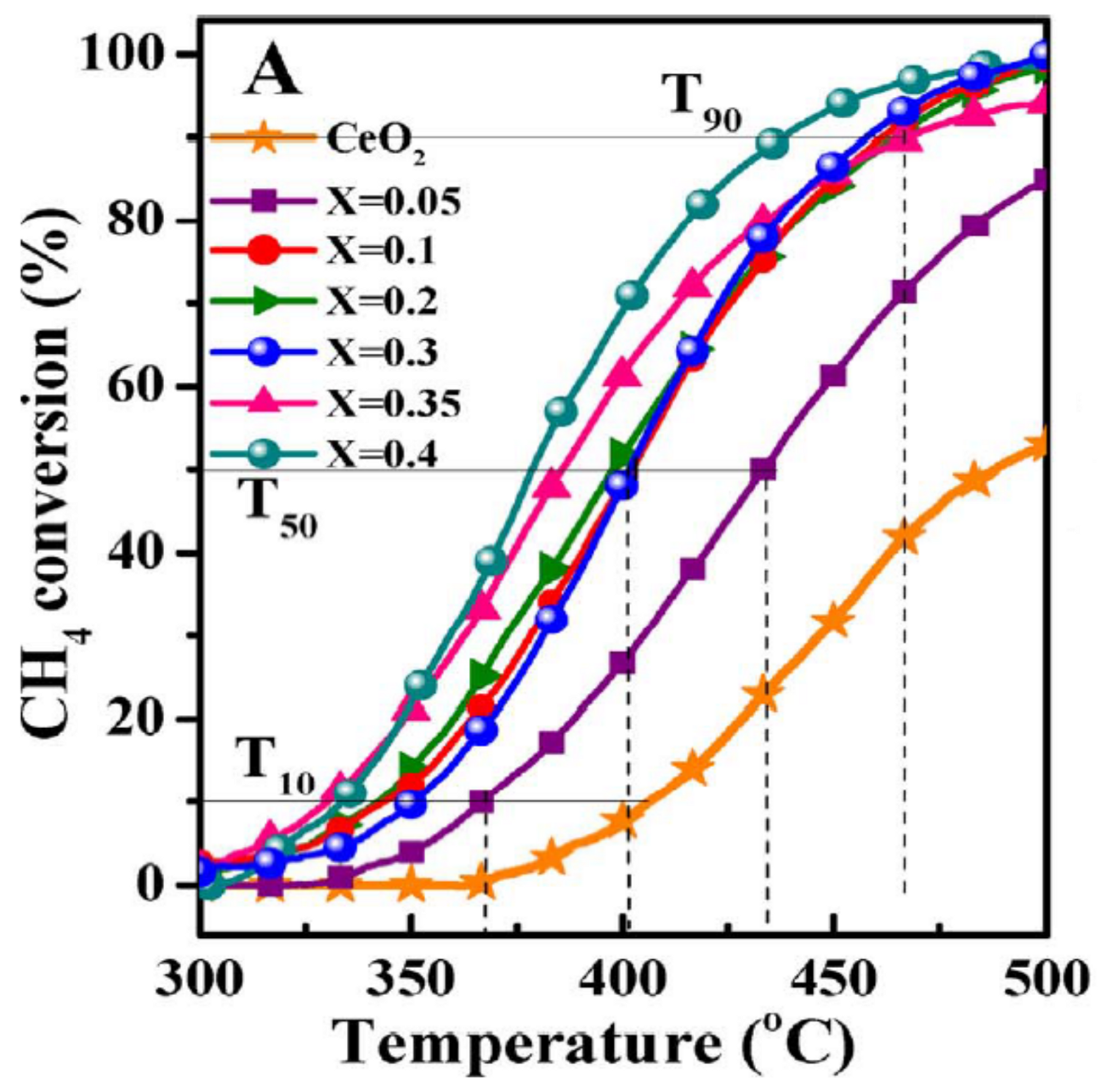
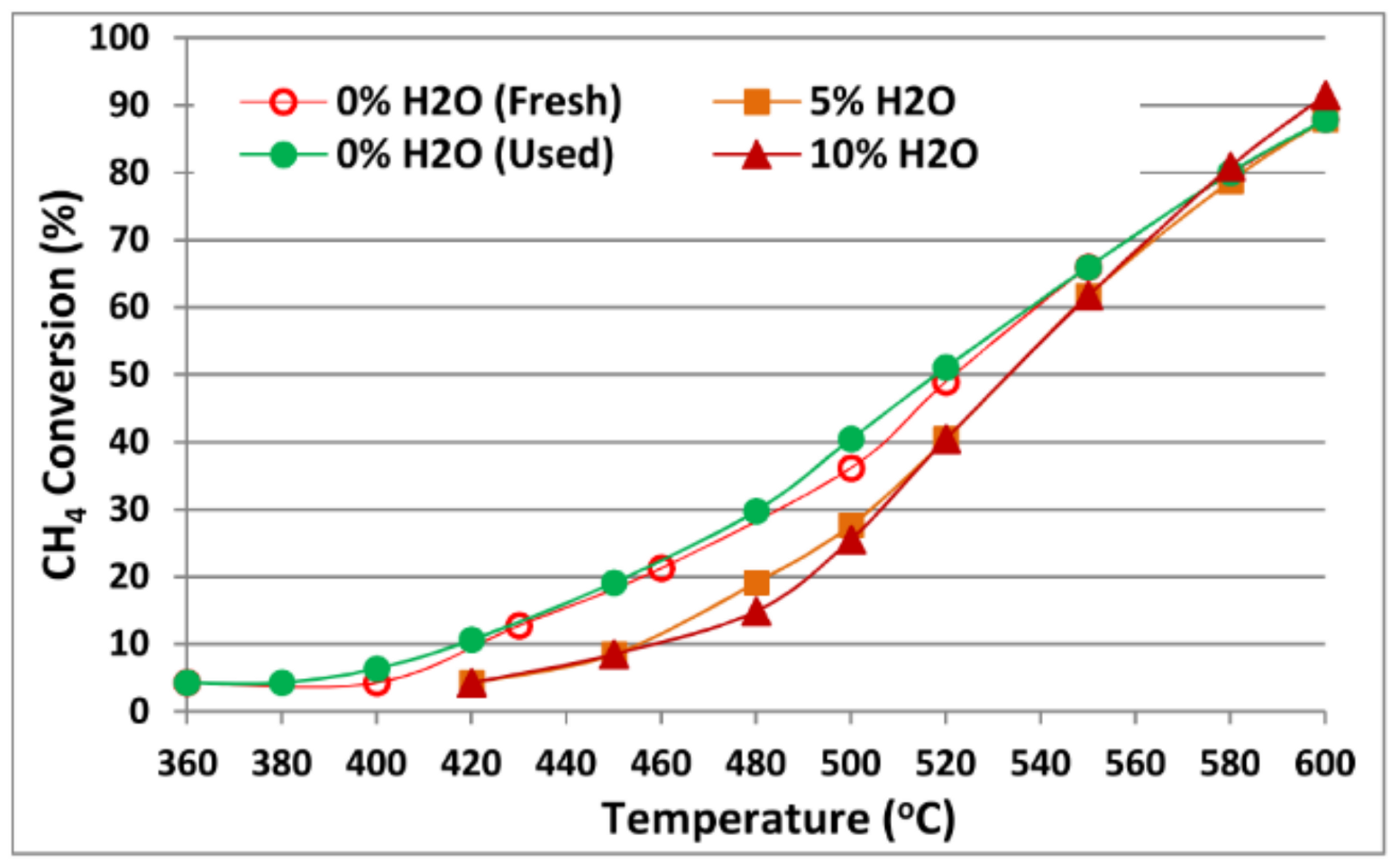
4.1. Monometallic Modified Ceria Catalysts
4.2. Bimetallic Modified Ceria Catalysts
4.3. Multimetallic Modified Ceria Catalysts
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tucker, W.G. Volatile Organic Compounds. In Indoor Air Quality Handbook; Digital Engineering Library @McGraw-Hill: New York, NY, USA, 2004. [Google Scholar]
- Heck, R.M.; Farrauto, R.J. Catalytic Pollution Control, 2nd ed.; Wiley-Interscience: New York, NY, USA, 2002. [Google Scholar]
- Lewis, A.C.; Carslaw, N.; Marriott, P.J.; Kinghorn, R.M.; Morrison, P.; Lee, A.L.; Bartle, K.D.; Pilling, M.J. A larger pool of ozone-forming carbon compounds in urban atmospheres. Nature 2000, 405, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Marcu, I.C.; Urda, A.; Popescu, I.; Hulea, V. Layered double hydroxides-based materials as oxidation catalysts. In Sustainable Nanosystems Development, Properties, and Applications; Putz, M.V., Mirica, M.C., Eds.; IGI Global: Hershey, PA, USA, 2017; Chapter 3; pp. 59–121. [Google Scholar]
- Molina, M.J.; Rowland, F.S. Stratospheric sink for chlorofluoromethanes: Chlorine atom-catalysed destruction of ozone. Nature 1974, 249, 810–812. [Google Scholar] [CrossRef]
- Amann, M.; Lutz, M. The revision of the air quality legislation in the European Union related to ground-level ozone. J. Hazard. Mater. 2000, 78, 41–62. [Google Scholar] [CrossRef]
- Ascaso, S.; Gálvez, M.E.; Da Costa, P.; Moliner, R.; Jesús Lázaro Elorri, M. Influence of gas hourly space velocity on the activity of monolithic catalysts for the simultaneous removal of soot and NOx. Comptes Rendus Chimie 2015, 18, 1007–1012. [Google Scholar] [CrossRef]
- Lazar, L.; Koeser, H.; Fechete, I.; Balasanian, I. A modelling approach of the catalytic oxidation of volatile organic compounds in the SCR-DENOx monolithic reactor. Revista de Chimie 2020, 71, 79–87. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Tropospheric air pollution: Ozone, airborne toxics, polycyclic aromatic hydrocarbons, and particles. Science 1997, 276, 1045–1051. [Google Scholar] [CrossRef]
- Stoian, M.; Lazar, L.; Uny, F.; Fechete, I. Chemical vapour deposition (CVD) technique for abatement of volatile organic compounds (VOCs). Revista Chimie 2020, 71, 97–113. [Google Scholar] [CrossRef]
- Yang, H.; Xie, P.; Ni, L.; Flower, R.J. Underestimation of CH4 emission from freshwater lakes in China. Environ. Sci. Technol. 2011, 45, 4203–4204. [Google Scholar] [CrossRef]
- Ducamp, J.; Bengaouer, A.; Baurens, P.; Fechete, I.; Turek, P.; Garin, F. Status quo of the CO2 methanation. C.R. Chimie 2018, 21, 427–469. [Google Scholar] [CrossRef]
- Merkache, R.; Fechete, I.; Maamache, M.; Bernard, M.; Turek, P.; Al-Dalama, K.; Garin, F. 3D ordered mesoporous Fe-KIT-6 catalysts for methylcyclopentane (MCP) conversion and carbon dioxide (CO2) hydrogenation for energy and environmental applications. Appl. Catal. A Gen. 2015, 504, 672–681. [Google Scholar] [CrossRef]
- Cicerone, R.J.; Oremland, R.S. Biogeochemical aspects of atmospheric methane. Glob. Biogeochem. Cycle 1988, 2, 299–327. [Google Scholar] [CrossRef]
- Simpson, I.J.; Chen, T.Y.; Blake, D.R.; Rowland, F.S. Implications of the recent fluctuations in the growth rate of tropospheric methane. Geophys. Res. Lett. 2002, 29, 117–1–117–4. [Google Scholar] [CrossRef]
- Lambert, G.; Chappelaz, J.; Foucher, J.-P.; Ramstein, G. Le Méthane et le Destin de la Terre; EDP Science: Paris, France, 2006; pp. 1–150. [Google Scholar]
- Rapport de l’Academie des Technologies—Le Methane; EDP Sciences: Paris, France, 2013; pp. 1–170.
- Fernández, J.; Marín, P.; Díez, F.V.; Ordónez, S. Coal mine ventilation air methane combustion in a catalytic reverse flow reactor: Influence of emission humidity. Fuel Process. Technol. 2015, 133, 202–209. [Google Scholar] [CrossRef][Green Version]
- Yuan, B.; Wu, X.; Chen, Y.; Huang, J.; Luo, H.; Deng, S. Adsorption of CO2, CH4, and N2 on ordered mesoporous carbon: Approach for greenhouse gases capture and biogas upgrading. Environ. Sci. Technol. 2013, 47, 5474–5480. [Google Scholar] [CrossRef]
- Urda, A.; Popescu, I.; Cacciaguerra, T.; Tanchoux, N.; Tichit, D.; Marcu, I.-C. Total oxidation of methane over rare earth cation-containing mixed oxides derived from LDH precursors. Appl. Catal. A Gen. 2013, 464–465, 20–27. [Google Scholar] [CrossRef]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. Catalytic combustion of methane over palladium-based catalysts. Catal. Rev. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- Demoulin, O.; Le Clef, B.; Navez, M.; Ruiz, P. Combustion of methane, ethane and propane and of mixtures of methane with ethane or propane on Pd/γ-Al2O3 catalysts. Appl. Catal. A Gen. 2008, 344, 1–9. [Google Scholar] [CrossRef]
