Trends and Outlook of Computational Chemistry and Microkinetic Modeling for Catalytic Synthesis of Methanol and DME



Abstract
1. Introduction
2. Results and Discussion
2.1. Computational Chemistry
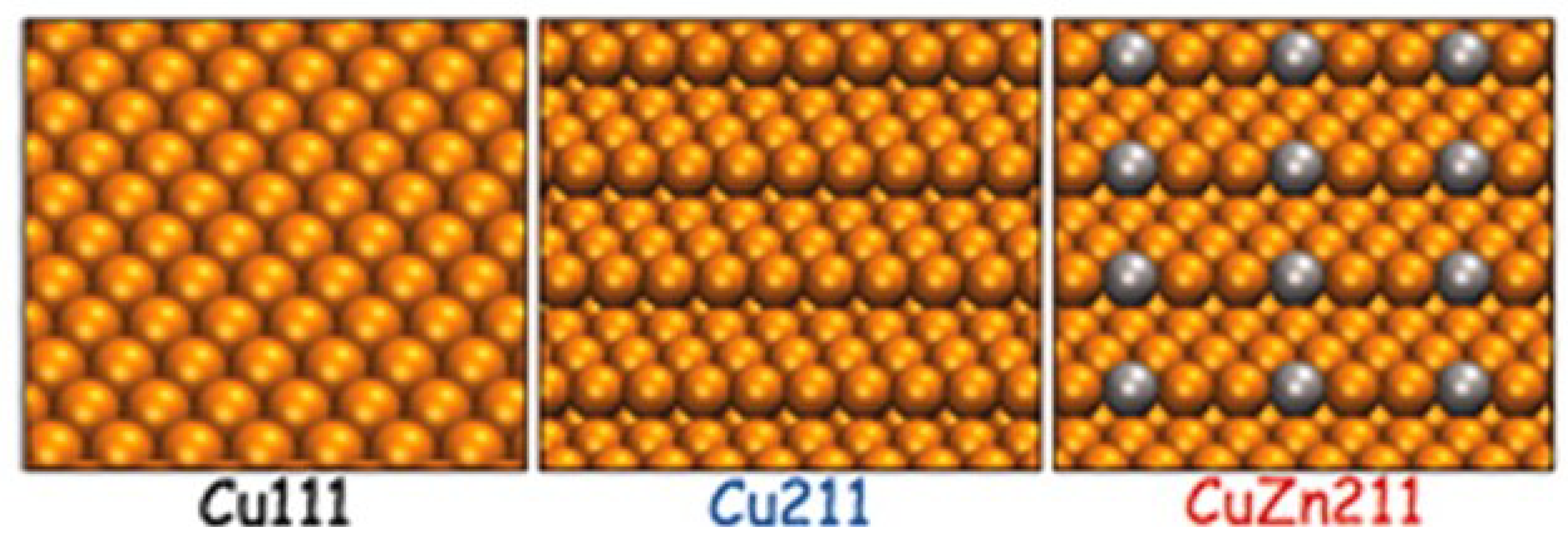
2.1.1. Methanol Synthesis over Cu-based Catalysts
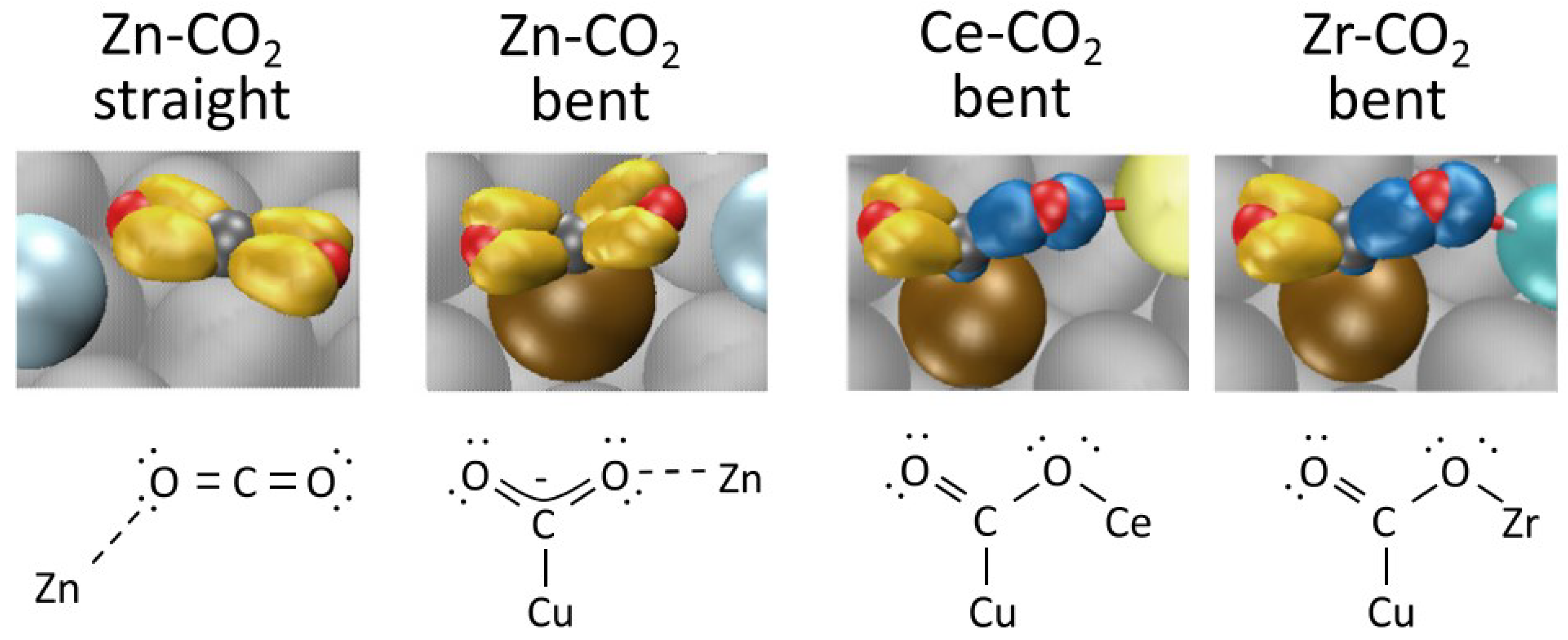
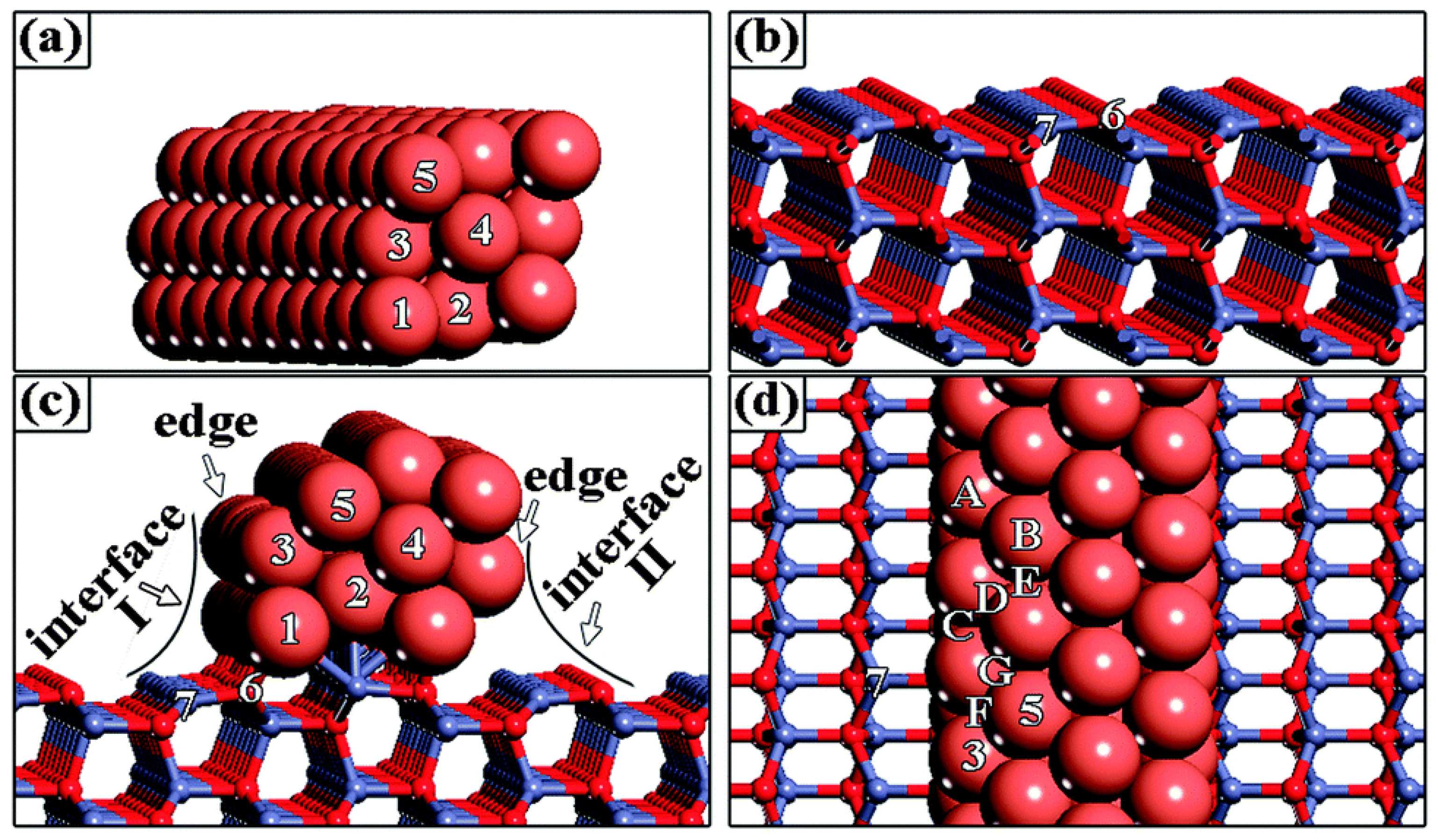
2.1.2. Methanol Synthesis over Other Catalysts
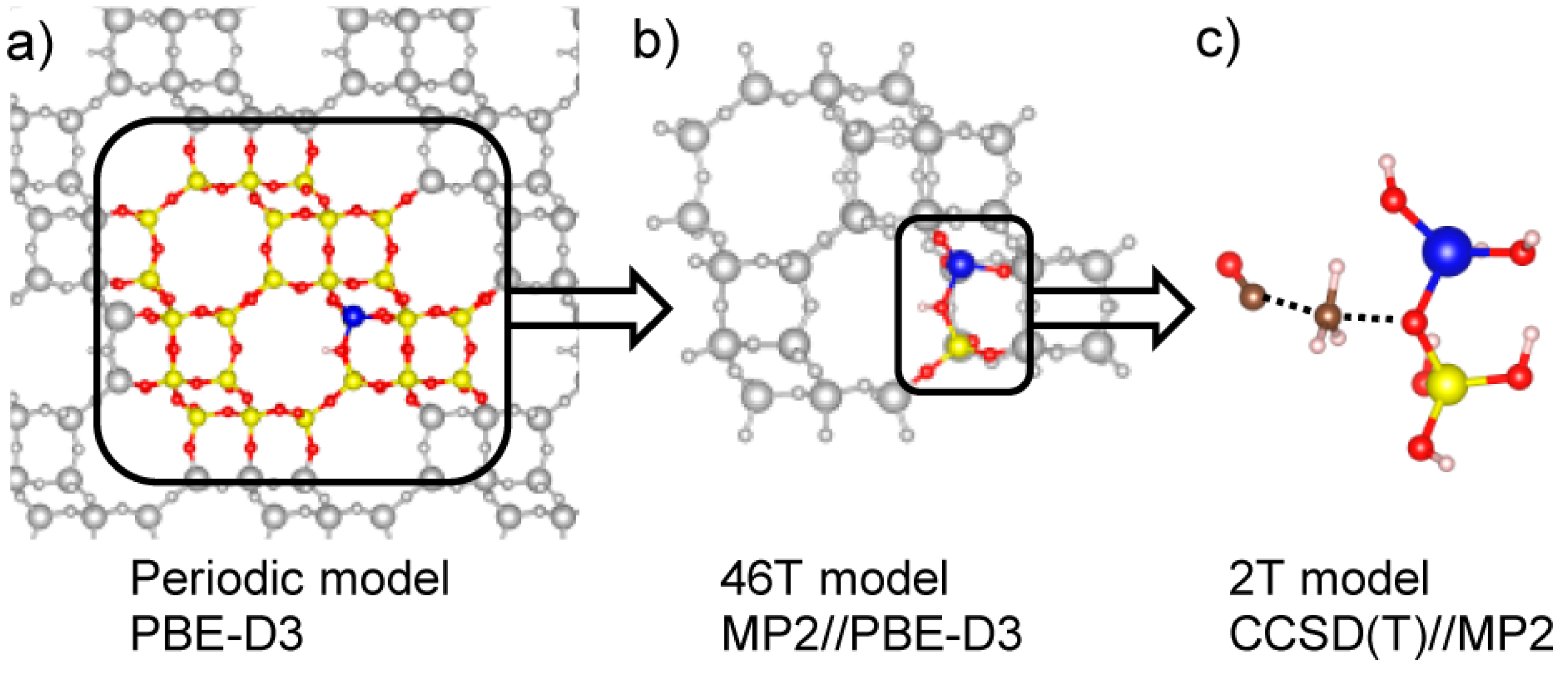
2.1.3. First-Principle Modeling on DME Synthesis
2.2. Microkinetic Modeling
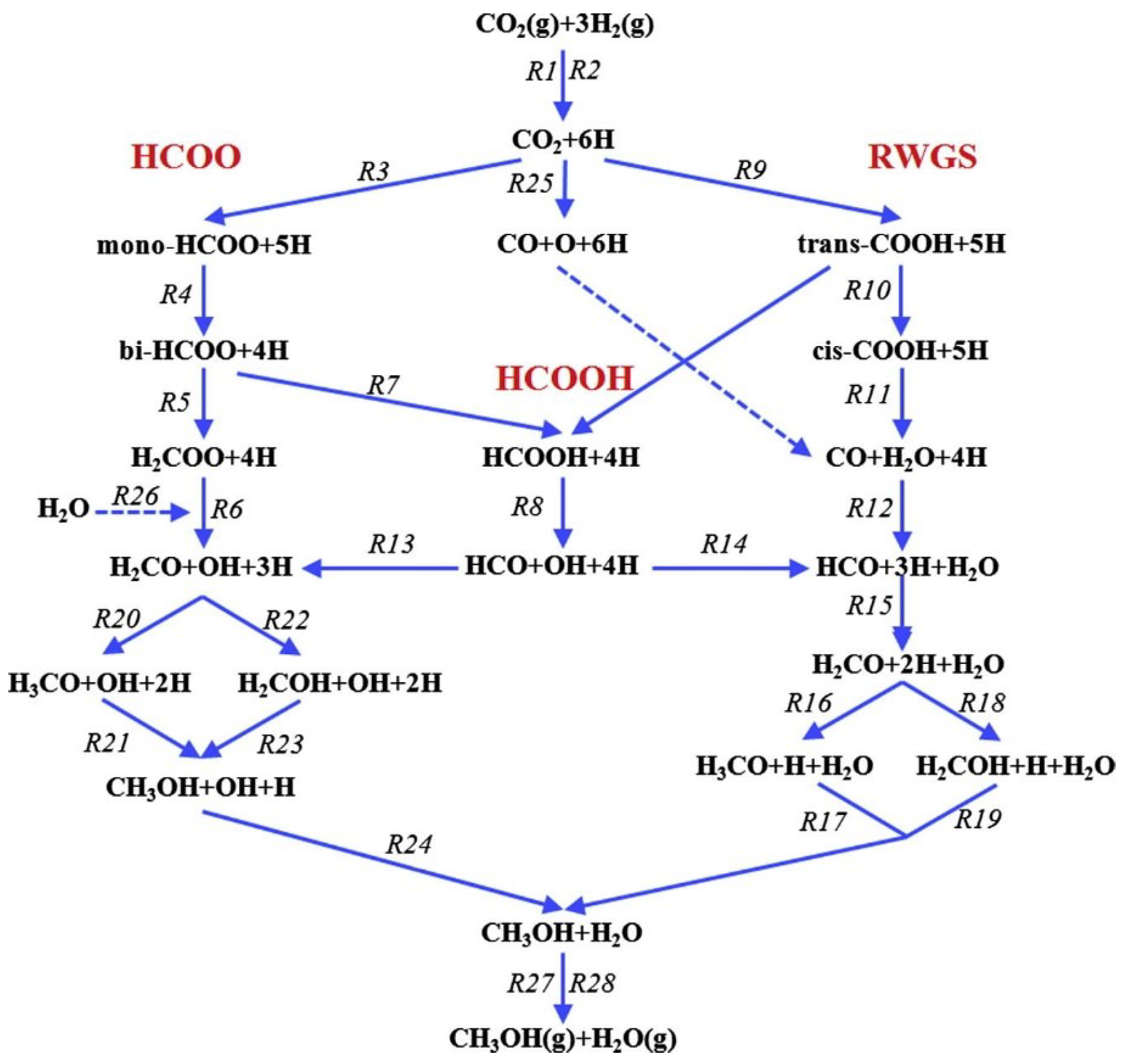
2.2.1. Methanol Synthesis over Cu-based Catalysts
2.2.2. MeOH Synthesis over Other Catalysts
2.2.3. DME Synthesis
3. Methods
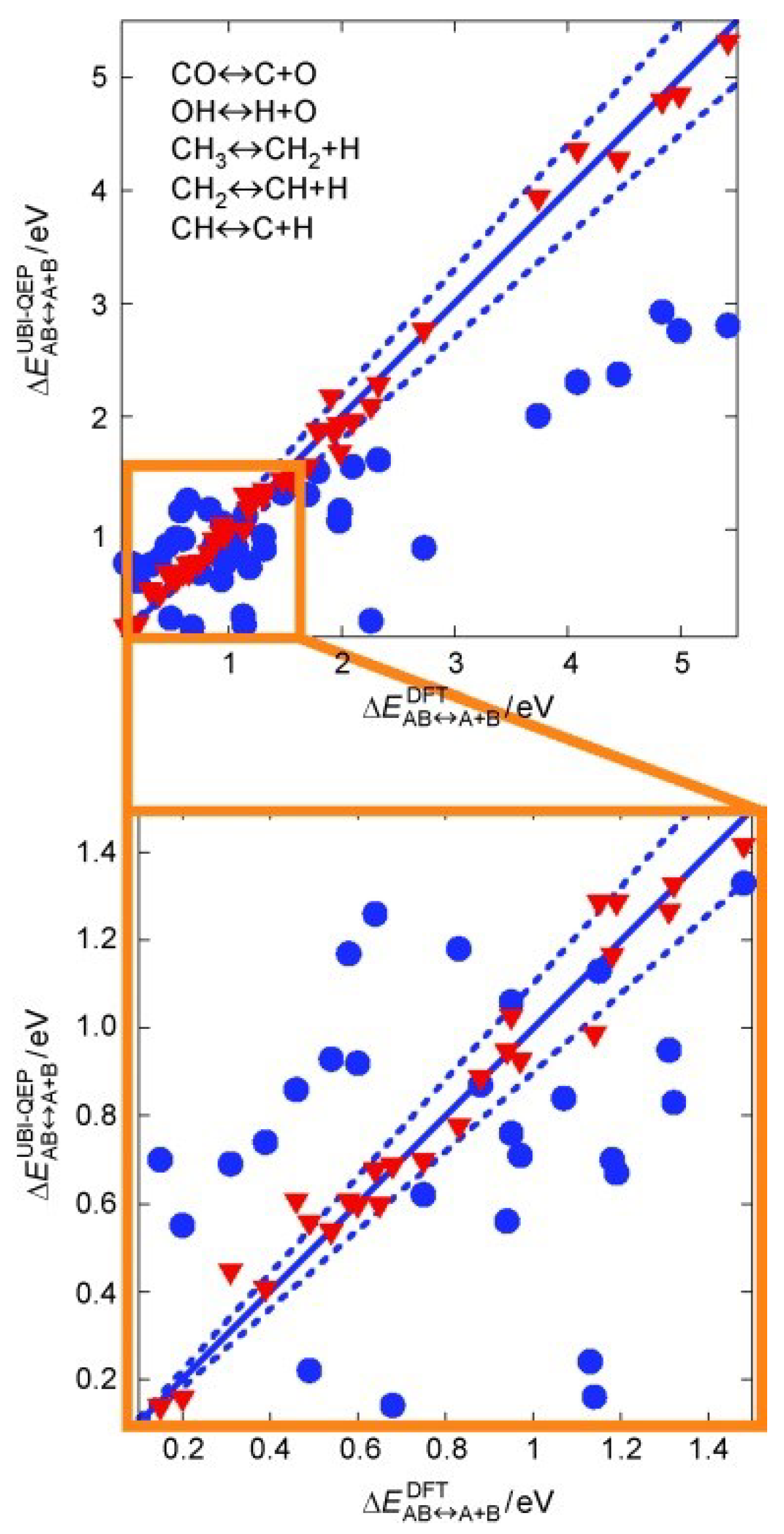
3.1. Computational Chemistry
3.1.1. Surface Modeling
3.1.2. Adsorption Energy
3.1.3. Activation Energy
3.2. Microkinetic Modeling
3.2.1. Kinetic Parameter
3.2.2. Microkinetic Model
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Norskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, V.; De Wispelaere, K.; Van Der Mynsbrugge, J.; Vandichel, M.; Hemelsoet, K.; Waroquier, M. First principle chemical kinetics in zeolites: The methanol-to-olefin process as a case study. Chem. Soc. Rev. 2014, 43, 7326–7357. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T. Future directions and industrial perspectives micro-and macro-kinetics: Their relationship in heterogeneous catalysis. Top. Catal. 1994, 1, 353–366. [Google Scholar] [CrossRef]
- Bush, S.F.; Dyer, P. The experimental and computational determination of complex chemical kinetics mechanisms. Proc. R. Soc. Lond. Ser. A 1976, 351, 33–53. [Google Scholar]
- Hickman, D.A.; Schmidt, L.D. Steps in CH4 oxidation on Pt and Rh surfaces: High-temperature reactor simulations. AIChE J. 1993, 39, 1164–1177. [Google Scholar] [CrossRef]
- Oh, S.H.; Fisher, G.B.; Carpenter, J.E.; Goodman, D.W. Comparative kinetic studies of CO–O2 and CO–NO reactions over single crystal and supported rhodium catalysts. J. Catal. 1986, 100, 360–376. [Google Scholar] [CrossRef]
- Dumesic, J.A. The Microkinetics of Heterogeneous Catalysis; American Chemical Society: Washington, DC, USA, 1993; pp. 1–315. [Google Scholar]
- Ali, K.A.; Abdullah, A.Z.; Mohamed, A.R. Recent development in catalytic technologies for methanol synthesis from renewable sources: A critical review. Renew. Sustain. Energy Rev. 2015, 44, 508–518. [Google Scholar] [CrossRef]
- Ateka, A.; Pérez-Uriarte, P.; Gamero, M.; Ereña, J.; Aguayo, A.T.; Bilbao, J. A comparative thermodynamic study on the CO2 conversion in the synthesis of methanol and of DME. Energy 2017, 120, 796–804. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef]
- Frusteri, F.; Bonura, G.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation. Appl. Catal. B Environ. 2015, 176, 522–531. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Oloman, C.; Li, H. Electrochemical processing of carbon dioxide. ChemSusChem 2008, 1, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Oloman, C. Development of a continuous reactor for the electro-reduction of carbon dioxide to formate—Part 1: Process variables. J. Appl. Electrochem. 2006, 36, 1105. [Google Scholar] [CrossRef]
