High Temperature Water Gas Shift Reactivity of Novel Perovskite Catalysts

 , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussions
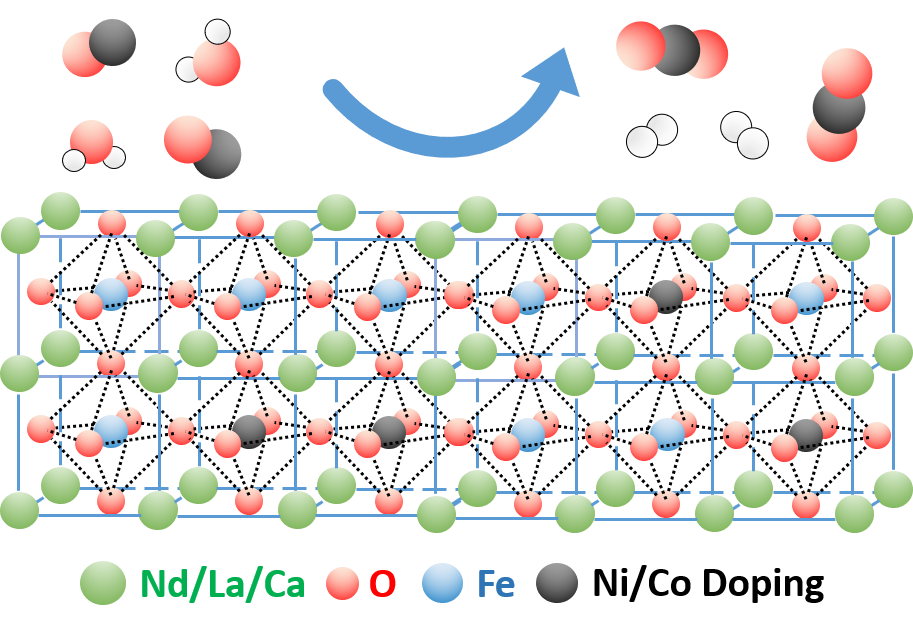
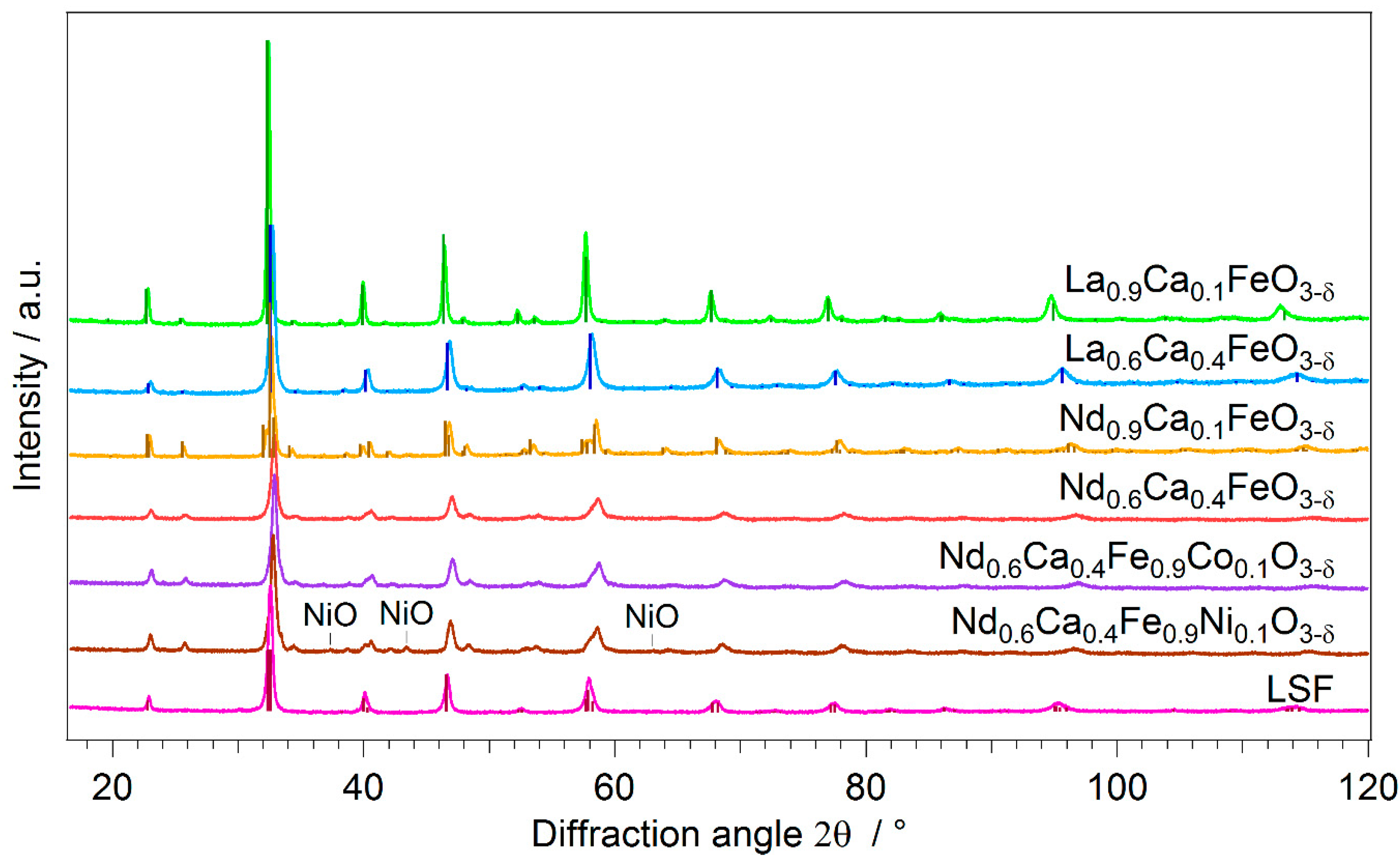
2.1. Novel Perovskite Materials
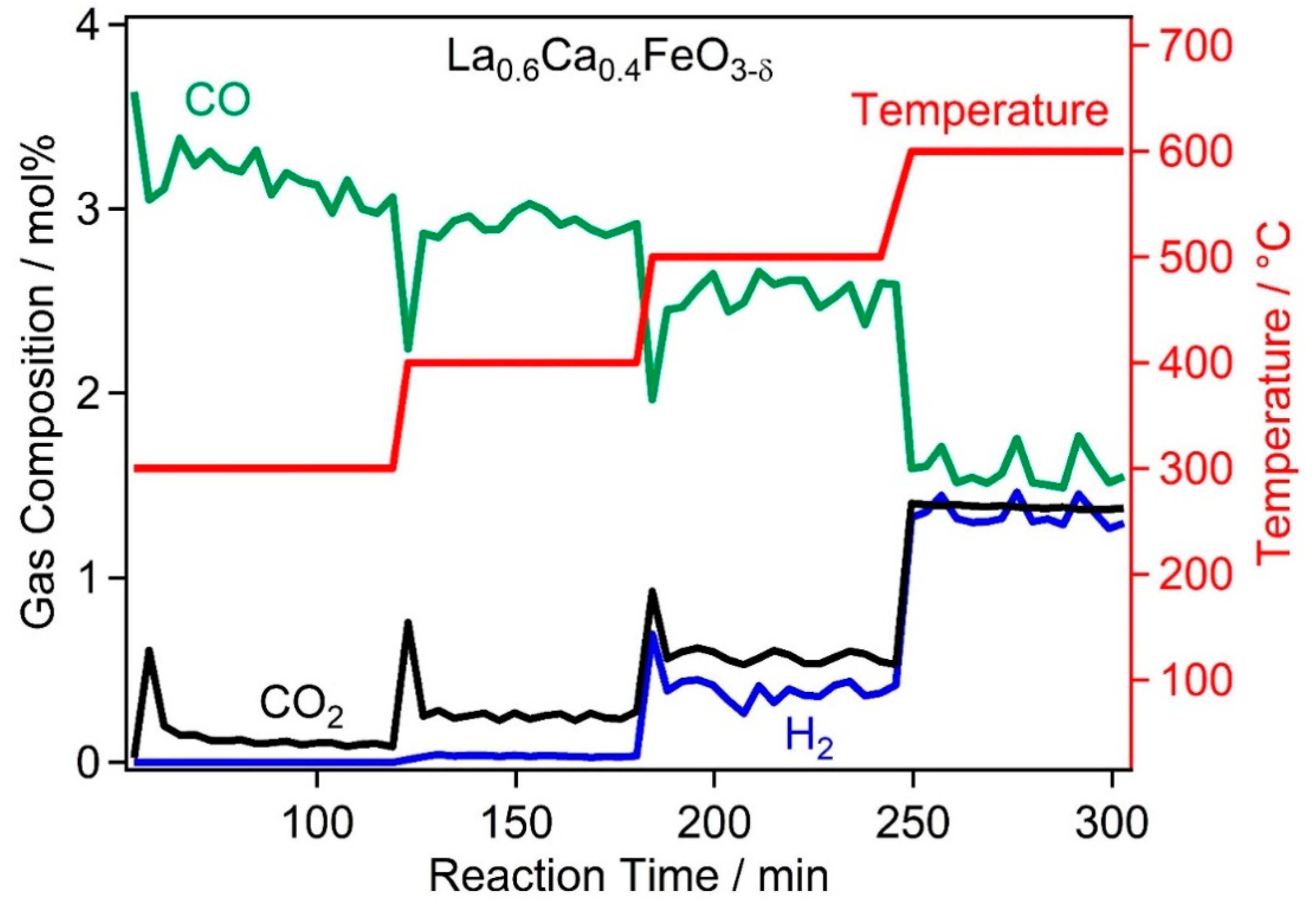
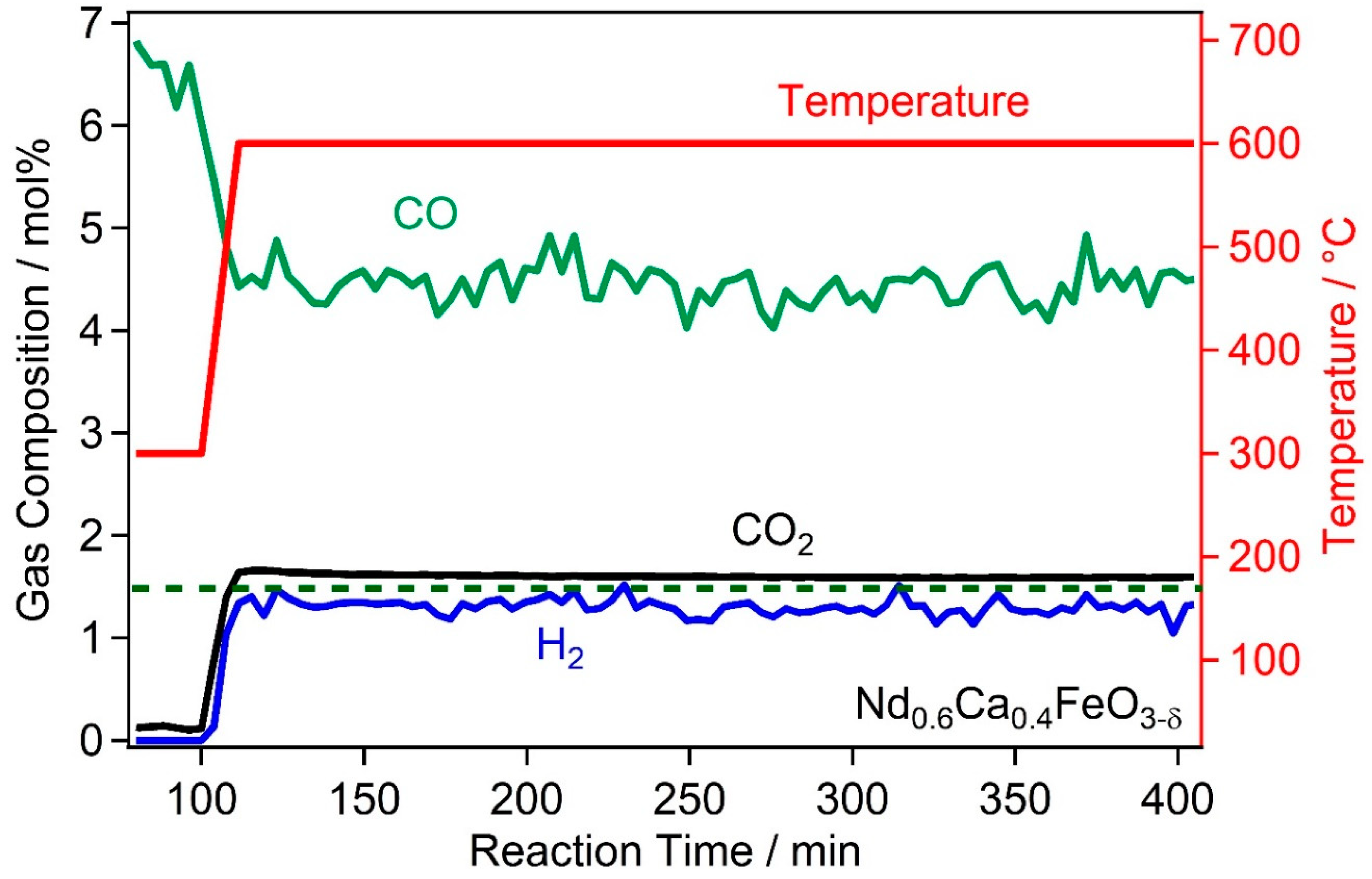
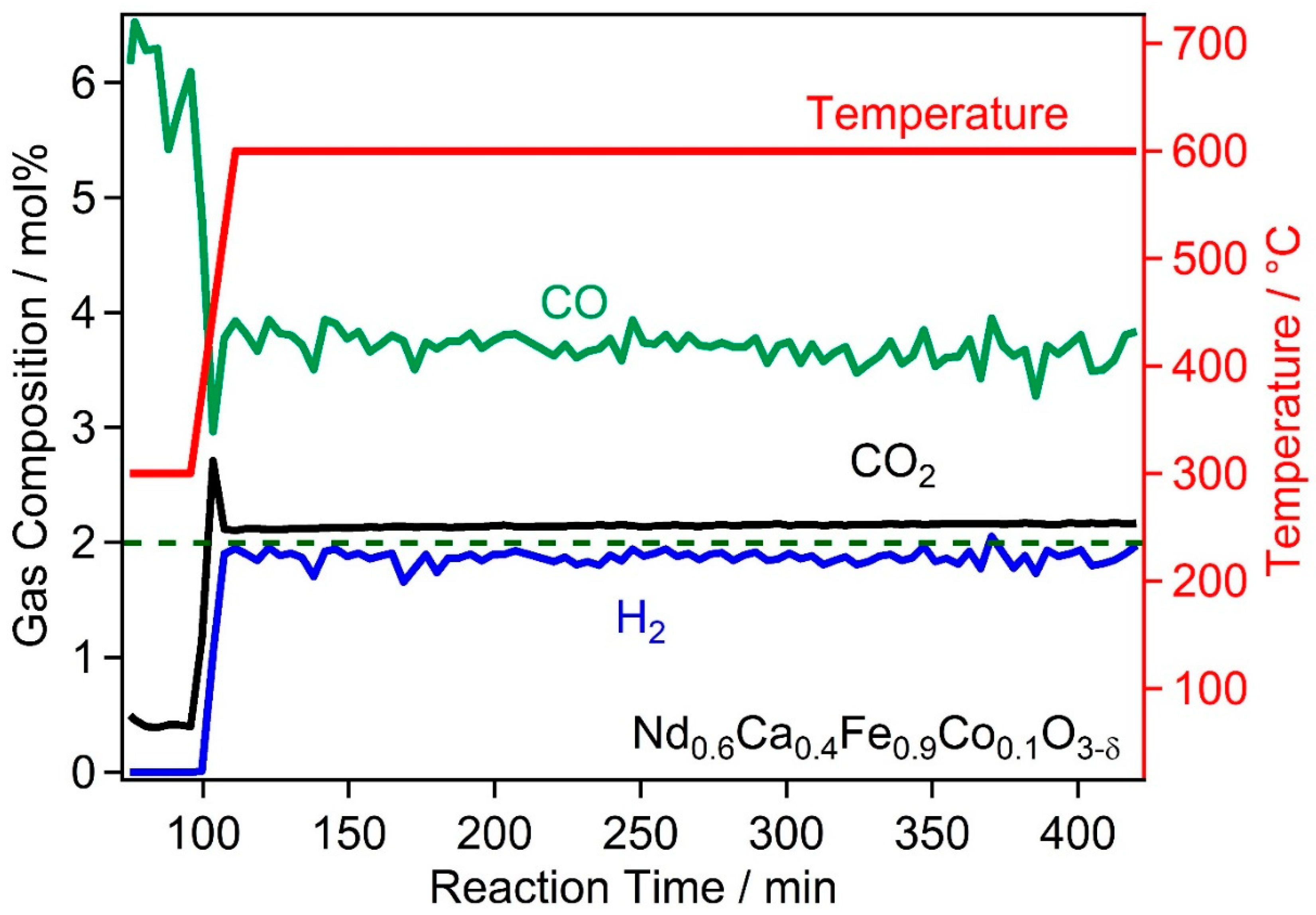
2.2. High Temperature Water Gas Shift Reactivity
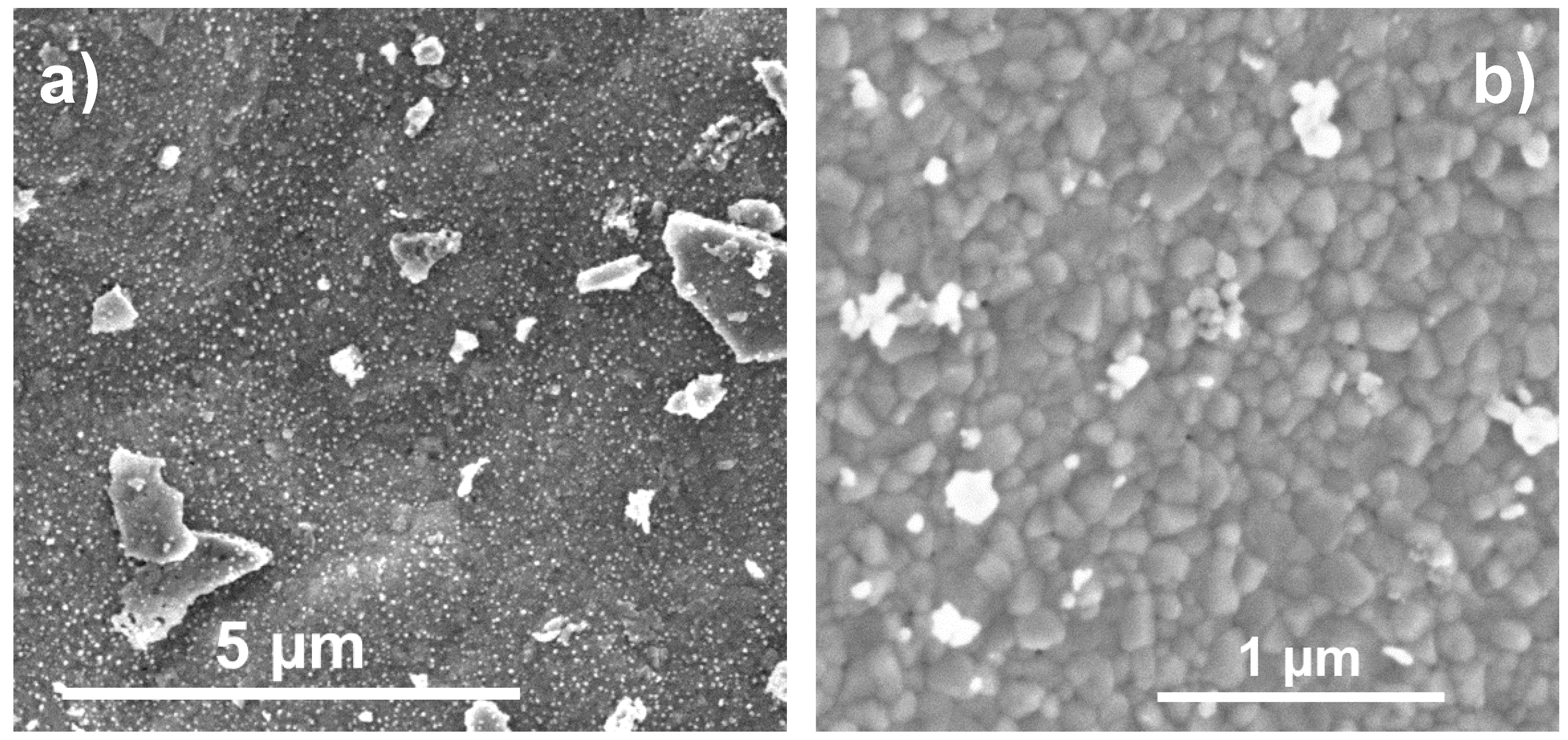
2.3. Electron Microscopy
2.4. Discussion of WGS Activity of the Catalyst Materials
2.5. Stability of Novel Perovskite Catalysts
3. Materials and Methods
3.1. Synthesis of Novel Perovskites
3.2. Materials Characterisation
3.3. Catalytic Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhu, M.H.; Wachs, I.E. Iron-Based Catalysts for the High-Temperature Water Gas Shift (HT-WGS) Reaction: A Review. ACS Catal. 2016, 6, 722–732. [Google Scholar] [CrossRef]
- LeValley, T.L.; Richard, A.R.; Fan, M.H. The progress in water gas shift and steam reforming hydrogen production technologies—A review. Int. J. Hydrog. Energy 2014, 39, 16983–17000. [Google Scholar] [CrossRef]
- Chen, C.C.; Tseng, H.H.; Lin, Y.L.; Chen, W.H. Hydrogen production and carbon dioxide enrichment from ethanol steam reforming followed by water gas shift reaction. J. Clean. Prod. 2017, 162, 1430–1441. [Google Scholar] [CrossRef]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Guczi, L.; Stefler, G.; Geszti, O.; Sajo, I.; Paszti, Z.; Tompos, A.; Schay, Z. Methane dry reforming with CO2: A study on surface carbon species. Appl. Catal. A Gen. 2010, 375, 236–246. [Google Scholar] [CrossRef]
- Bukur, D.B.; Todic, B.; Elbashir, N. Role of water-gas-shift reaction in Fischer-Tropsch synthesis on iron catalysts: A review. Catal. Today 2016, 275, 66–75. [Google Scholar] [CrossRef]
- Zhu, M.H.; Tian, P.F.; Kurtz, R.; Lunkenbein, T.; Xu, J.; Schlogl, R.; Wachs, I.E.; Han, Y.F. Strong Metal-Support Interactions between Copper and Iron Oxide during the High-Temperature Water-Gas Shift Reaction. Angew. Chem. Int. Ed. 2019, 58, 9083–9087. [Google Scholar] [CrossRef]