- Gelin, P.; Primet, M. Complete oxidation of methane at low temperature over noble metal based catalysts: A review. Appl. Catal. B Environ. 2002, 39, 1–37. [Google Scholar] [CrossRef]
- Okal, J.; Zawadzki, M. Catalytic combustion of methane over ruthenium supported on zinc aluminate spinel. Appl. Catal. A Gen. 2013, 453, 349–357. [Google Scholar] [CrossRef]
- Raciulete, M.; Layrac, G.; Tichit, D.; Marcu, I.-C. Comparison of CuxZnAlO mixed oxide catalysts derived from multicationic and hybrid LDH precursors for methane total oxidation. Appl. Catal. A Gen. 2014, 477, 195–204. [Google Scholar] [CrossRef]
- Persson, K.; Jansson, K.; Järås, S.G. Characterisation and microstructure of Pd and bimetallic Pd–Pt catalysts during methane oxidation. J. Catal. 2007, 245, 401–414. [Google Scholar] [CrossRef]
- Fujimoto, K.-I.; Ribeiro, F.H.; Avalos-Borja, M.; Iglesia, E. Structure and reactivity of PdOx/ZrO2 catalysts for methane oxidation at low temperatures. J. Catal. 1998, 179, 431–442. [Google Scholar] [CrossRef]
- Baldwin, T.R.; Burch, R. Catalytic combustion of methane over supported palladium catalysts: II. Support and possible morphological effects. Appl. Catal. 1990, 66, 359–381. [Google Scholar] [CrossRef]
- Wong, A.P.; Kyriakidou, E.A.; Toops, T.J.; Regalbuto, J.R. The catalytic behavior of precisely synthesized Pt–Pd bimetallic catalysts for use as diesel oxidation catalysts. Catal. Today 2016, 267, 145–156. [Google Scholar] [CrossRef]
- Schwartz, W.R.; Ciuparu, D.; Pfefferle, L.D. Combustion of methane over palladium-based catalysts: Catalytic deactivation and role of the support. J. Phys. Chem. C 2012, 116, 8587–8593. [Google Scholar] [CrossRef]
- Shao, C.; Li, W.; Lin, Q.; Huang, Q.; Pi, D. Low temperature complete combustion of lean methane over cobalt-nickel mixed-oxide catalysts. Energy Technol. 2016, 5, 604–610. [Google Scholar] [CrossRef]
- Prasad, R.; Kennedy, L.A.; Ruckenstein, E. Catalytic combustion. Catal. Rev. Sci. Eng. 1984, 26, 1–58. [Google Scholar] [CrossRef]
- Zwinkels, M.F.M.; Jaras, S.G.; Menon, P.G. Catalytic materials for high-temperature combustion. Catal. Rev. Sci. Eng. 1993, 35, 319–358. [Google Scholar] [CrossRef]
- Zwinkels, M.F.M.; Jaras, S.G.; Menon, P.G. Catalytic fuel combustion in honeycomb monolith reactors. Chem. Ind. 1998, 71, 149–177. [Google Scholar]
- Groppi, G.; Cristiani, C.; Forzatti, P. BaFexAl(12−x)O19System for High-Temperature Catalytic Combustion: Physico-Chemical Characterization and Catalytic Activity. J. Catal. 1997, 168, 95–103. [Google Scholar] [CrossRef]
- Farrauto, R.J.; Heck, R.M. Environmental catalysis into the 21st century. Catal. Today 2000, 55, 179–187. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for combustion of methane and lower alkanes. Appl. Catal. A Gen. 2002, 234, 1–23. [Google Scholar] [CrossRef]
- Mouaddib, N.; Feumi-Jantou, C.; Garbbowski, E.; Primet, M. Catalytic oxidation of methane over palladium supported on alumina: Influence of the oxygen-to-methane ratio. Appl. Catal. A Gen. 1992, 87, 129–144. [Google Scholar] [CrossRef]
- Mizushima, Y.; Hori, M. Alumina aerogel for support of a methane combustion catalyst. Appl. Catal. A Gen. 1992, 88, 137–148. [Google Scholar] [CrossRef]
- Yagita, H.; Ogata, A.; Obuchi, A.; Mizumo, K. Proceeding of First Russia–Japan joint Symposium on Petroleum. Nat. Gas Petrochem. 1993, 164, 28. [Google Scholar]
- Najbar, M.; Baranska, M.; Jura, W. Low temperature oxidation of light hydrocarbons over silica supported noble metal catalysts. Catal. Today 1993, 17, 201–208. [Google Scholar] [CrossRef]
- Li, Y.; Armor, J.N. Catalytic combustion of methane over palladium exchanged zeolites. Appl. Catal. B Environ. 1994, 3, 275–282. [Google Scholar] [CrossRef]
- Ishihara, T.; Sumi, H.; Takita, Y. Palladium ion-exchanged SAPO-5 as a catalyst for low Temperature combustion of methane. Chem. Lett. 1994, 23, 1499–1502. [Google Scholar] [CrossRef]
- Muto, K.I.; Katada, N.; Niwa, M. Complete oxidation of methane on supported palladium catalyst: Support effect. Appl. Catal. A Gen. 1996, 134, 203–215. [Google Scholar] [CrossRef]
- Shen, J.; Hayes, R.E.; Semagina, N. On the contribution of oxygen from Co3O4 to the Pd-catalyzed methane combustion. Catal. Today 2021, 360, 435–443. [Google Scholar] [CrossRef]
- Lampert, J.K.; Kazi, M.S.; Farrauto, R.J. Palladium catalyst performance for methane emissions abatement from lean burn natural gas vehicles. Appl. Catal. B 1997, 14, 211–223. [Google Scholar] [CrossRef]