- Li, H.; Oloman, C. Development of a continuous reactor for the electro-reduction of carbon dioxide to formate—Part 2: Scale-up. J. Appl. Electrochem. 2007, 37, 1107–1117. [Google Scholar] [CrossRef]
- Lerner, A.; Brear, M.J.; Lacey, J.S.; Gordon, R.L.; Webley, P.A. Life cycle analysis (LCA) of low emission methanol and di-methyl ether (DME) derived from natural gas. Fuel 2018, 220, 871–878. [Google Scholar] [CrossRef]
- Matzen, M.; Demirel, Y. Methanol and dimethyl ether from renewable hydrogen and carbon dioxide: Alternative fuels production and life-cycle assessment. J. Clean. Prod. 2016, 139, 1068–1077. [Google Scholar] [CrossRef]
- Bae, C.; Kim, J. Alternative fuels for internal combustion engines. Proc. Combust. Inst. 2017, 36, 3389–3413. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Denny, P.J.; Parker, D.G.; Spencer, M.S.; Whan, D.A. Mechanism of methanol synthesis from CO2/CO/H2 mixtures over copper/zinc oxide/alumina catalysts: Use of 14C-labelled reactants. Appl. Catal. 1987, 30, 333–338. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of Methanol Synthesis on Cu through CO2 and CO Hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Alharbi, W.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Dehydration of Methanol to Dimethyl Ether over Heteropoly Acid Catalysts: The Relationship between Reaction Rate and Catalyst Acid Strength. ACS Catal. 2015, 5, 7168–7193. [Google Scholar] [CrossRef]
- Din, I.U.; Shaharun, M.S.; Alotaibi, M.A.; Alharthi, A.I.; Naeem, A. Recent developments on heterogeneous catalytic CO2 reduction to methanol. J. CO2 Util. 2019, 34, 20–33. [Google Scholar] [CrossRef]
- Kakumoto, T. A theoretical study for the CO2 hydrogenation mechanism on Cu/ZnO catalyst. Energy Convers. Manag. 1995, 36, 661–664. [Google Scholar] [CrossRef]
- Kakumoto, T.; Watanabe, T. A theoretical study for methanol synthesis by CO2 hydrogenation. Catal. Today 1997, 36, 39–44. [Google Scholar] [CrossRef]
- Bauschlicher, C.W. A theoretical study of CO/Cu(100). J. Chem. Phys. 1994, 101, 3250–3254. [Google Scholar] [CrossRef]
- Tameh, M.S.; Dearden, A.K.; Huang, C. Accuracy of Density Functional Theory for Predicting Kinetics of Methanol Synthesis from CO and CO2 Hydrogenation on Copper. J. Phys. Chem. C 2018, 122, 17942–17953. [Google Scholar] [CrossRef]
- Wellendorff, J.; Lundgaard, K.T.; Møgelhøj, A.; Petzold, V.; Landis, D.D.; Nørskov, J.K.; Bligaard, T.; Jacobsen, K.W. Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation. Phys. Rev. B 2012, 85, 235149. [Google Scholar] [CrossRef]
- Studt, F.; Abild-Pedersen, F.; Varley, J.B.; Nørskov, J.K. CO and CO2 hydrogenation to methanol calculated using the BEEF-vdW functional. Catal. Lett. 2013, 143, 71–73. [Google Scholar] [CrossRef]
