Structure of Nanocrystalline, Partially Disordered MoS2+δ Derived from HRTEM—An Abundant Material for Efficient HER Catalysis

 ,
,
Abstract
1. Introduction
2. Results
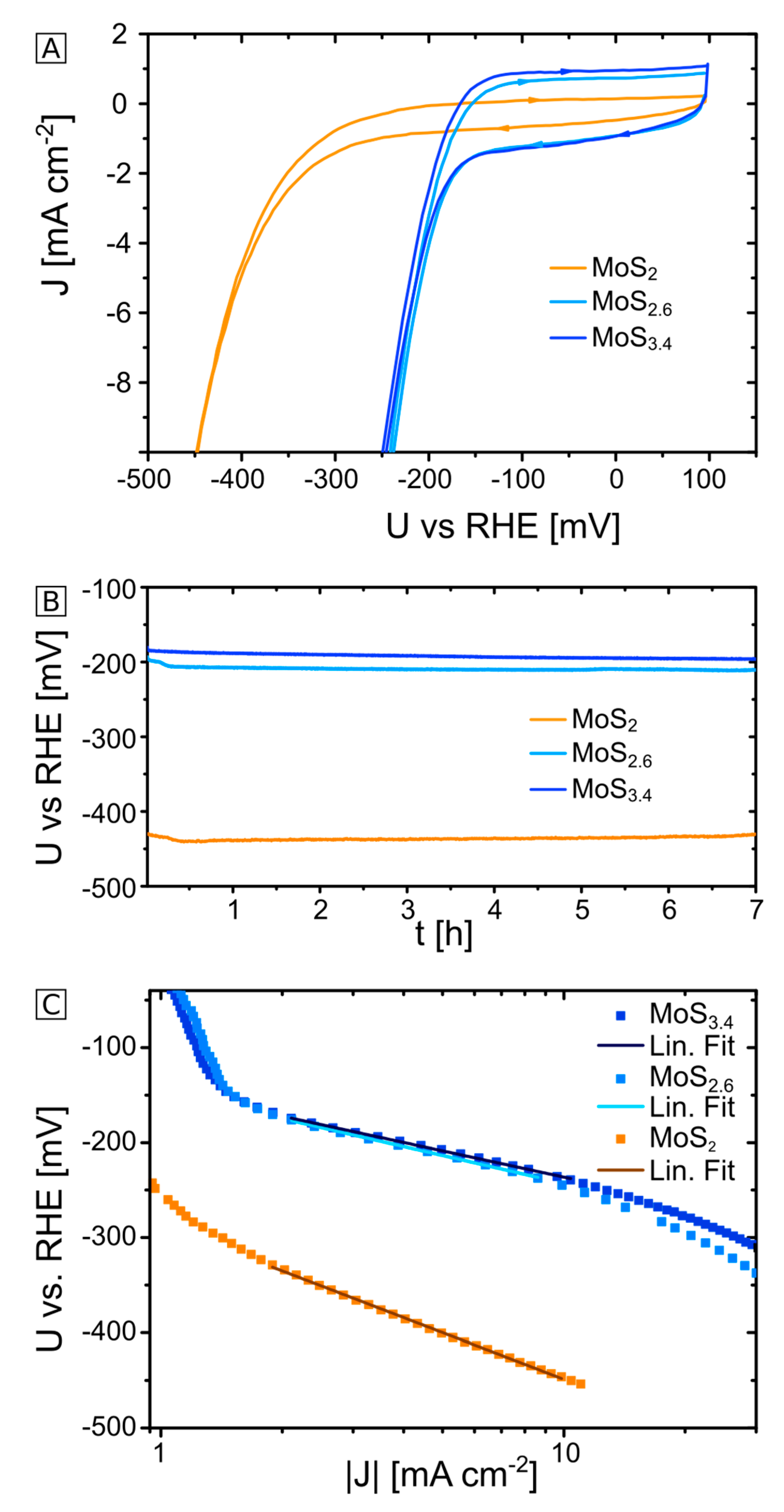
2.1. Electrochemical Characterization of MoS2+δ Electrodes
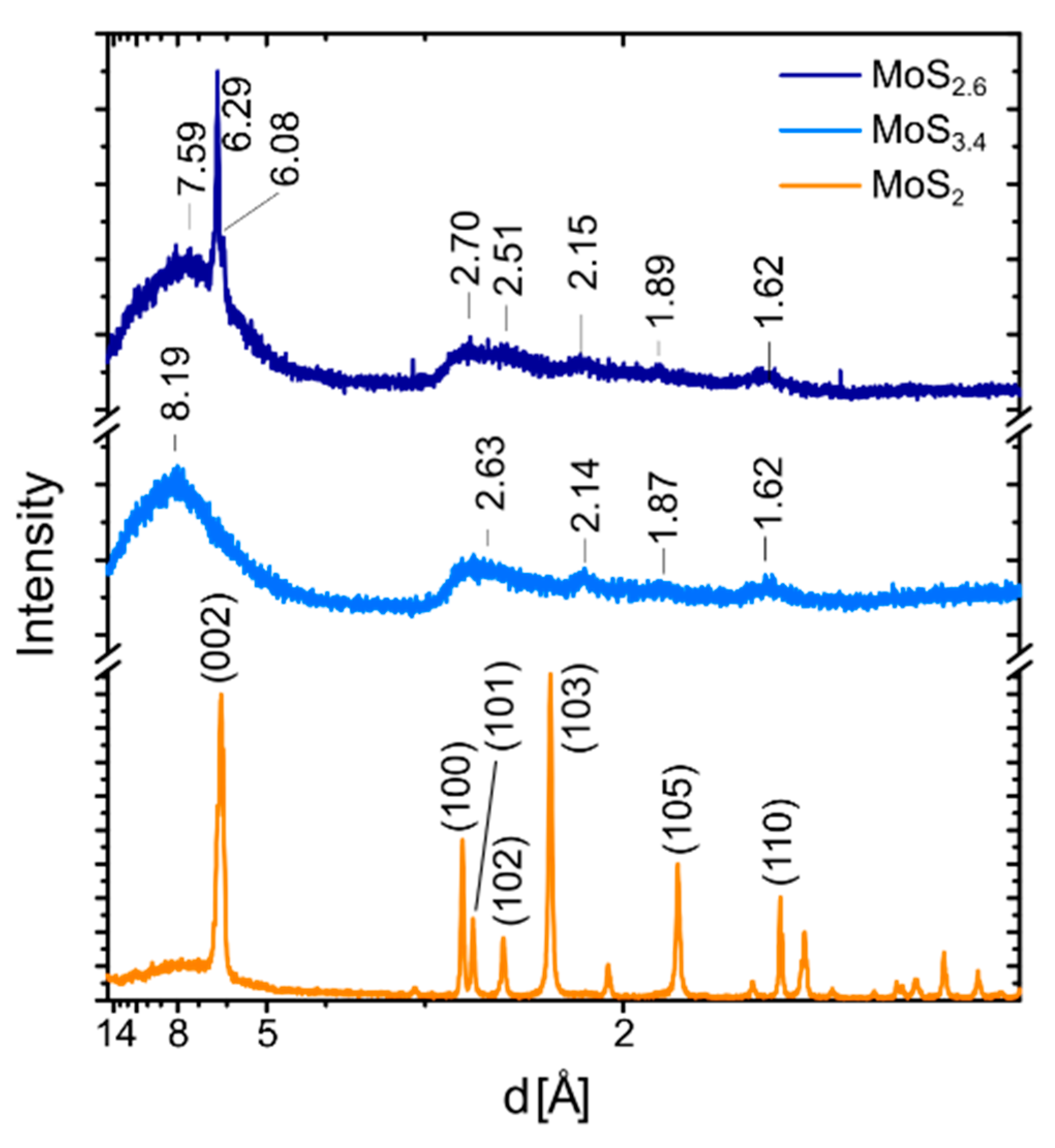
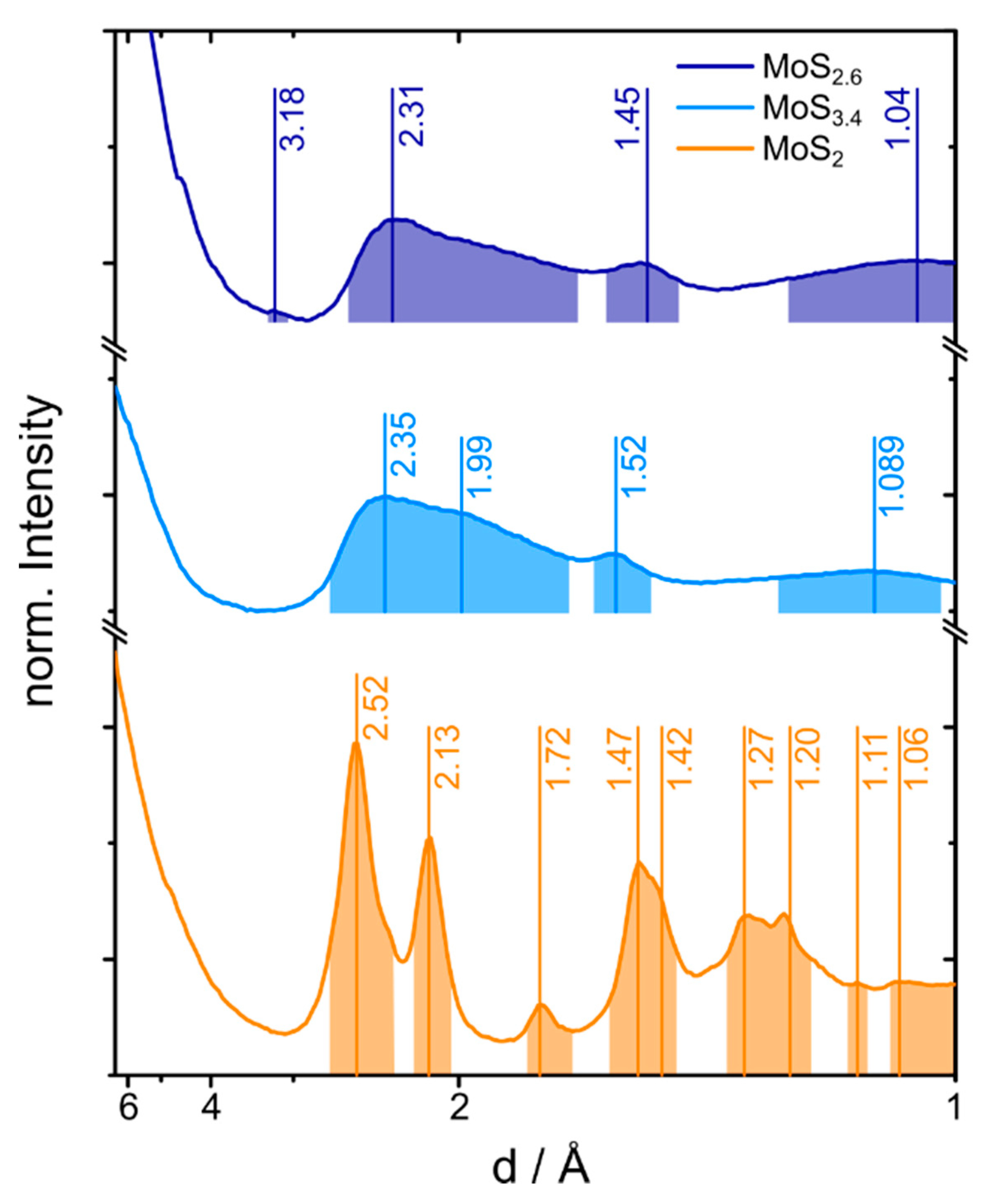
2.2. Electron and X-ray Diffraction Analysis of the MoS2+δ Structure
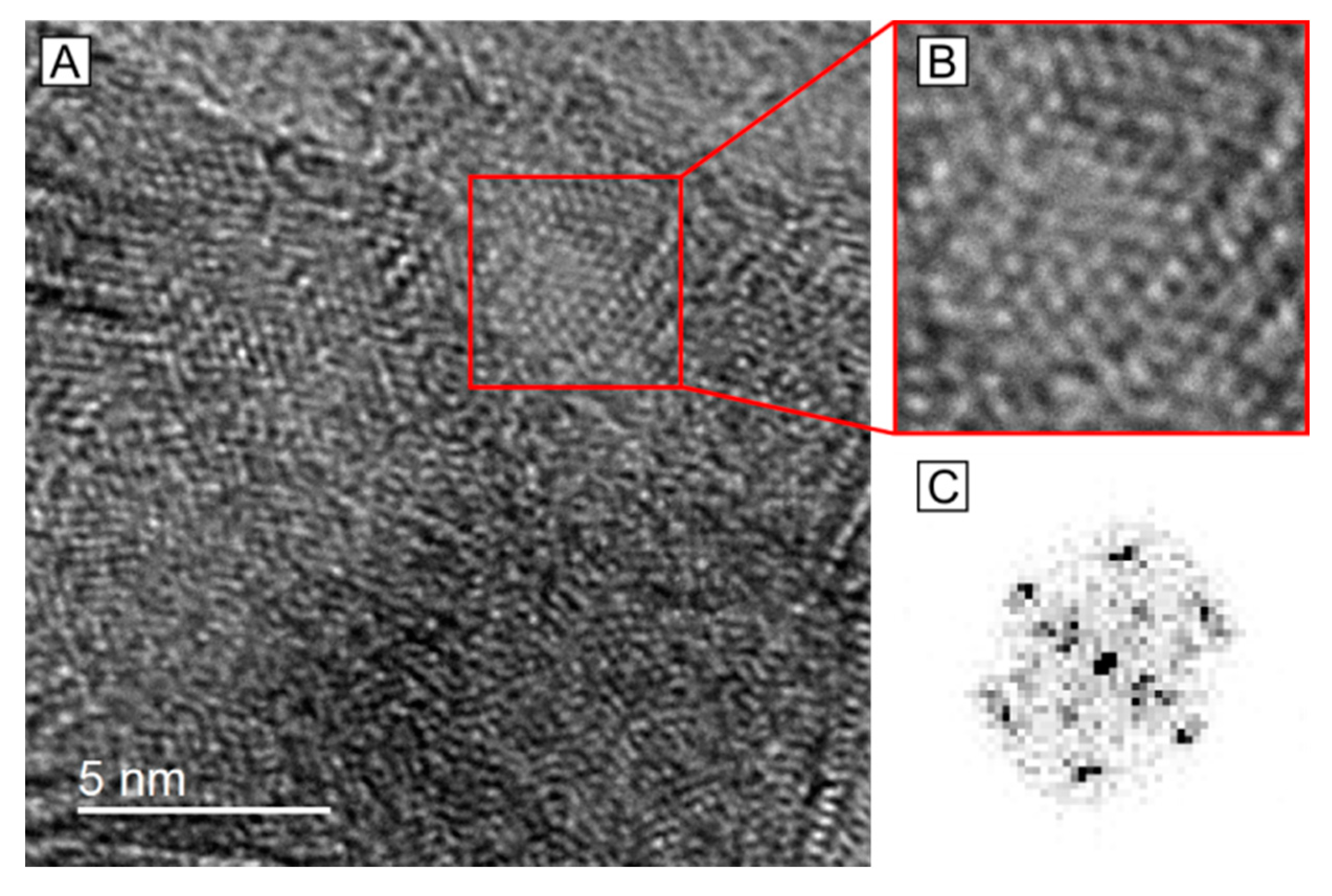
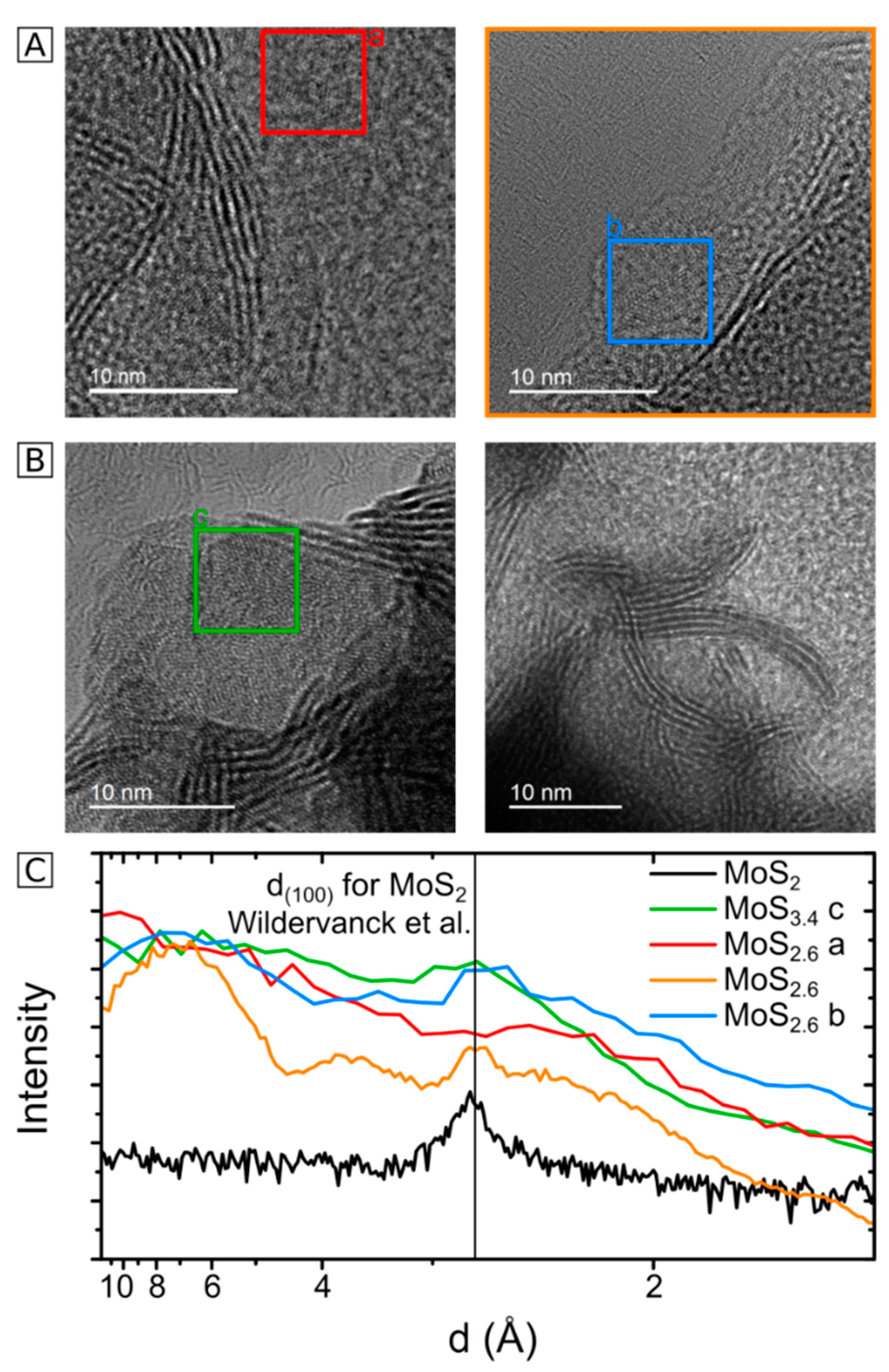
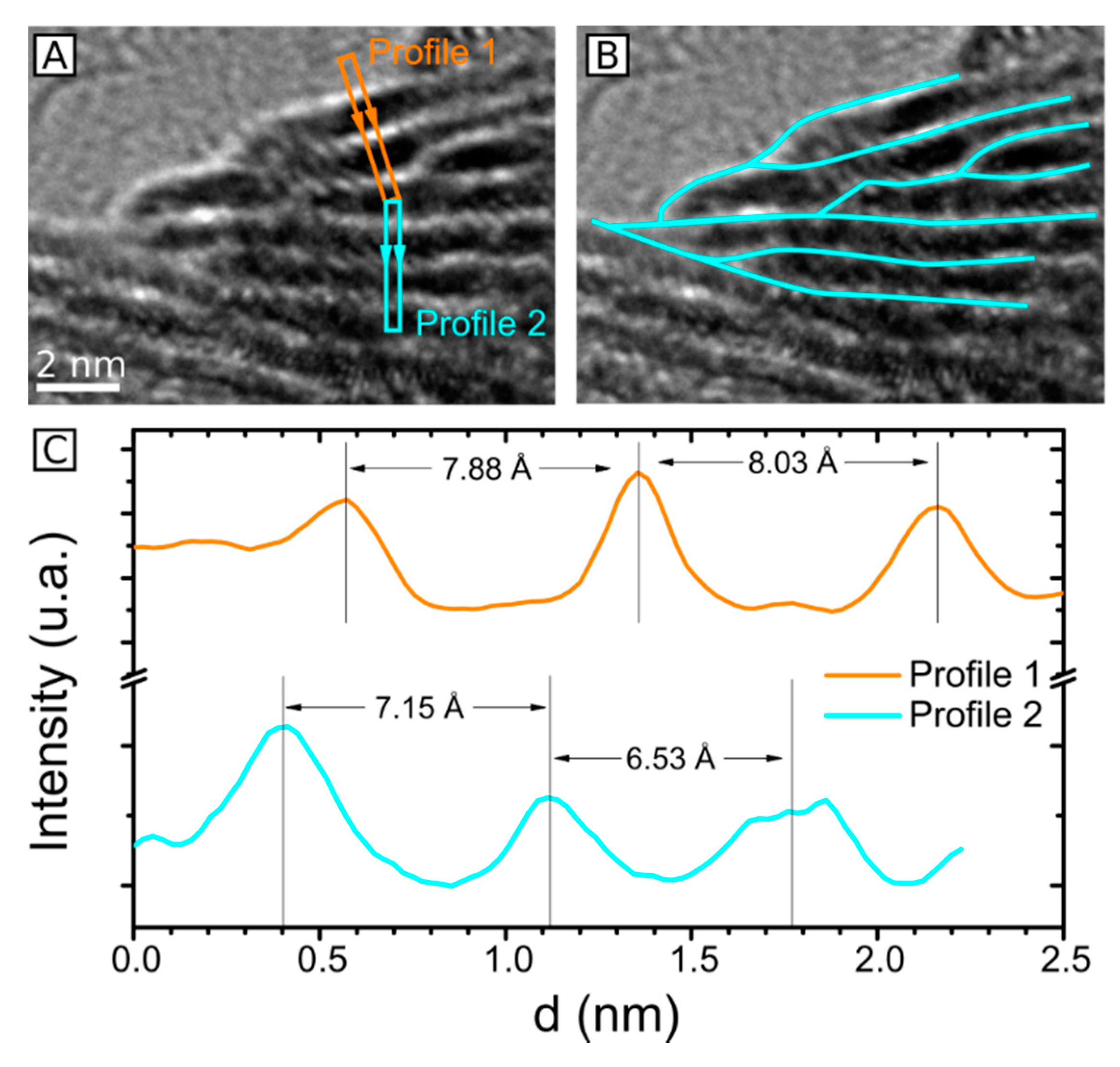
2.3. HRTEM Analysis of the Microstructure of MoS2+δ
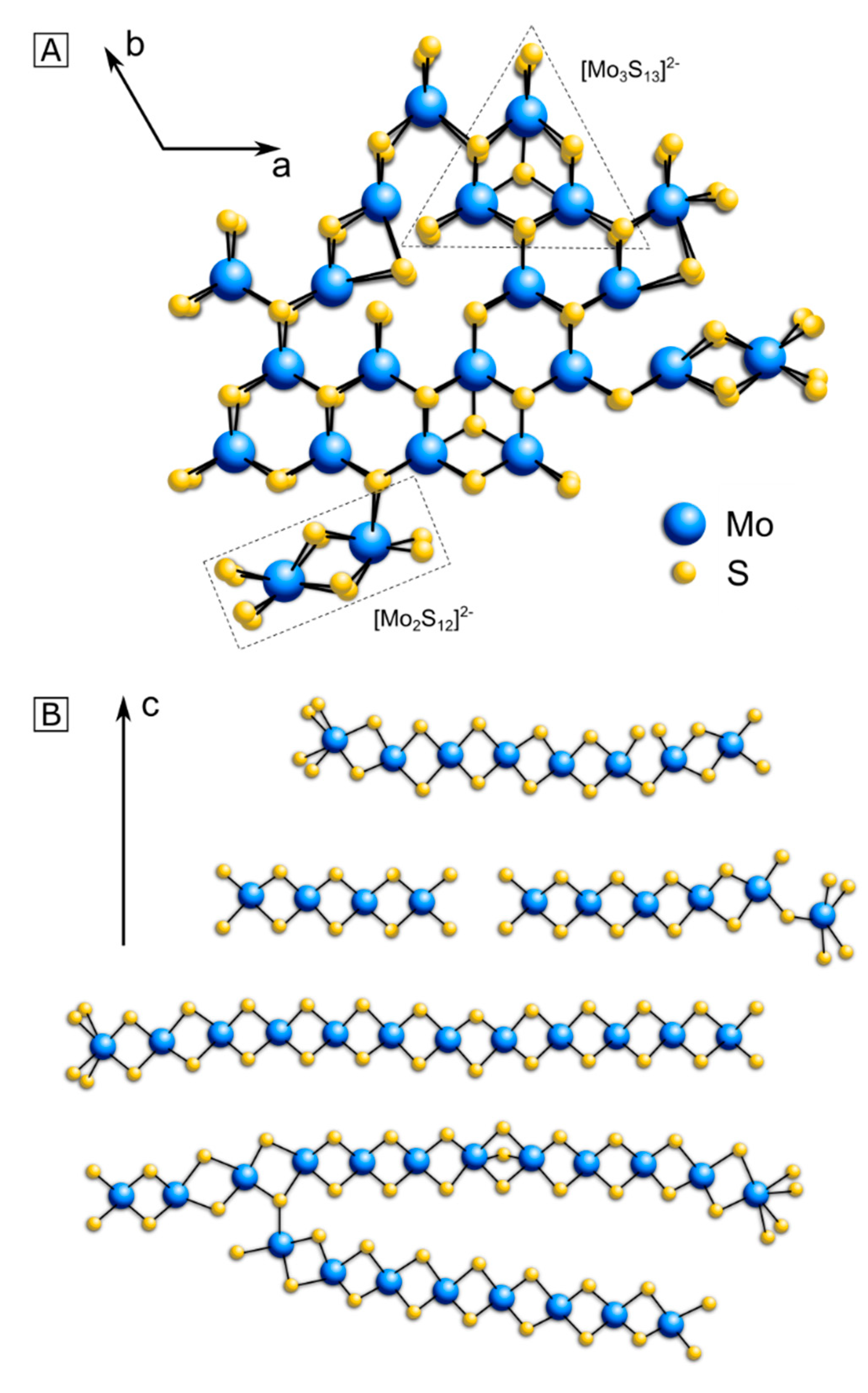
3. Structure Model
4. Materials and Methods
4.1. Synthesis of MoSx
4.2. Stoichiometry Determination
4.3. Electrochemistry
4.4. Electrode Preparation
4.5. XRD Analysis
4.6. TEM Lamella Preparation and TEM Analysis
4.7. Accuracy of Lattice Spacing Measurement in TEM
5. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pellow, M.A.; Emmott, C.J.M.; Barnhart, C.J.; Benson, S.M. Hydrogen or batteries for grid storage? A net energy analysis. Energy Environ. Sci. 2015, 8, 1938–1952. [Google Scholar] [CrossRef]
- Chalk, S.G.; Miller, J.F. Key challenges and recent progress in batteries, fuel cells, and hydrogen storage for clean energy systems. J. Power Sources 2006, 159, 73–80. [Google Scholar] [CrossRef]
- Edwards, P.P.; Kuznetsov, V.L.; David, W.I.F.; Brandon, N.P. Hydrogen and fuel cells: Towards a sustainable energy future. Energy Policy 2008, 36, 4356–4362. [Google Scholar] [CrossRef]
