Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study

 ,
,  , and
, and
Abstract
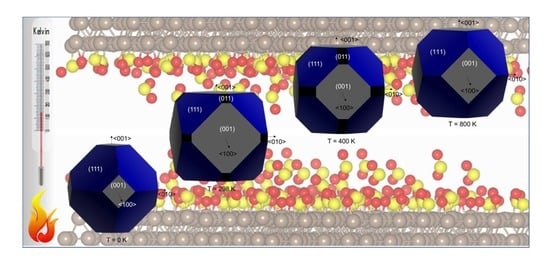
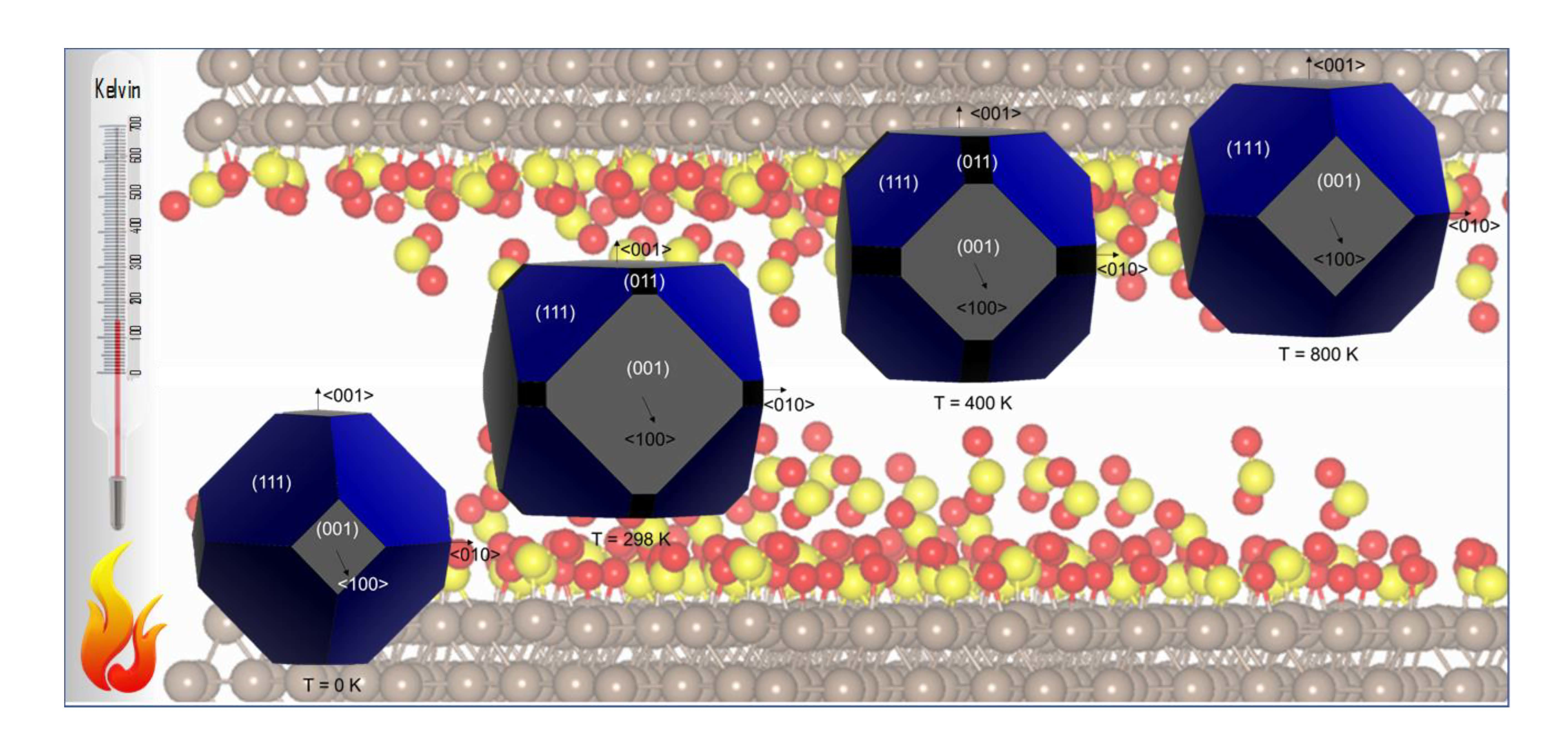
1. Introduction
2. Results and Discussion
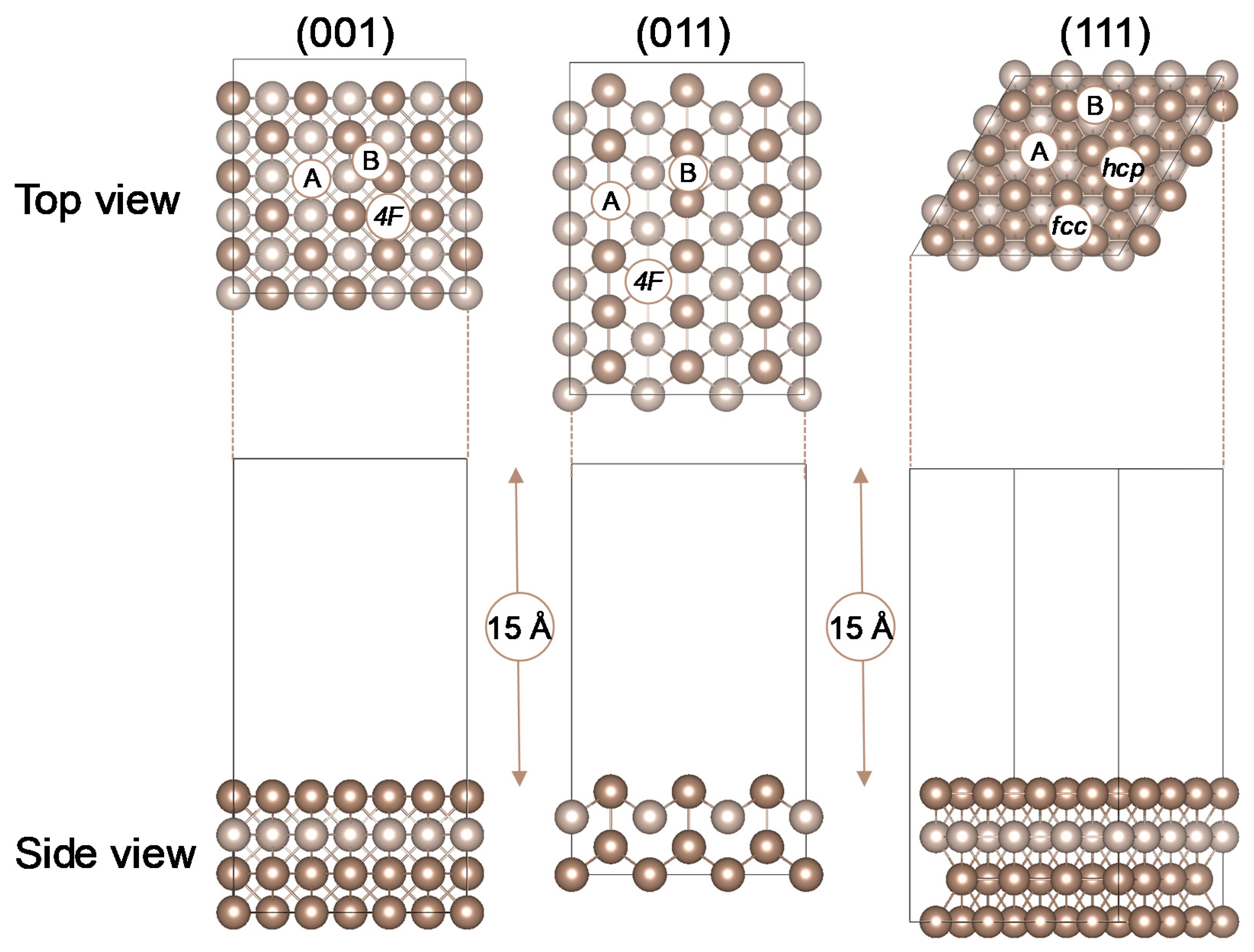
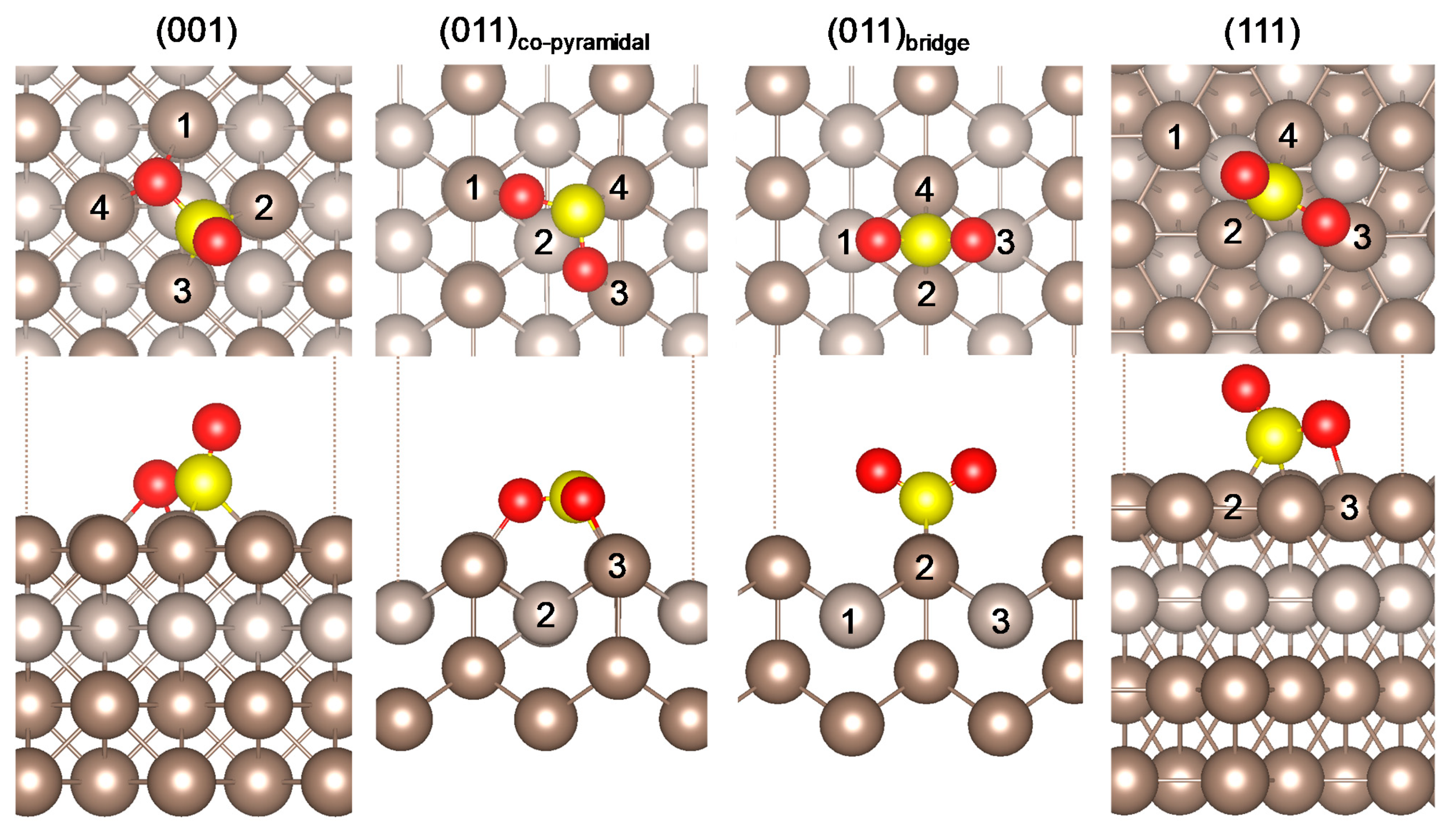
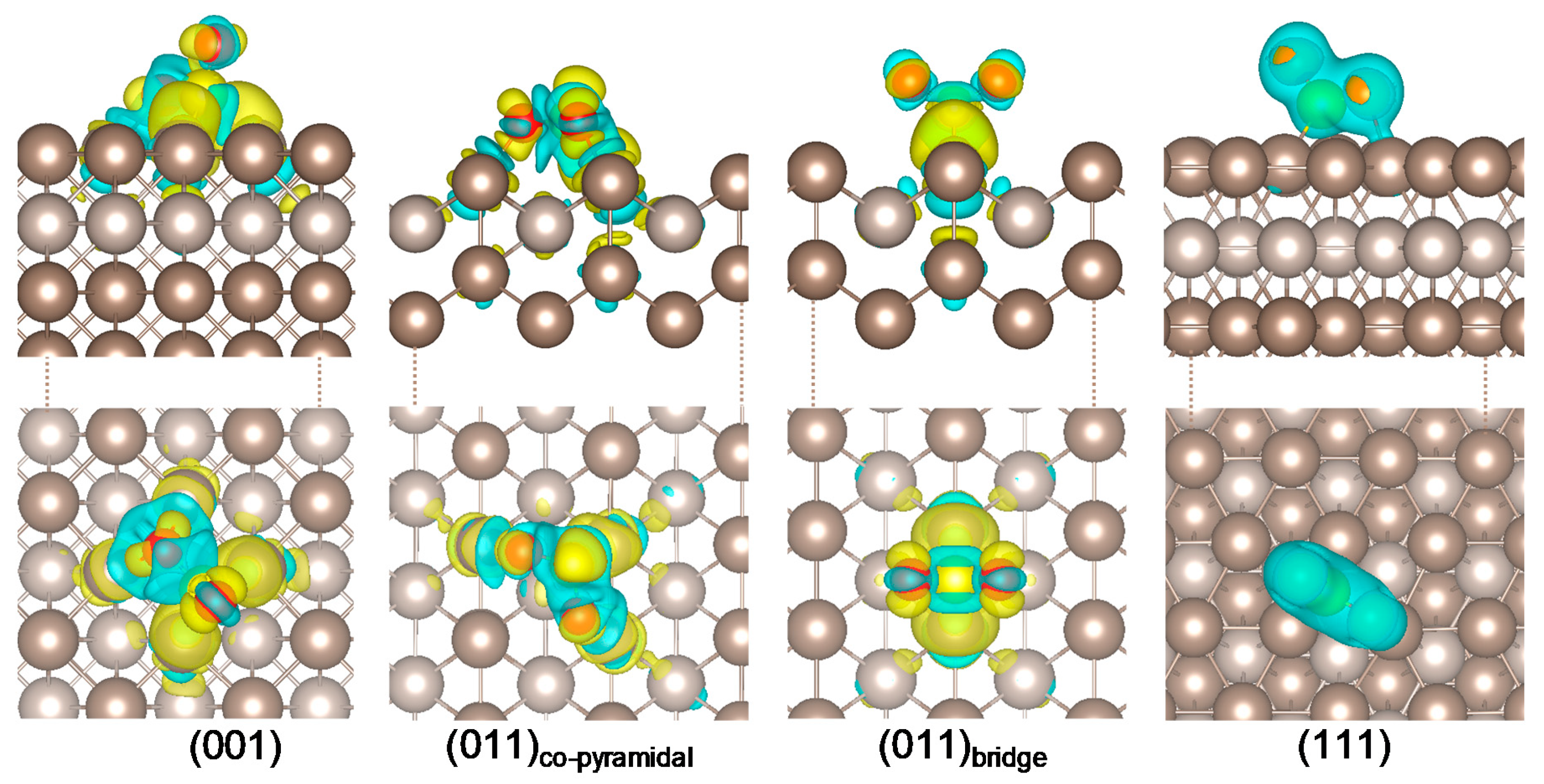
2.1. SO2 Adsorption
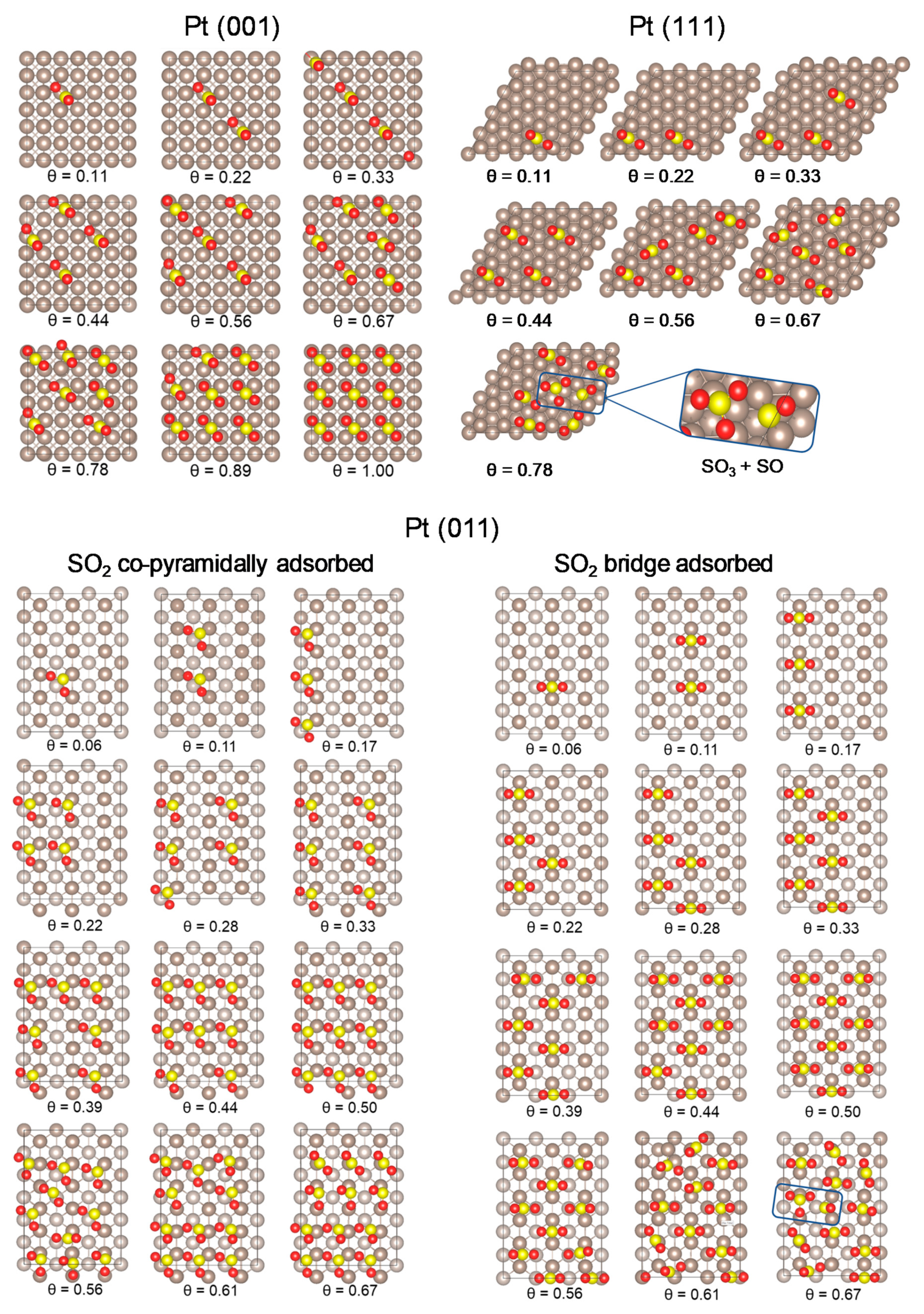
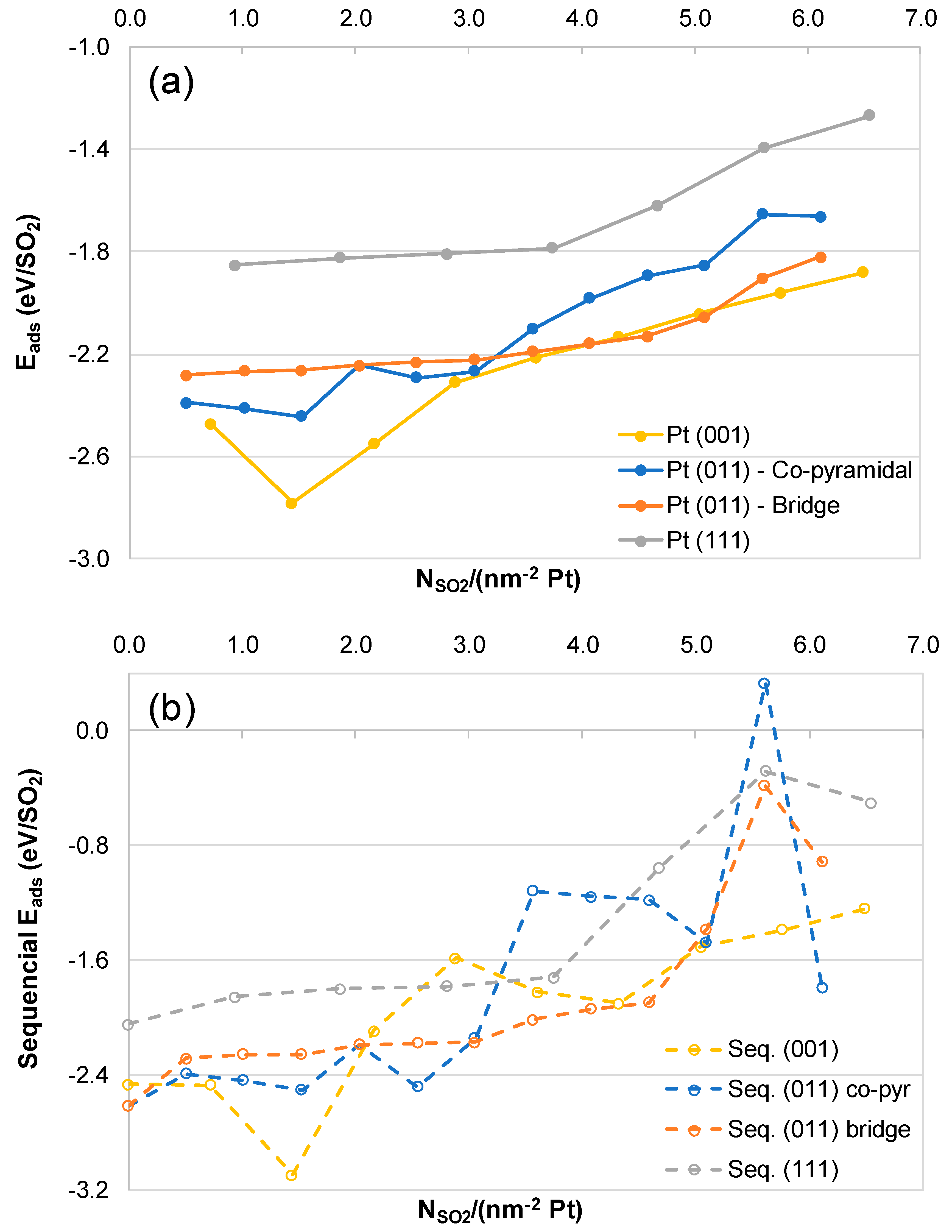
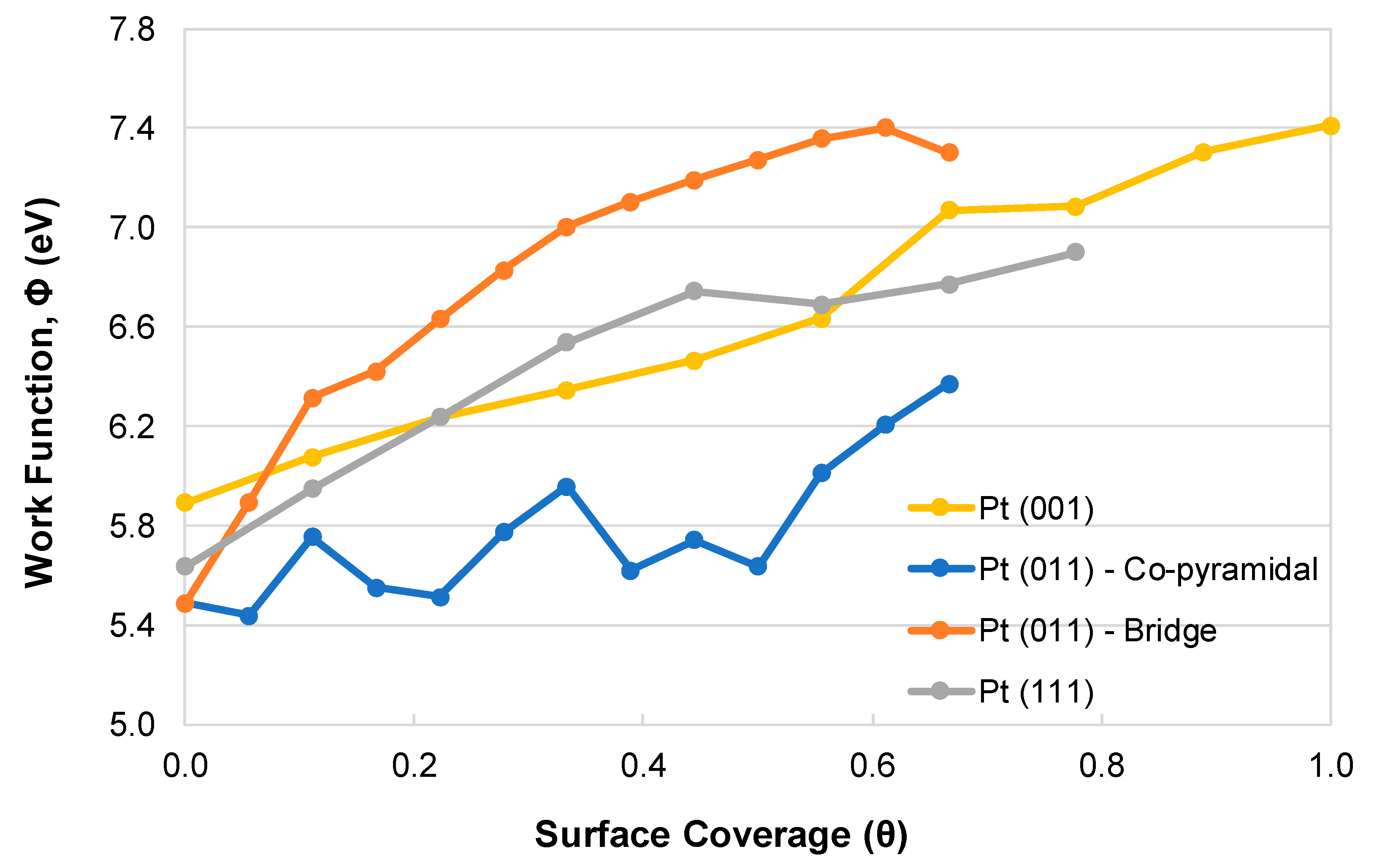
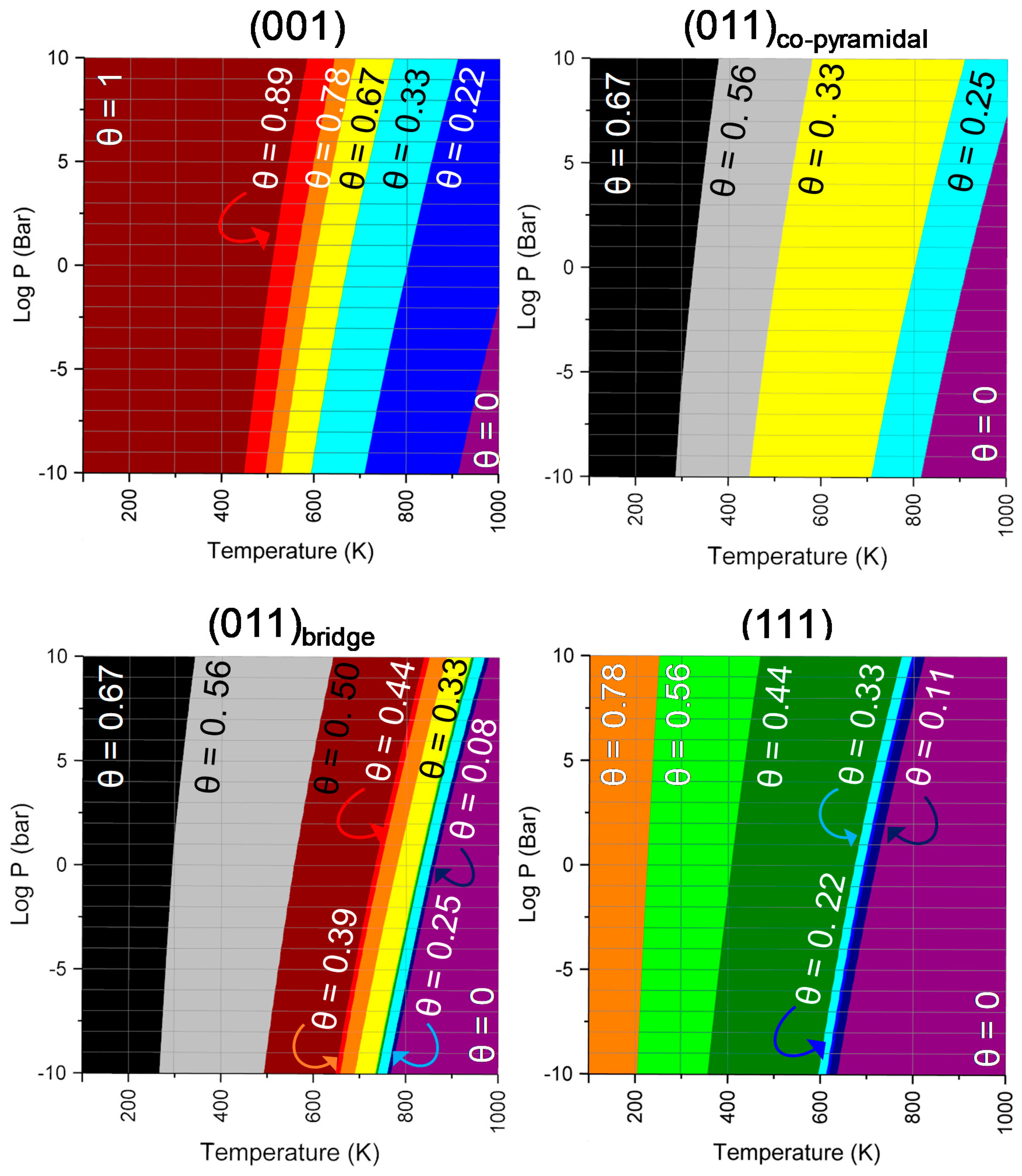
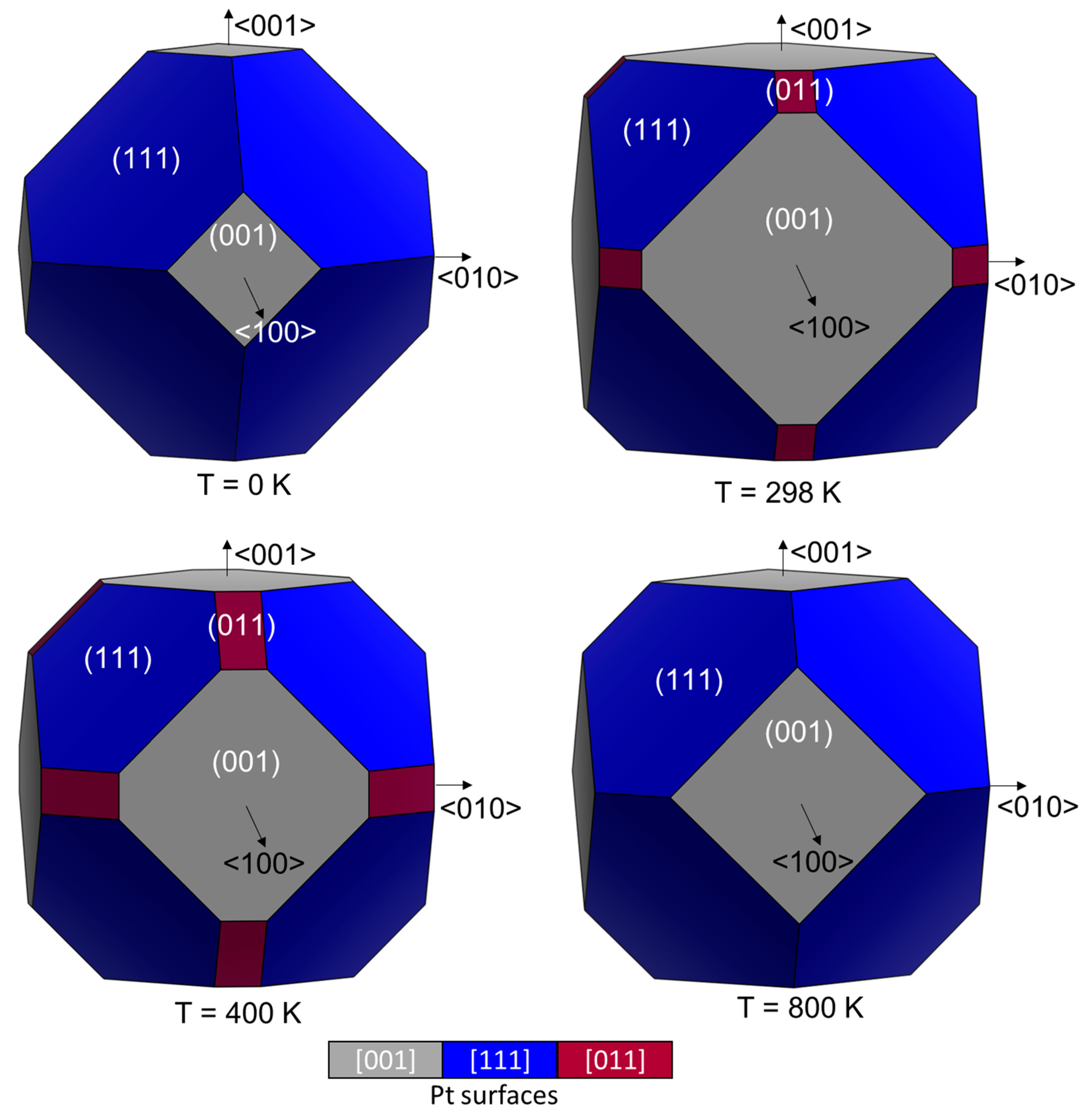
2.2. SO2 Surface Coverage
3. Computational Methods
3.1. Calculation Methods
3.2. Coverage-Dependent Surface Energies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cormos, C.-C. Hydrogen production from fossil fuels with carbon capture and storage based on chemical looping systems. Int. J. Hydrog. Energy 2011, 36, 5960–5971. [Google Scholar] [CrossRef]
- Ni, M.; Leung, D.Y.C.; Leung, M.K.H.; Sumathy, K. An overview of hydrogen production from biomass. Fuel Process. Technol. 2006, 87, 461–472. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I.; Naterer, G.F. Review of photocatalytic water-splitting methods for sustainable hydrogen production. Int. J. Energy Res. 2016, 40, 1449–1473. [Google Scholar] [CrossRef]
- Ursúa, A.; Gandía, L.M.; Sanchis, P. Hydrogen Production From Water Electrolysis: Current Status and Future Trends. Proc. IEEE 2012, 100, 410–426. [Google Scholar] [CrossRef]
- De Bruijn, F. The current status of fuel cell technology for mobile and stationary applications. Green Chem. 2005, 7, 132–150. [Google Scholar] [CrossRef]
- Baharudin, L.; James Watson, M. Hydrogen applications and research activities in its production routes through catalytic hydrocarbon conversion. Rev. Chem. Eng. 2017, 34, 43–72. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. In Materials for Sustainable Energy; Co-Published with Macmillan Publishers Ltd.: Basingstoke, UK, 2010; pp. 265–270. [Google Scholar]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef]