- Reichenbach, T.; Mondal, K.; Jäger, M.; Vent-Schmidt, T.; Himmel, D.; Dybbert, V.; Bruix, A.; Krossing, I.; Walter, M.; Moseler, M. Ab initio study of CO2 hydrogenation mechanisms on inverse ZnO/Cu catalysts. J. Catal. 2018, 360, 168–174. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Van Den Berg, R.; Prieto, G.; Korpershoek, G.; Van Der Wal, L.I.; Van Bunningen, A.J.; Lægsgaard-Jørgensen, S.; De Jongh, P.E.; De Jong, K.P. Structure sensitivity of Cu and CuZn catalysts relevant to industrial methanol synthesis. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wu, P.; Yang, B. Essential role of water in the autocatalysis behavior of methanol synthesis from CO2 hydrogenation on Cu: A combined DFT and microkinetic modeling study. J. Phys. Chem. C 2019, 123, 5966–8959. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Lee, Y.; Park, M.J.; Lee, W.B. Practical microkinetic modeling approach for methanol synthesis from syngas over a Cu-based catalyst. Ind. Eng. Chem. Res. 2019, 58, 8663–8673. [Google Scholar] [CrossRef]
- Huš, M.; Kopač, D.; Štefančič, N.S.; Jurković, D.L.; Dasireddy, V.D.B.C.; Likozar, B. Unravelling the mechanisms of CO2 hydrogenation to methanol on Cu-based catalysts using first-principles multiscale modelling and experiments. Catal. Sci. Technol. 2017, 7, 5900–5913. [Google Scholar] [CrossRef]
- Cheng, Z.; Lo, C.S. Mechanistic and microkinetic analysis of CO2 hydrogenation on ceria. Phys. Chem. Chem. Phys. 2016, 18, 7987–7996. [Google Scholar] [CrossRef] [PubMed]
- Sakahara, S.; Yajima, K.; Belosludov, R.; Takami, S.; Kubo, M.; Miyamoto, A. Combinatorial computational chemistry approach to the design of methanol synthesis catalyst. Appl. Surf. Sci. 2002, 189, 253–259. [Google Scholar] [CrossRef]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 25 May 2020).
- Yarulina, I.; Chowdhury, A.D.; Meirer, F.; Weckhuysen, B.M.; Gascon, J. Recent trends and fundamental insights in the methanol-to-hydrocarbons process. Nat. Catal. 2018, 1, 398–411. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Yu, J. Recent advances in zeolite chemistry and catalysis. Chem. Soc. Rev. 2015, 44, 7022–7024. [Google Scholar] [CrossRef]
- Haase, F.; Sauer, J. Interaction of Methanol with Brønsted Acid Sites of Zeolite Catalysts: An ab Initio Study. J. Am. Chem. Soc. 1995, 117, 3780–3789. [Google Scholar] [CrossRef]
- Plessow, P.N.; Studt, F. Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations. ACS Catal. 2017, 7, 7987–7994. [Google Scholar] [CrossRef]
- Waugh, K.C. Methanol synthesis. Catal. Lett. 2012, 142, 1153–1166. [Google Scholar] [CrossRef]
- Medford, A.J.; Sehested, J.; Rossmeisl, J.; Chorkendorff, I.; Studt, F.; Nørskov, J.K.; Moses, P.G. Thermochemistry and micro-kinetic analysis of methanol synthesis on ZnO (0 0 0 1). J. Catal. 2014, 309, 397–407. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Denny, P.J.; Jennings, J.R.; Spencer, M.S.; Waugh, K.C. Synthesis of Methanol. Part 1. Catalysts and Kinetics. Appl. Catal. 1988, 36, 1–65. [Google Scholar] [CrossRef]
