Highly Efficient and Reusable Alkyne Hydrosilylation Catalysts Based on Rhodium Complexes Ligated by Imidazolium-Substituted Phosphine



Abstract
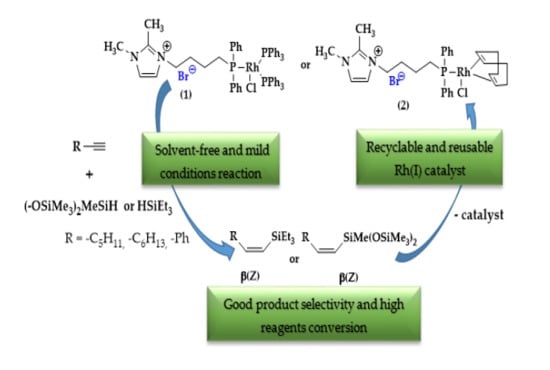
1. Introduction
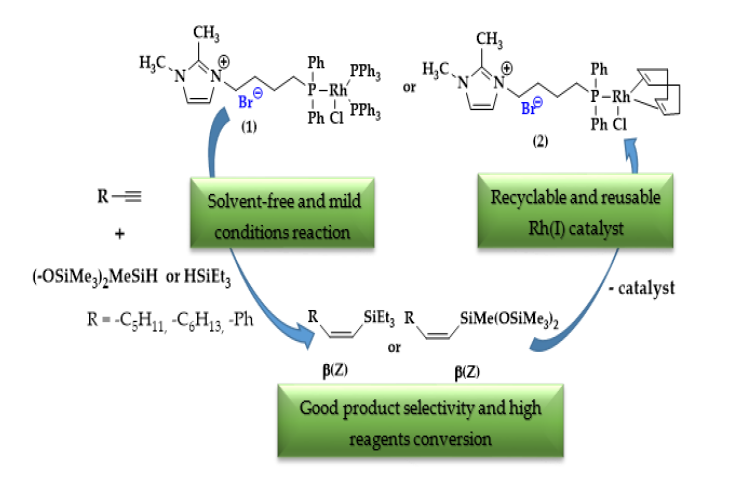
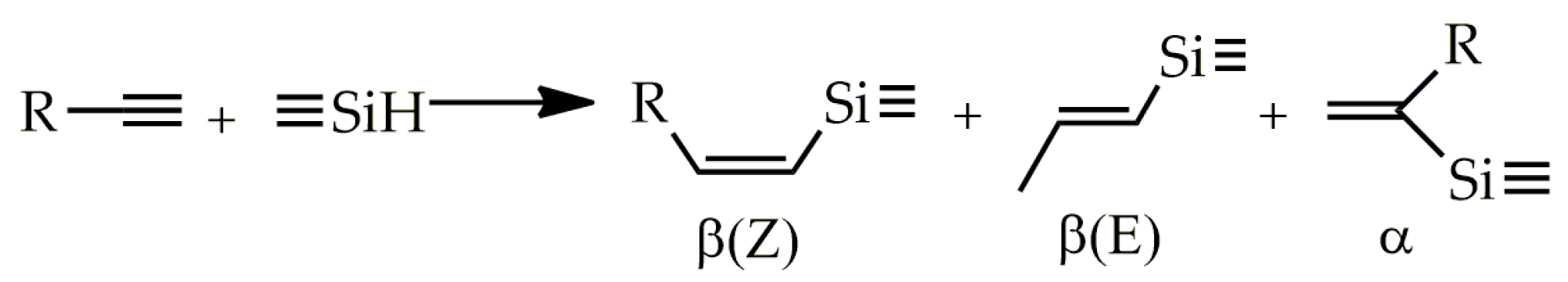
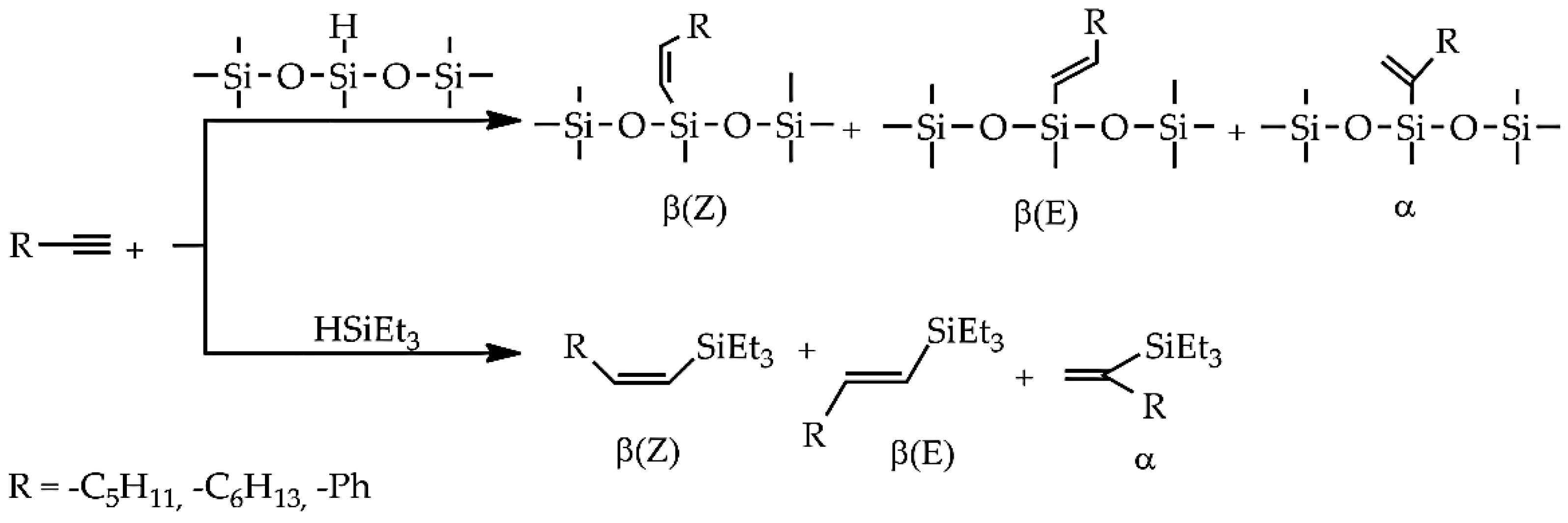
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Analytical Techniques
3.3. Synthesis of Transition-Metal Based Complexes
3.4. General Procedure for Catalytic Tests
1,1,1,3,5,5,5-Heptamethyl-3-[(1Z)Hept-1-Enyl)]Trisiloxane
1,1,1,3,5,5,5-Heptamethyl-3-[(1Z)Oct-1-Enyl)]Trisiloxane
1,1,1,3,5,5,5-Heptamethyl-3-[(1Z)-2-Phenylethenyl]Trisiloxane
(Z)-Triethyl(Hept-1-Enyl)Silane
(Z)-Triethyl(Oct-1-Enyl)Silane
(E)-Triethyl(Phenyl-1-Ethene)Silane
3.5. General Procedure for Catalyst Isolation and Tests with Subsequent Catalytic Runs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Luh, T.-Y.; Liu, S.-T. Synthetic Applications of Allylsilanes and Vinylsilanes. In The Chemistry in Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Willey: New York, NY, USA, 1998. [Google Scholar]
- Fleming, I.; Barbero, A.; Walter, D. Stereochemical Control in Organic Synthesis Using Silicon-Containing Compounds. Chem. Rev. 1997, 97, 2063–2192. [Google Scholar] [CrossRef]
- Nakao, Y.; Hiyama, T. Silicon-based cross-coupling reaction: An environmentally benign version. Chem. Soc. Rev. 2011, 40, 4893–4901. [Google Scholar] [CrossRef]
- Hiyama, T.; Diederich, F.; Stang, P.J. Metal-Catalyzed Cross-Coupling Reactions; Willey-VCH: New York, NY, USA, 1998; pp. 421–453. [Google Scholar]
- Lim, D.S.W.; Anderson, E.A. Synthesis of Vinylsilanes. Synthesis 2012, 44, 983–1010. [Google Scholar]
- Marciniec, B.; Maciejewski, H.; Pawluć, P. Hydrosilylation of Carbon-Carbon Multiple Bonds—Applications in Synthesis and Materials Science. In Organosilicon Compounds; Lee, V.Y., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 2, Chapter 5. [Google Scholar]
- Marciniec, B.; Maciejewski, H.; Pietraszuk, C.; Pawluć, P. Hydrosilylation. A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Takeuchi, R.; Tanouchi, N. Solvent-controlled Stereoselectivity in the Hydrosilylation of Alk-1-ynes Catalysed by Rhodium Complexes. J. Chem. Soc. Perkin Trans. 1994, 1, 2909–2913. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, X.Y.; Kang, S.X.; Gao, Y.M. Tri(t-butyl)phosphine-assisted selective hydrosilylation of terminal alkynes. Chin. Chem. Lett. 2010, 21, 312–316. [Google Scholar] [CrossRef]
