1. Introduction
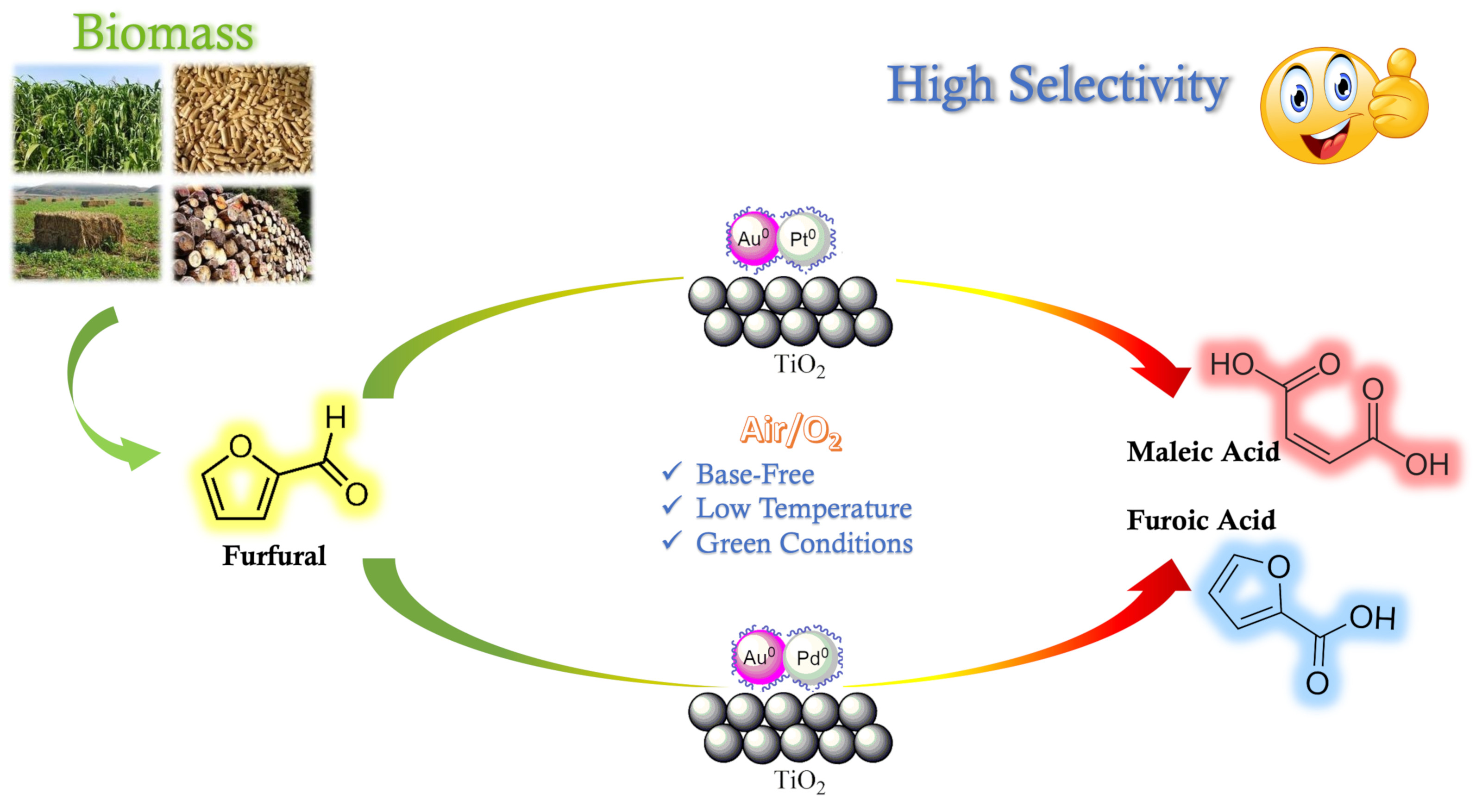
The catalytic oxidation of bio-based molecules in general and of furanics in particular is a highly attractive process. In recent years, the production of biofuels via the hydrogenation of furfurals such as tetrahydrofuran and 2-methyltetrahydrofuran has been reported [
1]. However, the oxidation of furfural can also lead to the formation of many interesting molecules such as furoic acid (FA), maleic acid (MA), and succinic acid (SA) [
2]. Moreover, the oxidation of furfural to furoic acid is not easy because the overoxidation of products can also be obtained. The formation of SA is possible by passing 2(3H)-furanone as an intermediate, while MA can be produced by using 2(5H)-furanone as an intermediate. However, very often the rate of these competitive reactions is likely to be limited by the decarboxylation of furoic acid [
3]. Furfural oxidation reactions can proceed by using different methods such as chemical oxidation, biochemical transformation, and homogeneous or heterogeneous catalytic conversions [
4,
5,
6]. Although the presence of a base allows for higher reaction rates, higher feed concentrations, better product solubility, and lower adsorption of the products on the catalyst surface [
7], many disadvantages arise when controlling for the selectivity to the desired product and in avoiding forming other byproducts. Moreover, there are also several difficulties in the separation process that can occur.
In previous research, the use of inorganic bases such as KOH or NaOH were commonly used for the oxidation reaction of furfural. It has already been proven by Besson et al. [
8] and Wojcieszak et al. [
9] that the use of a base in the oxidation of furfural results in its degradation and in the formation of humins, which present as a black precipitate. Moreover, the basic medium facilitates the C–C bond cleavage and leads to the formation of levulinic acid and formic acid (compounds with a low molar weight). Thus, the design of heterogeneous catalytic systems capable of maintaining a high activity and selectivity without the use of a homogeneous base is still a huge challenge in the oxidation of furfural under noncontrolled pH [
10,
11]. It is well-known that, contrary to the conventional catalysts based on Pt and Pd, Au-based catalysts can offer a better resistance to water and O
2, and thus present more stability and selectivity in the oxidation of organic compounds in water. However, due to the absence of a base promoter and the formation of organic products or intermediates such as carboxylic acid, Au could be more easily deactivated. The introduction of a second metal to form the bimetallic nanoparticles has been successfully demonstrated in the oxidation of furfural under base-free conditions [
12]. The alloying of metals such as Au and Pd, and Au and Pt in the catalysts used combines the advantages of different components at the atomic level, and as a result enhances the activity and the selectivity to the desired products. Moreover, it may help to slow or prevent catalyst deactivation, as well as to prevent the leaching of metals from the catalyst support in bimetallic Au-Pd-based catalysts [
13]. The arrangement of the metal NPs influence the catalytic performance of the bimetallic catalytic systems. Thus, the design of the catalyst by controlling the structure of the bimetallic NPs is needed to obtain high catalytic performance [
12]. The advantage of an addition of a second metal was clearly shown when working under an uncontrolled pH. To this end, Pt showed a better effect when compared to Pd, which means that Pt had a boosting effect wherein only gluconic acid was produced [
14]. Furthermore, this high catalytic activity was attributed to the presence of a core–shell structure, as charges could be easily transferred between the core and the shell; thus oxygen was activated, leading to the attack of the desired functional group. Therefore, the catalytic activity changed according to the composition of the system. The synergy and the interaction between the two metals in the catalyst was mainly due to the geometric and the electronic effects. This synergistic effect was proven to be one of the major factors that strongly influences the catalytic performance, which has already been reported [
14,
15]. Moreover, the choice to use bimetallic Au-Pd and Au-Pt was made in order to avoid using a base that was mandatory when a monometallic gold catalyst was used as a catalyst in a water/oxygen system. In this article, preliminary studies applying a smart methodology based on the experimental design (
Table S1) was used to better understand the role of the noble bimetallic catalyst on the oxidation of the furfural orientation. Sol immobilization method was applied for the synthesis. This method proved its capacity to produce metal particles with random alloy structures [
16,
17].
3. Discussion
To determine the specific surface area of the analyzed catalysts, nitrogen adsorption and desorption analyses on monometallic catalysts (2% Au/TiO
2, 2% Au
1Pd
1/TiO
2, and 2% Au
1Pt
1/TiO
2) were performed. The pore volume, pore size, and the values of the surface areas of the catalysts were calculated. The results are shown in
Table 4.