- Taylor, P.A.; Rasmussen, P.B.; Ovesen, C.V.; Stoltze, P.; Chorkendorff, I. Formate synthesis on Cu(100). Surf. Sci. 1992, 261, 191–206. [Google Scholar] [CrossRef]
- Askgaard, T.S.; Norskov, J.K.; Ovesen, C.V.; Stoltze, P. A Kinetic Model of Methanol Synthesis. J. Catal. 1995, 156, 229–242. [Google Scholar] [CrossRef]
- Rasmussen, P.B.; Holmblad, P.M.; Askgaard, T.; Ovesen, C.V.; Stoltze, P.; Nørskov, J.K.; Chorkendorff, I. Methanol synthesis on Cu(100) from a binary gas mixture of CO2 and H2. Catal. Lett. 1994, 26, 373–381. [Google Scholar] [CrossRef]
- Ovesen, C.V.; Clausen, B.S.; Schiøtz, J.; Stoltze, P.; Topsøe, H.; Nørskov, J.K. Kinetic implications of dynamical changes in catalyst morphology during methanol synthesis over Cu/ZnO catalysts. J. Catal. 1997, 168, 133–142. [Google Scholar] [CrossRef]
- Clausen, B.S.; Schiøtz, J.; Gråbæk, L.; Ovesen, C.V.; Jacobsen, K.W.; Nørskov, J.K.; Topsøe, H. Wetting/ non-wetting phenomena during catalysis: Evidence from in situ on-line EXAFS studies of Cu-based catalysts. Top. Catal. 1994, 1, 367–376. [Google Scholar] [CrossRef]
- Clausen, B.S.; Steffensen, G.; Fabius, B.; Villadsen, J.; Feidenhans’l, R.; Topsøe, H. In situ cell for combined XRD and on-line catalysis tests: Studies of Cu-based water gas shift and methanol catalysts. J. Catal. 1991, 132, 524–535. [Google Scholar] [CrossRef]
- Clausen, B.S.; Gråbæk, L.; Steffensen, G.; Hansen, P.L.; Topsøe, H. A combined QEXAFS/XRD method for on-line, in situ studies of catalysts: Examples of dynamic measurements of Cu-based methanol catalysts. Catal. Lett. 1993, 20, 23–36. [Google Scholar] [CrossRef]
- Gokhale, A.A.; Dumesic, J.A.; Mavrikakis, M. On the mechanism of low-temperature water gas shift reaction on copper. J. Am. Chem. Soc. 2008, 130, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Fichtl, M.B.; Ruland, H.; Kaluza, S.; Muhler, M.; Hinrichsen, O. Detailed kinetic modeling of methanol synthesis over a ternary copper catalyst. Chem. Eng. J. 2012, 203, 480–491. [Google Scholar] [CrossRef]
- Rubert-Nason, P.; Mavrikakis, M.; Maravelias, C.T.; Grabow, L.C.; Biegler, L.T. Advanced solution methods for microkinetic models of catalytic reactions: A methanol synthesis case study. AIChE J. 2014, 60, 1336–1346. [Google Scholar] [CrossRef]
- Tang, Q.L.; Zou, W.T.; Huang, R.K.; Wang, Q.; Duan, X.X. Effect of the components’ interface on the synthesis of methanol over Cu/ZnO from CO2/H2: A microkinetic analysis based on DFT + U calculations. Phys. Chem. Chem. Phys. 2015, 17, 7317–7333. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, W.J.; Petersen, M.A.; Datt, M.S.; Van Den Berg, J.A.; Van Helden, P. On the kinetic interpretation of DFT-derived energy profiles: Cu-catalyzed methanol synthesis. Catal. Lett. 2015, 145, 559–568. [Google Scholar] [CrossRef]
