Concatenated Batch and Continuous Flow Procedures for the Upgrading of Glycerol-Derived Aminodiols via N-Acetylation and Acetalization Reactions




Abstract
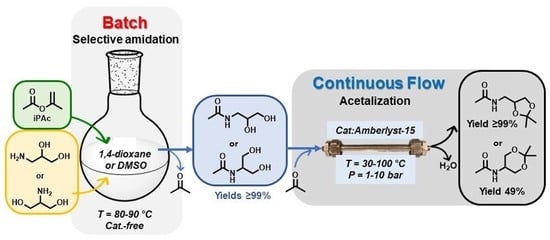
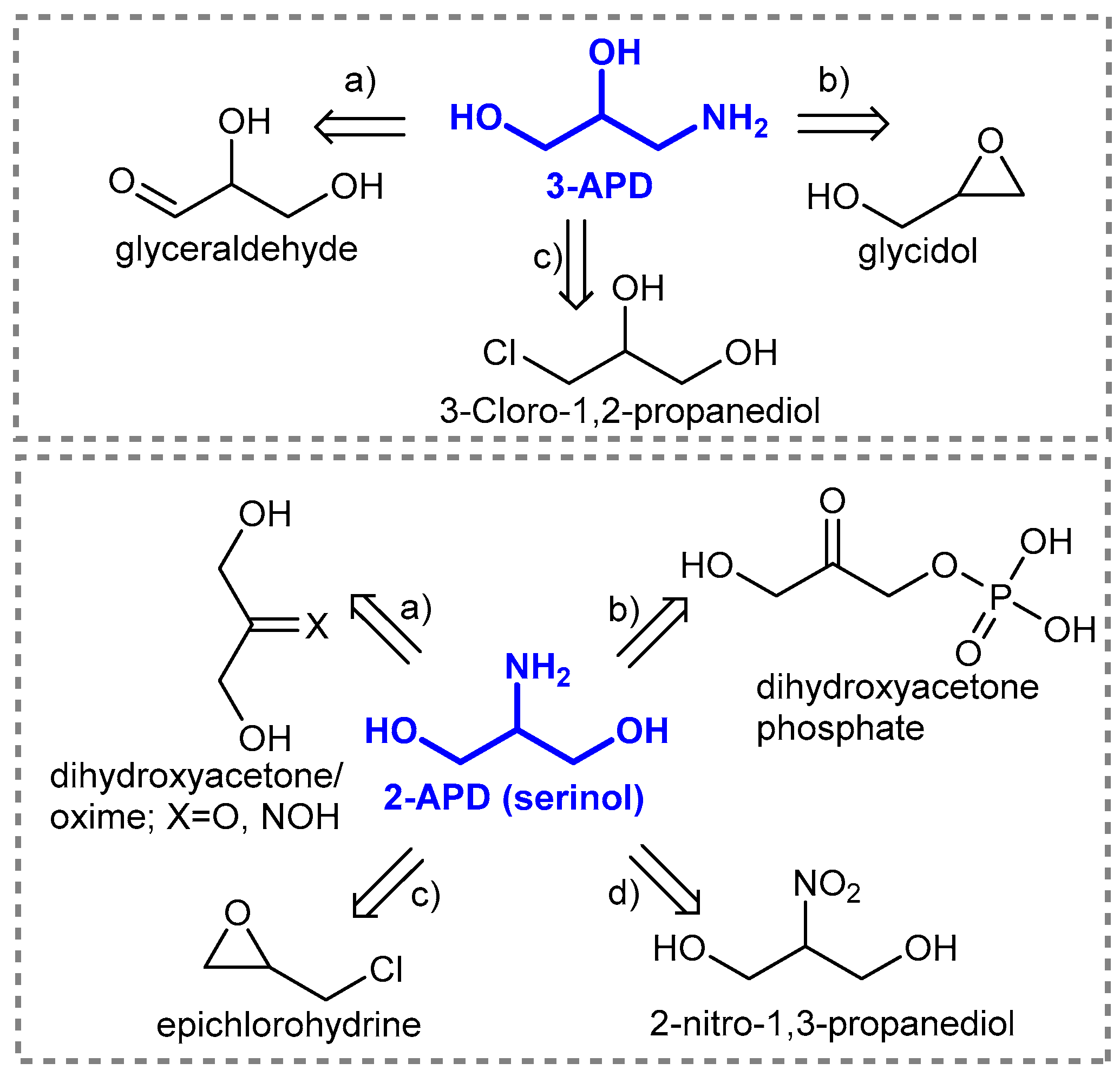
1. Introduction
2. Results and Discussion
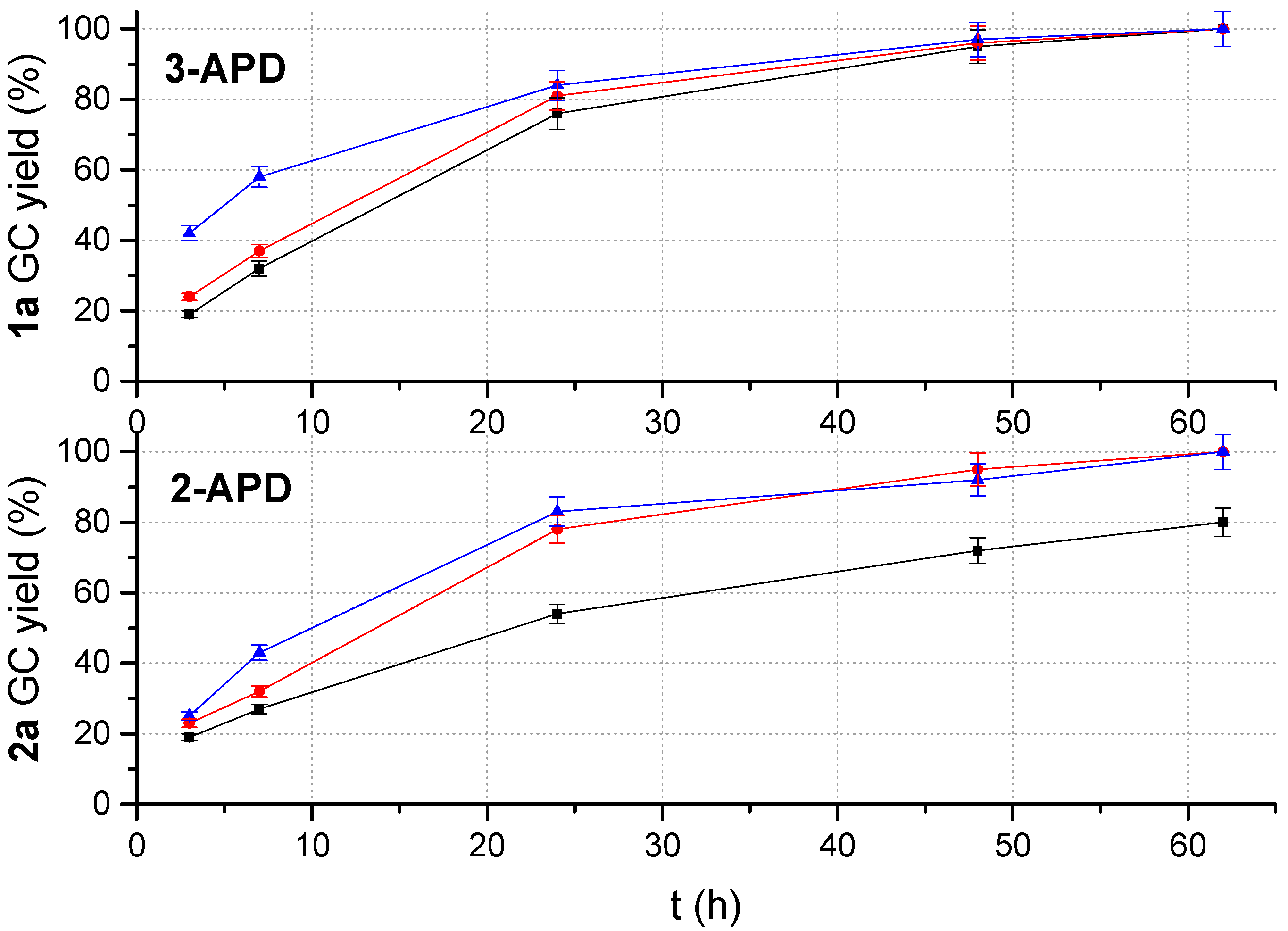
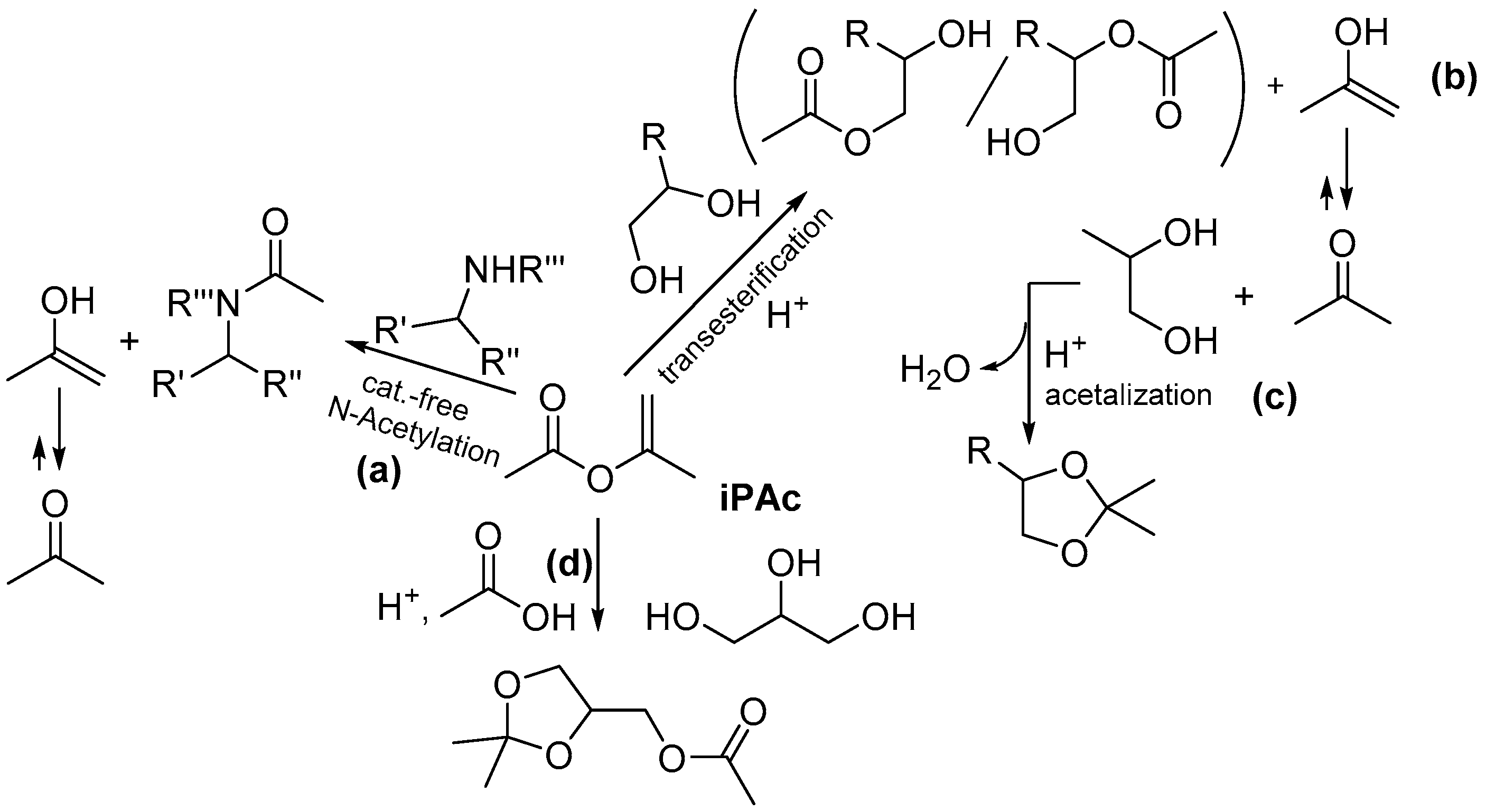
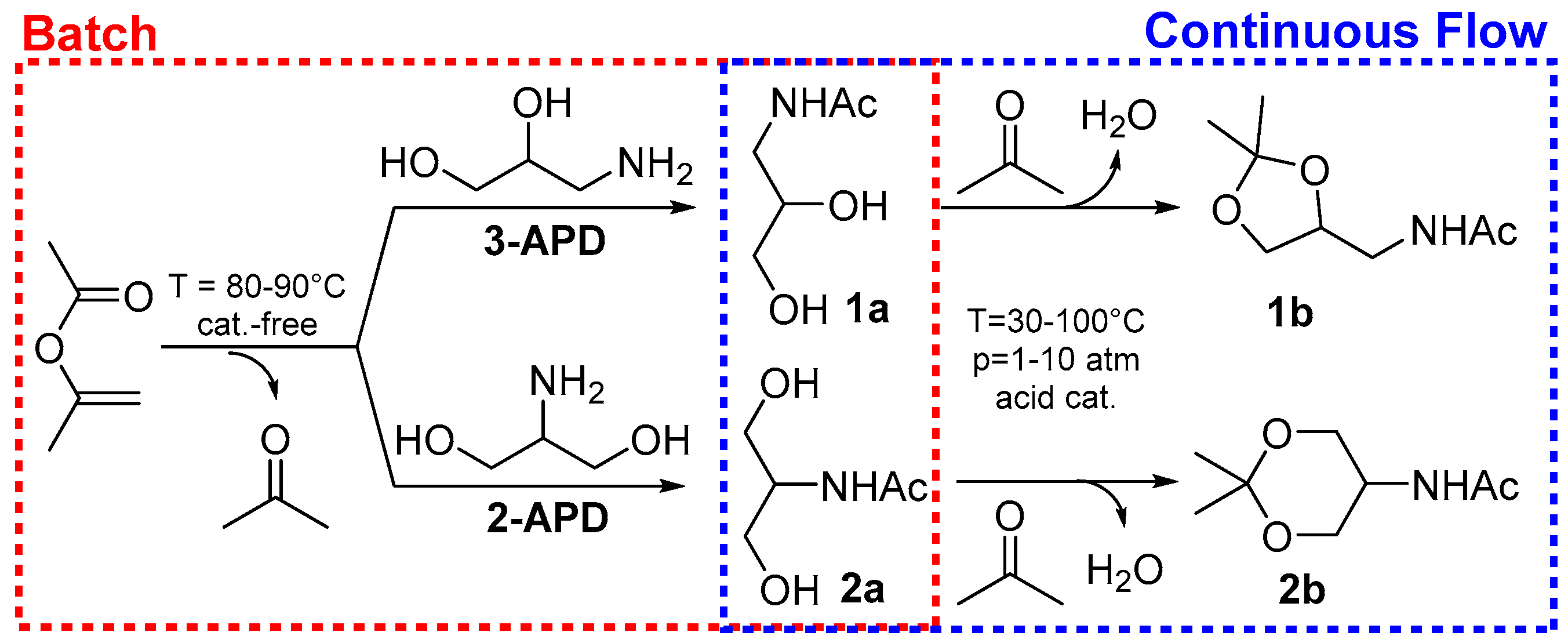
2.1. Batch Catalyst-Free N-Acetylation of 2- and 3-APD with iPAc
2.2. The Continuous Flow (CF) Acetalization of APD-Amides under Acid Catalysis
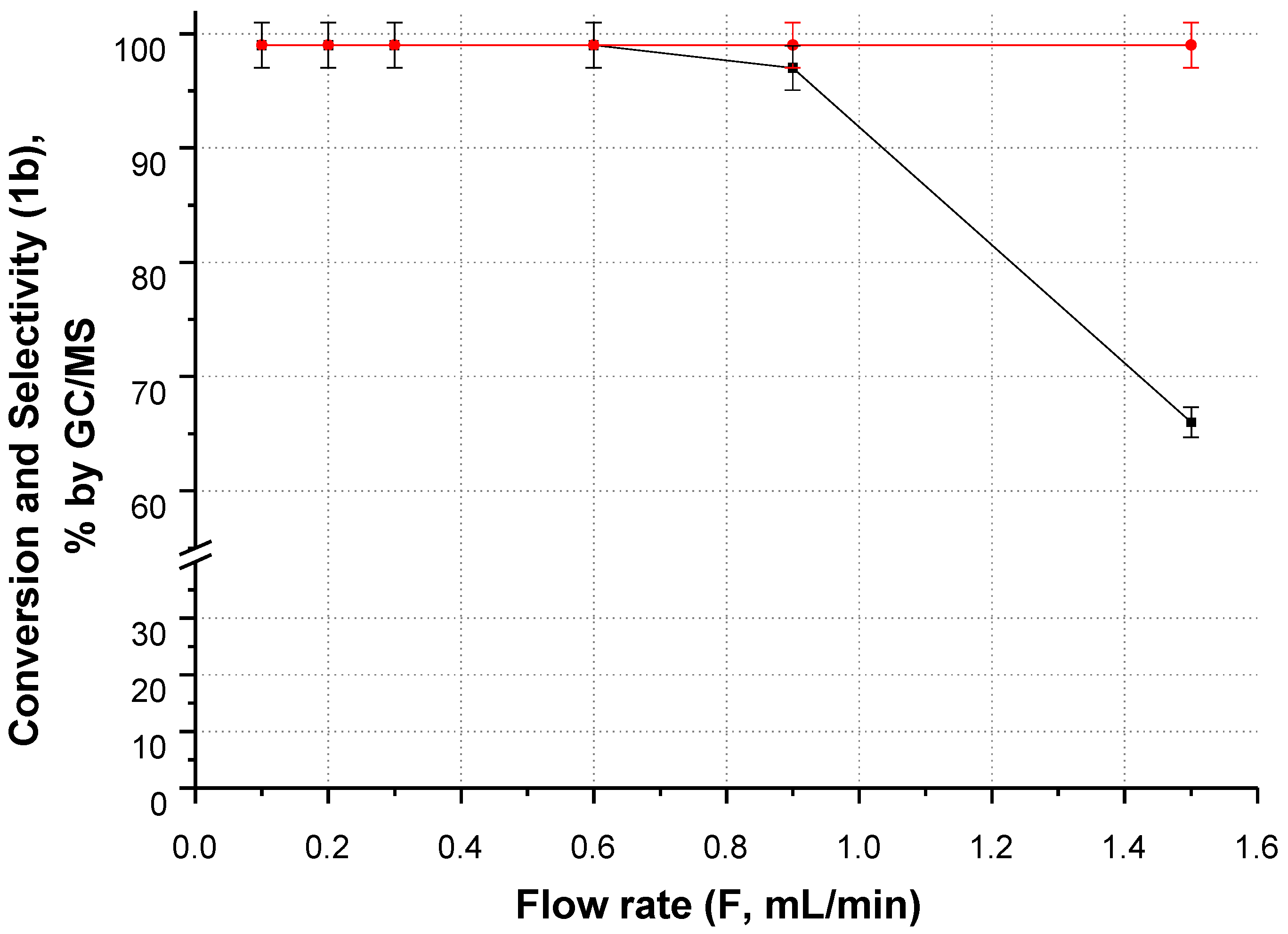
2.2.1. CF-Synthesis of N-((2,2-Dimethyl-1,3-Dioxolan-4-yl)Methyl)Acetamide (1b)
Acetone as a Reagent and a Solvent/Carrier
Dioxane and DMSO as Solvents/Carriers
2.2.2. CF-Synthesis of N-(2,2-Dimethyl-1,3-Dioxan-5-yl)Acetamide (2b)
3. Materials and Methods
3.1. General
3.2. General Procedure for the Batch N-Acetylation of 3-APD and 2-APD (Serinol)
3.3. General Procedure for the CF Acetalization of Amides 1a, 2a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, G.; Wang, A.; Li, L.; Xu, G.; Yan, N.; Zhang, T. Production of Primary Amines by Reductive Amination of Biomass-Derived Aldehydes/Ketones. Angew. Chem.