- Dierick, S.; Vercruysse, E.; Berthon-Gelloz, G.; Marko, I.E. User-Friendly Platinum Catalysts for the Highly Stereoselective Hydrosilylation of Alkynes and Alkenes. Chem. Eur. J. 2015, 21, 17073–17078. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, Y.; Mohara, Y.; Saito, S. (Z)-Selective Hydrosilylation of Terminal Alkynes with HSiMe(OSiMe3)2 Catalyzed by Ruthenium Complex Containing an N-Heterocyclic Carbene. Org. Lett. 2017, 19, 5204–5207. [Google Scholar] [CrossRef] [PubMed]
- Chaulagain, M.R.; Mahandru, G.M.; Montgomery, J. Alkyne hydrosilylation catalysed by nickel complexes of N-heterocyclic carbenes. Tetrahedron 2006, 62, 7560–7566. [Google Scholar] [CrossRef]
- Field, L.D.; Ward, A.J. Catalytic hydrosilylation of acetylenes mediated by phosphine complexes of cobalt (I), rhodium(I) and iridium(I). J. Organomet. Chem. 2003, 681, 91–97. [Google Scholar] [CrossRef]
- Lewis, N.L.; Sy, K.G.; Bryant, G.L.; Donahue, P.E. Platinum-catalyzed hydrosilylation of alkynes. Organometallics 1991, 10, 3750–3759. [Google Scholar] [CrossRef]
- Takeuchi, R.; Nitta, S.; Watanabe, D. Cationic Rhodium Complex-catalysed Highly Selective Hydrosilylation of Propynylic Alcohols: A Convenient Synthesis of (a-y-Silyl Allylic Alcohols. J. Chem. Soc. Chem. Commun. 1994, 1777–1778. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tanouchi, N. Complete reversal of stereoselectivity in rhodium complex-catalysed hydrosilylation of alk-1-yne. J. Chem. Soc. Chem. Commun. 1993, 1319–1320. [Google Scholar] [CrossRef]
- Sato, A.; Kinoshita, H.; Shinokubo, H.; Oshima, K. Hydrosilylation of Alkynes with a Cationic Rhodium Species Formed in an Anionic Micellar System. Org. Lett. 2004, 6, 2217–2220. [Google Scholar] [CrossRef] [PubMed]
- Hamze, A.; Provot, O.; Brion, J.D.; Alami, M. Xphos ligand and platinum catalysts: A versatile catalyst for synthesis of functionalized β(E)-vinylsilanes for terminal alkynes. J. Org. Chem. 2008, 693, 2789–2797. [Google Scholar] [CrossRef]