- Yildiz, B.; Kazimi, M. Efficiency of hydrogen production systems using alternative nuclear energy technologies. Int. J. Hydrog. Energy 2006, 31, 77–92. [Google Scholar] [CrossRef]
- Alves, H.J.; Bley Junior, C.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.S.; Coimbra-Araújo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrog. Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, P.; Chen, S.; Wang, L. Pt-based bimetallic catalysts for SO2-depolarized electrolysis reaction in the hybrid sulfur process. Int. J. Hydrog. Energy 2014, 39, 14196–14203. [Google Scholar] [CrossRef]
- O’Brien, J.A.; Hinkley, J.T.; Donne, S.W.; Lindquist, S.E. The electrochemical oxidation of aqueous sulfur dioxide: A critical review of work with respect to the hybrid sulfur cycle. Electrochim. Acta 2010, 55, 573–591. [Google Scholar] [CrossRef]
- Colón-Mercado, H.R.; Hobbs, D.T. Catalyst evaluation for a sulfur dioxide-depolarized electrolyzer. Electrochem. Commun. 2007, 9, 2649–2653. [Google Scholar] [CrossRef]
- Lu, P.W.T.; Ammon, R.L. An Investigation of Electrode Materials for the Anodic Oxidation of Sulfur Dioxide in Concentrated Sulfuric Acid. J. Electrochem. Soc. 1980, 127, 2610. [Google Scholar] [CrossRef]
- Appleby, A.J.; Pinchon, B. Electrochemical aspects of the H2SO4SO2 thermoelectrochemical cycle for hydrogen production. Int. J. Hydrog. Energy 1980, 5, 253–267. [Google Scholar] [CrossRef]
- Falch, A.; Lates, V.; Kriek, R.J. Combinatorial Plasma Sputtering of PtxPdy Thin Film Electrocatalysts for Aqueous SO2 Electro-oxidation. Electrocatalysis 2015, 6, 322–330. [Google Scholar] [CrossRef]
- Polcik, M.; Wilde, L.; Haase, J.; Brena, B.; Cocco, D.; Comelli, G.; Paolucci, G. Adsorption and temperature-dependent decomposition of SO2 on Cu(100) and Cu(111): A fast and high-resolution core-level spectroscopy study. Phys. Rev. B 1996, 53, 13720–13724. [Google Scholar] [CrossRef]