-Int. Ed. 2017, 56, 3050–3054. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Takabe, A.; Takemoto, E. A Process for the Preparation of a Dihydroxyamino Compound. EP1201644 (A2), 2 May 2002. [Google Scholar]
- Felder, E.; Roemer, M.; Bardonner, H.; Haertner, H.; Fruhstorfer, W. Process for the Preparation of Hydroxyamines. US5023379 (A), 11 June 1991. [Google Scholar]
- Borregaard, A.; Tjosaas, F. Process for the Production of Amino Alcohols. WO2012108777 (A1), 16 August 2012. [Google Scholar]
- Baum, K.; Maurice, W.T. The Mannich Condensation of 3-amino-1,2-propanediol with 2,2-dinitropropanol and the nitration of the product. J. Org. Chem. 1962, 27, 2231–2233. [Google Scholar] [CrossRef]
- Andreeßen, B.; Steinbüchel, A. Serinol: Small molecule-big impact. AMB Express 2011, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cerioli, L.; Planchestainer, M.; Cassidy, J.; Tessaro, D.; Paradisi, F. Characterization of a novel amine transaminase from Halomonas elongata. J. Mol. Catal. B Enzym. 2015, 120, 141–150. [Google Scholar] [CrossRef]
- Hegde, S.; Zhao, L. Process for Production of Serinol and Its Bis-Adduct. US8653306 (B1), 18 February 2014. [Google Scholar]
- Cespi, D.; Cucciniello, R.; Ricciardi, M.; Capacchione, C.; Vassura, I.; Passarini, F.; Proto, A. A simplified early stage assessment of process intensification: Glycidol as a value-added product from epichlorohydrin industry wastes. Green Chem. 2016, 18, 4559–4570. [Google Scholar] [CrossRef]
- Fedorov, A.L.; Davidenko, T.I.; Kotlyar, I.I.; Kuznetsov, V.V. Reduction of nitro groups in aliphatic compounds. Russ. J. Org. Chem. 1982, 18, 1161. [Google Scholar]
- Felder, E.; Bianchi, S.; Bollinger, H. Process for the Preparation of Serinol and of Serinol Derivatives, and Products Obtained Therefrom. US Patent 4503252 (A), 5 March 1985. [Google Scholar]
- Nardi, A.; Villa, M. Process for the Preparation of 2 amino-1,3-propanediol. US Patent 5922917 (A), 13 July 1999. [Google Scholar]
- Guidi, S.; Calmanti, R.; Noè, M.; Perosa, A.; Selva, M. Thermal (catalyst-free) transesterification of diols and glycerol with dimethyl carbonate: A flexible reaction for batch and continuous-flow applications. ACS Sustain. Chem. Eng. 2016, 4, 6144–6151. [Google Scholar] [CrossRef]