- Mori, A.; Takahisa, E.; Kajiro, H.; Nishihara, Y.; Hiyama, T. Stereodivergent hydrosilylation of 1-alkynes catalysed by RhI(PPh3)3 leading to (E)- and (Z)-alkenylsilanes and the application to polymer. Polyhedron 2000, 19, 567–568. [Google Scholar] [CrossRef]
- Ojima, I.; Clos, N.; Donovan, R.J.; Ingallina, P. Hydrosilylation of 1-hexyne catalyzed by rhodium and cobalt-rhodium mixed-metal complexes. Mechanism of apparent trans addition. Organometallics 1990, 9, 3127–3133. [Google Scholar] [CrossRef]
- Ojima, I.; Kumagai, M.; Nagai, Y. The stereochemistry of the addition of hydrosilanes to alkyl acetylenes catalyzed by tris(triphenylphosphine)-chlororhodium. J. Organomet. Chem. 1974, 66, C14–C16. [Google Scholar] [CrossRef]
- Jun, C.H.; Crabtree, R.H. Dehydrogenative Silation, Isomerization and the Control of Syn- vs. Antiaddition in the Hydrosilation of Alkynes. J. Organomet. Chem. 1993, 447, 177–187. [Google Scholar] [CrossRef]
- Jimenes, M.V.; Perez-Torrente, J.J.; Bartolome, M.I.; Gierz, V.; Lahoz, F.J.; Oro, L.A. Rhodium(I) Complexes with Hemilabile N-heterocyclic Carbenes: Efficient Alkyne Hydrosilylation Catalysts. Organometallics 2008, 27, 224–234. [Google Scholar] [CrossRef]
- Andavan, G.T.S.; Bauer, E.B.; Letko, C.S.; Hollis, T.K.; Tham, F.S. Synthesis and Characterization of a Free Phenylene Bis(N-heterocyclic Carbene) and Its Di-Rh Complex: Catalytic Activity of the Di-Rh and CCC_NHC Rh Pincer Complexes in Intermolecular Hydrosilylation of Alkynes. J. Organomet. Chem. 2005, 690, 5938–5947. [Google Scholar] [CrossRef]
- Iglesias, M.; Sanz Miguel, P.J.; Polo, V.; Fernandez-Alvarez, F.J.; Perez-Torrente, J.J.; Oro, L.A. An Alternative Mechanistic Paradigm for the β-Z Hydrosilylation of Terminal Alkynes: The Role of Acetone as a Silane Shuttle. Chem. Eur. J. 2013, 19, 17559–17566. [Google Scholar] [CrossRef] [PubMed]
- Busetto, L.; Cassani, M.C.; Femoni, C.; Mancinelli, M.; Mazzanti, A.; Mazzoni, R.; Solinas, G. N-Heterocyclic Carbene-Amide Rhodium(I) Complexes: Structures, Dynamics, and Catalysis. Organometallics 2011, 30, 5258–5272. [Google Scholar] [CrossRef]
- De Bo, G.; Berthon-Gelloz, G.; Tinant, B.; István, E.; Markó, I.E. Hydrosilylation of Alkynes Mediated by N-Heterocyclic Carbene Platinum(0) Complexes. Organometallics 2006, 25, 1881–1890. [Google Scholar] [CrossRef]