- Morales-Guio, C.G.; Hu, X. Amorphous Molybdenum Sulfides as Hydrogen Evolution Catalysts. Acc. Chem. Res. 2014, 47, 2671–2681. [Google Scholar] [CrossRef]
- Benck, J.D.; Chen, Z.; Kuritzky, L.Y.; Forman, A.J.; Jaramillo, T.F. Amorphous molybdenum sulfide catalysts for electrochemical hydrogen production: Insights into the origin of their catalytic activity. ACS Catal. 2012, 2, 1916–1923. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Zhang, J.; Lei, Z.; Liang, X.; Chen, B. MoS2 with tunable surface structure directed by thiophene adsorption toward HDS and HER. Sci. China Mater. 2016, 59, 1051–1061. [Google Scholar] [CrossRef]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jørgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef]
- Jaramillo, T.F.; Jørgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef]
- Seo, B.; Joo, S.H. Recent advances in unveiling active sites in molybdenum sulfide-based electrocatalysts for the hydrogen evolution reaction. Nano Converg. 2017, 4, 19. [Google Scholar] [CrossRef]
- Voiry, D.; Salehi, M.; Silva, R.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; Chhowalla, M. Conducting MoS2 nanosheets as catalysts for hydrogen evolution reaction. Nano Lett. 2013, 13, 6222–6227. [Google Scholar] [CrossRef]
- Ting, L.R.L.; Deng, Y.; Ma, L.; Zhang, Y.-J.; Peterson, A.A.; Yeo, B.S. Catalytic Activities of Sulfur Atoms in Amorphous Molybdenum Sulfide for the Electrochemical Hydrogen Evolution Reaction. ACS Catal. 2016, 6, 861–867. [Google Scholar] [CrossRef]