- Fiorani, G.; Perosa, A.; Selva, M. Dimethyl carbonate: A versatile reagent for a sustainable valorization of renewables. Green Chem. 2018, 20, 288–322. [Google Scholar] [CrossRef]
- Pelagalli, R.; Chiarotto, I.; Feroci, M.; Vecchio, S. Isopropenyl acetate, a remarkable, cheap and acylating agent of amines under solvent- and catalyst-free conditions: A systematic investigation. Green Chem. 2012, 14, 2251–2255. [Google Scholar] [CrossRef]
- Rigo, D.; Fiorani, G.; Perosa, A.; Selva, M. Acid-Catalyzed Reactions of Isopropenyl Esters and Renewable. Diols: A 100% Carbon Efficient Transesterification/Acetalization Tandem Sequence, from Batch to Continuous Flow. ACS Sustain. Chem. Eng. 2019, 7, 18810–18818. [Google Scholar] [CrossRef]
- Rigo, D.; Calmanti, R.; Perosa, A.; Selva, M. A transesterification–acetalization catalytic tandem process for the functionalization of glycerol: The pivotal role of isopropenyl acetate. Green Chem. 2020, 22, 5487–5496. [Google Scholar] [CrossRef]
- Rui Faria, P.V.; Pereira, C.S.M.; Silva, V.M.T.M.; Loureiro, J.M.; Rodrigues, E.A. Glycerol valorisation as biofuels: Selection of a suitable solvent for an innovative process for the synthesis of GEA. Chem. Eng. J. 2013, 233, 159–167. [Google Scholar] [CrossRef]
