
An Electrocatalytic Activity of AuCeO2/Carbon Catalyst in Fuel Cell Reactions: Oxidation of Borohydride and Reduction of Oxygen


 ,
,  , ,
, ,  and
and
Abstract
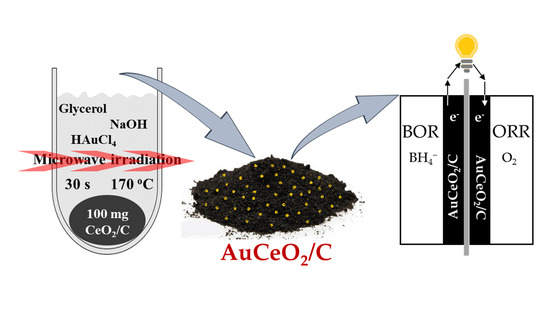
1. Introduction
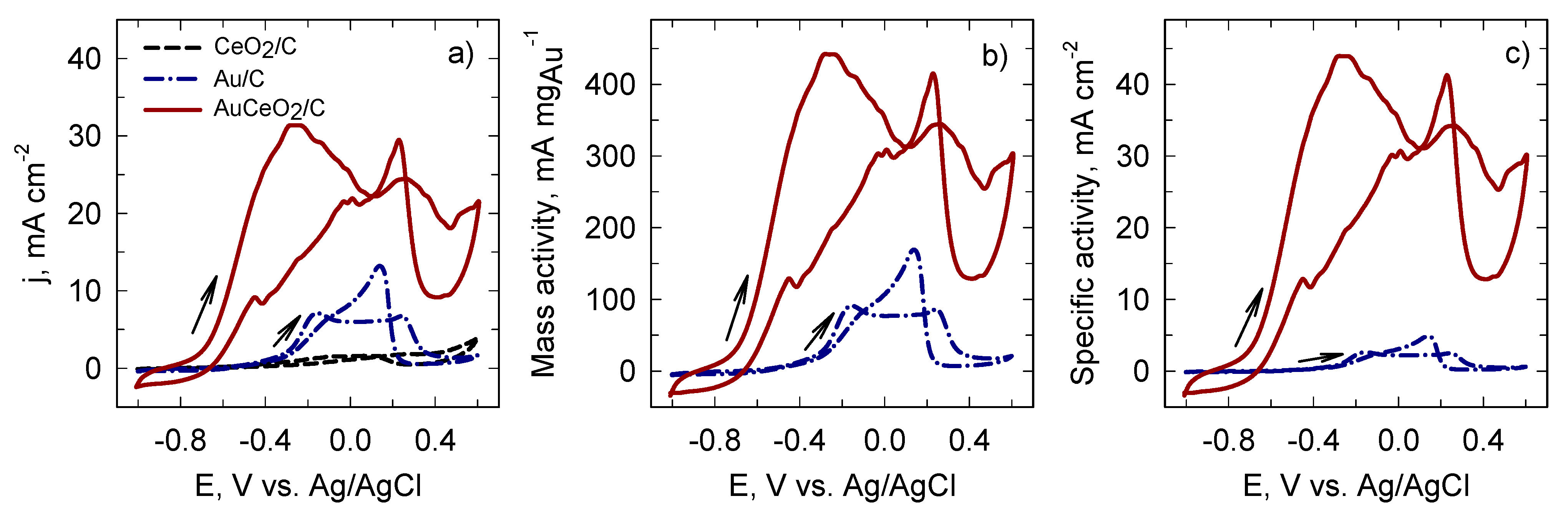
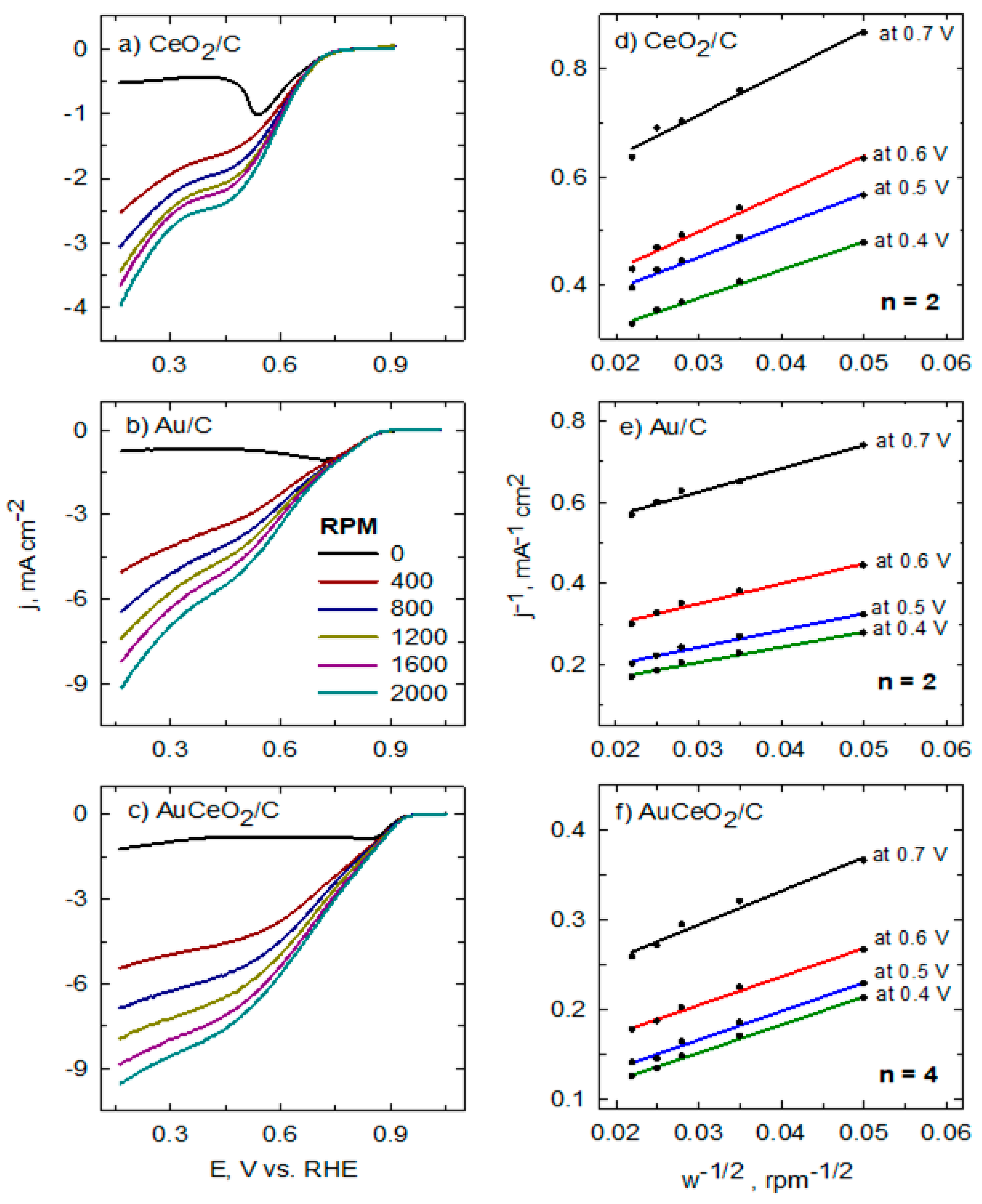