- Hsu, C.L.; Chang, Y.H.; Chen, T.Y.; Tseng, C.C.; Wei, K.H.; Li, L.J. Enhancing the electrocatalytic water splitting efficiency for amorphous MoSx. Int. J. Hydrogen Energy 2014, 39, 4788–4793. [Google Scholar] [CrossRef]
- Kokko, M.; Bayerköhler, F.; Erben, J.; Zengerle, R.; Kurz, P.; Kerzenmacher, S. Molybdenum sulphides on carbon supports as electrocatalysts for hydrogen evolution in acidic industrial wastewater. Appl. Energy 2017, 190, 1221–1233. [Google Scholar] [CrossRef]
- Yan, Y.; Ge, X.; Liu, Z.; Wang, J.Y.; Lee, J.M.; Wang, X. Facile synthesis of low crystalline MoS2 nanosheet-coated CNTs for enhanced hydrogen evolution reaction. Nanoscale 2013, 5, 7768–7771. [Google Scholar] [CrossRef]
- Vrubel, H.; Merki, D.; Hu, X. Hydrogen evolution catalyzed by MoS3 and MoS2 particles. Energy Environ. Sci. 2012, 5, 6136. [Google Scholar] [CrossRef]
- Tran, P.D.; Tran, T.V.; Orio, M.; Torelli, S.; Truong, Q.D.; Nayuki, K.; Sasaki, Y.; Chiam, S.Y.; Yi, R.; Honma, I.; et al. Coordination polymer structure and revisited hydrogen evolution catalytic mechanism for amorphous molybdenum sulfide. Nat. Mater. 2016, 15, 640–646. [Google Scholar] [CrossRef]
- Ye, Z.; Yang, J.; Li, B.; Shi, L.; Ji, H.; Song, L.; Xu, H. Amorphous Molybdenum Sulfide/Carbon Nanotubes Hybrid Nanospheres Prepared by Ultrasonic Spray Pyrolysis for Electrocatalytic Hydrogen Evolution. Small 2017, 13, 1700111. [Google Scholar] [CrossRef]
- Grutza, M.-L.; Rajagopal, A.; Streb, C.; Kurz, P. Hydrogen evolution catalysis by molybdenum sulfides (MoSx): Are thiomolybdate clusters like [Mo3S13]2− suitable active site models? Sustain. Energy Fuels 2018, 2, 1893–1904. [Google Scholar] [CrossRef]