- Maciejewski, H.; Szubert, K.; Marciniec, B.; Pernak, J. Hydrosilylation of functionalised olefins catalysed by rhodium siloxide complexes in ionic liquids. Green Chem. 2009, 11, 1045–1051. [Google Scholar] [CrossRef]
- Maciejewski, H.; Wawrzynczak, A.; Dutkiewicz, M.; Fiedorow, R. Silicone waxes—synthesis via hydrosilylation in homo- and heterogeneous systems. J. Mol. Catal. Chem. 2006, 257, 141–148. [Google Scholar] [CrossRef]
- Zielinski, W.; Kukawka, R.; Maciejewski, H.; Smiglak, M. Ionic Liquids as Solvents for Rhodium and Platinum Catalysts Used in Hydrosilylation Reaction. Molecules 2016, 21, 1115. [Google Scholar] [CrossRef]
- Jankowska-Wajda, M.; Kukawka, R.; Smiglak, M.; Maciejewski, H. The effect of the catalyst and the type of ionic liquid on the hydrosilylation process under batch and continuous reaction conditions. New J. Chem. 2018, 42, 5229–5236. [Google Scholar] [CrossRef]
- Maciejewski, H.; Jankowska-Wajda, M.; Dabek, I.; Fiedorow, R. The effect of the morpholinium ionic liquid anion on the catalytic activity of Rh (or Pt) complex–ionic liquid systems in hydrosilylation processes. RSC Adv. 2018, 8, 26922–26927. [Google Scholar]
- Pernak, J.; Swierczynska, A.; Kot, M.; Walkiewicz, F.; Maciejewski, H. Pyrylium sulfonate based ionic liquids. Tetrahedron Lett. 2011, 52, 4342–4345. [Google Scholar] [CrossRef]
- Maciejewski, H.; Szubert, K.; Fiedorow, R.; Giszter, R.; Niemczak, M.; Pernak, J.; Klimas, W. Diallyldimethylammonium and trimethylvinylammonium ionic liquids—Synthesis and application to catalysis. Appl. Catal. 2013, 451, 168–175. [Google Scholar] [CrossRef]
- Luska, K.L.; Demmans, K.Z.; Stratton, S.A.; Moores, A. Rhodium complexes stabilized by phosphine-functionalized phosphonium ionic liquids used as higher alkene hydroformylation catalysts: Influence of the phosphonium headgroup on catalytic activity. Dalton Trans. 2012, 41, 13533–13540. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Xu, X.-f.; Zhao, K. Amino acid- and imidazolium-tagged chiral pyrrolidinodiphosphine ligands and their applications in catalytic asymmetric hydrogenations in ionic liquid systems. Tetrahedron Asymm. 2012, 23, 1058–1067. [Google Scholar] [CrossRef]