- Polo-Garzon, F.; Fung, V.; Nguyen, L.; Tang, Y.; Tao, F.; Cheng, Y.Q.; Daemen, L.L.; Ramirez-Cuesta, A.J.; Foo, G.S.; Zhu, M.H.; et al. Elucidation of the Reaction Mechanism for High-Temperature Water Gas Shift over an Industrial-Type Copper-Chromium-Iron Oxide Catalyst. J. Am. Chem. Soc. 2019, 141, 7990–7999. [Google Scholar] [CrossRef]
- Pastor-Perez, L.; Gu, S.; Sepulveda-Escribano, A.; Reina, T.R. Bimetallic Cu-Ni catalysts for the WGS reaction—Cooperative or uncooperative effect? Int. J. Hydrog. Energy 2019, 44, 4011–4019. [Google Scholar] [CrossRef]
- Liu, N.; Xu, M.; Yang, Y.S.; Zhang, S.M.; Zhang, J.; Wang, W.L.; Zheng, L.R.; Hong, S.; Wei, M. Auδ--Ov-Ti3+ Interfacial Site: Catalytic Active Center toward Low-Temperature Water Gas Shift Reaction. ACS Catal. 2019, 9, 2707–2717. [Google Scholar] [CrossRef]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Ovesen, C.V.; Stoltze, P.; Norskov, J.K.; Campbell, C.T. A Kinetic-Model of the Water Gas Shift Reaction. J. Catal. 1992, 134, 445–468. [Google Scholar] [CrossRef]
- Koryabkina, N.A.; Phatak, A.A.; Ruettinger, W.F.; Farrauto, R.J.; Ribeiro, F.H. Determination of kinetic parameters for the water-gas shift reaction on copper catalysts under realistic conditions for fuel cell applications. J. Catal. 2003, 217, 233–239. [Google Scholar] [CrossRef]
- Rhodes, C.; Williams, B.P.; King, F.; Hutchings, G.J. Promotion of Fe3O4/Cr2O3 high temperature water gas shift catalyst. Catal. Commun. 2002, 3, 381–384. [Google Scholar] [CrossRef]
- Lee, D.W.; Lee, M.S.; Lee, J.Y.; Kim, S.; Eom, H.J.; Moon, D.J.; Lee, K.Y. The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions. Catal. Today 2013, 210, 2–9. [Google Scholar] [CrossRef]
- Liu, B.N.; Zhao, L.; Wu, Z.J.; Zhang, J.; Zong, Q.Y.; Almegren, H.; Wei, F.; Zhang, X.H.; Zhao, Z.; Gao, J.S.; et al. Recent Advances in Industrial Sulfur Tolerant Water Gas Shift Catalysts for Syngas Hydrogen Enrichment: Application of Lean (Low) Steam/Gas Ratio. Catalysts 2019, 9. [Google Scholar] [CrossRef]