- Weber, T.; Muijsers, J.C.; Niemantsverdriet, J.W. Structure of Amorphous MoS3. J. Phys. Chem. 1995, 99, 9194–9200. [Google Scholar] [CrossRef]
- Huntley, D.R.; Parham, T.G.; Merrill, R.P.; Sienko, M.J. An EXAFS study of the thermal decomposition of molybdenum trisulfide. Inorg. Chem. 1983, 22, 4144–4146. [Google Scholar] [CrossRef]
- Hibble, S.J.; Wood, G.B. Modeling the Structure of Amorphous MoS3: A Neutron Diffraction and Reverse Monte Carlo Study. J. Am. Chem. Soc. 2004, 126, 959–965. [Google Scholar] [CrossRef]
- Mabayoje, O.; Wygant, B.R.; Wang, M.; Liu, Y.; Mullins, C.B. Sulfur-Rich MoS6 as an Electrocatalyst for the Hydrogen Evolution Reaction. ACS Appl. Energy Mater. 2018, 1, 4453–4458. [Google Scholar] [CrossRef]
- Müller, A.; Fedin, V.; Hegetschweiler, K.; Amrein, W. Characterization of amorphous substances by studying isotopically labelled compounds with FAB-MS: Evidence for extrusion of triangular Mo3IV clusters from a mixture of 92MoS3 and 100MoS3. J. Chem. Soc. Chem. Commun. 1992, 1795–1796. [Google Scholar] [CrossRef]
- Hibble, S.J.; Feaviour, M.R.; Almond, M.J. Chemical excision from amorphous MoS3; a quantitative EXAFS study. J. Chem. Soc. Dalton Trans. 2001, 0, 935–940. [Google Scholar] [CrossRef]
- Li, Y.; Yu, Y.; Huang, Y.; Nielsen, R.A.; Goddard, W.A.; Li, Y.; Cao, L. Engineering the Composition and Crystallinity of Molybdenum Sulfide for High-Performance Electrocatalytic Hydrogen Evolution. ACS Catal. 2015, 5, 448–455. [Google Scholar] [CrossRef]