- Polčik, M.; Wilde, L.; Haase, J. SO2-induced surface reconstruction of Cu(111): An x-ray-absorption fine-structure study. Phys. Rev. B 1998, 57, 1868–1874. [Google Scholar] [CrossRef]
- Wilburn, M.S.; Epling, W.S. SO2 adsorption and desorption characteristics of Pd and Pt catalysts: Precious metal crystallite size dependence. Appl. Catal. A Gen. 2017, 534, 85–93. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ricart, J.M.; Clotet, A.; Illas, F. Density functional studies on the adsorption and decomposition of SO2 on Cu(100). J. Chem. Phys. 2001, 115, 454–465. [Google Scholar] [CrossRef]
- Yokoyama, T.; Terada, S.; Yagi, S.; Imanishi, A.; Takenaka, S.; Kitajima, Y.; Ohta, T. Surface structures and electronic properties of SO2 adsorbed on Ni(111) and Ni(100) studied by S K-edge X-ray absorption fine structure spectroscopy. Surf. Sci. 1995, 324, 25–34. [Google Scholar] [CrossRef]
- Terada, S.; Imanishi, A.; Yokoyama, T.; Takenaka, S.; Kitajima, Y.; Ohta, T. Surface structure of SO2 adsorbed on Ni(110) studied by S K-edge X-ray absorption fine structure spectroscopy. Surf. Sci. 1995, 336, 55–62. [Google Scholar] [CrossRef]
- Zebisch, P.; Weinelt, M.; Steinrück, H.-P. Sulphur dioxide adsorption on the Ni(110) surface. Surf. Sci. 1993, 295, 295–305. [Google Scholar] [CrossRef]
- Ahner, J.; Effendy, A.; Vajen, K.; Wassmuth, H.-W. Chemisorption and multilayer adsorption of SO2 on Ag(111) and Ag(110). Vacuum 1990, 41, 98–101. [Google Scholar] [CrossRef]
- Solomon, J.L.; Madix, R.J.; Wurth, W.; Stohr, J. NEXAFS and EELS study of the orientation of sulfur dioxide on silver(110). J. Phys. Chem. 1991, 95, 3687–3691. [Google Scholar] [CrossRef]
- Ku, R.C.; Wynblatt, P. SO2 adsorption on Rh(110) and Pt(110) surfaces. Appl. Surf. Sci. 1981, 8, 250–259. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Jirsak, T.; Chaturvedi, S. Reaction of S2 and SO2 with Pd/Rh(111) surfaces: Effects of metal–metal bonding on sulfur poisoning. J. Chem. Phys. 1999, 110, 3138–3147. [Google Scholar] [CrossRef]