- Angulo, G.; Brucka, M.; Gerecke, M.; Grampp, G.; Jeannerat, D.; Milkiewicz, J.; Mitrev, Y.; Radzewicz, C.; Rosspeintner, A.; Vautheyf, E.; et al. Characterization of dimethylsulfoxide/glycerol mixtures: A binary solvent system for the study of “friction-dependent” chemical reactivity. Phys. Chem. Chem. Phys. 2016, 18, 18460–18469. [Google Scholar] [CrossRef] [PubMed]
- Guidi, S.; Noè, M.; Riello, P.; Perosa, A.; Selva, M. Towards a Rational Design of a Continuous-Flow Method for the Acetalization of Crude Glycerol: Scope and Limitations of Commercial Amberlyst 36 and AlF3·3H2O as Model Catalysts. Molecules 2016, 21, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Huang, C.; Zhang, H.; Xiang, S.; Liu, S.; Xu, D.; Li, H. Alkylation of phenol with tert-butyl alcohol catalysed by zeolite Hβ. Appl. Catal. 1998, 166, 89–95. [Google Scholar] [CrossRef]
- Mun, D.; Huynh, N.T.T.; Shin, S.; Kim, Y.J.; Kim, S.; Shul, Y.-J.; Cho, J.K. Facile isomerization of glucose into fructose using anion-exchange resins in organic solvents and application to direct conversion of glucose into furan compounds. Res. Chem. Intermed. 2017, 43, 5495–5506. [Google Scholar] [CrossRef]