- Dave, M.; Rajagopal, A.; Damm-Ruttensperger, M.; Schwarz, B.; Nägele, F.; Daccache, L.; Fantauzzi, D.; Jacob, T.; Streb, C. Understanding homogeneous hydrogen evolution reactivity and deactivation pathways of molecular molybdenum sulfide catalysts. Sustain. Energy Fuels 2018, 2, 1020–1026. [Google Scholar] [CrossRef]
- Luo, Z.; Ouyang, Y.; Zhang, H.; Xiao, M.; Ge, J.; Jiang, Z.; Wang, J.; Tang, D.; Cao, X.; Liu, C.; et al. Chemically activating MoS2 via spontaneous atomic palladium interfacial doping towards efficient hydrogen evolution. Nat. Commun. 2018, 9, 2120. [Google Scholar] [CrossRef]
- Hu, J.; Huang, B.; Zhang, C.; Wang, Z.; An, Y.; Zhou, D.; Lin, H.; Leung, M.K.H.; Yang, S. Engineering stepped edge surface structures of MoS2 sheet stacks to accelerate the hydrogen evolution reaction. Energy Environ. Sci. 2017, 10, 593–603. [Google Scholar] [CrossRef]
- Yin, Y.; Han, J.; Zhang, Y.; Zhang, X.; Xu, P.; Yuan, Q.; Samad, L.; Wang, X.; Wang, Y.; Zhang, Z.; et al. Contributions of Phase, Sulfur Vacancies, and Edges to the Hydrogen Evolution Reaction Catalytic Activity of Porous Molybdenum Disulfide Nanosheets. J. Am. Chem. Soc. 2016, 138, 7965–7972. [Google Scholar] [CrossRef]
- Wildervanck, J.C.; Jellinek, F. Preparation and Crystallinity of Molybdenum and Tungsten Sulfides. Z. für Anorg. und Allg. Chemie 1964, 328, 309–318. [Google Scholar] [CrossRef]