The Brunauer–Emmett–Teller (BET) analysis showed that different porosities were obtained for the tested samples. The BET surface area of the Au
1Pd
1/TiO
2 and Au
1Pt
1/TiO
2 samples were twice as large as that of the Au/TiO
2 sample. The very low surface area of this catalyst is probably due to the pore blockage, as could be deduced from the significant decrease in the pore volume when compared to the TiO
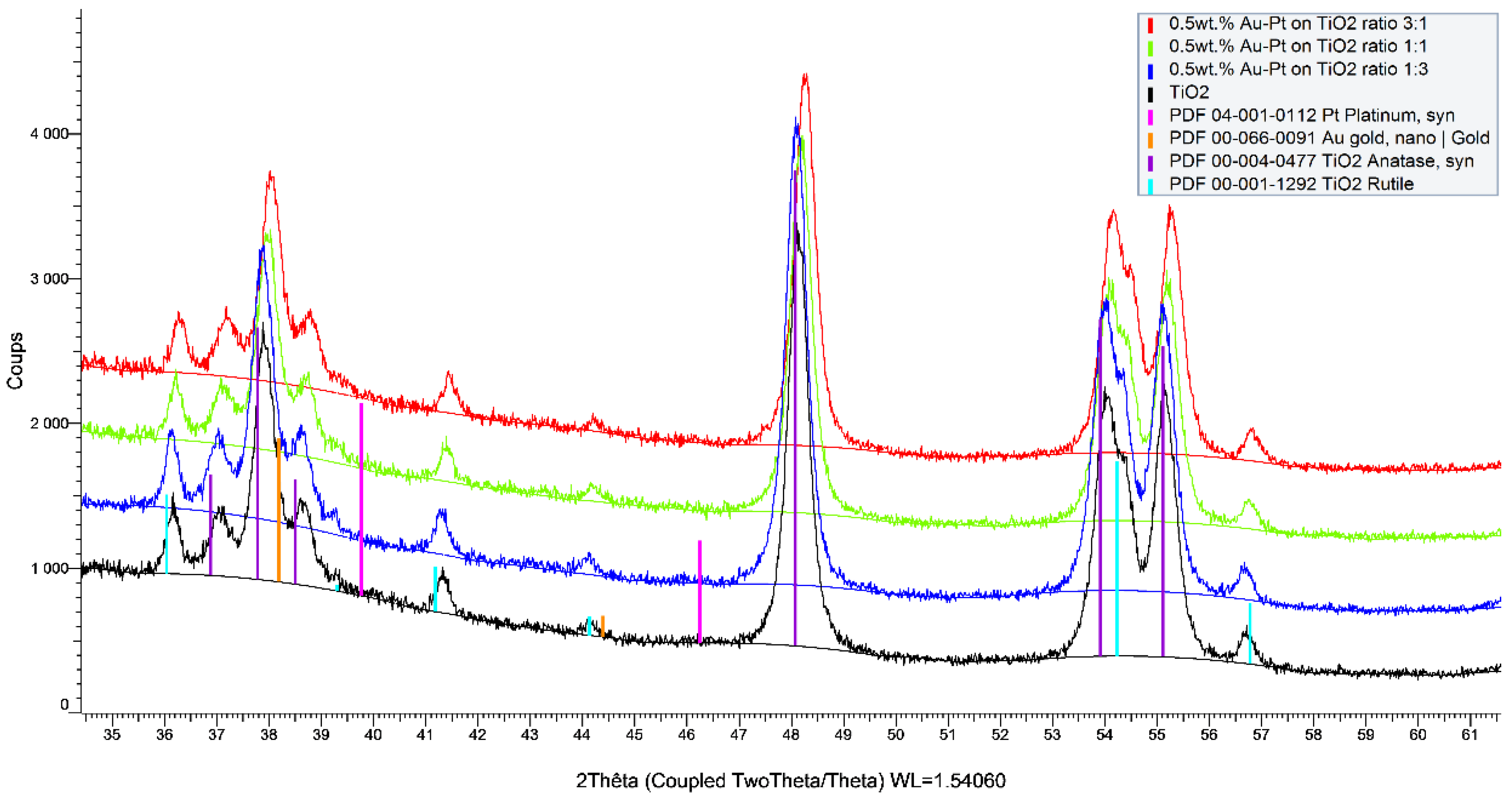
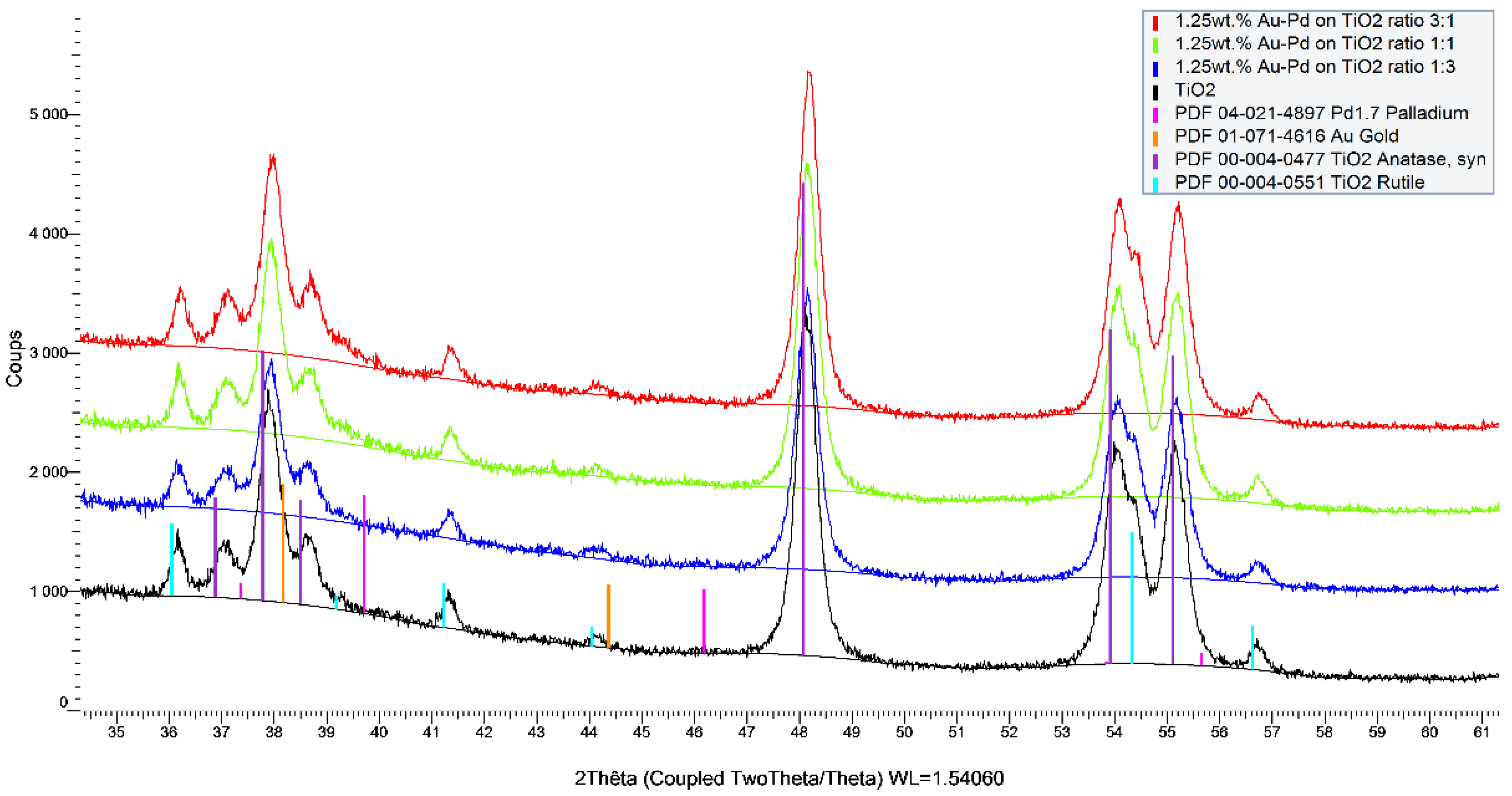
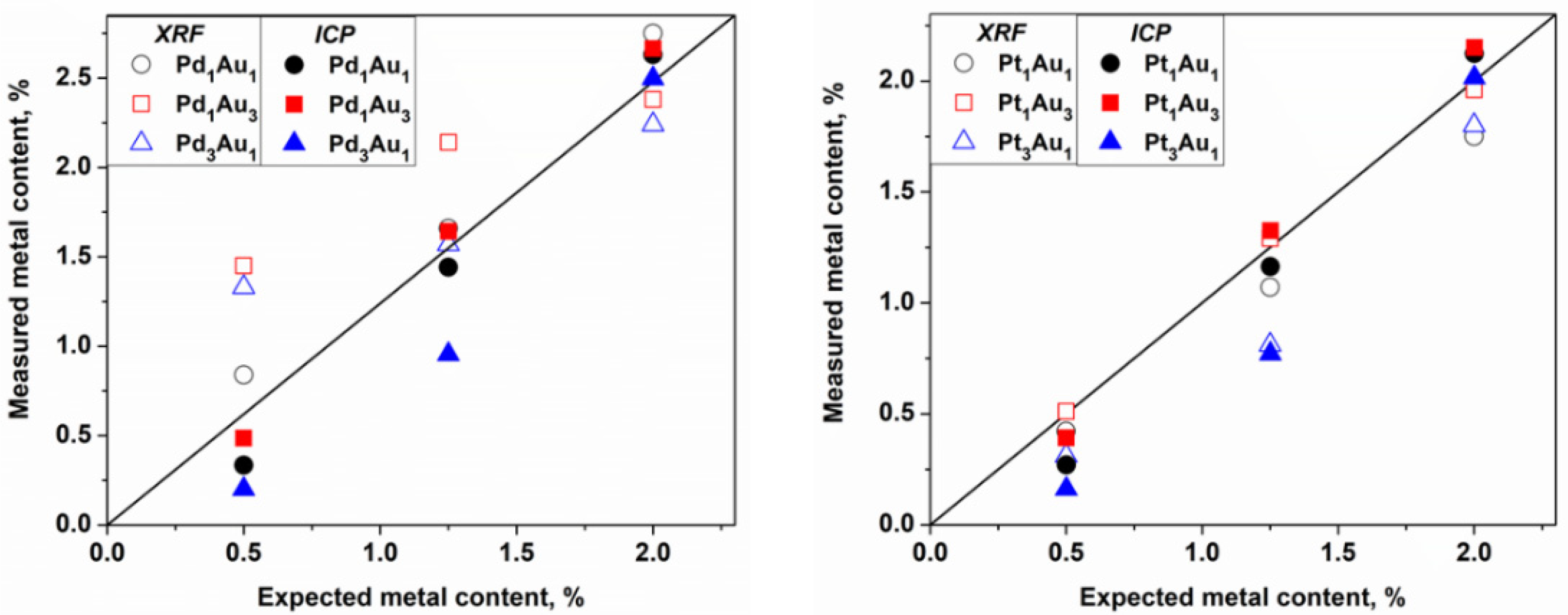
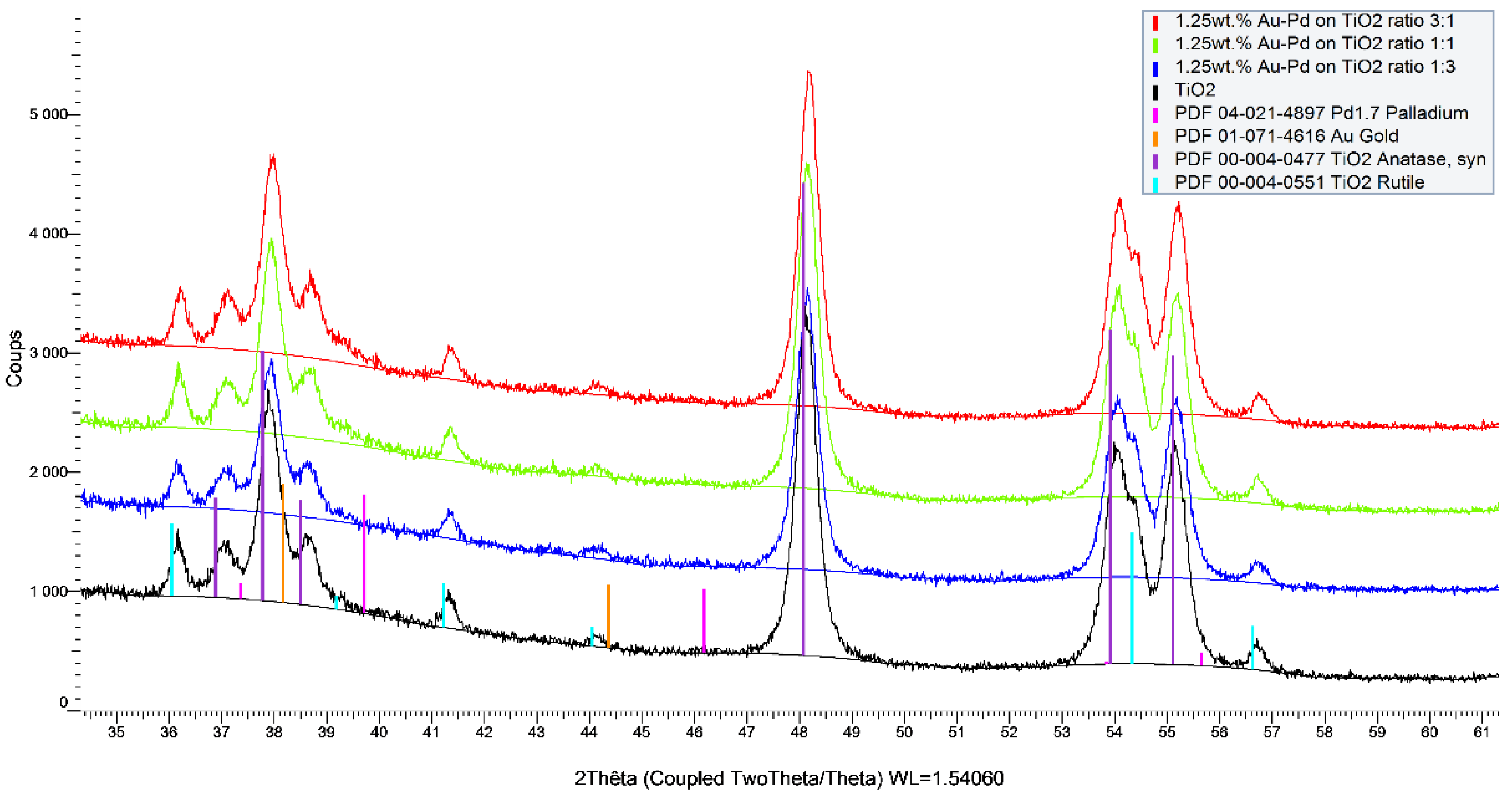
2 support. The differences between the catalysts were also observed in the ICP and XRF analyses (
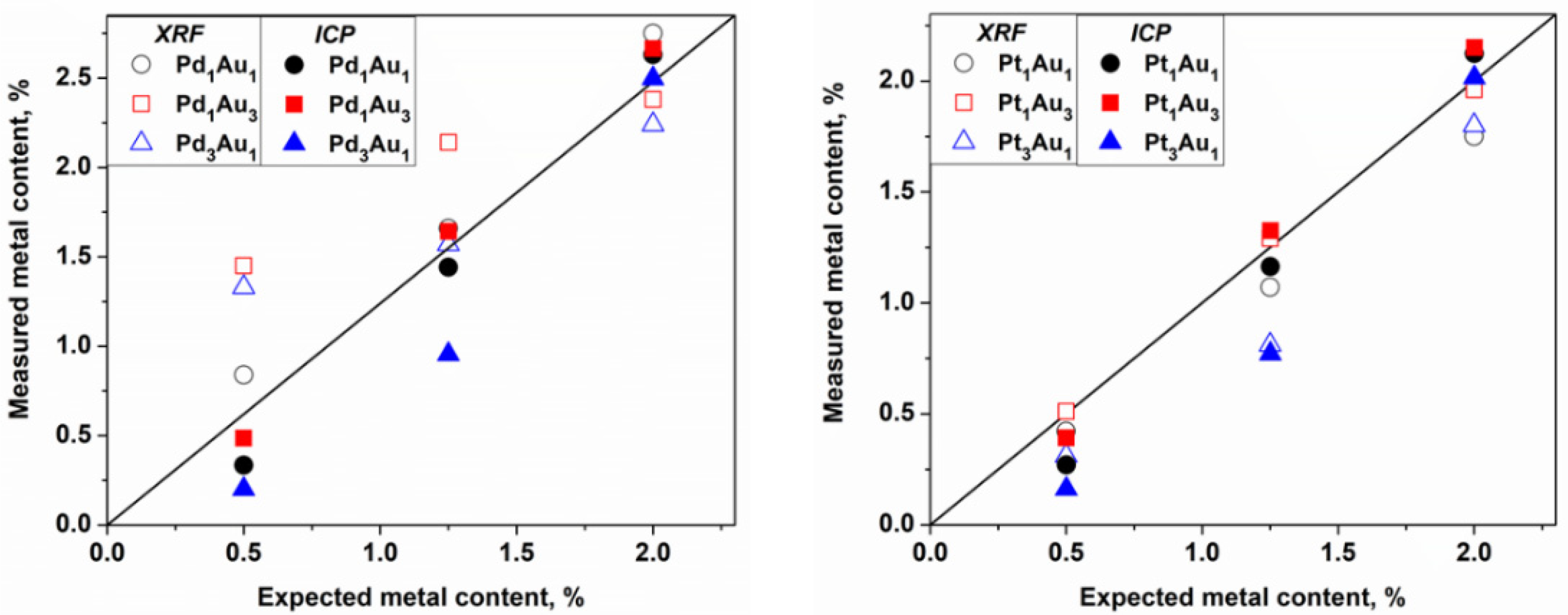
Figure 6).
Observing the ICP values, which are quantitative data, some differences were observed between the theoretical and measured values that could be due to several factors. However, in considering the intrinsic error of the robotic system in the distribution of reagents and support (error estimated at 5%), values very close to the expected values for the catalysts were obtained. The exceptions were the catalysts rich in Pd and Pt at 0.5 and 1.0 wt.%, respectively, which always had a lower metal loading than was expected. The same observation was made already for the monometallic Pt/TiO
2 catalyst prepared on the bench (
Table 1). The XRF analysis is a semiquantitative and nondestructive technique that can be very useful for the quantification of metals in catalysts, and especially in this case where the catalysts were synthesized in an automated way. Thus, the values were reasonably similar to the values obtained by the ICP for the Au-Pt catalysts but not for the Au-Pd catalysts with loadings of 0.5 and 1.25 wt.%, respectively. The overestimation of the Pd concentration in the Au-Pd catalysts using the XRF in comparison with that of the ICP is due to the Rh tube that is used as the X-ray source, which has an energy close to that of Pd and which induces an interference of lines on the spectra while quantifying this element. No differences observed in the TEM analysis confirmed that the sol-immobilization method permits us to obtain small metal particle sizes and a good particle distribution. All these parameters significantly affected the catalytic properties of the catalysts.