- Terada, S.; Sakano, M.; Kitajima, Y.; Yokoyama, T.; Ohta, T. Adsorption of SO2 on Pd(100) Studied by S K-Edge XAFS. Le J. Phys. IV 1997, 7, C2-703–C2-704. [Google Scholar]
- Burke, M.L.; Madix, R.J. Hydrogen on Pd(100)-S: The effect of sulfur on precursor mediated adsorption and desorption. Surf. Sci. 1990, 237, 1–19. [Google Scholar] [CrossRef]
- Saleh, J.M. Interaction of sulphur compounds with palladium. Trans. Faraday Soc. 1970, 66, 242. [Google Scholar] [CrossRef]
- Burke, M.L.; Madix, R.J. SO2 structure and reactivity on clean and sulfur modified Pd(100). Surf. Sci. 1988, 194, 223–244. [Google Scholar] [CrossRef]
- Astegger, S.; Bechtold, E. Adsorption of sulfur dioxide and the interaction of coadsorbed oxygen and sulfur on Pt(111). Surf. Sci. 1982, 122, 491–504. [Google Scholar] [CrossRef]
- Köhler, U.; Wassmuth, H.-W. SO2 adsorption and desorption kinetics on Pt(111). Surf. Sci. 1983, 126, 448–454. [Google Scholar] [CrossRef]
- Sun, Y.-M.; Sloan, D.; Alberas, D.J.; Kovar, M.; Sun, Z.-J.; White, J.M. SO2 adsorption on Pt(111): HREELS, XPS and UPS study. Surf. Sci. 1994, 319, 34–44. [Google Scholar] [CrossRef]
- Polčik, M.; Wilde, L.; Haase, J.; Brena, B.; Comelli, G.; Paolucci, G. High-resolution XPS and NEXAFS study of SO2 adsorption on Pt(111): Two surface SO2 species. Surf. Sci. 1997, 381, L568–L572. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, Z.; Wang, C.; Gao, Y. Shape Evolution of Metal Nanoparticles in Water Vapor Environment. Nano Lett. 2016, 16, 2628–2632. [Google Scholar] [CrossRef]
- Ungerer, M.J.; Santos-Carballal, D.; Cadi-Essadek, A.; van Sittert, C.G.C.E.; de Leeuw, N.H. Interaction of H2O with the Platinum Pt (001), (011) and (111) Surfaces: A Density Functional Theory Study with Long-Range Dispersion Corrections. J. Phys. Chem. C 2019, 123, 27465–27476. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Arblaster, J.W. Crystallographic properties of platinum. Platin. Met. Rev. 1997, 41, 12–21. [Google Scholar] [CrossRef]