- Farrauto, R.J.; Hobson, M.C.; Kennelly, T.; Waterman, E.M. Catalytic chemistry of supported palladium for combustion of methane. Appl. Catal. A Gen. 1992, 81, 227–237. [Google Scholar] [CrossRef]
- Domingos, D.; Simplicio, L.M.T.; Estrela, G.S.; dos Prazeres, M.A.G.; Brandao, S.T. Catalytic combustion of methane over PdOCeO2/Al2O3 and PdO-CeO2/ZrO2 catalysts. Stud. Surf. Sci. Catal. 2007, 167, 7–12. [Google Scholar] [CrossRef]
- Lapisardi, G.; Urfels, L.; Gelin, P.; Primet, M.; Kaddouri, A.; Garbowski, E.; Toppi, S.; Tena, E. Superior catalytic behaviour of Pt-doped Pd catalysts in the complete oxidation of methane at low-temperature. Catal. Today 2006, 117, 564–568. [Google Scholar] [CrossRef]
- Narui, K.; Yata, H.; Furuta, K.; Nishida, A.; Kohtoku, Y.; Matsuzaki, T. Effects of addition of Pt to PdO/Al2O3 catalyst on catalytic activity for methane combustion and TEM observations of supported particles. Appl. Catal. A Gen. 1999, 179, 165–173. [Google Scholar] [CrossRef]
- Okumura, K.; Shinohara, E.; Niwa, M. Pd loaded on high silica beta support active for the total oxidation of diluted methane in the presence of water vapor. Catal. Today 2006, 117, 577–583. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, B.; Shin, C.H.; Seo, G.; Kim, S.H.; Hong, S.B. Methane combustion over Pd catalysts loaded on medium and large pore zeolites. Top. Catal. 2009, 52, 27–34. [Google Scholar] [CrossRef]
- Hicks, R.F.; Qi, H.; Young, M.L.; Lee, R.G. Effect of catalyst structure on methane oxidation over palladium on alumina. J. Catal. 1990, 122, 295–306. [Google Scholar] [CrossRef]
- Colussi, S.; Gayen, A.; Camellone, M.F.; Boaro, M.; Llorca, J.; Fabris, S.; Trovarelli, A. Nanofaceted Pd-O sites in Pd-Ce surface superstructures: Enhanced activity in catalytic combustion of methane. Angew. Chem. Int. Ed. 2009, 48, 8481–8484. [Google Scholar] [CrossRef]
- Dissanayake, D.P. Applications of Metal Oxides for Volatile Organic Compound Combustion. In Metal Oxides Chemistry and Applications; Fierro, J.L.G., Ed.; Taylor & Francis Group: Boca Raton, FL, USA, 2006; pp. 543–568. [Google Scholar]
- Schwartz, W.R.; Pfefferle, L.D. Combustion of methane over Palladium-based catalysts: Support interactions. J. Phys. Chem. C 2012, 116, 8571–8578. [Google Scholar] [CrossRef]
- Ersson, A.; Kušar, H.; Carroni, R.; Griffin, T.; Järås, S. Catalytic combustion of methane over bimetallic catalysts: A comparison between a novel annular reactor and a high-pressure reactor. Catal. Today 2003, 83, 265–277. [Google Scholar] [CrossRef]
- Persson, K.; Ersson, A.; Carrera, A.M.; Jayasuriya, J.; Fakhrai, R.; Fransson, T.; Järås, S. Supported palladium-platinum catalyst for methane combustion at high pressure. Catal. Today 2005, 100, 479–483. [Google Scholar] [CrossRef]
- Okumura, K.; Matsumoto, S.; Nishiaki, N.; Niwa, M. Support effect of zeolite on methane combustion activity of palladium. Appl. Catal. B 2003, 40, 151–159. [Google Scholar] [CrossRef]
- Matam, S.K.; Aguirre, M.H.; Weidenkaff, A.; Ferri, D. Revisiting the problem of active sites for methane combustion on Pd/Al2O3 by operando XANES in a lab-scale fixed-bed reactor. J. Phys. Chem. C 2010, 114, 9439–9443. [Google Scholar] [CrossRef]
- Chen, J.; Arandiyan, H.; Gao, X.; Li, J. Recent advances in catalysts for methane combustion. Catal. Surv. Asia 2015, 19, 140–171. [Google Scholar] [CrossRef]
- Yoshida, H.; Nakajima, T.; Yazawa, Y.; Hattori, T. Support effect on methane combustion over palladium catalysts. Appl. Catal. B Environ. 2007, 71, 70–79. [Google Scholar] [CrossRef]
- Corro, G.; Cano, C.; Fierro, J.L.G. Improved activity of Pt/γ-Al2O3 catalysts for CH4-O2 reaction under lean conditions due to sulfation and catalyst loading. Catal. Commun. 2008, 9, 2601–2605. [Google Scholar] [CrossRef]
- Kinnunen, N.M.; Suvanto, M.; Moreno, M.A.; Savimaki, A.; Kallinen, K.; Kinnunen, T.J.J.; Pakkanen, T.A. Methane oxidation on alumina supported palladium catalysts: Effect of Pd precursor and solvent. Appl. Catal. A Gen. 2009, 370, 78–87. [Google Scholar] [CrossRef]
- Lee, C.Y.; Han, K.H.; Ha, B.H. Characteristics and combustion/decomposition activities of CuO/mordenite. Microporous Mater. 1997, 11, 227–235. [Google Scholar] [CrossRef]
- Neyestanaki, A.K.; Kumar, N.; Lindfors, L.E. Catalytic combustion of propane and natural-gas over Cu and Pd modified ZSM zeolite catalysts. Appl. Catal. B Environ. 1995, 7, 95–111. [Google Scholar] [CrossRef]
- M’Ramadj, O.; Zhang, B.; Li, D.; Wang, X.; Lu, G. Catalytic combustion of methane over high copper-loading ZSM-5 catalysts. J. Nat. Gas. Chem. 2007, 16, 258–265. [Google Scholar] [CrossRef]
