Current State of the Art of the Solid Rh-Based Catalyzed Hydroformylation of Short-Chain Olefins



Abstract
1. Introduction
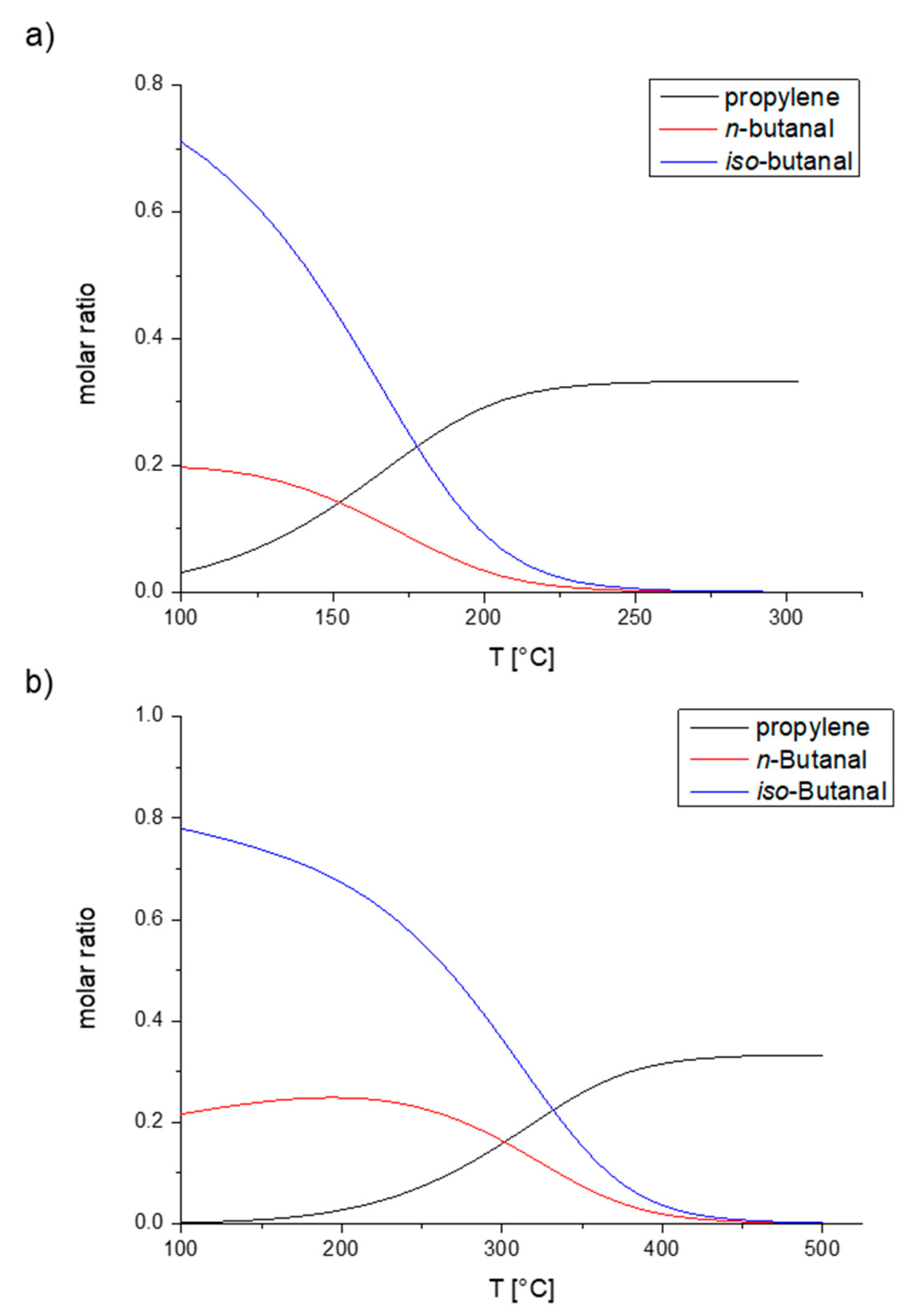