- Jeong, D.W.; Jang, W.J.; Shim, J.O.; Han, W.B.; Roh, H.S.; Jung, U.H.; Yoon, W.L. Low-temperature water-gas shift reaction over supported Cu catalysts. Renew. Energy 2014, 65, 102–107. [Google Scholar] [CrossRef]
- Damma, D.; Smirniotis, P.G. Recent advances in iron-based high-temperature water-gas shift catalysis for hydrogen production. Curr. Opin. Chem. Eng. 2018, 21, 103–110. [Google Scholar] [CrossRef]
- Damma, D.; Boningari, T.; Smirniotis, P.G. High-temperature water-gas shift over Fe/Ce/Co spinel catalysts: Study of the promotional effect of Ce and Co. Mol. Catal. 2018, 451, 20–32. [Google Scholar] [CrossRef]
- Oemar, U.; Bian, Z.; Hidajat, K.; Kawi, S. Sulfur resistant LaxCe1-xNi0.5Cu0.5O3 catalysts for an ultra-high temperature water gas shift reaction. Catal. Sci. Technol. 2016, 6, 6569–6580. [Google Scholar] [CrossRef]
- Saw, E.T.; Oemar, U.; Tan, X.R.; Du, Y.; Borgna, A.; Hidajat, K.; Kawi, S. Bimetallic Ni-Cu catalyst supported on CeO2 for high-temperature water-gas shift reaction: Methane suppression via enhanced CO adsorption. J. Catal. 2014, 314, 32–46. [Google Scholar] [CrossRef]
- Roh, H.S.; Potdar, H.S.; Jeong, D.W.; Kim, K.S.; Shim, J.O.; Jang, W.J.; Koo, K.Y.; Yoon, W.L. Synthesis of highly active nano-sized (1 wt% Pt/CeO2) catalyst for water gas shift reaction in medium temperature application. Catal. Today 2012, 185, 113–118. [Google Scholar] [CrossRef]
- Buitrago, R.; Ruiz-Martinez, J.; Silvestre-Albero, J.; Sepulveda-Escribano, A.; Rodriguez-Reinoso, F. Water gas shift reaction on carbon-supported Pt catalysts promoted by CeO2. Catal. Today 2012, 180, 19–24. [Google Scholar] [CrossRef]
- Lindenthal, L.; Rameshan, R.; Summerer, H.; Ruh, T.; Popovic, J.; Nenning, A.; Löffler, S.; Opitz, A.K.; Blaha, P.; Rameshan, C. Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts 2020, 10, 268. [Google Scholar] [CrossRef]
- Sun, Y.; Hla, S.S.; Duffy, G.J.; Cousins, A.J.; French, D.; Morpeth, L.D.; Edwards, J.H.; Roberts, D.G. Effect of Ce on the structural features and catalytic properties of La(0.9-x)CexFeO3 perovskite-like catalysts for the high temperature water-gas shift reaction. Int. J. Hydrog. Energy 2011, 36, 79–86. [Google Scholar] [CrossRef]