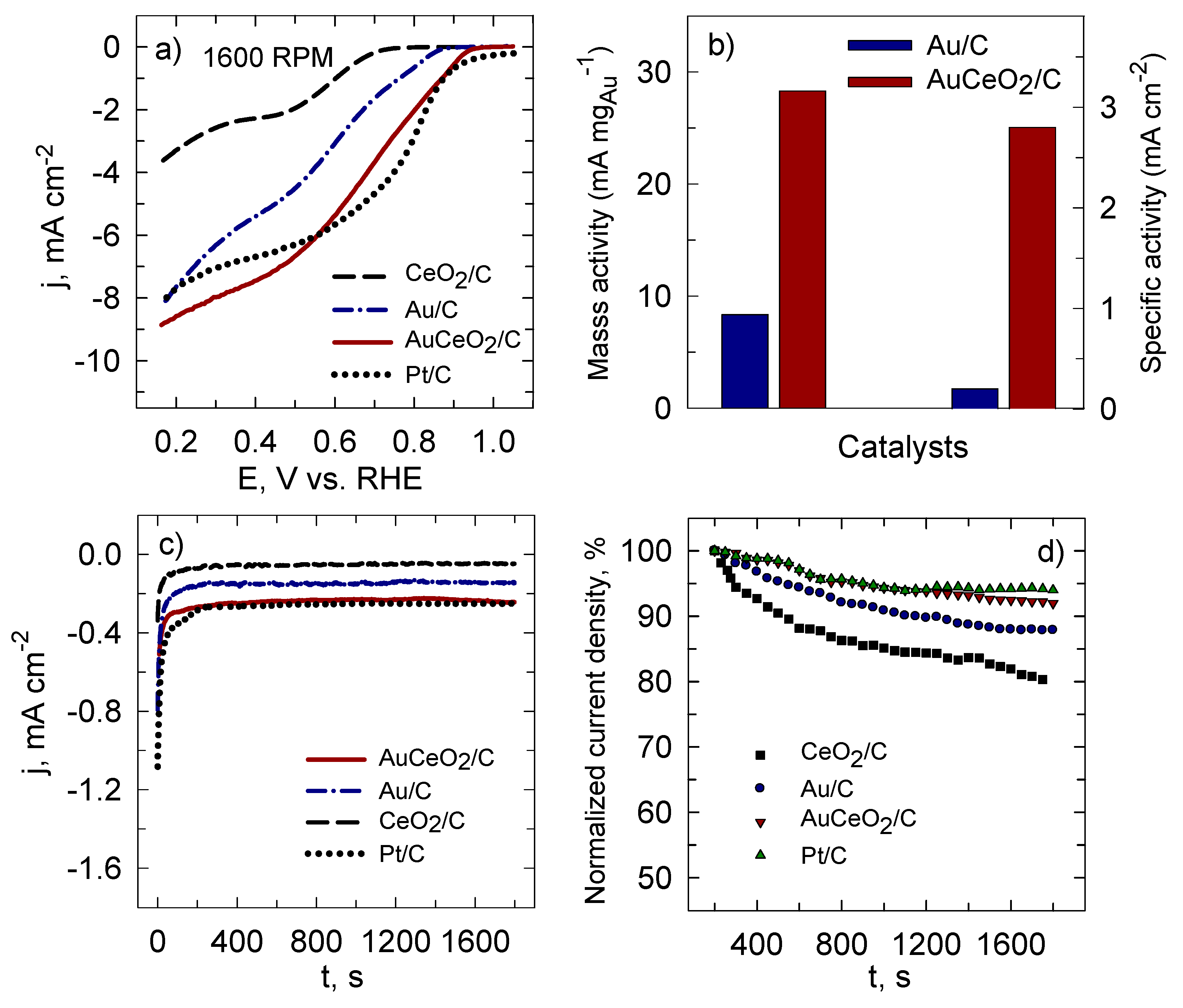
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Catalysts
3.3. Characterization of Catalysts
3.4. Electrochemical Measurements
3.5. Fuel Cell Test Measurements
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fuel Cells. Available online: https://www.energy.gov/eere/fuelcells/fuel-cells (accessed on 13 December 2020).
- Akay, R.G.; Yurtcan, A.B. Direct Liquid Fuel Cells, 1st ed.; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Fuel Cell Basics. Available online: https://americanhistory.si.edu/fuelcells/basics.htm (accessed on 13 December 2020).