- Védrine, J.C.; Millet, J.M.M.; Volta, J.C. Molecular description of active sites in oxidation reactions: Acid-base and redox properties, and role of water. Catal. Today 1996, 32, 115–123. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Rane, V.H. Acidity/basicity of rare-earth oxides and their catalytic activity in oxidative coupling of methane to C2-hydrocarbons. J. Catal. 1991, 130, 411–422. [Google Scholar] [CrossRef]
- Liotta, L.F.; Wu, H.; Pantaleo, G.; Venezia, A.M. Co3O4 nanocrystals and Co3O4-MOx binary oxides for CO, CH4 and VOC oxidation at low temperatures: A review. Catal. Sci. Technol. 2013, 3, 3085–3102. [Google Scholar] [CrossRef]
- Hu, L.; Peng, Q.; Li, Y. Selective synthesis of Co3O4 nanocrystal with different shape and crystal plane effect on catalytic property for methane combustion. J. Am. Chem. Soc. 2008, 130, 16136–16137. [Google Scholar] [CrossRef]
- Teng, F.; Chen, M.; Li, G.; Teng, Y.; Xu, T.; Hang, Y.; Yao, W.; Santhanagopalan, S.; Meng, D.D.; Zhu, Y. High combustion activity of CH4 and cataluminescence properties of CO oxidation over porous Co3O4 nanorods. Appl. Catal. B Environ. 2011, 110, 133–140. [Google Scholar] [CrossRef]
- Solsona, B.; Davies, T.E.; Garcia, T.; Vazquez, I.; Dejoz, A.; Taylor, S. Total oxidation of propane using nanocrystalline cobalt oxide and supported cobalt oxide catalysts. Appl. Catal. B Environ. 2008, 84, 176–184. [Google Scholar] [CrossRef]
- Batiot-Dupeyrat, C.; Martinez-Ortega, F.; Ganne, M.; Tatibouet, J.M. Methane catalytic combustion on La-based perovskite type catalysts in high temperature isothermal conditions. Appl. Catal. A Gen. 2001, 206, 205–215. [Google Scholar] [CrossRef]
- Kirchnerova, J.; Alifanti, M.; Delmon, B. Evidence of phase cooperation in the LaCoO3-CeO2-Co3O4 catalytic system in relation to activity in methane combustion. Appl. Catal. A Gen. 2002, 231, 65–80. [Google Scholar] [CrossRef]
- Ciambelli, P.; Cimino, S.; De Rossi, S.; Faticanti, M.; Lisi, L.; Minelli, G.; Pettiti, I.; Porta, P.; Russo, G.; Turco, M. AMnO3 (A=La, Nd, Sm) and Sm1-xSrxMnO3 perovskites as combustion catalysts: Structural, redox and catalytic properties. Appl. Catal. B Environ. 2000, 24, 243–253. [Google Scholar] [CrossRef]
- Campagnoli, E.; Tavares, A.; Fabbrini, L.; Rossetti, I.; Dubitsky, Y.A.; Zaopo, A.; Forni, L. Effect of preparation method on activity and stability of LaMnO3 and LaCoO3 catalysts for the flameless combustion of methane. Appl. Catal. B Environ. 2005, 55, 133–139. [Google Scholar] [CrossRef]
- Park, S.; Hwang, H.J.; Moon, J. Catalytic combustion of methane over rare earth stannate pyrochlore. Catal. Lett. 2003, 87, 219–223. [Google Scholar] [CrossRef]
- Sohn, J.M.; Kim, M.R.; Woo, S.I. The catalytic activity and surface characterization of Ln2B2O7 (Ln=Sm, Eu, Gd and Tb; B=Ti or Zr) with pyrochlore structure as novel CH4 combustion catalyst. Catal. Today 2003, 83, 289–297. [Google Scholar] [CrossRef]
- Tanasoi, S.; Mitran, G.; Tanchoux, N.; Cacciaguerra, T.; Fajula, F.; Săndulescu, I.; Tichit, D.; Marcu, I.-C. Transition metal-containing mixed oxides catalysts derived from LDH precursors for short-chain hydrocarbons oxidation. Appl. Catal. A Gen. 2011, 395, 78–86. [Google Scholar] [CrossRef]
- Tanasoi, S.; Tanchoux, N.; Urdă, A.; Tichit, D.; Săndulescu, I.; Fajula, F.; Marcu, I.-C. New Cu-based mixed oxides obtained from LDH precursors, catalysts for methane total oxidation. Appl. Catal. A Gen. 2009, 363, 135–142. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, J.; Cheng, J.; Xiao, T.; Jones, M.O.; Hao, Z.; Edwards, P.P. Catalytic combustion of methane over mixed oxides derived from Co–Mg/Al ternary hydrotalcites. Fuel Process. Technol. 2010, 91, 97–102. [Google Scholar] [CrossRef]
- Jiang, Z.; Hao, Z.; Yu, J.; Hou, H.; Hu, C.; Su, J. Catalytic combustion of methane on novel catalysts derived from Cu-Mg/Al-hydrotalcites. Catal. Lett. 2005, 99, 157–163. [Google Scholar] [CrossRef]
- Jirátova, K.; Čuba, P.; Kovanda, F.; Hilaire, L.; Pitchon, V. Preparation and characterisation of activated Ni (Mn)/Mg/Al hydrotalcites for combustion catalysis. Catal. Today 2002, 76, 43–53. [Google Scholar] [CrossRef]
- Stamate, A.-E.; Pavel, O.D.; Zavoianu, R.; Marcu, I.-C. Polyoxometalate-Intercalated Layered Double Hydroxides: A Review. Catalysts 2020, 10, 57. [Google Scholar] [CrossRef]