- Van Helden, P.; Van Den Berg, J.A.; Weststrate, C.J. Hydrogen adsorption on co surfaces: A density functional theory and temperature programmed desorption study. ACS Catal. 2012, 2, 1097–1107. [Google Scholar] [CrossRef]
- Medford, A.J.; Shi, C.; Hoffmann, M.J.; Lausche, A.C.; Fitzgibbon, S.R.; Bligaard, T.; Nørskov, J.K. CatMAP: A software package for descriptor-based microkinetic mapping of catalytic trends. Catal. Lett. 2015, 145, 794–807. [Google Scholar] [CrossRef]
- Yang, N.; Medford, A.J.; Liu, X.; Studt, F.; Bligaard, T.; Bent, S.F.; Nørskov, J.K. Intrinsic selectivity and structure sensitivity of rhodium catalysts for C2+ oxygenate production. J. Am. Chem. Soc. 2016, 138, 3705–3714. [Google Scholar] [CrossRef]
- Bonivardi, A.L.; Chiavassa, D.L.; Querini, C.A.; Baltanás, M.A. Enhancement of the catalytic performance to methanol synthesis from CO2/H2 by gallium addition to palladium/silica catalysts. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2000; Volume 130, pp. 3747–3752. [Google Scholar]
- Fujitani, T.; Nakamura, I. Methanol synthesis from CO and CO2 hydrogenations over supported palladium catalysts. Bull. Chem. Soc. Jpn. 2002, 75, 1393–1398. [Google Scholar] [CrossRef]
- Fujitani, T.; Saito, M.; Kanai, Y.; Watanabe, T.; Nakamura, J.; Uchijima, T. Development of an active Ga2O3 supported palladium catalyst for the synthesis of methanol from carbon dioxide and hydrogen. Appl. Catal. A Gen. 1995, 125, L199–L202. [Google Scholar] [CrossRef]
- Chiavassa, D.L.; Collins, S.E.; Bonivardi, A.L.; Baltanás, M.A. Methanol synthesis from CO2/H2 using Ga2O3-Pd/silica catalysts: Kinetic modeling. Chem. Eng. J. 2009, 150, 204–212. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.J.; Mei, D.; Ge, Q. Methanol synthesis from CO2 hydrogenation over a Pd4/In2O3 model catalyst: A combined DFT and kinetic study. J. Catal. 2014, 317, 44–53. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Logadottir, A.; Rod, T.H.; Nørskov, J.K.; Hammer, B.; Dahl, S.; Jacobsen, C.J.H. The Brønsted-Evans-Polanyi relation and the volcano plot for ammonia synthesis over transition metal catalysts. J. Catal. 2001, 197, 229–231. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.B.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.; Mavrikakis, M.; et al. Universality in heterogeneous catalysis. J. Catal. 2002, 209, 275–278. [Google Scholar] [CrossRef]
- Bligaard, T.; Nørskov, J.K.; Dahl, S.; Matthiesen, J.; Christensen, C.H.; Sehested, J. The Brønsted-Evans-Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Frei, M.S.; Capdevila-Cortada, M.; García-Muelas, R.; Mondelli, C.; López, N.; Stewart, J.A.; Curulla Ferré, D.; Pérez-Ramírez, J. Mechanism and microkinetics of methanol synthesis via CO2 hydrogenation on indium oxide. J. Catal. 2018, 361, 313–321. [Google Scholar] [CrossRef]
- Frei, M.S.; Mondelli, C.; García-Muelas, R.; Kley, K.S.; Puértolas, B.; López, N.; Safonova, O.V.; Stewart, J.A.; Curulla Ferré, D.; Pérez-Ramírez, J. Atomic-scale engineering of indium oxide promotion by palladium for methanol production via CO2 hydrogenation. Nat. Commun. 2019, 10, 3377. [Google Scholar] [CrossRef]