- Arblaster, J.W. Crystallographic properties of platinum (Errata). Platin. Met. Rev. 2006, 50, 118–119. [Google Scholar] [CrossRef]
- Tyson, W.R.; Miller, W.A. Surface free energies of solid metals: Estimation from liquid surface tension measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Ma, F.; Xu, K.-W. Calculation of the surface energy of fcc metals with modified embedded-atom method. Chin. Phys. 2004, 13, 1082–1090. [Google Scholar] [CrossRef]
- Singh-Miller, N.E.; Marzari, N. Surface energies, work functions, and surface relaxations of low-index metallic surfaces from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 80, 235407. [Google Scholar] [CrossRef]
- Kubas, G.J. Diagnostic Features of Transition-Metal-S02 Coordination Geometries. Inorg. Chem. 1979, 18, 182. [Google Scholar] [CrossRef]
- Happel, M.; Luckas, N.; Viñes, F.; Sobota, M.; Laurin, M.; Libuda, J. SO2 Adsorption on Pt(111) and Oxygen Precovered Pt(111): A Combined Infrared Reflection Absorption Spectroscopy and Density Functional Study. J. Phys. Chem. C 2011, 115, 479–491. [Google Scholar] [CrossRef]
- Lin, X.; Hass, K.C.; Schneider, W.F.; Trout, B.L. Chemistry of Sulfur Oxides on Transition Metals I: Configurations, Energetics, Orbital Analyses, and Surface Coverage Effects of SO2 on Pt(111). J. Phys. Chem. B 2002, 106, 12575–12583. [Google Scholar] [CrossRef]
- Briggs, A.G. Vibrational frequencies of sulfur dioxide. Determination and application. J. Chem. Educ. 1970, 47, 391–393. [Google Scholar] [CrossRef]
- Ryan, R.R.; Kubas, G.J.; Moody, D.C.; Eller, P.G. Structure and bonding of transition metal-sulfur dioxide complexes. In Structural Bonding; Springer: Berlin/Heidelberg, Germany, 1981; Volume 46, pp. 47–100. [Google Scholar]
- Ahner, J.; Wassmuth, H.-W. Molecular and dissociative chemisorption and condensation of SO2 on Cu(111). Surf. Sci. 1993, 287–288, 125–129. [Google Scholar] [CrossRef]