- Liu, B.H.; Li, Z.P.; Arai, K.; Suda, S. Performance improvement of a micro borohydride fuel cell operating at ambient conditions. Electrochim. Acta 2005, 50, 3719–3725. [Google Scholar] [CrossRef]
- Li, Z.P.; Liu, B.H.; Arai, K.; Suda, S. Development of the direct borohydride fuel cell. J. Alloys Compd. 2005, 404–406, 648–652. [Google Scholar] [CrossRef]
- Jamard, R.; Latour, A.; Salomon, J.; Capron, P.; Martinent-Beaumont, A. Study of fuel efficiency in a direct borohydride fuel cell. J. Power Sour. 2008, 176, 287–292. [Google Scholar] [CrossRef]
- Liu, B.H.; Li, Z.P.; Suda, S. Development of high-performance planar borohydride fuel cell modules for portable applications. J. Power Sour. 2008, 175, 226–231. [Google Scholar] [CrossRef]
- Mirkin, M.V.; Yang, H.; Bard, A.J. Borohydride oxidation at a gold electrode. J. Electrochem. Soc. 1992, 139, 2212–2217. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Cyclic voltammetry investigation of borohydride oxidation at a gold electrode. Electrochim. Acta 2010, 55, 6775–6781. [Google Scholar] [CrossRef]
- Pinto, A.M.R.R.; Oliveira, V.B.; Falcao, D.S. Direct Alcohol Fuel Cells for Portable Applications: Fundamentals, Engineering and Advances, 1st ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 209–244. [Google Scholar]
- Yu, E.H.; Wang, X.; Krewer, U.; Li, L.; Scott, K. Direct oxidation alkaline fuel cells: From materials to systems. Energy Environ. Sci. 2011, 5, 5668–5680. [Google Scholar] [CrossRef]
- Vielstich, A.W.; Lamm, A.; Gasteiger, H. Handbook of Fuel Cells: Fundamentals, Technology, Applications, 1st ed.; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Milikić, J.; Martins, M.; Dobrota, A.S.; Bozkurt, G.; Soylu, G.S.P.; Yurtcane, A.B.; Skorodumova, N.V.; Paštia, I.A.; Šljukić, B.; Santos, D.M.F. A Pt/MnV2O6 nanocomposite for the borohydride oxidation reaction. J. Energy Chem. 2021, 55, 428–436. [Google Scholar] [CrossRef]
- Braesch, G.; Bonnefont, A.; Martin, V.; Savinova, E.R.; Chatenet, M. Borohydride oxidation reaction mechanisms and poisoning effects onAu, Pt and Pd bulk electrodes: From model (low) to direct borohydridefuel cell operating (high) concentrations. Electrochim. Acta 2018, 273, 483–494. [Google Scholar] [CrossRef]
- Backović, G.; Sljukić, B.; Kanberoglu, G.S.; Yurderi, M.; Bulut, A.; Zahmakiran, M.; Santos, D.M.F. Ruthenium(0) nanoparticles stabilized by metal-organic framework as an efficient electrocatalyst for borohydride oxidation reaction. Int. J. Hydrogen Energy 2020, 45, 27056–27066. [Google Scholar] [CrossRef]
- Bortoloti, F.; Angelo, A.C.D. Ordered PtSn/C electrocatalyst: A high performance material for the borohydride electrooxidation reaction. Catalysts 2017, 7, 198. [Google Scholar] [CrossRef]
- Jurzinsky, T.; Kintzel, B.; Bär, R.; Cremers, C.; Tübke, J. Methanol oxidation on PdRh/C electrocatalyst in alkaline media: Temperature and methanol concentration dependencies. J. Electroanal. Chem. 2016, 766, 49–52. [Google Scholar] [CrossRef]