- Maluf, S.S.; Nascente, P.A.P.; Afonso, C.R.M.; Assaf, E.M. Study of La2-xCaxCuO4 perovskites for the low temperature water gas shift reaction. Appl. Catal. A Gen. 2012, 413, 85–93. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef]
- Shin, T.H.; Myung, J.H.; Verbraeken, M.; Kim, G.; Irvine, J.T.S. Oxygen deficient layered double perovskite as an active cathode for CO2 electrolysis using a solid oxide conductor. Faraday Discuss. 2015, 182, 227–239. [Google Scholar] [CrossRef]
- Stöger, B.; Hieckel, M.; Mittendorfer, F.; Wang, Z.M.; Fobes, D.; Peng, J.; Mao, Z.Q.; Schmid, M.; Redinger, J.; Diebold, U. High Chemical Activity of a Perovskite Surface: Reaction of CO with Sr3Ru2O7. Phys. Rev. Lett. 2014, 113. [Google Scholar] [CrossRef]
- Katz, M.B.; Zhang, S.Y.; Duan, Y.W.; Wang, H.J.; Fang, M.H.; Zhang, K.; Li, B.H.; Graham, G.W.; Pan, X.Q. Reversible precipitation/dissolution of precious-metal clusters in perovskite-based catalyst materials: Bulk versus surface re-dispersion. J. Catal. 2012, 293, 145–148. [Google Scholar] [CrossRef]
- Thalinger, R.; Opitz, A.K.; Kogler, S.; Heggen, M.; Stroppa, D.; Schmidmair, D.; Tappert, R.; Fleig, J.; Klotzer, B.; Penner, S. Water-Gas Shift and Methane Reactivity on Reducible Perovskite-Type Oxides. J. Phys. Chem. C 2015, 119, 11739–11753. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.N.; Machala, M.L.; Bluhm, H.; Chueh, W.C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, C.; Jhalani, A.; Klein, E.J.; Tummala, S.; Schmidt, L.D. The water-gas-shift reaction at short contact times. J. Catal. 2004, 223, 191–199. [Google Scholar] [CrossRef]
- Wang, X.Q.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martinez-Arias, A.; Fernandez-Garcia, M. In situ studies of the active sites for the water gas shift reaction over Cu-CeO2 catalysts: Complex interaction between metallic copper and oxygen vacancies of ceria. J. Phys. Chem. B 2006, 110, 428–434. [Google Scholar] [CrossRef]