- García-Trenco, A.; Regoutz, A.; White, E.R.; Payne, D.J.; Shaffer, M.S.P.; Williams, C.K. PdIn intermetallic nanoparticles for the hydrogenation of CO2 to methanol. Appl. Catal. B Environ. 2018, 220, 9–18. [Google Scholar] [CrossRef]
- Wu, P.; Yang, B. Intermetallic PdIn catalyst for CO2 hydrogenation to methanol: Mechanistic studies with a combined DFT and microkinetic modeling method. Catal. Sci. Technol. 2019, 9, 6102–6113. [Google Scholar] [CrossRef]
- Carr, R.T.; Neurock, M.; Iglesia, E. Catalytic consequences of acid strength in the conversion of methanol to dimethyl ether. J. Catal. 2011, 278, 78–93. [Google Scholar] [CrossRef]
- Moses, P.G.; Nørskov, J.K. Methanol to dimethyl ether over ZSM-22: A periodic density functional theory study. ACS Catal. 2013, 3, 735–745. [Google Scholar] [CrossRef]
- Jones, A.J.; Iglesia, E. Kinetic, spectroscopic, and theoretical sssessment of sssociative and dissociative methanol dehydration routes in zeolites. Angew. Chem. Int. Ed. 2014, 53, 12177–12181. [Google Scholar] [CrossRef]
- Lee, K.; Murray, É.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Park, M.J.; Lee, W.B. Microkinetic modeling of DME synthesis from methanol over H-zeolite catalyst: Associative vs. dissociative pathways. Catal. Today 2020. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Sabbe, M.K.; Reyniers, M.F.; Reuter, K. First-principles kinetic modeling in heterogeneous catalysis: An industrial perspective on best-practice, gaps and needs. Catal. Sci. Technol. 2012, 2, 2010–2024. [Google Scholar] [CrossRef]
- Li, H.; Roy, P.N.; Le Roy, R.J. Analytic Morse/long-range potential energy surfaces and predicted infrared spectra for CO2—H2. J. Chem. Phys. 2010, 132, 214309. [Google Scholar] [CrossRef]
- Feibelman, P.J.; Hammer, B.; Norskov, J.K.; Wagner, F.; Scheffler, M.; Stump, R.; Watwe, R.; Dumesic, J. The CO/Pt(111) puzzle. J. Phys. Chem. B 2001, 105, 4018–4025. [Google Scholar] [CrossRef]
- Kresse, G.; Gil, A.; Sautet, P. Significance of single-electron energies for the description of CO on Pt(111). Phys. Rev. B. 2003, 68, 073401. [Google Scholar] [CrossRef]
- Soini, T.M.; Genest, A.; Rösch, N. Assessment of hybrid density functionals for the adsorption of carbon monoxide on platinum model clusters. J. Phys. Chem. A 2015, 119, 4051–4056. [Google Scholar] [CrossRef] [PubMed]
- Stroppa, A.; Termentzidis, K.; Paier, J.; Kresse, G.; Hafner, J. CO adsorption on metal surfaces: A hybrid functional study with plane-wave basis set. Phys. Rev. B. 2007, 76, 195440. [Google Scholar] [CrossRef]
- Rohrbach, G.; Hafner, J.; Kresse, G. Molecular adsorption on the surface of strongly correlated transition-metal oxides: A case study for CO/NiO(100). Phys. Rev. B. 2004, 69, 075413. [Google Scholar] [CrossRef]