- Li, X.; Qin, X.; Yan, B.; Huang, H.; Zhang, W.; Piao, Y. Pt Nanoclusters anchored on hollow Ag-Au nanostructures for electrochemical oxidation of methanol. Catalysts 2020, 10, 1440. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, D.; Du, C.; Zou, Z.; Zhang, X.; Xia, B.; Yang, H.; Akins, D.L. Carbon-supported Pd-Co bimetallic nanoparticles as electrocatalysts for the oxygen reduction reaction. J. Power Sour. 2007, 167, 243–249. [Google Scholar] [CrossRef]
- Tamašauskaitė-Tamašiūnaitė, L.; Balčiūnaitė, A.; Zabielaitė, A.; Stankevičienė, I.; Kepenienė, V.; Selskis, A.; Juškėnas, R.; Norkus, E. Investigation of electrocatalytic activity of the nanostructured Au-Cu catalyst deposited on the titanium surface towards borohydride oxidation. J. Electroanal. Chem. 2013, 700, 1–7. [Google Scholar] [CrossRef]
- Kepenienė, V.; Tamašauskaitė-Tamašiūnaitė, L.; Jablonskienė, J.; Vaičiūnienė, J.; Kondrotas, R.; Juškėnas, R.; Norkus, E. Investigation of graphene supported platinum-cobalt nanocomposites as electrocatalysts for ethanol oxidation. J. Electrochem. Soc. 2014, 161, F1354–F1359. [Google Scholar] [CrossRef]
- Yongprapat, S.; Therdthianwong, A.; Therdthianwong, S. Au/C catalysts promoted with metal oxides for ethylene glycol electro-oxidation in alkaline solution. J. Electroanal. Chem. 2013, 697, 46–52. [Google Scholar] [CrossRef]
- Van Dao, D.; Adilbish, G.; Le Duc, T.; Nguyen, T.T.D.; Lee, I.H.; Yu, Y.T. Au@CeO2 nanoparticles supported Pt/C electrocatalyst to improve the removal of CO in methanol oxidation reaction. J. Catal. 2019, 377, 589–599. [Google Scholar] [CrossRef]
- Gao, H.; Cao, Y.; Chen, Y.; Lai, X.; Ding, S.; Tu, J.; Qi, J. Au nanoparticle-decorated NiCo2O4 nanoflower with enhanced electrocatalytic activity toward methanol oxidation. J. Alloy Compd. 2018, 732, 460–469. [Google Scholar] [CrossRef]
- Karuppasamy, L.; Chen, C.Y.; Anandan, S.; Wu, J.J. High index surfaces of Au-nanocrystals supported on one-dimensional MoO3-nanorod as a bifunctional electrocatalyst for ethanol oxidation and oxygen reduction. Electrochim. Acta 2017, 246, 75–88. [Google Scholar] [CrossRef]
- Kaskow, I.; Decyk, P.; Sobczak, I. The effect of copper and silver on the properties of Au-ZnO catalyst and its activity in glycerol oxidation. Appl. Surf. Sci. 2018, 444, 197–207. [Google Scholar] [CrossRef]
- Ostojic, N.; Duan, Z.; Galyamova, A.; Henkelman, G.; Crooks, R.M. Electrocatlytic study of the oxygen reduction reaction at gold nanoparticles in the absence and presence of interactions with SnOx supports. J. Am. Chem. Soc. 2018, 140, 13775–13785. [Google Scholar] [CrossRef]
- Kepenienė, V.; Tamašauskaitė-Tamašiūnaitė, L.; Vaičiūnienė, J.; Kondrotas, R.; Pakštas, V.; Norkus, E. Platinum-niobium(V) oxide/carbon nanocomposites prepared by microwave synthesis for ethanol oxidation. Mater. Sci. 2016, 22, 243–248. [Google Scholar] [CrossRef]
- Kepenienė, V.; Stagniūnaitė, R.; Tamašauskaitė-Tamašiūnaitė, L.; Pakštas, V.; Jasulaitienė, V.; Léger, B.; Rousseau, J.; Ponchel, A.; Monflier, E.; Norkus, E. Co3O4/C and Au supported Co3O4/C nanocomposites—Peculiarities of fabrication and application towards oxygen reduction reaction. Mat. Chem. Phys. 2020, 241, 122332–122340. [Google Scholar] [CrossRef]