- Lanzani, G.; Laasonen, K. SO2 and its fragments on a Cu(110) surface. Surf. Sci. 2008, 602, 321–344. [Google Scholar] [CrossRef]
- Minachev, K.M.; Shuikin, N.I.; Rozhdestivenskaya, I.D. Poisoning of platinum catalysts with a low content of active metal on a carrier, under conditions of dehydrogenation catalysis. Bull. Acad. Sci. USSR Div. Chem. Sci. 1952, 1, 567–575. [Google Scholar] [CrossRef]
- Somorjai, G. On the mechanism of sulfur poisoning of platinum catalysts. J. Catal. 1972, 27, 453–456. [Google Scholar] [CrossRef][Green Version]
- Lamy-Pitara, E.; Bencharif, L.; Barbier, J. Effect of sulphur on the properties of platinum catalysts as characterized by cyclic voltammetry. Appl. Catal. 1985, 18, 117–131. [Google Scholar] [CrossRef]
- Nasri, N.S.; Jones, J.M.; Dupont, V.A.; Williams, A. A Comparative Study of Sulfur Poisoning and Regeneration of Precious-Metal Catalysts. Energy Fuels 1998, 12, 1130–1134. [Google Scholar] [CrossRef]
- Oudar, J. Sulfur Adsorption and Poisoning of Metallic Catalysts. Catal. Rev. 1980, 22, 171–195. [Google Scholar] [CrossRef]
- Wulff, G. XXV. Zur Frage der Geschwindigkeit des Wachsthums und der Auflösung der Krystallflächen. Z. Krist. Cryst. Mater. 1901, 34, 449–530. [Google Scholar] [CrossRef]
- Shi, Q.; Sun, R. Adsorption manners of hydrogen on Pt(1 0 0), (1 1 0) and (1 1 1) surfaces at high coverage. Comput. Theor. Chem. 2017, 1106, 43–49. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metalamorphous- semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Posada-Pérez, S.; Santos-Carballal, D.; Terranova, U.; Roldan, A.; Illas, F.; de Leeuw, N.H. CO2 interaction with violarite (FeNi2S4) surfaces: A dispersion-corrected DFT study. Phys. Chem. Chem. Phys. 2018, 20, 20439–20446. [Google Scholar] [CrossRef]
- Tafreshi, S.S.; Roldan, A.; Dzade, N.Y.; de Leeuw, N.H. Adsorption of hydrazine on the perfect and defective copper (111) surface: A dispersion-corrected DFT study. Surf. Sci. 2014, 622, 1–8. [Google Scholar] [CrossRef]