- Schimka, L.; Harl, J.; Stroppa, A.; Grüneis, A.; Marsman, M.; Mittendorfer, F.; Kresse, G. Accurate surface and adsorption energies from many-body perturbation theory. Nat. Mater. 2010, 9, 741–744. [Google Scholar] [CrossRef]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 134106. [Google Scholar] [CrossRef]
- Raimondeau, S.; Vlachos, D.G. Recent developments on multiscale, hierarchical modeling of chemical reactors. Chem. Eng. J. 2002, 90, 3–12. [Google Scholar] [CrossRef]
- Salciccioli, M.; Stamatakis, M.; Caratzoulas, S.; Vlachos, D.G. A review of multiscale modeling of metal-catalyzed reactions: Mechanism development for complexity and emergent behavior. Chem. Eng. Sci. 2011, 66, 4319–4355. [Google Scholar] [CrossRef]
- Brönsted, J.N. Acid and basic catalysis. Chem. Rev. 1928, 5, 231–338. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Further considerations on the thermodynamics of chemical equilibria and reaction rates. Trans. Faraday Soc. 1936, 32, 1333–1360. [Google Scholar] [CrossRef]
- Shustorovich, E.; Sellers, H. The UBI-QEP method: A practical theoretical approach to understanding chemistry on transition metal surfaces. Surf. Sci. Rep. 1998, 31, 1–119. [Google Scholar] [CrossRef]
- Maestri, M.; Reuter, K. Semiempirical Rate Constants for Complex Chemical Kinetics: First-Principles Assessment and Rational Refinement. Angew. Chem. Int. Ed. 2011, 50, 1194–1197. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Redox Pathway | Carboxyl Pathway |
|---|---|
| CO + * ⇌ CO* | |
| H2O + * ⇌ H2O* H2O* + * ⇌ H* + OH* | |
| OH* + * ⇌ O* + H* | CO* + OH* ⇌ COOH* + * |
| 2OH* ⇌ H2O* + O* | COOH* + * ⇌ CO2* + H* |
| CO* + O* ⇌ CO2* + * | COOH* + OH* ⇌ CO2* + H2O* |
| CO2* ⇌ CO2 + * | |
| 2H* ⇌ H2 + 2* | |
| Researcher | Year | Reaction § | # of Steps | Catalyst Model | Kinetic Parameter | Rate-Limiting Step | Dominant Pathway § |
|---|---|---|---|---|---|---|---|
| Askgaard et al. [47] | 1995 | CO2, WGS | 16 | Cu(100) | Estimation | Assumed | None |
| Ovesen et al. [49] | 1997 | CO2, WGS | 13 | Cu(100), Cu(110), Cu(111) | Calculation with partition functions, estimation | Assumed | None |
| Grabow and Mavrikakis [20] | 2011 | CO, CO2, WGS | 49 | Cu(111) | DFT, estimation | H3CO* + H* → CH3OH* + * | CO (1/3) and CO2 (2/3) |
| Tang et al. [56] | 2015 | CO2, WGS | 38 | Cu(111)/ZnO(100) | DFT + U | None | None |
| Janse Van Rensburg et al. [57] | 2015 | CO, CO2 | 11 | Cu(111), Cu(211), CuZn(211) | DFT | CO2 hydrogenation: HCOOH (g) + H* → H2COOH* + * | CO |
| Tameh et al. [26] | 2018 | CO, CO2 | 14 | Cu(211) | DFT | CO hydrogenation: HCO* + H* → H2CO* + * CO2 hydrogenation: HCOOH* + H* → H2COOH* + * | CO2 |
| Xu et al. [33] | 2019 | CO2 | 7 | Cu(211) | DFT | None | None |
| Park et al. [34] | 2019 | CO, CO2, WGS | 28 | Cu(111) | DFT, UBI-QEP, estimation | H3CO* + H* → CH3OH* + * | None |
| Researcher | Year | Reaction § | # of step | Catalyst Model | Kinetic Parameter | Rate-Limiting Step | Dominant Pathway § |
|---|---|---|---|---|---|---|---|