- Kepenienė, V.; Stagniūnaitė, R.; Upskuvienė, D.; Tamašauskaitė-Tamašiūnaitė, L.; Pakštas, V.; Drabavičius, A.; Andrulevičius, M.; Norkus, E. Electrocatalytic activity of AuCeO2/C towards ethylene glycol oxidation and oxygen reduction reactions. Chemija 2020, 31, 57–68. [Google Scholar] [CrossRef][Green Version]
- Kepenienė, V.; Stagniūnaitė, R.; Balčiūnaitė, A.; Tamašauskaitė-Tamašiūnaitė, L.; Norkus, E. Microwave-assisted synthesis of AuCeO2/C catalyst and its application for oxidation of alcohols in an alkaline medium. New J. Chem. 2020, 44, 18308–18318. [Google Scholar] [CrossRef]
- Yuan, W.; Zhang, J.; Shen, P.K.; Li, C.M.; Jiang, S.P. Self-assembled CeO2 on carbon nanotubes supported Au nanoclusters as superior electrocatalysts for glycerol oxidation reaction of fuel cells. Electrochim. Acta 2016, 190, 817–828. [Google Scholar] [CrossRef]
- Fuggle, J.C.; Kallne, E.; Watson, L.M.; Fabian, D.J. Electronic structure of aluminum and aluminum-noble-metal alloys studied by soft-x-ray and x-ray photoelectron spectroscopies. Phys. Rev. B 1977, 16, 750–761. [Google Scholar] [CrossRef]
- Battistoni, C.; Mattogno, G.; Zanoni, R.; Naldini, L.J. Characterisation of some gold clusters by X-ray photoelectron spectroscopy. Electron. Spectrosc. Relat. Phenom. 1982, 28, 23–31. [Google Scholar] [CrossRef]
- Yuan, Q.; Qin, C.; Wu, J.; Xu, A.; Zhang, Z.; Liao, J.; Lin, S.; Ren, X.; Zhang, P. Synthesis and characterization of Cerium-doped hydroxyapatite/polylactic acid composite coatings on metal substrates. Mat. Chem. Phys. 2016, 182, 365–371. [Google Scholar] [CrossRef]
- Beche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Jeong, H.K.; Echeverria, E.; Chakraborti, P.; Le, H.T.; Dowben, P.A. Electronic structure of cyclodextrin-carbon nanotube composite films. RSC Adv. 2017, 7, 10968–10972. [Google Scholar] [CrossRef]
- Baker, M.A.; Hammer, P. A study of the chemical bonding and microstructure if ion beam-deposited cnxfilms including an XPS C 1s peak simulation. Surf. Interface Anal. 1998, 25, 629–642. [Google Scholar] [CrossRef]
- Angerstein-Kozlowska, H.; Conway, B.E.; Hamelin, A.; Stoicoviciu, L. Elementary steps of electrochemical oxidation of single-crystal planesof Au-I. Chemical basis of processes involving geometry of anions and the electrode surfaces. Electrochim. Acta 1986, 31, 1051–1061. [Google Scholar] [CrossRef]
- Paulus, U.; Schmidt, T.; Gasteiger, H.; Behm, R. Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: A thin-film rotating ring-disc electrode study. J. Electroanal. Chem. 2001, 495, 134–145. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. PEM Fuel Cell Electrocatalysts and Catalyst Layers; Springer: London, UK, 2008. [Google Scholar]
- Ge, X.; Sumboja, A.; Wuu, D.; An, T.; Li, B.; Thomas Goh, F.W.; Andy Hor, T.S.; Zong, Y.; Liu, Z. Oxygen reduction in alkaline media: From mechanisms to recent advances of catalysts. ACS Catal. 2015, 5, 4643–4667. [Google Scholar] [CrossRef]
- He, Q.; Li, Q.; Khene, S.; Ren, X.; López-Suárez, F.E.; Lozano-Castelló, D.; Bueno-López, A.; Wu, G. High-loading cobalt oxide coupled with nitrogen-doped graphene for oxygen reduction in anion-exchange-membrane alkaline fuel cells. J. Phys. Chem. C 2013, 117, 8697–8707. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, S.; Zheng, M.; Tang, J.; Liu, L.; Chen, J.; Wang, X. Graphitic-N-rich N-doped graphene as a high performance catalyst for oxygen reduction reaction in alkaline solution. Int. J. Hydrogen Energy 2020, 45, 32402–32412. [Google Scholar] [CrossRef]