- Jankowska-Wajda, M.; Bartlewicz, O.; Szpecht, A.; Zając, A.; Śmiglak, M.; Maciejewski, H. Platinum and rhodium complexes ligated by imidazolium- substituted phosphine as the efficient and recyclable catalysts for hydrosilylation. RSC Adv. 2019, 9, 29396–29404. [Google Scholar] [CrossRef]
- Stefanowska, K.; Franczyk, A.; Szyling, J.; Salamon, K.; Marciniec, B.; Walkowiak, J. An effective hydrosilylation of alkenyles in supracritical CO2—A green approach to alkenyl silanes. J. Catal. 2017, 356, 206–213. [Google Scholar] [CrossRef]
- Berthon-Gelloz, G.; Schumers, J.M.; De Bo, G.; Marko, I.E. Highly β-(E)-Selective Hydrosilylation of Terminal and Internal Alkynes Catalyzed by a (IPr)Pt(diene) Complex. J. Org. Chem. 2008, 73, 4190–4197. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Simmons, E.M.; Hartwig, J.F. Iridium-Catalyzed, Diastereoselective Dehydrogenative Silylation of Terminal Alkenes with (TMSO)2MeSiH. Angew. Chem. 2013, 125, 9154–9159. [Google Scholar] [CrossRef]
- Bokka, A.; Jeon, J. Regio- and Stereoselective Dehydrogenative Silylation and Hydrosilylation of Vinylarenes Catalyzed by Ruthenium Alkylidenes. Org. Lett. 2016, 18, 5324–5327. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, D.; Zhang, Y.; Wang, B.; Qu, J. Highly β(Z)-Selective Hydrosilylation of Terminal Alkynes Catalyzed by Thiolate-Bridged Dirhodium Complexes. Org. Lett. 2018, 20, 5357–5361. [Google Scholar] [CrossRef]
- Faller, J.W.; D’Alliessi, D.G. Tunable Stereoselective Hydrosilylation of PhC≡CH Catalyzed by Cp*Rh Complexes. Organometallics 2002, 21, 1743–1746. [Google Scholar] [CrossRef]
- Li, J.; Peng, J.; Bai, Y.; Zhang, G.; Lai, G.; Li, X. Phosphines with 2-imidazolium ligands enhance the catalytic activity and selectivity of rhodium complexes for hydrosilylation reactions. J. Org. Chem. 2010, 695, 431–436. [Google Scholar] [CrossRef]
- Chen, S.J.; Wang, Y.Y.; Yao, W.M.; Zhao, X.L.; Thanh, G.V. An ionic phosphine-ligated rhodium (III) complexes as the efficient and recyclable catalyst for biphasic hydroformylation of 1-octene. J. Mol. Catal. 2013, 378, 293–298. [Google Scholar] [CrossRef]
- Consorti, C.S.; Aydos, G.L.P.; Ebeling, G.; Dupont, J. Ionophilic phosphines: Versatile ligands for ionic liquid biphasic catalysis. Org. Lett. 2008, 10, 237–240. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | R | |||||
|---|---|---|---|---|---|---|