- Gibbons, W.T.; Liu, T.H.; Gaskell, K.J.; Jackson, G.S. Characterization of palladium/copper/ceria electrospun fibers for water-gas shift catalysis. Appl. Catal. B Environ. 2014, 160, 465–479. [Google Scholar] [CrossRef]
- Keiski, R.L.; Salmi, T.; Niemisto, P.; Ainassaari, J.; Pohjola, V.J. Stationary and transient kinetics of the high temperature water-gas shift reaction. Appl. Catal. A Gen. 1996, 137, 349–370. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Wang, X.Q.; Zhang, L.Z.; Ozkan, U.S. Development of chromium-free iron-based catalysts for high-temperature water-gas shift reaction. J. Mol. Catal. A Chem. 2006, 260, 82–94. [Google Scholar] [CrossRef]
- Nagai, M.; Matsuda, K. Low-temperature water-gas shift reaction over cobalt-molybdenum carbide catalyst. J. Catal. 2006, 238, 489–496. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.S.; Miller, D.N.; Menard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Oh, T.S.; Rahani, E.K.; Neagu, D.; Irvine, J.T.S.; Shenoy, V.B.; Gorte, R.J.; Vohs, J.M. Evidence and Model for Strain-Driven Release of Metal Nanocatalysts from Perovskites during Exsolution. J. Phys. Chem. Lett. 2015, 6, 5106–5110. [Google Scholar] [CrossRef] [PubMed]
- Neagu, D.; Tsekouras, G.; Miller, D.N.; Menard, H.; Irvine, J.T.S. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 2013, 5, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Han, J.W.; Chen, Y.; Cai, Z.; Yildiz, B. Cation Size Mismatch and Charge Interactions Drive Dopant Segregation at the Surfaces of Manganite Perovskites. J. Am. Chem. Soc. 2013, 135, 7909–7925. [Google Scholar] [CrossRef] [PubMed]
- Geller, S. Crystallographic Studies of Perovskite-like Compounds V: Relative Ionic Sizes. Acta Crystallogr. 1957, 10, 248–251. [Google Scholar] [CrossRef]
- Nenning, A.; Opitz, A.K.; Rameshan, C.; Rameshan, R.; Blume, R.; Haevecker, M.; Knop-Gericke, A.; Rupprechter, G.; Kloetzer, B.; Fleigt, J. Ambient Pressure XPS Study of Mixed Conducting Perovskite-Type SOFC Cathode and Anode Materials under Well-Defined Electrochemical Polarization. J. Phys. Chem. C 2016, 120, 1461–1471. [Google Scholar] [CrossRef]