- Yi, L.; Song, Y.; Liu, X.; Wang, X.; Zou, G.; He, P.; Yi, W. High activity of Au-Cu/C electrocatalyst as anodic catalyst for direct borohydride-hydrogen peroxide fuel cell. Int. J. Hydrogen Energy 2011, 36, 15775–15782. [Google Scholar] [CrossRef]
- Ma, J.; Li, J.; Yang, S.; Lu, H.; Liu, L.; Wang, R. Ultrathin veil-like SnO2 supported Co3O4 nanoparticles for direct borohydride fuel cell anode. J. Power Sour. 2020, 453, 227866. [Google Scholar] [CrossRef]
- Yi, L.; Wei, W.; Zhao, C.; Tian, L.; Liu, J.; Wang, X. Enhanced activity of Au-Fe/C anodic electrocatalyst for direct borohydride-hydrogen peroxide fuel cell. J. Power Sour. 2015, 285, 325–333. [Google Scholar] [CrossRef]
- Shi, K.M.; Cheng, X.; Jia, Z.Y.; Guo, J.W.; Wang, C.; Wang, J. Oxygen reduction reaction of Fe-polyaniline/carbon nanotube and Pt/C catalysts in alkali media. Int. J. Hydrogen Energy 2016, 41, 16903–16912. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | T(°C) | Peak Power Density(mW cm−2) | j at Peak Power Density (mA cm−2) | E at Peak Power Density (V) | Specific Peak Power Density (mW mgAu−1) |
|---|---|---|---|---|---|
| Au/C | 25 | 31.6 | 79 | 0.40 | 405.1 |
| 35 | 43.0 | 86.0 | 0.50 | 551.3 | |
| 45 | 47.8 | 86.6 | 0.55 | 612.8 | |
| 55 | 59.0 | 90.7 | 0.65 | 756.4 | |
| AuCeO2/C | 25 | 69.8 | 116.2 | 0.60 | 983.1 |
| 35 | 78.9 | 143.3 | 0.55 | 1111.3 | |
| 45 | 90.3 | 164.0 | 0.55 | 1271.8 | |
| 55 | 96.4 | 175.1 | 0.55 | 1357.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stagniūnaitė, R.; Kepenienė, V.; Balčiūnaitė, A.; Drabavičius, A.; Pakštas, V.; Jasulaitienė, V.; Tamašauskaitė-Tamašiūnaitė, L.; Norkus, E. An Electrocatalytic Activity of AuCeO2/Carbon Catalyst in Fuel Cell Reactions: Oxidation of Borohydride and Reduction of Oxygen. Catalysts 2021, 11, 342. https://doi.org/10.3390/catal11030342
Stagniūnaitė R, Kepenienė V, Balčiūnaitė A, Drabavičius A, Pakštas V, Jasulaitienė V, Tamašauskaitė-Tamašiūnaitė L, Norkus E. An Electrocatalytic Activity of AuCeO2/Carbon Catalyst in Fuel Cell Reactions: Oxidation of Borohydride and Reduction of Oxygen. Catalysts. 2021; 11(3):342. https://doi.org/10.3390/catal11030342
Chicago/Turabian StyleStagniūnaitė, Raminta, Virginija Kepenienė, Aldona Balčiūnaitė, Audrius Drabavičius, Vidas Pakštas, Vitalija Jasulaitienė, Loreta Tamašauskaitė-Tamašiūnaitė, and Eugenijus Norkus. 2021. "An Electrocatalytic Activity of AuCeO2/Carbon Catalyst in Fuel Cell Reactions: Oxidation of Borohydride and Reduction of Oxygen" Catalysts 11, no. 3: 342. https://doi.org/10.3390/catal11030342
APA StyleStagniūnaitė, R., Kepenienė, V., Balčiūnaitė, A., Drabavičius, A., Pakštas, V., Jasulaitienė, V., Tamašauskaitė-Tamašiūnaitė, L., & Norkus, E. (2021). An Electrocatalytic Activity of AuCeO2/Carbon Catalyst in Fuel Cell Reactions: Oxidation of Borohydride and Reduction of Oxygen. Catalysts, 11(3), 342. https://doi.org/10.3390/catal11030342