- Trifoia, A.R.; Agachib, P.S.; Pap, T. Glycerol acetals and ketals as possible diesel additives. A review of their synthesis protocols. Renew. Sustain. Energy Rev. 2016, 62, 804–814. [Google Scholar] [CrossRef]
- Available online: https://webbook.nist.gov/ (accessed on 30 November 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | T (°C) | Q a (mol/mol) | Solvent (mL) | t (h) | Amide Yield b (%) |
|---|---|---|---|---|---|---|
| 1 | 3-APD | 80 | 1.1 | 1,4-dioxane (5) | 48 | 1a: 94% c |
| 2 | 2.2 | 1,4-dioxane (5) | 24 | 1a: ≥99% | ||
| 3 c | 4.4 | 1,4-dioxane (5) | 16 | 1a: ≥99% Isolated: ≥99% | ||
| 4 | 4.4 | DMSO (5) | 3 | 1a: ≥99% | ||
| 5 | 4.4 | DMSO (1) | 1 | 1a: ≥99% | ||
| 6 | 4.4 | DMSO (0.5) | 0.75 | 1a: ≥99% | ||
| 7 c | 2-APD | 90 | 1.1 | 1,4-dioxane (5) | 62 | 2a: ≥99% c |
| 8 | 2.2 | 1,4-dioxane (5) | 48 | 2a: ≥99% | ||
| 9 | 4.4 | 1,4-dioxane (5) | 24 | 2a: ≥99% Isolated: ≥99% | ||
| 10 | 4.4 | DMSO (5) | 8 | 2a: ≥99% | ||
| 11 | 4.4 | DMSO (1) | 2.5 | 2a: ≥99% | ||
| 12 | 4.4 | DMSO (0.5) | 2 | 2a: ≥99% |