- Dzade, N.Y.; Roldan, A.; de Leeuw, N.H. Activation and dissociation of CO2 on the (001), (011), and (111) surfaces of mackinawite (FeS): A dispersion-corrected DFT study. J. Chem. Phys. 2015, 143, 094703. [Google Scholar] [CrossRef]
- Mishra, A.K.; Roldan, A.; de Leeuw, N.H. CuO Surfaces and CO2 Activation: A Dispersion-Corrected DFT+ U Study. J. Phys. Chem. C 2016, 120, 2198–2214. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Corbel, G.; Topić, M.; Gibaud, A.; Lang, C.I. Selective dry oxidation of the ordered Pt-11.1 at.% v alloy surface evidenced by in situ temperature-controlled X-ray diffraction. J. Alloys Compd. 2011, 509, 6532–6538. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zon integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Watson, G.W.; Kelsey, E.T.; de Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic simulation of dislocations, surfaces and interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 1–8. [Google Scholar] [CrossRef]
- Santos-Carballal, D.; Roldan, A.; Grau-Crespo, R.; de Leeuw, N.H. A DFT study of the structures, stabilities and redox behaviour of the major surfaces of magnetite Fe3O4. Phys. Chem. Chem. Phys. 2014, 16, 21082–21097. [Google Scholar] [CrossRef]
- Postica, V.; Vahl, A.; Strobel, J.; Santos-Carballal, D.; Lupan, O.; Cadi-Essadek, A.; de Leeuw, N.H.; Schütt, F.; Polonskyi, O.; Strunskus, T.; et al. Tuning doping and surface functionalization of columnar oxide films for volatile organic compound sensing: Experiments and theory. J. Mater. Chem. A 2018, 6, 23669–23682. [Google Scholar] [CrossRef]
- Chase, M. NIST-JANAF Thermochemical Tables 4th ed. J. Phys. Chem. Ref. Data 1998, 1529–1564. [Google Scholar]
- Fleming, S.; Rohl, A. GDIS: A vizualization program for molecular and periodic systems. Z. Krist. 2005, 220, 580–584. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (001) | (011)co-pyramidal | (011)bridge | (111) | Literature | ||