| Chiavassa et al. [64] | 2009 | CO, CO2, WGS | 12 | Ga2O3-Pd/silica | Estimation | HCOO* + H* → H2COO* + * | None |
| Medford et al. [44] | 2014 | CO, CO2, WGS | 19 | ZnO(0001) | DFT | Industrial condition: H2CO* + H* → H3CO* + *High T and low P: H3CO* + H* → CH3OH* + * | CO |
| Ye et al. [65] | 2014 | CO2 | 28 | Pd4/In2O3 | DFT | H2COO* + H* → H2CO* + OH* | HCOO pathway of CO2 |
| Cheng and Lo [36] | 2016 | CO2 | 21 | CeO2(110) | DFT, BEP relation | H2COO* + H* → H2CO* + OH* | HCOO pathway of CO2 |
| Huš et al. [35] | 2017 | CO2 | 33 | Zn3O3/Cu, Mg3O3/Cu, Cr3O3/Cu, Fe3O3/Cu, | DFT | None | HCOO pathway of CO2 |
| Frei et al. [70] | 2018 | CO2, WGS | 26 | In2O3(111) | DFT | None | None |
| Frei et al. [71] | 2019 | CO2, WGS | 19 | Pd-promoted In2O3 | DFT | None | None |
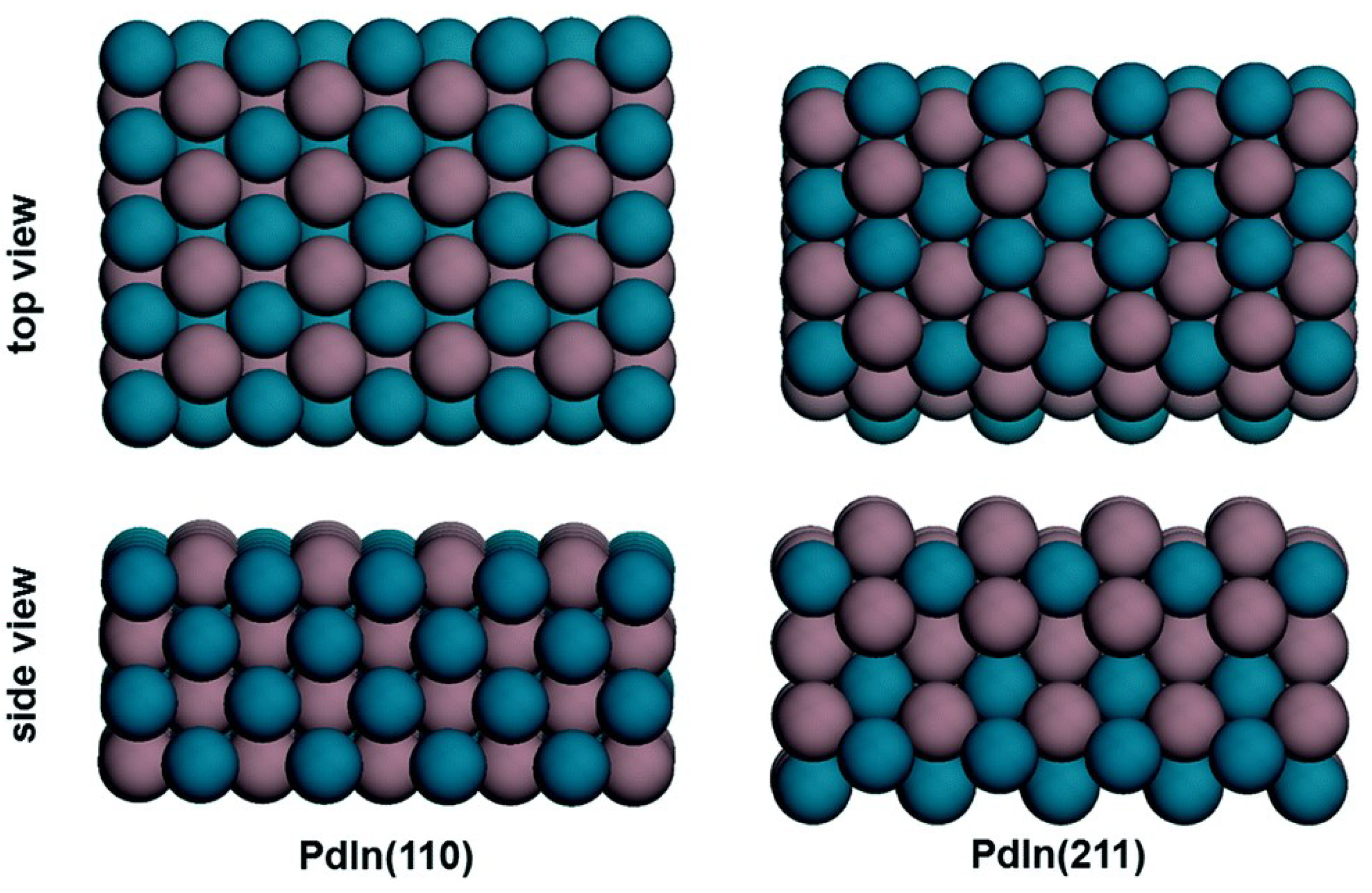
| Wu and Yang [73] | 2019 | CO2, WGS | 24 | PdIn(110), PdIn(211) | DFT | PdIn(110) and (211) at high T: HCOOH* + H* → H2COOH* + * PdIn(211) at low T: H2COOH* + * → H2CO* + OH* | HCOOH pathway of CO2 |
| Researcher | Year | Catalyst Model | Kinetic Parameter | Rate-Limiting Step | Dominant Pathway |
|---|---|---|---|---|---|
| Carr et al. [74] | 2011 | Tungsten Keggin POM cluster | DFT | None | Associative pathway |
| Moses and Nørskov [75] | 2013 | ZSM-22 | DFT | DME formation step of the dissociative pathway | Dissociative pathway |
| Jones and Iglesia [76] | 2014 | H-MFI | DFT | None | Dependent on T and P |
| Park et al. [78] | 2020 | H-zeolite cluster | MP2, Estimation | DME formation step of the dissociative pathway | Dissociative pathway |
| COOH Pathway | HCOO Pathway |
|---|---|
| CO2 + * ⇌ CO2* | |
| CO2* + H* ⇌ COOH* + * | CO2* + H* ⇌ HCOO* + * |
| COOH* + * ⇌ CO* + OH* | HCOO* + H* ⇌ H2CO2* + * |
| CO* + H* ⇌ HCO* + * | H2CO2* + H* ⇌ H2COOH* + * |
| HCO* + H* ⇌ HCOH* + * | H2COOH* + * ⇌ CH2O* + OH* |
| HCOH* + H* ⇌ H2COH* + * | CH2O* + H* ⇌ CH3O* + * |
| H2COH* + H* ⇌ H3COH* + * | CH3O* + H* ⇌ H3COH* + * |
| H3COH* ⇌ CH3OH + * | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Kim, H.S.; Lee, W.B.; Park, M.-J. Trends and Outlook of Computational Chemistry and Microkinetic Modeling for Catalytic Synthesis of Methanol and DME. Catalysts 2020, 10, 655. https://doi.org/10.3390/catal10060655
Park J, Kim HS, Lee WB, Park M-J. Trends and Outlook of Computational Chemistry and Microkinetic Modeling for Catalytic Synthesis of Methanol and DME. Catalysts. 2020; 10(6):655. https://doi.org/10.3390/catal10060655
Chicago/Turabian StylePark, Jongmin, Hyo Seok Kim, Won Bo Lee, and Myung-June Park. 2020. "Trends and Outlook of Computational Chemistry and Microkinetic Modeling for Catalytic Synthesis of Methanol and DME" Catalysts 10, no. 6: 655. https://doi.org/10.3390/catal10060655
APA StylePark, J., Kim, H. S., Lee, W. B., & Park, M.-J. (2020). Trends and Outlook of Computational Chemistry and Microkinetic Modeling for Catalytic Synthesis of Methanol and DME. Catalysts, 10(6), 655. https://doi.org/10.3390/catal10060655