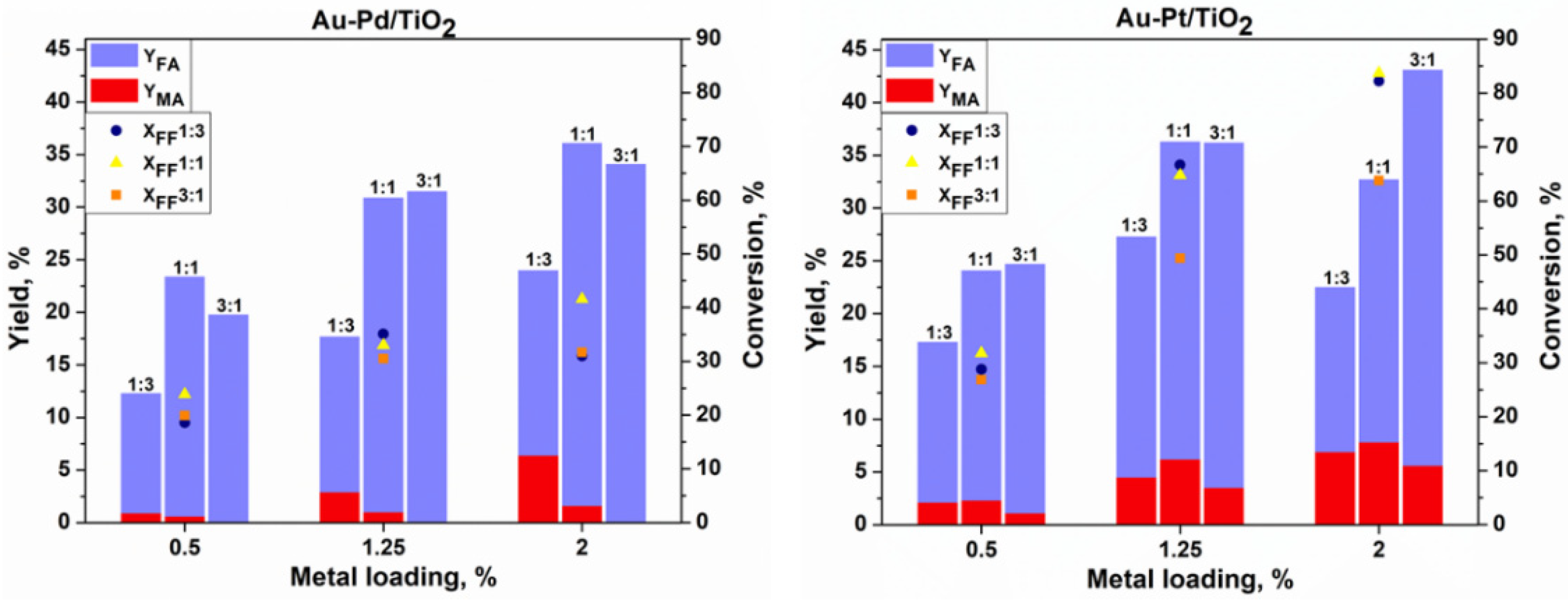
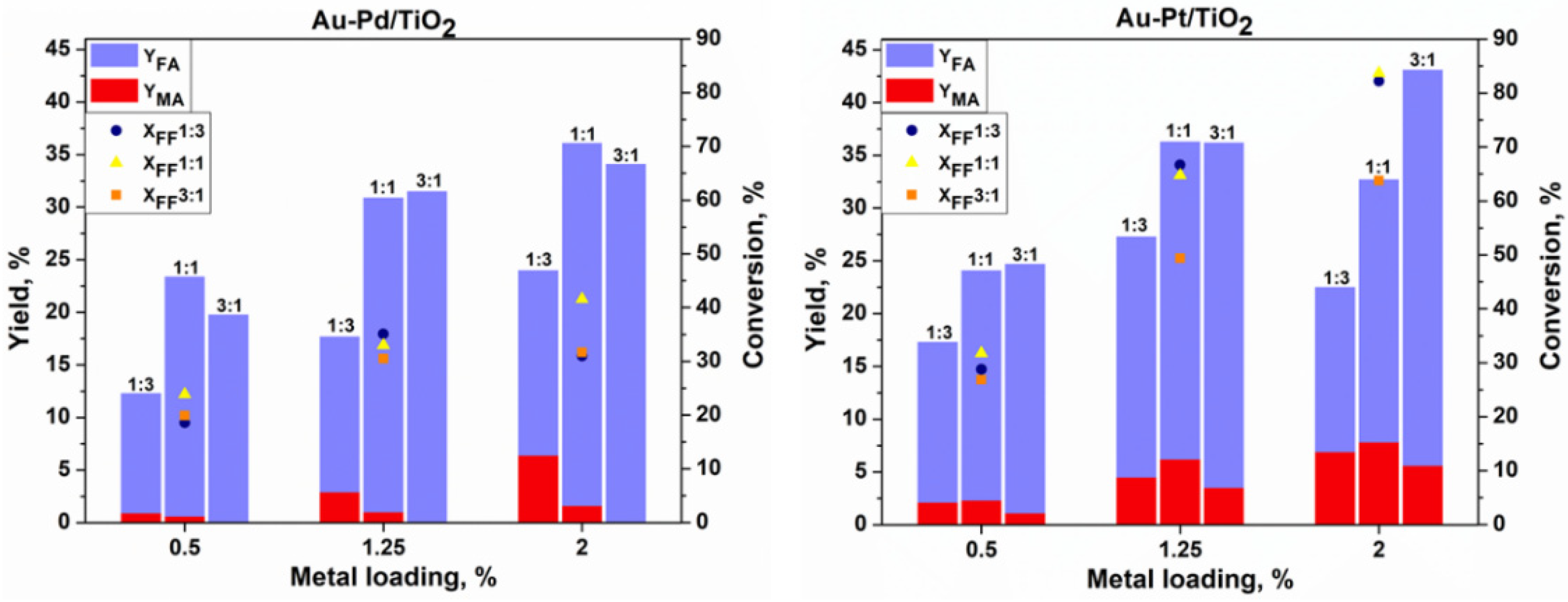
All the tested catalysts were active in the base-free oxidation of furfural at 80 °C, as shown in
Figure 5. It could be seen that generally the increase in the catalyst metal loading increases the conversion of furfural. However, this increase depends on the composition of the catalyst. As expected, the higher catalytic activity was observed for catalysts with a lower Au content. The activity of the bimetallic catalysts with a high Au content is comparable to the results observed for the monometallic Au/TiO
2 catalyst (
Figure 4). Much higher activity was observed for the Au-Pt catalysts when compared to the Au-Pd catalysts. This boosting effect was already observed for the Au-Pt catalysts in the glucose oxidation reaction, where a better effect of Pt was observed in comparison to Pd [
14].
An increase in the metal loading from 0.5% to 2% using the Au
3Pt
1/TiO
2 catalyst leads to the increase in furfural conversion by 37%, reaching 64% with a 38% and 6% yield of FA and MA, respectively. The same behavior could be seen in terms of the FF conversion using Au
3Pd
1/TiO
2, which increases to reach only 32% but with 100% carbon balance and selectivity to FA, with neither MA nor 2-furanone (FAO) being formed. This is a remarkable result, as for the first time 100% selectivity to furoic acid with 100% carbon balance was observed in the base-free oxidation of furfural. Indeed, the increase in the catalytic activity in the oxidation reactions was well established using Au-Pd bimetallic catalysts [
16,
17,
18]. The Au-Pd nanoparticles were found to be two times more active than Pd alone for the oxidation reaction of alcohols, where it was proposed that Au is an electronic promoter of Pd [
19,
20].
The effect of the catalyst’s composition was also reported, whereby increasing the Pd content to an optimum ratio of Au:Pd in ca. 1:3 leads to the increase in the catalytic activity, while there is a progressive decrease in the activity with a further increase in the Pd content [
21]. It may be concluded that there should be an optimum ratio between both metals that will maintain a high activity as well as a high selectivity. It is highly probable that with the changing concentrations of metals relative to the support material, the electronic and the geometric structures of the individual clusters change significantly, thereby affecting the bonding between the reactant and the catalyst, and as a result altering the catalytic performance. By considering this, it can be concluded that studying the composition and the structure of the Au-Pd and Au-Pt bimetallic catalysts is a key factor for designing more active catalysts and to find the relationships between their structures and the activities.
The catalytic activity changed significantly depending on the composition of the catalytic system.