- Wilkes, M.F.; Hayden, P.; Bhattacharya, A.K. Catalytic studies on ceria lanthana solid solutions I: Oxidation of methane. J. Catal. 2003, 219, 286–294. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Deganello, G. Co3O4/CeO2 and Co3O4/CeO2–ZrO2 composite catalysts for methane combustion: Correlation between morphology reduction properties and catalytic activity. Catal. Commun. 2005, 6, 329–336. [Google Scholar] [CrossRef]
- Mayernick, A.D.; Janik, M.J. Methane oxidation on Pd–Ceria: A DFT study of the mechanism over PdxCe1−xO2, Pd, and PdO. J. Catal. 2011, 278, 16–25. [Google Scholar] [CrossRef]
- Primet, M.; Garbowski, E. Catalysis by Ceria and Related Materials; Catalytic Science Series; Trovarelli, A., Hutchings, G.J., Eds.; Imperial College Press: London, UK, 2005; Volume 2, pp. 407–429. [Google Scholar]
- Thevenin, P.O.; Alcade, A.; Pettersson, L.J.; Jaras, S.G.; Fierro, J.L.G. Catalytic combustion of methane over cerium-doped palladium catalysts. J. Catal. 2003, 215, 78–86. [Google Scholar] [CrossRef]
- Specchia, S.; Finocchio, E.; Busca, G.; Palmisano, P.; Specchia, V. Surface chemistry and reactivity of ceria–zirconia-supported palladium oxide catalysts for natural gas combustion. J. Catal. 2009, 263, 134–145. [Google Scholar] [CrossRef]
- Colussi, S.; Trovarelli, A.; Cristiani, C.; Lietti, L.; Groppi, G. The influence of ceria and other rare earth promoters on palladium-based methane combustion catalysts. Catal. Today 2012, 180, 124–130. [Google Scholar] [CrossRef]
- Zarur, A.Y.; Ying, J.Y. Reverse microemulsion synthesis of nanostructured complex oxides for catalytic combustion. Nature 2000, 403, 65–67. [Google Scholar] [CrossRef]
- Li, J.; Liang, X.; Xu, S.; Hao, J. Catalytic performance of manganese cobalt oxides on methane combustion at low temperature. Appl. Catal. B Environ. 2009, 90, 307–312. [Google Scholar] [CrossRef]
- Gil, A.; Gandia, L.M.; Korili, S.A. Effect of the temperature of calcination on the catalytic performance of manganese- and samarium-manganese-based oxides in the complete oxidation of acetone. Appl. Catal. A Gen. 2004, 274, 229–235. [Google Scholar] [CrossRef]
- Yue, B.; Zhou, R.; Wang, Y.; Zheng, X. Effect of rare earths (La, Pr, Nd, Sm and Y) on the methane combustion over Pd/Ce–Zr/Al2O3 catalysts. Appl. Catal. A Gen. 2005, 295, 31–39. [Google Scholar] [CrossRef]
- Gupta, C.K.; Krishnamurthy, N. Extractive Metallurgy of Rare Earths; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Trovarelli, A. Catalysis by Ceria and Related Materials; World Scientific: London, UK, 2013; Volume 12, pp. 1–888. [Google Scholar]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and catalytic applications of CeO2-based materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Beckers, J.; Rothenberg, G. Sustainable selective oxidations using ceria-based materials. Green Chem. 2010, 12, 939–948. [Google Scholar] [CrossRef]
- Óvári, L.; Calderon, S.; Krick Lykhach, Y.; Libuda, J.; Erdőhelyi, A.; Papp, C.; Kiss, J.; Steinrück, H.-P. Near ambient pressure XPS investigation of the interaction of ethanol with Co/CeO2(1 1 1). J. Catal. 2013, 307, 132–139. [Google Scholar] [CrossRef]
- Ferencz, Z.S.; Erdőhelyi, A.; Baán, K.; Oszkó, A.; Óvári, L.; Kónya, Z.; Papp, C.; Steinrück, H.-P.; Kiss, J. Effects of support and Rh additive on Co-based catalysts in the ethanol steam reforming reaction. ACS Catal. 2014, 4, 1205–1218. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J.P. Water gas shift catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Heck, R.M.; Farrauto, R.J. Automobile exhaust catalysts. Appl. Catal. A Gen. 2001, 221, 443–457. [Google Scholar] [CrossRef]
- Shelef, M.; McCabe, R.W. Twenty-five years after introduction of automotive catalysts: What next? Catal. Today 2000, 62, 35–50. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic properties of ceria and CeO2-containing materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Zhou, Y.; Lawrence, N.J.; Wang, L.; Kong, L.; Wu, T.-S.; Liu, J.; Gao, Y.; Brewer, J.R.; Lawrence, V.K.; Sabirianov, R.F.; et al. Resonant photoemission observations and DFT study of s-d hybridization in catalytically active gold clusters on ceria nanorods. Angew. Chem. Int. Ed. 2013, 52, 6936–6939. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Menendez, C.L.; Guinel, M.J.F.; Needels, E.C.; Gonzalez-Gonzalez, I.; Jackson, D.L.; Lawrence, N.J.; Cabrera, C.R.; Cheung, C.L. Influence of nanostructured ceria support on platinum nanoparticles for methanol electrooxidation in alkaline media. RSC Adv. 2014, 4, 1270–1275. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Lawrence, N.J.; Wu, T.S.; Liu, J.; Kent, P.; Soo, Y.L.; Cheung, C.L. Pd/CeO2−x nanorod catalysts for CO oxidation: Insights into the origin of their regenerative ability at room temperature. ChemCatChem 2014, 6, 2937–2946. [Google Scholar] [CrossRef]