- Colliex, C.; Cowley, J.M.; Dudarev, S.L.; Fink, M.; Gjønnes, J.; Hilderbrandt, R.; Howie, A.; Lynch, D.F.; Peng, L.M.; Ren, G.; et al. Electron diffraction. In International Tables for Crystallography; International Union of Crystallography: Chester, UK, 2006; pp. 259–429. [Google Scholar]
- Michel, F.M.; Antao, S.M.; Chupas, P.J.; Lee, P.L.; Parise, J.B.; Schoonen, M.A.A. Short- To medium-range atomic order and crystallite size of the initial FeS precipitate from pair distribution function analysis. Chem. Mater. 2005, 17, 6246–6255. [Google Scholar] [CrossRef]
- Shyam, B.; Stone, K.H.; Bassiri, R.; Fejer, M.M.; Toney, M.F.; Mehta, A. Measurement and Modeling of Short and Medium Range Order in Amorphous Ta2O5 Thin Films. Sci. Rep. 2016, 6, 32170. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.D.; Zanchet, D.; Ugarte, D. Estimating nanoparticle size from diffraction measurements. J. Appl. Crystallogr. 2000, 33, 1335–1341. [Google Scholar] [CrossRef]
- Srolovitz, D.J.; Safran, S.A.; Homyonfer, M.; Tenne, R. Morphology of Nested Fullerenes. Phys. Rev. Lett. 1995, 74, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Nguyen, L.N.; Nguyen, P.D.; Thu, T.V.; Nguyen, A.D.; Tran, P.D. Crystallization of Amorphous Molybdenum Sulfide Induced by Electron or Laser Beam and Its Effect on H2—Evolving Activities. J. Phys. Chem. C 2016, 120, 28789–28794. [Google Scholar] [CrossRef]
- Xi, F.; Bogdanoff, P.; Harbauer, K.; Plate, P.; Höhn, C.; Rappich, J.; Wang, B.; Han, X.; Van De Krol, R.; Fiechter, S. Structural Transformation Identification of Sputtered Amorphous MoSx as an Efficient Hydrogen-Evolving Catalyst during Electrochemical Activation. ACS Catal. 2019, 9, 2368–2380. [Google Scholar] [CrossRef]