Table 5 shows that the selectivities to FA and MA depend on the ratio of metals in each catalyst. At the same FF conversion (about 30%), gold-rich catalysts promote the oxidation of FF to FA, reaching 100% of selectivity using 1.25% of the Au
3Pd
1-based catalyst and a maximum of 88% using 0.5% of Au
3Pt
1 catalyst (
Table 5). These values decrease with the increase in the quantity of the second added metal, but at the same time the selectivity to MA increases to 7% and 8% using 1.25% of the Au
3Pd
1 and 0.5% of Au
3Pt
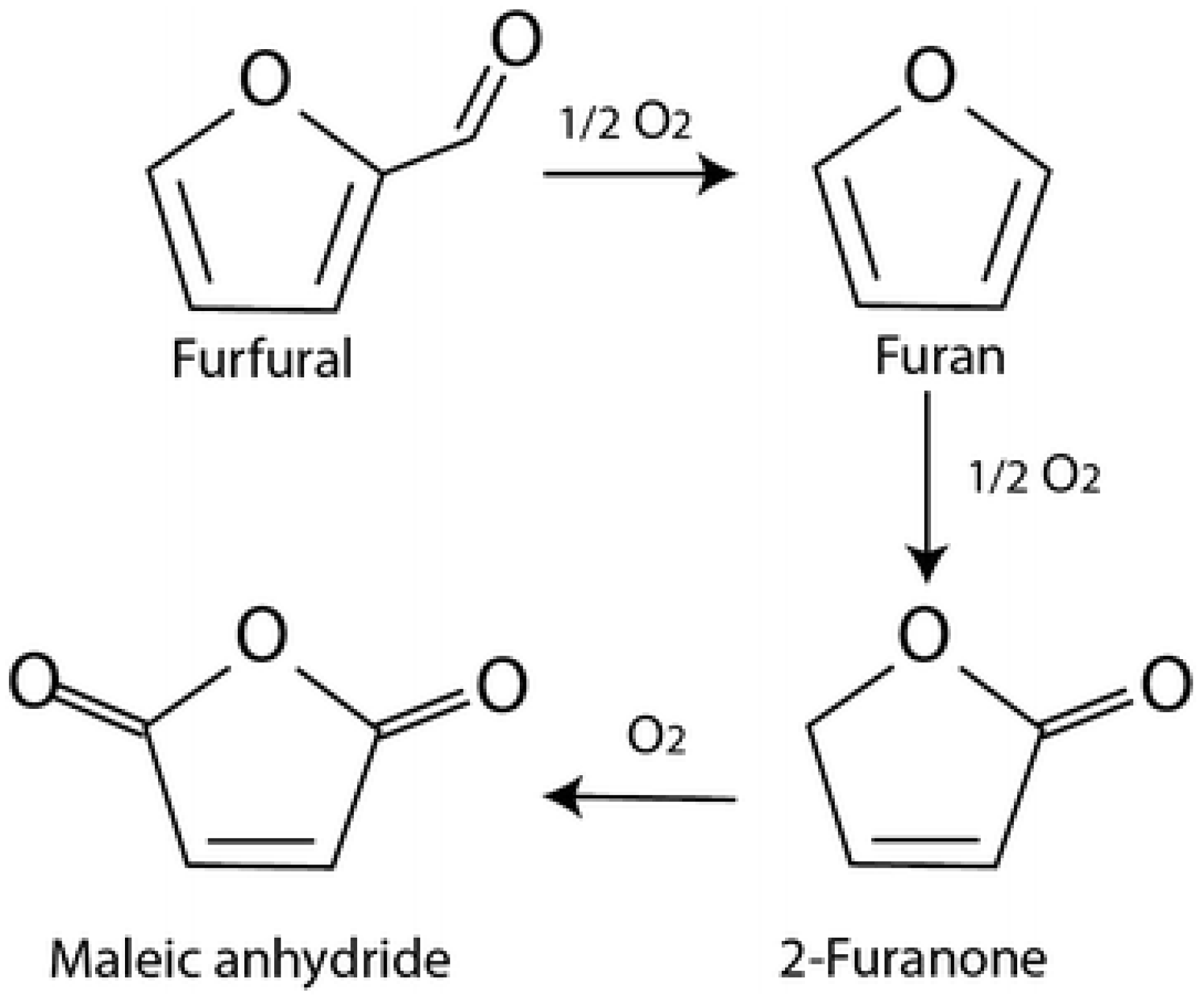
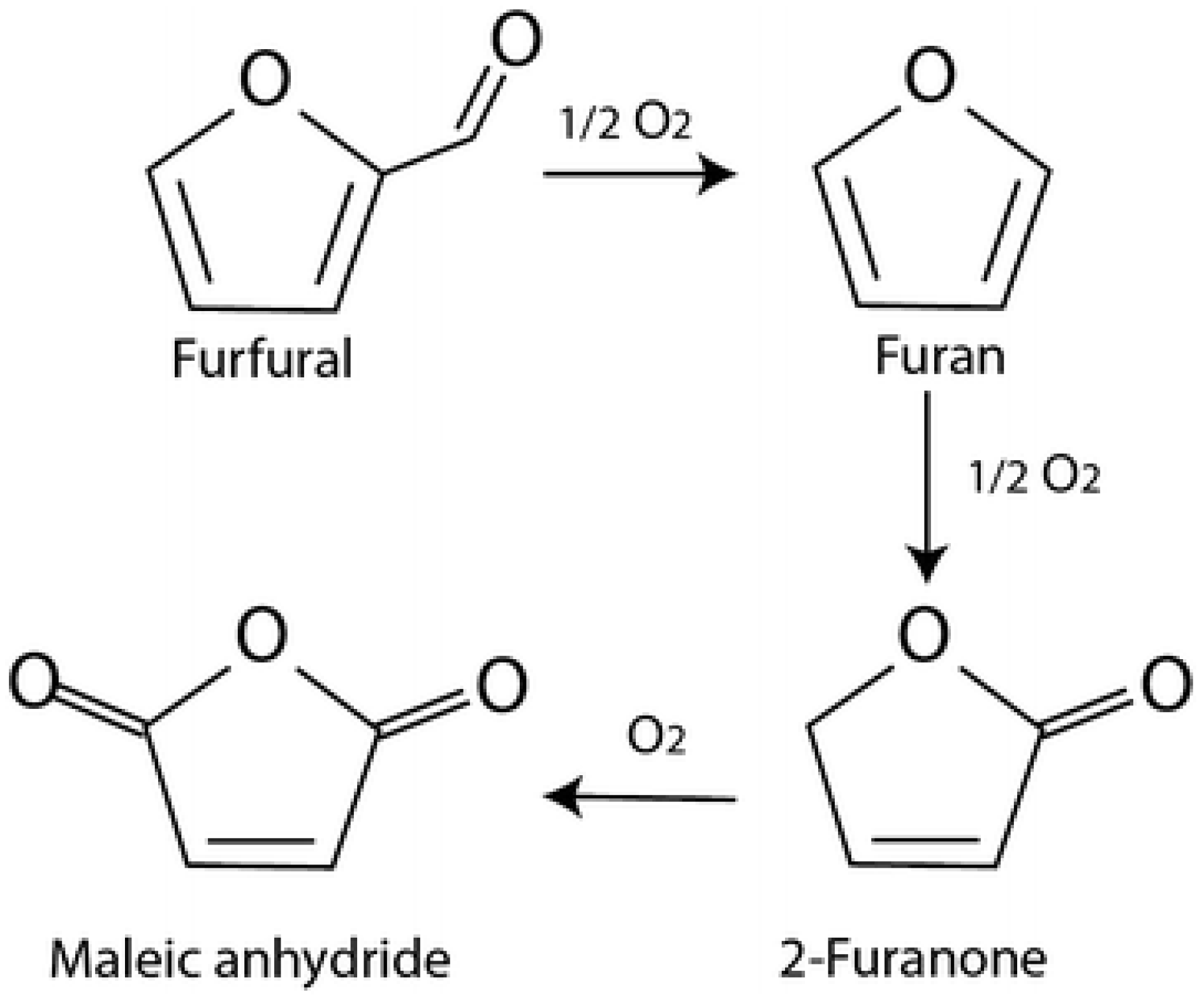
1 catalysts, respectively. This suggests that the selectivity of FA is strongly dependent on the ratio of Au, and that MA is dependent on the ratio of Pt or Pd. Therefore, Au has a beneficial effect on maintaining a high selectivity to furoic acid and limits the formation of byproducts. Moreover, the addition of platinum and palladium presents a boosting effect toward the ring-opening reactions and the formation of maleic acid. In addition, the formation of products such as FAO intermediate and two other unknown products were observed in small quantities, which could explain the decrease in the carbon balance. These observations clearly indicate a tandem pathway for the conversion of FF to MA via FA and FAO. Moreover, as has been proposed, maleic acid could be formed by the decarboxylation of furfural to furan, which then gives a 2-furanone intermediate (
Figure 7) [
22].