- Fechete, I.; Masseron, A.; Dumitriu, E.; Lutic, D.; Caullet, P.; Kessler, H. The role of acidity of CeH-EMT-type zeolite for catalyzing toluene alkylation with methanol to xylenes. Rev. Roum.Chim. 2008, 53, 55–61. [Google Scholar]
- Maksimchuk, N. Nanocrystalline Cerium Oxide Films for Microelectronic Biosensor Transducers. Proc. NAP. 2012, 1, 02NNBM281-3. [Google Scholar]
- Wang, G.; Wang, L.; Fei, X.; Zhou, Y.; Sabirianov, R.F.; Mei, W.N.; Cheung, C.L. Probing the bifunctional catalytic activity of ceria nanorods towards the cyanosilylation reaction. Catal. Sci. Technol. 2013, 3, 2602–2609. [Google Scholar] [CrossRef]
- Tamura, M.; Honda, M.; Nakagawa, Y.; Tomishige, K. Direct conversion of CO2 with diols, aminoalcohols and diamines to cyclic carbonates, cyclic carbamates and cyclic ureas using heterogeneous catalysts. J. Chem. Technol. Biotechnol. 2014, 89, 19–33. [Google Scholar] [CrossRef]
- Boaro, M.; Vicario, M.; de Leitenburg, C.; Dolcetti, G.; Trovarelli, A. The use of temperature-programmed and dynamic/transient methods in catalysis: Characterization of ceria-based, model three-way catalysts. Catal. Today 2003, 77, 407–417. [Google Scholar] [CrossRef]
- Fornasiero, P.; Dimonte, R.; Rao, G.; Kašpar, J.; Meriani, S.; Trovarelli, A.; Graziani, M. Rh-loaded CeO2-ZrO2 solid-solutions as highly efficient oxygen exchangers: Dependence of the reduction behavior and the oxygen storage capacity on the structural-properties. J. Catal. 1995, 151, 168–177. [Google Scholar] [CrossRef]
- Kašpar, J.; Fornasiero, P.; Graziani, M. Use of CeO2-based oxides in the three-way catalysis. Catal. Today 1999, 50, 285–298. [Google Scholar] [CrossRef]
- Kreuzer, T.; Lox, E.S.; Lindner, D.; Leyrer, J. Exhaust gas treatment advanced exhaust gas aftertreatment systems for gasoline and diesel fuelled vehicles. Catal. Today 1996, 29, 17–27. [Google Scholar] [CrossRef]
- Taylor, K.C. Automobile Catalytic Converters. In Catalysis—Science and Technology; Boudart, M., Anderson, J.R., Eds.; Springer: Berlin/Heidelberg, Germany, 1984; Volume 5, pp. 119–170. [Google Scholar]
- Zhang, Y.; Andersson, S.; Muhammed, M. Nanophase catalytic oxides: I. Synthesis of doped cerium oxides as oxygen storage promoters. Appl. Catal. B Environ. 1995, 6, 325–337. [Google Scholar] [CrossRef]
- Persson, A.E.; Zhang, Y.; Muhammed, M. Catalyst Materials for High Temperature Processes, Ceramic Transactions; Ramesh, K.S., Misono, M., Gai, P.L., Eds.; American Ceramic Society: Westerville, OH, USA, 1997; Volume 73, p. 85. [Google Scholar]
- Khaladji, J.; Peltier, M. Rare Earth Polishing Compositions. Google Patents US4942697A, 1990. [Google Scholar]
- Wang, J.F. System and Method. Google Patents US20100317420A1, 1996. [Google Scholar]
- Hanawa, K.; Mochizuki, N.; Ueda, N. Cerium oxide ultrafine particles and method for preparing the same. Google Patents , US5938837A, Patent Number: 5,938,837, 1999. [Google Scholar]
- Tsai, M.-S. Powder synthesis of nano grade cerium oxide via homogenous precipitation and its polishing performance. Mater. Sci. Eng. B 2004, 110, 132–134. [Google Scholar] [CrossRef]
- Sabot, J.-L.; Maestro, P. Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Hedrick, J.B. Rare Earth Minerals and Metals 1990; United States Bureau of Mines: Washington, DC, USA, 1992. [Google Scholar]
- Kaspar, J.; Graziani, M.; Fornasiero, P. Ceria-Containing Three-Way Catalyts. In Handbook on the Physics and Chemistry of Rare Earths: The Role of Rare Earths in Catalysis; Gschneidner, K.A., Jr., Eyring, L., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2000; Volume 29, Chapter 184; pp. 159–267. [Google Scholar] [CrossRef]
- Otsuka-Yao-Matsuo, S.; Omata, T.; Izu, N.; Kishimoto, H. Oxygen release behavior of CeZrO4 powders and appearance of new compoundsκand t*. J. Solid State Chem. 1998, 138, 47–54. [Google Scholar] [CrossRef]
- Izu, N.; Omata, T.; Otsuka-Yao-Matsuo, S. Oxygen release behaviour of Ce(1−x)ZrxO2 powders and appearance of Ce(8−4y)Zr4yO(14−δ) solid solution in the ZrO2–CeO2–CeO1.5 system. J. Alloy. Compd. 1998, 270, 107–114. [Google Scholar] [CrossRef]
- Bevan, D.J.M. Ordered intermediate phases in the system CeO2-Ce2O3. J. Inorg. Nucl. Chem. 1955, 1, 49–59. [Google Scholar] [CrossRef]
- Brauer, G.; Gingerich, K.A. Über die oxyde des cers–V: Hochtemperatur-Röntgenuntersuchungen an ceroxyden. J. Inorg. Nucl. Chem. 1960, 16, 87–99. [Google Scholar] [CrossRef]
- Averill, B.A. General Chemistry: Principles, Patterns, and Application. 2012. Available online: http://www.saylor.org/books (accessed on 23 February 2021).