- Wu, L.; Longo, A.; Dzade, N.Y.; Sharma, A.; Hendrix, M.M.R.M.; Bol, A.A.; de Leeuw, N.H.; Hensen, E.J.M.; Hofmann, J.P. The Origin of High Activity of Amorphous MoS2 in the Hydrogen Evolution Reaction. ChemSusChem 2019, 12, 4383–4389. [Google Scholar] [CrossRef]
- McDonald, J.W.; Friesen, G.D.; Rosenhein, L.D.; Newton, W.E. Syntheses and characterization of ammonium and tetraalkylammonium thiomolybdates and thiotungstates. Inorg. Chim. Acta 1983, 72, 205–210. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Xie, L.; Liang, Y.; Hong, G.; Dai, H. MoS2 nanoparticles grown on graphene: An advanced catalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2011, 133, 7296–7299. [Google Scholar] [CrossRef]
- Cui, W.; Liu, Q.; Xing, Z.; Asiri, A.M.; Alamry, K.A.; Sun, X. MoP nanosheets supported on biomass-derived carbon flake: One-step facile preparation and application as a novel high-active electrocatalyst toward hydrogen evolution reaction. Appl. Catal. B Environ. 2015, 164, 144–150. [Google Scholar] [CrossRef]
- Biskupek, J.; Kaiser, U. Practical considerations on the determination of the accuracy of the lattice parameters measurements from digital recorded diffractograms. J. Electron Microsc. (Tokyo) 2004, 53, 601–610. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tafel Slope [mV dec−1] | η @ 10 mA cm−2 [mV] | η @ 10 mA μmol(Mo)−1 [mV] | |
|---|---|---|---|
| MoS2.6 | 100 | 235 | 250 |
| MoS3.4 | 90 | 245 | 255 |
| MoS2 | 160 | 450 | - |
| MoS2 [27] | 160 | - | - |