| –C5H11 | –C6H13 | –Ph | ||||
| Conv. [%] | α/β(Z)/β(E) [%] | Conv. [%] | α/β(Z)/β(E) [%] | Conv. [%] | α/β(Z)/β(E) [%] | |
| [Rh(PPh3)3Cl] | 100 | 5/80/15 | 94 | 0/95/5 | 99 | 0/94/6 |
| 1 | 100 | 0/91/9 | 100 | 0/96/4 | 70 | 0/100/0 |
| [{Rh(µ-Cl)(cod)}2] | 100 | 0/54/46 | 95 | 12/50/38 | 0 | 0 |
| 2 | 100 | 0/88/12 | 100 | 0/89/11 | 87 | 0/86/14 |
| Catalyst | R | |||||
|---|---|---|---|---|---|---|
| –C5H11 | –C6H13 | –Ph | ||||
| Conv. [%] | α/β(Z)/β(E) [%] | Conv. [%] | α/β(Z)/β(E) [%] | Conv. [%] | α/β(Z)/β(E) [%] | |
| [Rh(PPh3)3Cl] | 87 | 0/95/5 | 79 | 0/97/3 | 88 | 0/25/75 |
| 1 | 100 | 0/93/7 | 95 | 0/95/5 | 89 | 0/17/83 |
| [{Rh(µ-Cl)(cod)}2] | 85 | 20/76/4 | 97 | 21/79/0 | 0 | 0 |
| 2 | 100 | 0/89/11 | 100 | 0/92/8 | 96 | 0/53/47 |
| No. of Catalytic Run | Conv. of Alkyne [%] | Selectivity α/β(Z)/β(E) [%] |
|---|---|---|
| 1 | 100 | 0/96/4 |
| 2 | 95 | 0/91/9 |
| 3 | 90 | 0/88/12 |
| 4 | 70 | 0/86/14 |
| 5 | 65 | 0/86/14 |
| No. of Catalytic Run | Conv. of Alkyne [%] | Selectivity α/β(Z)/β(E) [%] |
|---|---|---|
| 1 | 100 | 0/96/4 |
| 2 | 98 | 0/94/6 |
| 3 | 98 | 0/94/6 |
| 4 | 95 | 0/92/8 |
| 5 | 93 | 0/90/10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartlewicz, O.; Jankowska-Wajda, M.; Maciejewski, H. Highly Efficient and Reusable Alkyne Hydrosilylation Catalysts Based on Rhodium Complexes Ligated by Imidazolium-Substituted Phosphine. Catalysts 2020, 10, 608. https://doi.org/10.3390/catal10060608
Bartlewicz O, Jankowska-Wajda M, Maciejewski H. Highly Efficient and Reusable Alkyne Hydrosilylation Catalysts Based on Rhodium Complexes Ligated by Imidazolium-Substituted Phosphine. Catalysts. 2020; 10(6):608. https://doi.org/10.3390/catal10060608
Chicago/Turabian StyleBartlewicz, Olga, Magdalena Jankowska-Wajda, and Hieronim Maciejewski. 2020. "Highly Efficient and Reusable Alkyne Hydrosilylation Catalysts Based on Rhodium Complexes Ligated by Imidazolium-Substituted Phosphine" Catalysts 10, no. 6: 608. https://doi.org/10.3390/catal10060608
APA StyleBartlewicz, O., Jankowska-Wajda, M., & Maciejewski, H. (2020). Highly Efficient and Reusable Alkyne Hydrosilylation Catalysts Based on Rhodium Complexes Ligated by Imidazolium-Substituted Phosphine. Catalysts, 10(6), 608. https://doi.org/10.3390/catal10060608