For comparing the effect of each metal, the two catalysts (2% Au
3Pt
1 and 2% Au
3Pd
1) were chosen. The results showed that the Pt-based catalyst was able to convert 64% of FF after 4 h with a 6% yield of MA, while only 32% of the conversion was obtained using the Au-Pd catalyst with no formation of MA. However, the selectivity to FA reached 100% with the Au-Pd catalyst instead of 60% for the Au-Pt sample. Both Pd and Pt in the bimetallic catalyst produced furoic acid as a major product. These results further demonstrate that 2% Au
3Pd
1/TiO
2 is a promising catalyst for the base-free aerobic oxidation of FF to FA in water. It could also be seen that with the Au-Pt catalysts the maximum yield of MA (8%) was achieved using the 2% Au
1-Pt
1/TiO
2 catalyst while only 1.6% was achieved using the 2% Au
1-Pd
1/TiO
2 catalyst. It seems that the reaction temperature strongly affects the catalytic properties of the Au-Pt and Au-Pd nanoparticles, whereby each catalyst behaved differently during the oxidation of FF. Therefore, when performing the same tests at 110 °C, a full conversion (100%) of furfural was observed. However, no products were detected using the Au-Pt based catalysts, with a very low carbon balance (in some cases reaching 0%). This indicates the overoxidation of the furfural and the formation of low molecular weight molecules or condensation products (via the ring opening reaction and the formation of maleic acid). Regarding the Au-Pd-based catalysts, a maximum yield of MA (14%) and (5%) of FAO was obtained using the 2% Au
1-Pd
1/TiO
2 catalyst at 110 °C, while the carbon balance was very low with a complete conversion of FF, also indicating the overoxidation and the degradation of FF, but at a lower extent when compared to the Au-Pt catalyst. It was previously reported that the overoxidation and the poisoning from byproducts could be responsible for the deactivation of Pd- or Pt-based catalysts when they are used in the liquid phase with oxygen as an oxidant [
23]. By comparing the monometallic Au-, Pt-, and Pd-based catalysts to the bimetallic catalysts, the monometallic catalysts were by far less active than the bimetallic catalysts in terms of furfural conversion, and the formation of FA, MA, and FAO intermediates. From a functional point of view, it is important to state that better catalytic performances could be obtained with the highly active bimetallic catalysts than with the gold-based monometallic catalysts when working under certain conditions (such as 80 °C, air as oxidant) and with specific chemical compositions (such as Au-Pd and Au-Pt). A good correlation appeared between the performances of these catalysts, and in the chemical composition and the loading of both metals. The nature of the second metal (Pd or Pt) which mainly governs the catalytic activity seems to also have a strong impact on the oxidation in base-free conditions. Relatively high yields of MA observed for the Au-Pd catalysts indicate a radical mechanism of the reaction which favors the ring opening pathway.
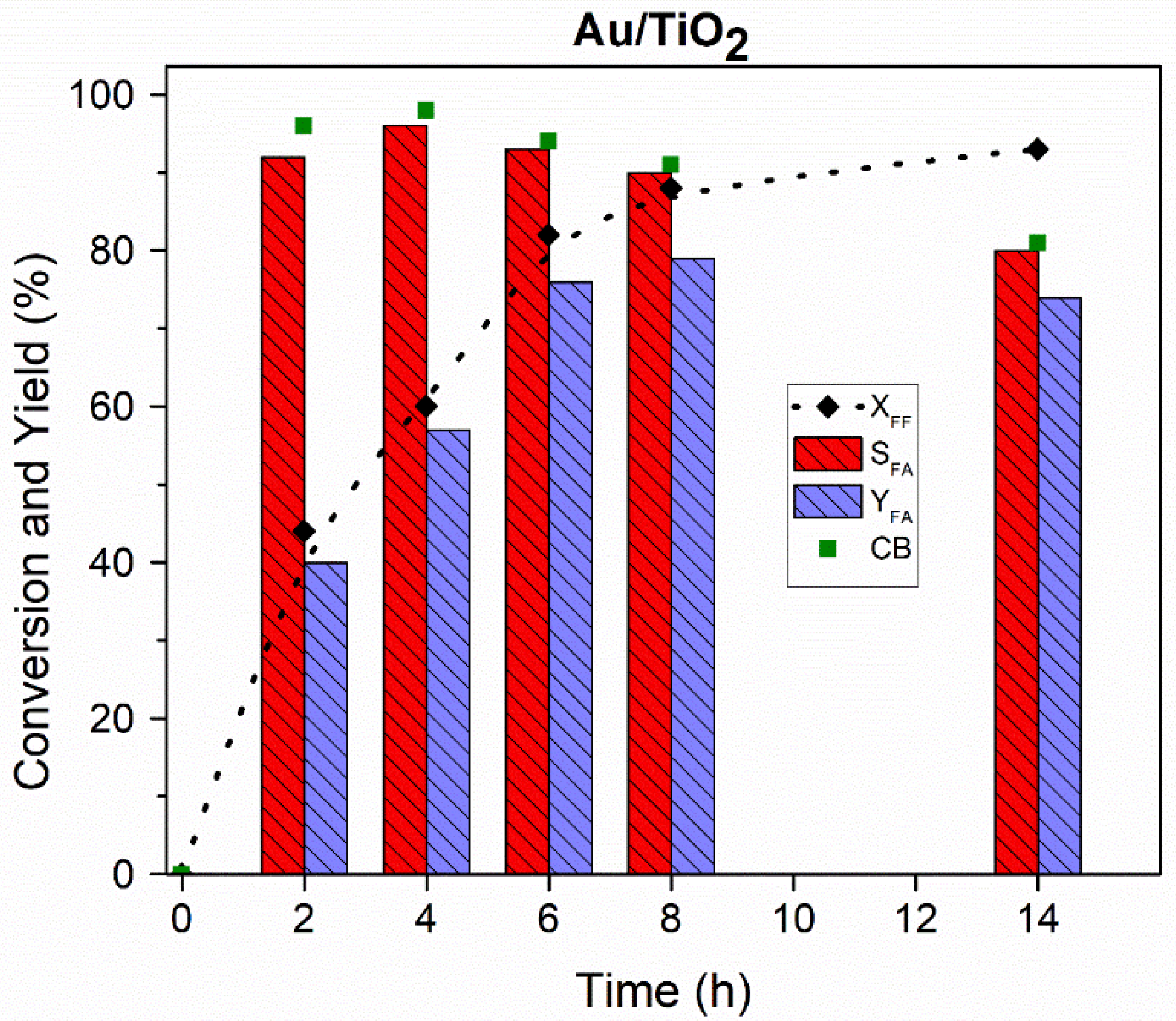
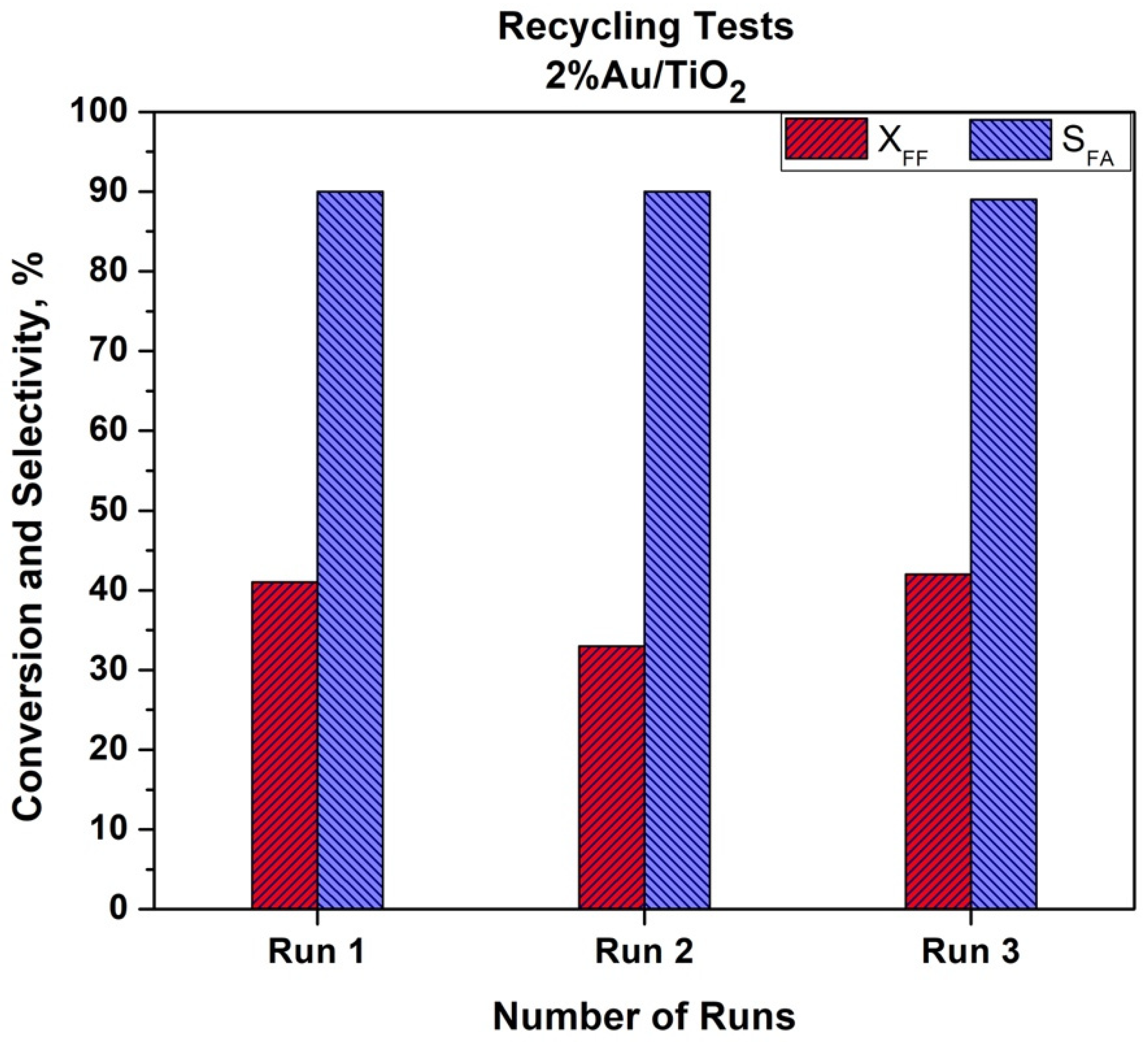