| MoS2 (step-edged stacks) [28] | 59 | 104 | - |
| 1T MoS2 (porous) [29] | 43 | 153 | - |
| MoS2+x [4] | 40 | 160 | - |
| MoS3.5 [5] | ≈60 | ≈200 | - |
| Literature [30] | HRTEM MoS2+δ | XRD | |||
|---|---|---|---|---|---|
| MoS2 | Mean | Min | Max | ||
| d(100) [Å] | 2.7368 | 2.69 ± 0.21 | 2.45 ± 0.18 | 2.79 ± 0.03 | |
| d(001) [Å] | 12.294 | 12.9 ± 0.8 | 11.15 ± 0.19 | 15.93 ± 0.61 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronge, E.; Hildebrandt, S.; Grutza, M.-L.; Klein, H.; Kurz, P.; Jooss, C. Structure of Nanocrystalline, Partially Disordered MoS2+δ Derived from HRTEM—An Abundant Material for Efficient HER Catalysis. Catalysts 2020, 10, 856. https://doi.org/10.3390/catal10080856
Ronge E, Hildebrandt S, Grutza M-L, Klein H, Kurz P, Jooss C. Structure of Nanocrystalline, Partially Disordered MoS2+δ Derived from HRTEM—An Abundant Material for Efficient HER Catalysis. Catalysts. 2020; 10(8):856. https://doi.org/10.3390/catal10080856
Chicago/Turabian StyleRonge, Emanuel, Sonja Hildebrandt, Marie-Luise Grutza, Helmut Klein, Philipp Kurz, and Christian Jooss. 2020. "Structure of Nanocrystalline, Partially Disordered MoS2+δ Derived from HRTEM—An Abundant Material for Efficient HER Catalysis" Catalysts 10, no. 8: 856. https://doi.org/10.3390/catal10080856
APA StyleRonge, E., Hildebrandt, S., Grutza, M.-L., Klein, H., Kurz, P., & Jooss, C. (2020). Structure of Nanocrystalline, Partially Disordered MoS2+δ Derived from HRTEM—An Abundant Material for Efficient HER Catalysis. Catalysts, 10(8), 856. https://doi.org/10.3390/catal10080856