- Vyas, S. Simulation of Ceria: Bulk and Surface Defects; University of London: London, UK, 2005. [Google Scholar]
- Steele, B.C.H.; Floyd, J.M. Oxygen self-diffusion and electrical transport properties of nonstoichiometric ceria and ceria solid solutions. Proc. British. Ceram. Trans. 1971, 72, 55–76. [Google Scholar]
- Faber, J., Jr.; Seitz, M.A.; Mueller, M.H. Defect characterization in CeO2−x at elevated temperatures—I: X-Ray diffraction. J. Phys. Chem. Solids 1976, 37, 903–907. [Google Scholar] [CrossRef]
- Tuller, H.L.; Nowick, A.S. Defect Structure and Electrical Properties of Nonstoichiometric CeO2 Single Crystals. J. Electrochem. Soc. 1979, 126, 209–217. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalysis by Ceria and Related Materials; World Scientific: London, UK, 2001; Volume 2, pp. 15–50. [Google Scholar]
- Fronzi, M.; Soon, A.; Delley, B.; Traversa, E.; Stampfl, C. Stability and morphology of cerium oxide surfaces in an oxidizing environment: A first-principles investigation. J. Chem. Phys. 2009, 131, 104701. [Google Scholar] [CrossRef]
- Sayle, T.X.T.; Parker, S.C.; Catlow, C.R.A. The role of oxygen vacancies on ceria surfaces in the oxidation of carbon monoxide. Surf. Sci. 1994, 316, 329–336. [Google Scholar] [CrossRef]
- Kaneko, K.; Inoke, K.; Freitag, B.; Hungria, A.B.; Midgley, P.A.; Hansen, T.W.; Zhang, J.; Ohara, S.; Adschiri, T. Structural and Morphological Characterization of Cerium Oxide Nanocrystals Prepared by Hydrothermal Synthesis. Nano Lett. 2007, 7, 421–425. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Vannice, M.A. An analysis of the Mars-van Krevelen rate expression. Catal. Today 2007, 123, 18–22. [Google Scholar] [CrossRef]
- Ladavos, A.; Pomonis, P. Methane Combustion on Perovskites. In Perovskites and Related Mixed Oxides: Concepts and Applications; Granger, P., Parvulescu, V.I., Kaliaguine, S., Prellier, W., Eds.; Wiley VCH Publications: Hoboken, NJ, USA, 2016. [Google Scholar]
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as substitutes of noble metals for heterogeneous catalysis: Dream or reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef]
- Keav, S.; Kumar Matam, S.; Ferri, D.; Weidenkaff, A. Structured perovskite-based catalysts and their application as three-way catalytic converters-a review. Catalysts 2014, 4, 226–255. [Google Scholar] [CrossRef]
- Toniolo, F.S.; Schmal, M. Improvement of catalytic performance of perovskites by partial substitution of cations and supporting on highsurface area materials. In Perovskite Materials—Synthesis, Characterisation, Properties, and Applications; Pan, L., Zhu, G., Eds.; InTech Publications: Rijeka, Croatia, 2016. [Google Scholar]
- Liang, C.-J.; Fang, J.W. Predicting the kinetics of catalytic oxidation of multicomponent organic waste gases. Chem. Eng. Sci. 2016, 144, 101–107. [Google Scholar] [CrossRef]
- Bion, N.; Can, F.; Courtois, X.; Duprez, D. Transition metal oxides for combustion and depollution processes. In Metal Oxides in Heterogeneous Catalysis; Védrine, J.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 287–354. [Google Scholar]
- Murzin, D.; Salmi, T. Catalytic Kinetics; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1–475. [Google Scholar]
- Zhong, L.S.; Hu, J.S.; Cao, A.M.; Liu, Q.; Song, W.G.; Wan, L.J. 3D flowerlike ceria micro/nanocomposite structure and its application for water treatment and CO removal. Chem. Mater. 2007, 19, 1648–1655. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Wang, Z.; Du, W.; Zhu, G. Morphological effect of CeO2 catalysts on their catalytic performance in lean methane combustion. Chem. Lett. 2020, 49, 461–464. [Google Scholar] [CrossRef]
- Ohtake, N.; Katoh, M.; Sugiyama, S. High thermal-stability ceria synthesized via thermal-hydrolysis route and methane-combustion performance. J. Ceram. Soc. Jpn. 2017, 125, 57–61. [Google Scholar] [CrossRef]
- Palmqvist, A.E.C.; Johansson, E.M.; Järås, S.G.; Muhammed, M. Total oxidation of methane over doped nanophase cerium oxides. Catal. Lett. 1998, 56, 69–75. [Google Scholar] [CrossRef]
- Dirstine, R.T.; Blumenthal, R.N.; Kuech, T.F. Ionic Conductivity of Calcia, Yttria, and Rare Earth-Doped Cerium Dioxide. J. Electrochem. Soc. 1979, 126, 264–269. [Google Scholar] [CrossRef]
- Yang, W.; Li, D.; Xu, D.; Wang, X. Effect of CeO2 preparation method and Cu loading on CuO/CeO2 catalysts for methane combustion. J. Nat. Gas. Chem. 2009, 18, 458–466. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chou, F.C.; Tsai, D.H. Mechanistic understanding of surface reduction of Cu–Ce–O hybrid nanoparticles for catalytic methane combustion. J. Taiwan Inst. Chem. Eng. 2018, 92, 80–90. [Google Scholar] [CrossRef]
- Zedan, A.F.; AlJaber, A.S. Combustion synthesis of non-precious CuO-CeO2 nanocrystalline catalysts with enhanced catalytic activity for methane oxidation. Materials 2019, 16, 878. [Google Scholar] [CrossRef]
- Chrzan, M.; Chlebda, D.; Jodłowski, P.; Salomon, E.; Kołodziej, A.; Gancarczyk, A.; Sitarz, M.; Łojewska, J. Towards Methane Combustion Mechanism on Metal Oxides Supported Catalysts: Ceria Supported Palladium Catalysts. Top. Catal. 2019, 62, 403–412. [Google Scholar] [CrossRef]
- Kundakovic, L.; Flytzani-Stephanopoulos, M. Cu- and Ag-modified cerium oxide catalysts for methane oxidation. J. Catal. 1998, 179, 203–221. [Google Scholar] [CrossRef]
- Bueno-López, A.; Krishna, K.; Makkee, M.; Moulijn, J.A. Enhanced soot oxidation by lattice oxygen via La3+-doped CeO2. J. Catal. 2005, 230, 237–248. [Google Scholar] [CrossRef]
- Zhang, B.; Li, D.; Wang, X. Catalytic performance of La-Ce-O mixed oxide for combustion of methane. Catal. Today 2010, 158, 348–353. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Liu, W.; Flytzanistephanopoulos, M. Total Oxidation of Carbon-Monoxide and Methane over Transition Metal Fluorite Oxide Composite Catalysts. J. Catal. 1995, 153, 317–332. [Google Scholar] [CrossRef]
- Zimicz, M.G.; Lamas, D.G.; Larrondo, S.A. Ce0.9Zr0.1O2 nanocatalyst: Influence of synthesis conditions in the reducibility and catalytic activity. Catal. Commun. 2011, 15, 68–73. [Google Scholar] [CrossRef]
- Li, D.; Li, K.; Xu, R.; Wang, H.; Tian, D.; Wei, Y.; Zhu, X.; Zeng, C.; Zeng, L. Ce1-xFexO2-δ catalysts for catalytic methane combustion: Role of oxygen vacancy and structural dependence. Catal. Today 2018, 318, 73–85. [Google Scholar] [CrossRef]
- Perez-Alonso, F.J.; Melián-Cabrera, I.; López Granados, M.; Kapteijn, F.; Fierro, J.L.G. Synergy of FexCe1-xO2 mixed oxides for N2O decomposition. J. Catal. 2006, 239, 340–346. [Google Scholar] [CrossRef]