| Entry | Catalyst a | T/p (°C/bar) | Flow (mL/min) | Conv. (%) b | Sel. (%) b | Mass Balance (%) c |
|---|---|---|---|---|---|---|
| 1 | A15 | 30/1 | 0.1 | >99 | >99 | >99 |
| 2 | 0.2 | >99 | >99 | >99 | ||
| 3 | AF | 100/20 | 0.2 | 40 | 96 | nd |
| 4 | 150/20 | 0.2 | 79 | 99 | ||
| 5 | HY | 100/20 | 0.2 | 27 | 92 | |
| 6 | 150/20 | 0.2 | 76 | 91 |

| Entry | Ace:1a, Q1 (mol:mol) b | Solvent | T/p (°C/bar) | Flow (mL/min) | Conv. (%) c | Sel. (%) c |
|---|---|---|---|---|---|---|
| 1 | 5 | 1,4-Dioxane | 30/1 | 0.1 | 33 | 94 |
| 2 | 50/1 | 0.1 | 73 | 91 | ||
| 3 | 20 | 1,4-Dioxane | 30/1 | 0.1 | 83 | >99 |
| 4 | 0.3 | 61 | >99 | |||
| 5 | 0.6 | 53 | >99 | |||
| 6 | 40 | 1,4-Dioxane | 30/1 | 0.1 | 95 | >99 |
| 7 | 0.3 | 86 | >99 | |||
| 8 | 40 | DMSO | 30/1 | 0.3 | 11 | 88 |
| 9 | 50/1 | 32 | 92 |
| Entry | Solvent | T/p (°C/bar) | Conv. (%) b | Sel. (%) b |
|---|---|---|---|---|
| 1 | acetone | 30/1 | 24 | 94 |
| 2 | 50/1 | 37 | 95 | |
| 3 | 100/10 | 62 | 92 | |
| 4 | 1,4-Dioxane | 30/1 | 15 | 96 |
| 5 | 50/1 | 24 | 97 | |
| 6 | 100/10 | 87 | 43 | |
| 7 | DMSO | 30/1 | 11 | 17 |