- Lorenz, H.; Zhao, Q.A.; Turner, S.; Lebedev, O.I.; van Tendeloo, G.; Klotzer, B.; Rameshan, C.; Penner, S. Preparation and structural characterization of SnO2 and GeO2 methanol steam reforming thin film model catalysts by (HR) TEM. Mater. Chem. Phys. 2010, 122, 623–629. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- ICDD. PDF-4 + 2019; Kabekkodu, S., Ed.; International Centre for Diffraction Data: Newtown Square, PA, USA, 2018. [Google Scholar]
- Yablonsky, G.S.; Pilasombat, R.; Breen, J.P.; Burch, R.; Hengrasmee, S. Cycles Across an Equilibrium: A Kinetic Investigation of the Reverse and Forward WGS Reaction over a 2% Pt/CeO2 Catalyst (Experimental Data and Qualitative Interpretation). Chem. Eng. Sci. 2010, 65, 2325–2332. [Google Scholar] [CrossRef]
- Kuhn, M.; Hashimoto, S.; Sato, K.; Yashiro, K.; Mizusaki, J. Oxygen nonstoichiometry, thermo-chemical stability and lattice expansion of La0.6Sr0.4FeO3-δ. Solid State Ion. 2011, 195, 7–15. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Rameshan, R.; Blume, R.; Haevecker, M.; Knop-Gericke, A.; Rupprechter, G.; Fleig, J.; Kloetzer, B. Enhancing Electrochemical Water-Splitting Kinetics by Polarization-Driven Formation of Near-Surface Iron (0): An In Situ XPS Study on Perovskite-Type Electrodes. Angew. Chem. Int. Ed. 2015, 54, 2628–2632. [Google Scholar] [CrossRef]
- Kuhn, M.; Fukuda, Y.; Hashimoto, S.; Sato, K.; Yashiro, K.; Mizusaki, J. Oxygen Nonstoichiometry and Thermo-Chemical Stability of Perovskite-Type La0.6Sr0.4Co1-yFeyO3-δ (y = 0, 0.2, 0.4, 0.5, 0.6, 0.8, 1) Materials. J. Electrochem. Soc. 2013, 160, F34–F42. [Google Scholar] [CrossRef]
- Dutta, G.; Waghmare, U.V.; Baidya, T.; Hegde, M.S.; Priolkar, K.R.; Sarode, P.R. Reducibility of Ce1-xZrxO2: Origin of enhanced oxygen storage capacity. Catal. Lett. 2006, 108, 165–172. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Kubicek, M.; Goetsch, T.; Blume, R.; Haevecker, M.; Knop-Gericke, A.; Rupprechter, G.; Kloetzer, B.; et al. Surface Chemistry of Perovskite-Type Electrodes During High Temperature CO2 Electrolysis Investigated by Operando Photoelectron Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 35847–35860. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Katayama, Y.; Lee, D.; Wang, X.R.; Crumlin, E.; Venkatesan, T.; Lee, H.N.; Shao-Horn, Y. CO2 Reactivity on Cobalt-Based Perovskites. J. Phys. Chem. C 2018, 122, 20391–20401. [Google Scholar] [CrossRef]
- Wu, X.Y.; Ghoniem, A.F. Hydrogen-assisted Carbon Dioxide Thermochemical Reduction on La0.9Ca0.1FeO3-δ Membranes: A Kinetics Study. Chemsuschem 2018, 11, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, G.; Ghoniem, A.F. Developing a multistep surface reaction mechanism to model the impact of H-2 and CO on the performance and defect chemistry of La0.9Ca0.1FeO3-δ mixed-conductors. J. Membr. Sci. 2017, 529, 114–132. [Google Scholar] [CrossRef]
- Shen, P.J.; Ding, W.Z.; Zhou, Y.D.; Huang, S.Q. Reaction mechanism on reduction surface of mixed conductor membrane for H2 production by coal-gas. Appl. Surf. Sci. 2010, 256, 5094–5101. [Google Scholar] [CrossRef]
- Dimitrakopoulos, G.; Ghoniem, A.F.; Yildiz, B. In situ catalyst exsolution on perovskite oxides for the production of CO and synthesis gas in ceramic membrane reactors. Sustain. Energy Fuels 2019, 3, 2347–2355. [Google Scholar] [CrossRef]
- Zhu, T.L.; Fowler, D.E.; Poeppelmeier, K.R.; Han, M.F.; Barnett, S.A. Hydrogen Oxidation Mechanisms on Perovskite Solid Oxide Fuel Cell Anodes. J. Electrochem. Soc. 2016, 163, F952–F961. [Google Scholar] [CrossRef]
- Koo, B.; Kim, K.; Kim, J.K.; Kwon, H.; Han, J.W.; Jung, W. Sr Segregation in Perovskite Oxides: Why It Happens and How It Exists. Joule 2018, 2, 1476–1499. [Google Scholar] [CrossRef]
- Rupp, G.M.; Limbeck, A.; Kubicek, M.; Penn, A.; Stoger-Pollach, M.; Friedbacher, G.; Fleig, J. Correlating surface cation composition and thin film microstructure with the electrochemical performance of lanthanum strontium cobaltite (LSC) electrodes. J. Mater. Chem. A 2014, 2, 7099–7108. [Google Scholar] [CrossRef]