|---|---|---|---|---|---|---|
| Eads (eV) | −2.47 | −2.39 | −2.28 | −1.85 | −1.099 a, −1.218 b | |
| d (Å) | O-Pt1 | 2.254 | 2.114 | 3.932 | 3.475 | - |
| O-Pt2 | 3.287 | 3.403 ± 0.003 | 3.148 | 3.254 (Oup) | - | |
| O-Pt3 | 3.289 | 2.120 | 3.934 | 2.419 | 2.30 b | |
| O-Pt4 | 2.255 | - | 3.144 | 3.102 (Oplane) | - | |
| S-O | 1.451 (Oup) 1.619 (Oplane) | 1.543 (Pt1) 1.563 (Pt3) | 1.458 | 1.450 (Oup) 1.500 (Oplane) | 1.47 (Oup) b 1.54 (Oplane) b | |
| S-Pt3 | 2.234 | 3.422 | 3.697 | 2.918 | 2.31 b | |
| S-Pt4 | 3.110 | 2.243 | 2.263 | 2.274 | - | |
| ∠ (°) | O-S-O | 110.05 | 111.01 | 118.88 | 115.27 | 155.5 b |
| S-O-Pt (O-S-Pt *) | 105.62 (Pt1/4) 124.98 ± 0.07 (Pt2/3) | 129.16 (Pt1) 112.16 (Pt3) | 113.57 ± 0.05 * | 93.22 (SOplane-Pt3) 120.21 * (Oplane-S-Pt2/4) | - | |
| (cm−1) | 1172.2 | 832.9 | 1211.8 | 1177.7 | 1153 c | |
| (cm−1) | 622.4 | 745.2 | 1033.1 | 875.9 | 1362 c | |
| (cm−1) | 452.1 | 414.3 | 495.8 | 502.6 | 508 c | |
| (e) | −0.349 | −0.432 | −0.198 | −0.240 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungerer, M.J.; Santos-Carballal, D.; Cadi-Essadek, A.; van Sittert, C.G.C.E.; de Leeuw, N.H. Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study. Catalysts 2020, 10, 558. https://doi.org/10.3390/catal10050558
Ungerer MJ, Santos-Carballal D, Cadi-Essadek A, van Sittert CGCE, de Leeuw NH. Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study. Catalysts. 2020; 10(5):558. https://doi.org/10.3390/catal10050558
Chicago/Turabian StyleUngerer, Marietjie J., David Santos-Carballal, Abdelaziz Cadi-Essadek, Cornelia G. C. E. van Sittert, and Nora H. de Leeuw. 2020. "Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study" Catalysts 10, no. 5: 558. https://doi.org/10.3390/catal10050558
APA StyleUngerer, M. J., Santos-Carballal, D., Cadi-Essadek, A., van Sittert, C. G. C. E., & de Leeuw, N. H. (2020). Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study. Catalysts, 10(5), 558. https://doi.org/10.3390/catal10050558