| 8 | 50/1 | 18 | 45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigo, D.; Carmo Dos Santos, N.A.; Perosa, A.; Selva, M. Concatenated Batch and Continuous Flow Procedures for the Upgrading of Glycerol-Derived Aminodiols via N-Acetylation and Acetalization Reactions. Catalysts 2021, 11, 21. https://doi.org/10.3390/catal11010021
Rigo D, Carmo Dos Santos NA, Perosa A, Selva M. Concatenated Batch and Continuous Flow Procedures for the Upgrading of Glycerol-Derived Aminodiols via N-Acetylation and Acetalization Reactions. Catalysts. 2021; 11(1):21. https://doi.org/10.3390/catal11010021
Chicago/Turabian StyleRigo, Davide, Nadia Alessandra Carmo Dos Santos, Alvise Perosa, and Maurizio Selva. 2021. "Concatenated Batch and Continuous Flow Procedures for the Upgrading of Glycerol-Derived Aminodiols via N-Acetylation and Acetalization Reactions" Catalysts 11, no. 1: 21. https://doi.org/10.3390/catal11010021
APA StyleRigo, D., Carmo Dos Santos, N. A., Perosa, A., & Selva, M. (2021). Concatenated Batch and Continuous Flow Procedures for the Upgrading of Glycerol-Derived Aminodiols via N-Acetylation and Acetalization Reactions. Catalysts, 11(1), 21. https://doi.org/10.3390/catal11010021