- Li, K.; Wang, H.; Wei, Y.; Yan, D. Syngas production from methane and air via a redox process using Ce-Fe mixed oxides as oxygen carriers. Appl. Catal. B Environ. 2010, 97, 361–372. [Google Scholar] [CrossRef]
- Zhong, L.; Fang, Q.; Li, X.; Li, Q.; Zhang, C.; Chen, G. Influence of preparation methods on the physicochemical properties and catalytic performance of Mn-Ce catalysts for lean methane combustion. Appl. Catal. A Gen. 2019, 579, 151–158. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, H.; Zhou, Y.; Zhang, L.; Wu, Z.; Yang, S.; Shi, H.; Zhu, Q.; Chen, Y.; Dai, S. Mesoporous MnCeOx solid solutions for low temperature and selective oxidation of hydrocarbons. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, W.; Li, D.; Wang, X. Influence of preparation method on the performance of Mn-Ce-O catalysts. React. Kinet. Catal. Lett. 2009, 97, 263–268. [Google Scholar] [CrossRef]
- Huang, X.; Li, J.; Wang, J.; Li, Z.; Xu, J. Catalytic combustion of methane over a highly active and stable NiO/CeO2 catalyst. Front. Chem. Sci. Eng. 2020, 14, 534–545. [Google Scholar] [CrossRef]
- Thaicharoensutcharittham, S.; Meeyoo, V.; Kitiyanan, B.; Rangsunvigit, P.; Rirksomboon, T. Catalytic combustion of methane over NiO/Ce0.75Zr0.25O2 catalyst. Catal. Commun. 2009, 10, 673–677. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Deganello, G. Catalytic performance of Co3O4/CeO2 and Co3O4/CeO2-ZrO2 composite oxides for methane combustion: Influence of catalyst pretreatment temperature and oxygen concentration in the reaction mixture. Appl. Catal. B Environ. 2007, 70, 314–322. [Google Scholar] [CrossRef]
- Darda, S.; Pachatouridou, E.; Lappas, A.; Iliopoulou, E. Effect of preparation method of Co-Ce catalysts on CH4 combustion. Catalysts 2019, 9, 219. [Google Scholar] [CrossRef]
- Chen, J.; Carlson, B.D.; Toops, T.J.; Li, Z.; Lance, M.J.; Karakalos, S.G.; Choi, J.S.; Kyriakidou, E.A. Methane Combustion Over Ni/CexZr1–xO2 Catalysts: Impact of Ceria/Zirconia Ratio. ChemCatChem 2020, 12, 5558–5568. [Google Scholar] [CrossRef]
- Toscani, L.M.; Curyk, P.A.; Zimicz, M.G.; Halac, E.B.; Saleta, M.E.; Lamas, D.G.; Larrondo, S.A. Methane catalytic combustion over CeO2-ZrO2-Sc2O3 mixed oxides. Appl. Catal. A Gen. 2019, 587, 117235. [Google Scholar] [CrossRef]
- Vickers, S.M.; Gholami, R.; Smith, K.J.; MacLachlan, M.J. Mesoporous Mn- and La-doped cerium oxide/cobalt oxide mixed metal catalysts for methane oxidation. ACS Appl. Mater. Interfaces 2015, 7, 11460–11466. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Xue, B. Catalytic combustion of methane over M (Ni, Co, Cu) supported on ceria-magnesia. Fuel Process. Technol. 2009, 90, 652–656. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Hu, Y.H. Methane partial oxidation over NiO/MgO solid solution catalysts. Appl. Catal. A Gen. 1999, 183, 85–92. [Google Scholar] [CrossRef]
- Fan, X.; Li, L.; Jing, F.; Li, J.; Chu, W. Effects of preparation methods on CoAlOx/CeO2 catalysts for methane catalytic combustion. Fuel 2018, 225, 588–595. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Oxidation of lean methane over cobalt catalysts supported on ceria/alumina. Appl. Catal. A Gen. 2020, 591, 117381. [Google Scholar] [CrossRef]
- Jiang, X.; Ma, Y.; Chen, Y.; Li, Y.; Ma, Q.; Zhang, Z.; Wang, C.; Yang, Y. Raman analysis of cobalt blue pigment in blue and white porcelain: A reassessment. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 61–67. [Google Scholar] [CrossRef]
- Al-Aani, H.M.S.; Iro, E.; Chirra, P.; Fechete, I.; Badea, M.; Negrilă, C.; Popescu, I.; Olea, M.; Marcu, I.C. CuxCeMgAlO mixed oxide catalysts derived from multicationic LDH precursors for methane total oxidation. Appl. Catal. A Gen. 2019, 586, 4–12. [Google Scholar] [CrossRef]
- Al-Aani, H.M.S.; Trandafir, M.M.; Fechete, I.; Leonat, L.N.; Badea, M.; Negrilă, C.; Popescu, I.; Florea, M.; Marcu, I.C. Highly Active Transition Metal-Promoted CuCeMgAlO Mixed Oxide Catalysts Obtained from Multicationic LDH Precursors for the Total Oxidation of Methane. Catalysts 2020, 10, 613. [Google Scholar] [CrossRef]
- Pengpanich, S.; Meeyoo, V.; Rirksomboon, T.; Bunyakiat, K. Catalytic oxidation of methane over CeO2-ZrO2 mixed oxide solid solution catalysts prepared via urea hydrolysis. Appl. Catal. A Gen. 2002, 234, 221–233. [Google Scholar] [CrossRef]
- Belessi, V.C.; Ladavos, A.K.; Armatas, G.S.; Pomonis, P.J. Kinetics of methane oxidation over La-Sr-Ce-Fe-O mixed oxide solids. Phys. Chem. Chem. Phys. 2001, 3, 3856–3862. [Google Scholar] [CrossRef]
- Zasada, F.; Piskorz, W.; Janas, J.; Grybos, J.; Indyka, P.; Sojka, Z. Reactive oxygen species on the (100) facet of cobalt spinel nanocatalyst and their relevance in 16O2/18O2 isotopic exchange, deN2O, and deCH4 processes. A theoretical and experimental account. ACS Catal. 2015, 5, 6879–6892. [Google Scholar] [CrossRef]
- Liu, C.; Xian, H.; Jiang, Z.; Wang, L.; Zhang, J.; Zheng, L.; Tan, Y.; Li, X. Insight into the improvement effect of the Ce doping into the SnO2 catalyst for the catalytic combustion of methane. Appl. Catal. B Environ. 2015, 176–177, 542–552. [Google Scholar] [CrossRef]
- Carlsson, P.A.; Skoglundh, M. Low-temperature oxidation of carbon monoxide and methane over alumina and ceria supported platinum catalysts. Appl. Catal. B Environ. 2011, 101, 669–675. [Google Scholar] [CrossRef]
- Alifanti, M.; Kirchnerova, J.; Delmon, B.; Klvana, D. Methane and propane combustion over lanthanum transition-metal perovskites: Role of oxygen mobility. Appl. Catal. A Gen. 2004, 262, 167–176. [Google Scholar] [CrossRef]































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoian, M.; Rogé, V.; Lazar, L.; Maurer, T.; Védrine, J.C.; Marcu, I.-C.; Fechete, I. Total Oxidation of Methane on Oxide and Mixed Oxide Ceria-Containing Catalysts. Catalysts 2021, 11, 427. https://doi.org/10.3390/catal11040427
Stoian M, Rogé V, Lazar L, Maurer T, Védrine JC, Marcu I-C, Fechete I. Total Oxidation of Methane on Oxide and Mixed Oxide Ceria-Containing Catalysts. Catalysts. 2021; 11(4):427. https://doi.org/10.3390/catal11040427
Chicago/Turabian StyleStoian, Marius, Vincent Rogé, Liliana Lazar, Thomas Maurer, Jacques C. Védrine, Ioan-Cezar Marcu, and Ioana Fechete. 2021. "Total Oxidation of Methane on Oxide and Mixed Oxide Ceria-Containing Catalysts" Catalysts 11, no. 4: 427. https://doi.org/10.3390/catal11040427
APA StyleStoian, M., Rogé, V., Lazar, L., Maurer, T., Védrine, J. C., Marcu, I.-C., & Fechete, I. (2021). Total Oxidation of Methane on Oxide and Mixed Oxide Ceria-Containing Catalysts. Catalysts, 11(4), 427. https://doi.org/10.3390/catal11040427