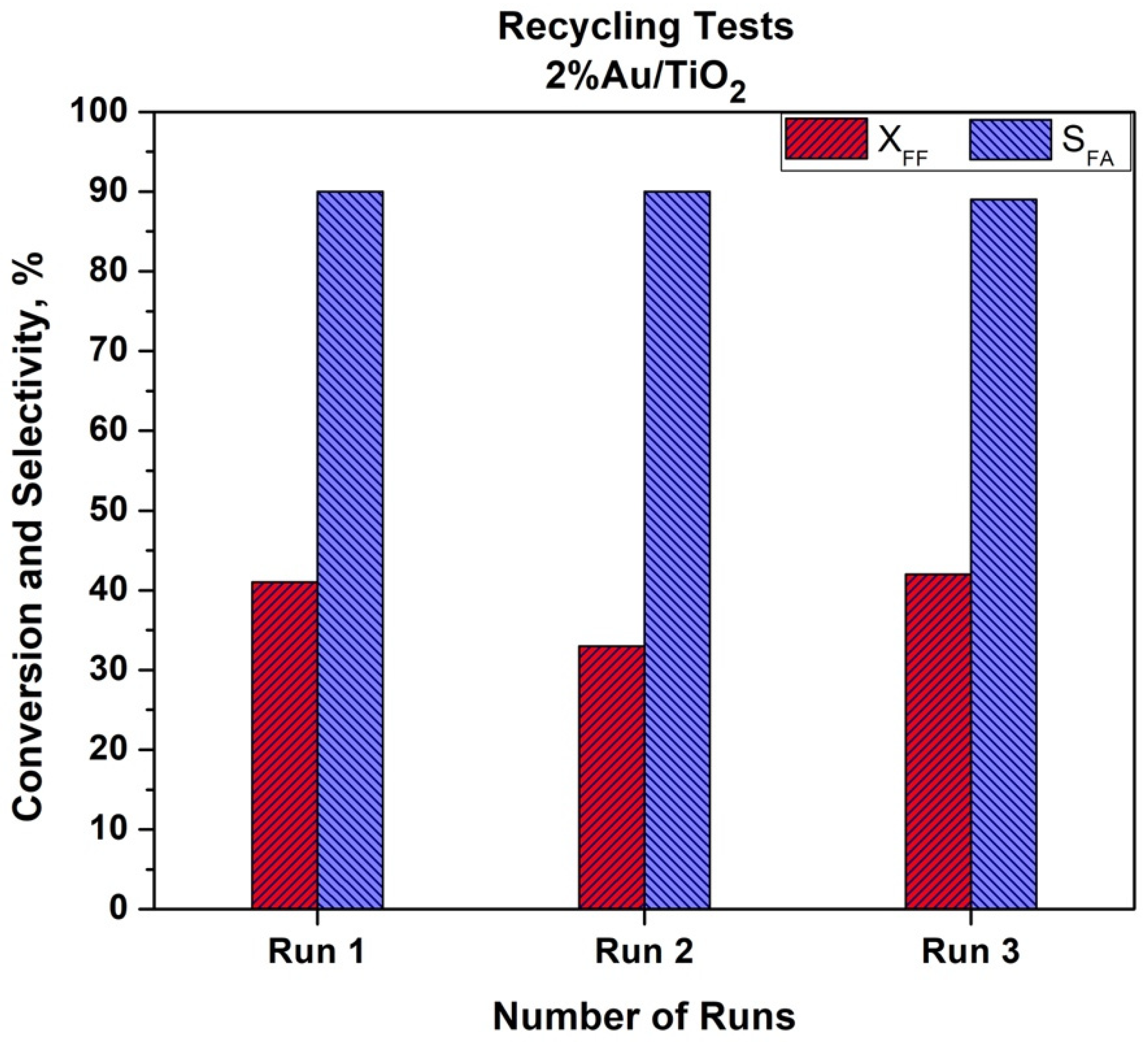
The recyclability of the Au/TiO
2 catalyst was also studied. The recycling tests were performed using the same methodology as described in the Experimental Part. The catalyst was dried under air atmosphere at 80 °C overnight after each run. The results are given in
Figure 8.
As can be seen from
Figure 8, a slight decrease in the catalytic activity was observed. After the first run, a 20% drop in furfural conversion was observed. This drop was not confirmed after the third run when initial activity was achieved. This could be due to the problem with the catalyst recovery after the first test. The ICP analysis of the post reactant solution did not show a leaching of gold. We can also expect that a temperature of 110 °C is quite low to observe Au particle size growth. It is worth mentioning that selectivity to furoic acid remained both stable and high (90%) during the three recycling tests. More in-depth studies are in progress to study the recyclability and stability of the bimetallic catalysts in the batch and in the flow conditions.
4. Materials and Methods
Gold(III) chloride solution (HAuCl4, 99.9%, Sigma–Aldrich, Saint Louis, MO, USA; 30%), potassium tetrachloropalladate(II) (K2PdCl4, 99.99%, Sigma–Aldrich, Saint Louis, MO, USA), chloroplatinic acid (H2PtCl6, 8 wt.% solution, Sigma–Aldrich, Saint Louis, MO, USA), sodium borohydride (NaBH4, 98%, Sigma–Aldrich, Saint Louis, MO, USA), poly(vinyl alcohol) (PVA, MW 9000–10,000, 80% hydrolyzed, Sigma–Aldrich, Saint Louis, MO, USA), 2-Furaldehyde (FF, C5H4O2, 99%, Sigma–Aldrich, Saint Louis, MO, USA), 2-furoic acid (FA, 98%, Sigma-Aldrich), and maleic acid (MA, >99%, Sigma-Aldrich). Supports: titanium(IV) oxide (TiO2 P25, 99.5%, Sigma–Aldrich, Saint Louis, MO, USA). Initially the monometallic gold, palladium, and platinum catalysts supported on titanium(IV) oxide were synthesized on the bench using the sol-immobilization method for comparison with their equimolar bimetallic catalysts (Au1Pd1/TiO2 and Au1Pt1/TiO2), and by using 2 wt.% total metal loading.
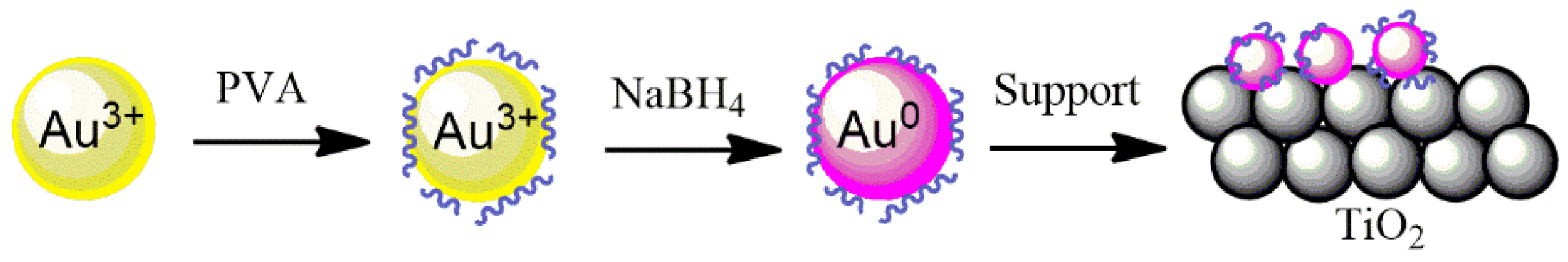
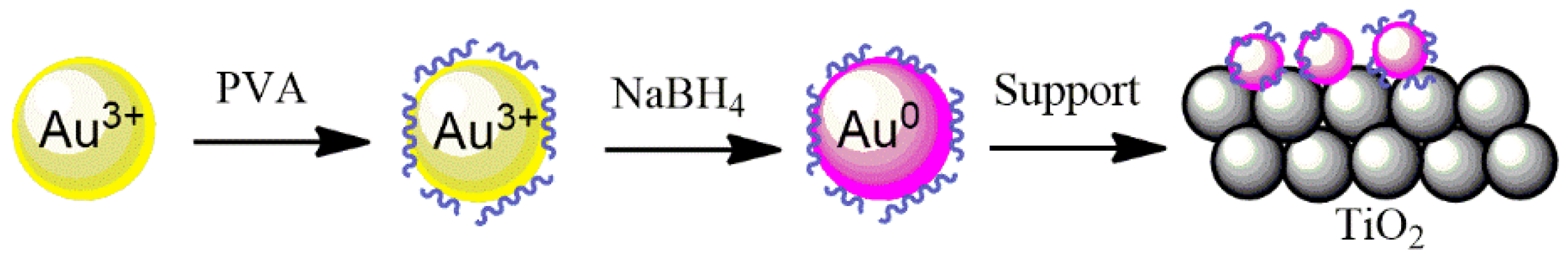
The 2 wt.% Au, Pd, and Pt supported on TiO
2 catalysts were prepared by a sol-immobilization method using NaBH
4 as a reducing agent and polyvinyl alcohol (PVA) as a stabilizing agent to prevent the NPs from aggregation, as well as to control the size of the particles being formed (
Figure 9). Briefly, 1.2 mL of a 2 wt.% aqueous solution of poly (vinyl alcohol) (PVA/Au (
w/
w) = 1.2) solution was dropped into a 200 mL metal precursor–water solution under stirring. In order to obtain the metallic nanoparticles, the freshly prepared solution of the reducing agent NaBH
4 (0.1 M, NaBH
4/metal (mol/mol) = 5) was added drop by drop to the PVA–metal solution and stirred for 30 min. TiO
2 support was then added and the pH was adjusted to 2 by the addition of H
2SO
4. Two hours later, the catalyst was filtered using a filter paper and a Büchner funnel, washed with hot water at 70 °C (3 times, 30 mL each) and ethanol (3 times, 30 mL each), and dried overnight at 80 °C.