- Rupp, G.M.; Opitz, A.K.; Nenning, A.; Limbeck, A.; Fleig, J. Real-time impedance monitoring of oxygen reduction during surface modification of thin film cathodes. Nat. Mater. 2017, 16, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Tsekouras, G.; Neagu, D.; Irvine, J.T.S. Step-change in high temperature steam electrolysis performance of perovskite oxide cathodes with exsolution of B-site dopants. Energy Environ. Sci. 2013, 6, 256–266. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to Form a Capacitor. U.S. Patent 3,330,697, 11 July 1967. [Google Scholar]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The HighScore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | PDF-Nr. | BET Area | Specific Activity |
|---|---|---|---|
| La0.9Ca0.1FeO3-δ | 01-082-9272 | 3.77 | 5.7 |
| La0.6Ca0.4FeO3-δ | 04-017-9772 | 2.76 | 9.1 |
| Nd0.9Ca0.1FeO3-δ | 04-014-5430 | 2.22 | 18.5 |
| Nd0.6Ca0.4FeO3-δ | - | 1.43 | 13.9 |
| Nd0.6Ca0.4Fe0.9Ni0.1O3-δ | - | 1.57 | 36.9 |
| Nd0.6Ca0.4Fe0.9Co0.1O3-δ | - | 1.24 | 28.4 |
| LSF | 04-007-6517 | 5.07 | 8.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popovic, J.; Lindenthal, L.; Rameshan, R.; Ruh, T.; Nenning, A.; Löffler, S.; Opitz, A.K.; Rameshan, C. High Temperature Water Gas Shift Reactivity of Novel Perovskite Catalysts. Catalysts 2020, 10, 582. https://doi.org/10.3390/catal10050582
Popovic J, Lindenthal L, Rameshan R, Ruh T, Nenning A, Löffler S, Opitz AK, Rameshan C. High Temperature Water Gas Shift Reactivity of Novel Perovskite Catalysts. Catalysts. 2020; 10(5):582. https://doi.org/10.3390/catal10050582
Chicago/Turabian StylePopovic, Janko, Lorenz Lindenthal, Raffael Rameshan, Thomas Ruh, Andreas Nenning, Stefan Löffler, Alexander Karl Opitz, and Christoph Rameshan. 2020. "High Temperature Water Gas Shift Reactivity of Novel Perovskite Catalysts" Catalysts 10, no. 5: 582. https://doi.org/10.3390/catal10050582
APA StylePopovic, J., Lindenthal, L., Rameshan, R., Ruh, T., Nenning, A., Löffler, S., Opitz, A. K., & Rameshan, C. (2020). High Temperature Water Gas Shift Reactivity of Novel Perovskite Catalysts. Catalysts, 10(5), 582. https://doi.org/10.3390/catal10050582