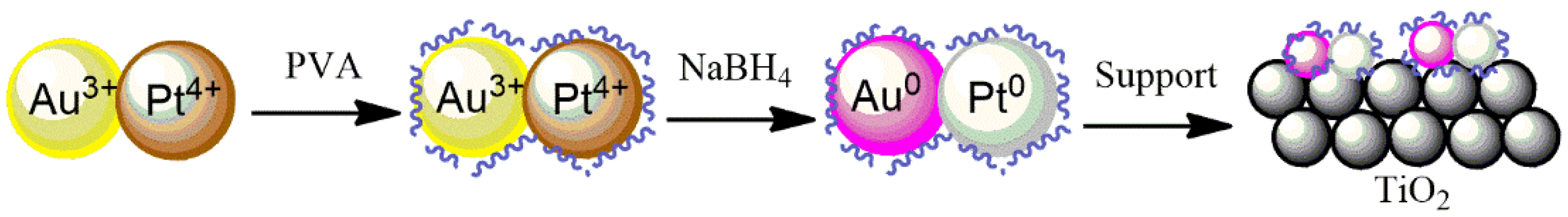
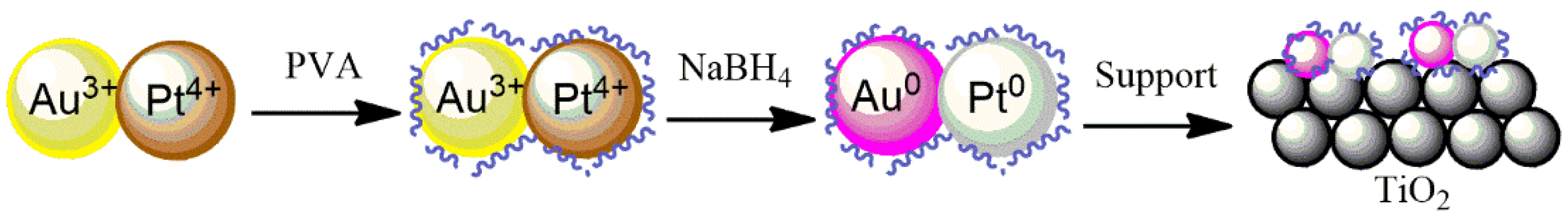
Bimetallic Au-Pd and Au-Pt systems have been synthesized using the REALCAT platform by applying the Design of Experiments (DoE) methodology (
Table S1) to study two different parameters (metal loading and metals ratios). The sol-immobilization method was applied (
Figure 10) to deposit Au, Pd, and Pt NPs onto the surface of TiO
2, in which the total metal loading and metal ratio (Au:Pd and Au:Pt) were varied.
Three different metal loadings were studied: 0.5 wt.%, 1.25 wt.%, and 2 wt.%. The molar ratios between gold and the second metal were varied at 3:1, 1:1, and 1:3, respectively. A Response Surface Design (Central Composite) was created. It was designed for three factors to model curvature data and identify the factor settings that optimize the response. The Central Composite Design (CCD) was used with two factors at three levels of evaluation: low (−1), high (+1), and central (0) levels. The central level was also repeated three times to evaluate the error of the DoE model. The aforementioned levels are used in the CCD to easily represent the minimum, the central, and the maximum values of a factor influencing the catalyst activity. In the CCD, all the possible combinations of high and low levels are studied for the two factors. Hence, the total number of conditions for the CCD was 11 by each metal as calculated: 2n + 3 repetitions at 0 level (where n = 2, the number of factors in this study, and 3 is the number of levels). As the metal are non-numerical factors, no zero level can be applied for this parameter. Therefore, two CCDs were conducted: one for Pt (11 experiences) and the second one for Pd (11 experiences), equaling 22 experiences for both metals. When varying all the parameters, a matrix was built using the “Minitab” software. The responses studied were the FF conversion, the MA and FA yield, and the MA and FA selectivity.
The prepared catalysts were characterized by using different techniques. X-Ray Diffraction (XRD) analysis was performed using a Bruker D8 Advance Powder X-ray diffractometer (Billerica, MA, USA) to determine the morphology of the prepared catalyst. X-Ray Fluorescence (XRF) (M4 Tornado) analysis was performed using an Energy Dispersive X-Ray Fluorescence (EDXRF) spectrometer provided from Bruker (Billerica, MA, USA) to study the chemical composition of the prepared catalysts. The elemental analysis for determining the real metal content in each catalyst was performed by Inductively Coupled Plasma-Optical Emission Spectroscopy 720-ES ICP-OES (Agilent, Santa Clara, CA, USA) with axial viewing and simultaneous CCD detection. The metal particle size in the supported catalysts was determined using Transmission Electron Microscopy (TEM) with a TEM/STEM FEI TECNAI F20 microscope (Hillsboro, Ore. USA) combined with an Energy Dispersive X-ray Spectrometer (EDS) (Hillsboro, OR, USA) at 200 kV. The surface area, pore volume, and distribution of the pore size were determined by nitrogen adsorption/desorption at 77.35 K using a TriStar II Plus and a 3Flex apparatus from Micromeritics (Norcross, GA, USA). The Brunauer–Emmett–Teller (BET) method was used for determining the specific surface area of the prepared materials.
The catalytic tests were performed in a Top Industry autoclave (batch reactor, Vaux le Penil, France). The reactant ((FF) = 24.7 mM) solutions were prepared by diluting a 43 µL of furfural in 21 mL of H2O and stirring the solution to dissolve the furfural before adding it into the vessel. Initially, 1 mL of furfural aqueous solution was taken off for HPLC analysis (t0) and the desired amount of catalyst was added in the autoclave. The reactor was purged three times with pure oxygen before reaching 15 bar. Then, the heating system was started, and the reaction was initiated after reaching 110 °C. The reaction was carried out at 15 bar, 600 rpm during 2, 4, 6, 8, or 14 h, respectively. At the end of the reaction, the catalyst was filtered off and 1 mL of the final solution was diluted for HPLC analysis in a Phenomenex column (ROA, organic acid H+; 300 × 7.8 mm). Sulfuric acid (5 mmol/L−1) was used as a mobile phase with a flow rate of 0.60 mL/min, and the products were detected on a UV-Vis detector at 253 nm. Catalytic furfural oxidation using the bimetallic catalysts were carried out on the REALCAT platform in a Screening Pressure Reactors system (SPR) from UnchainedLabs (UK). The required amounts of catalysts were placed in each reactor. Then, 2 mL of an aqueous solution of furfural ((FF) = 24.7 mM) was injected. The catalytic tests were performed under air pressure (15 bar) with stirring at 600 rpm for 4 h at 110 °C. After the reaction, the catalyst was filtered off and 1 mL of the final solution was diluted for HPLC analysis. The liquid products were analyzed by a High-Performance Liquid Chromatography (HPLC, Shimadzu, Japan) equipped with a UV detector SPD-20A operated at wavelengths of 210 nm and 253 nm, respectively, and a Bio-Rad Aminex HPX-87H column (7.8 × 300 mm) operated at 60 °C. Diluted H2SO4 (5 mM, 0.7 mL/min) was used as a mobile phase. Commercial standards (furfural, furoic acid, and maleic acid) were used for the calibration of the HPLC set up.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}