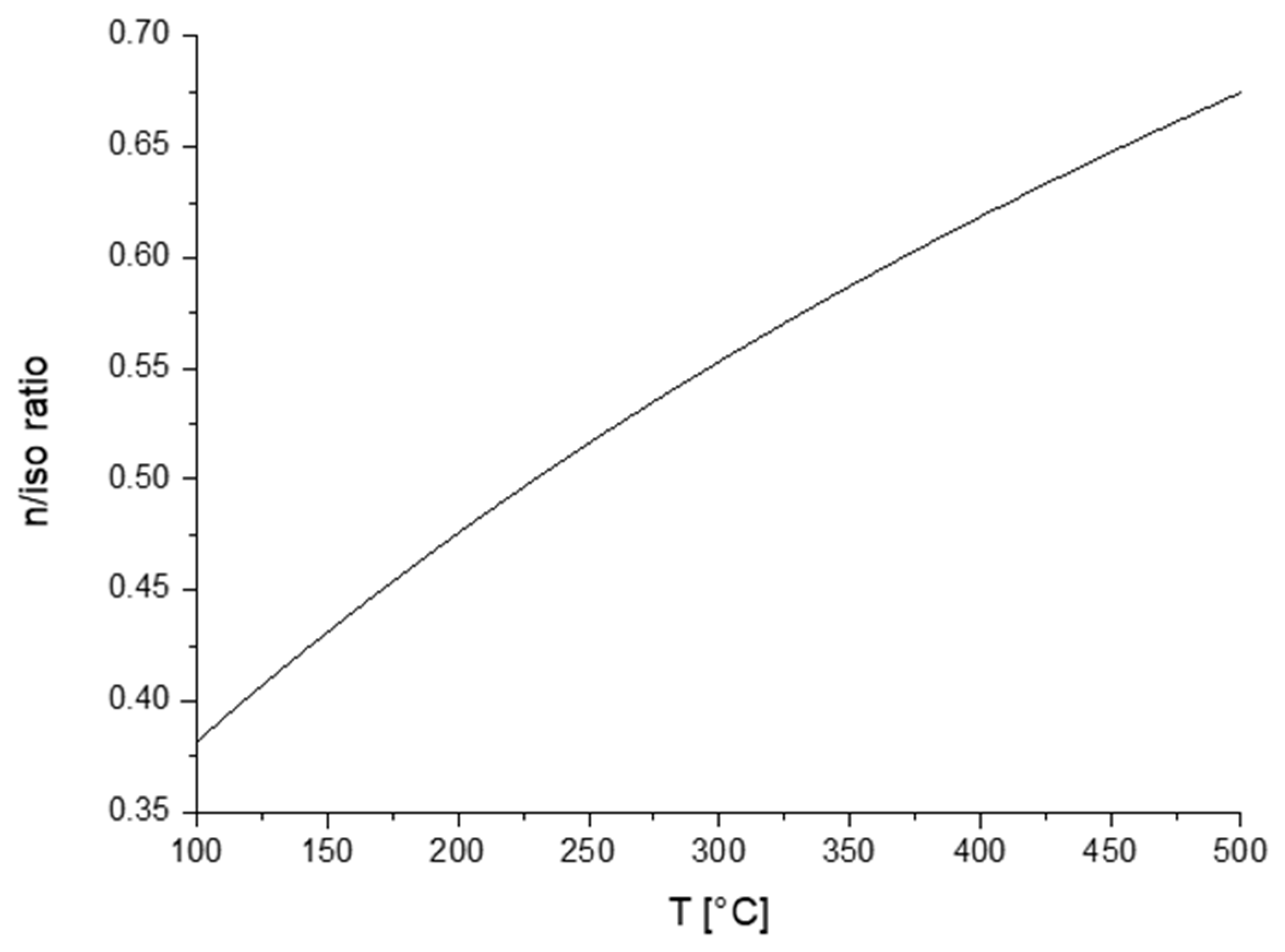
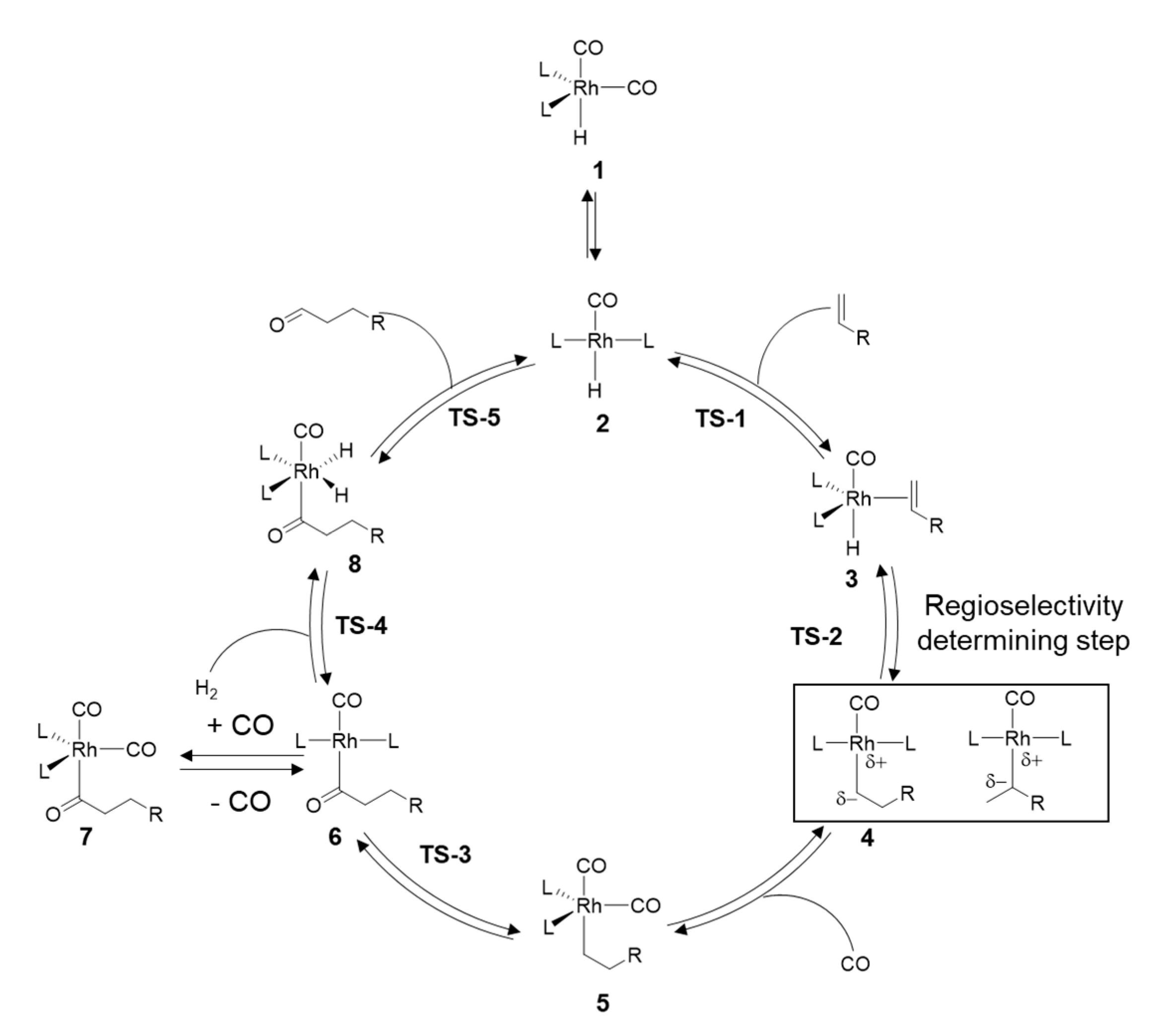
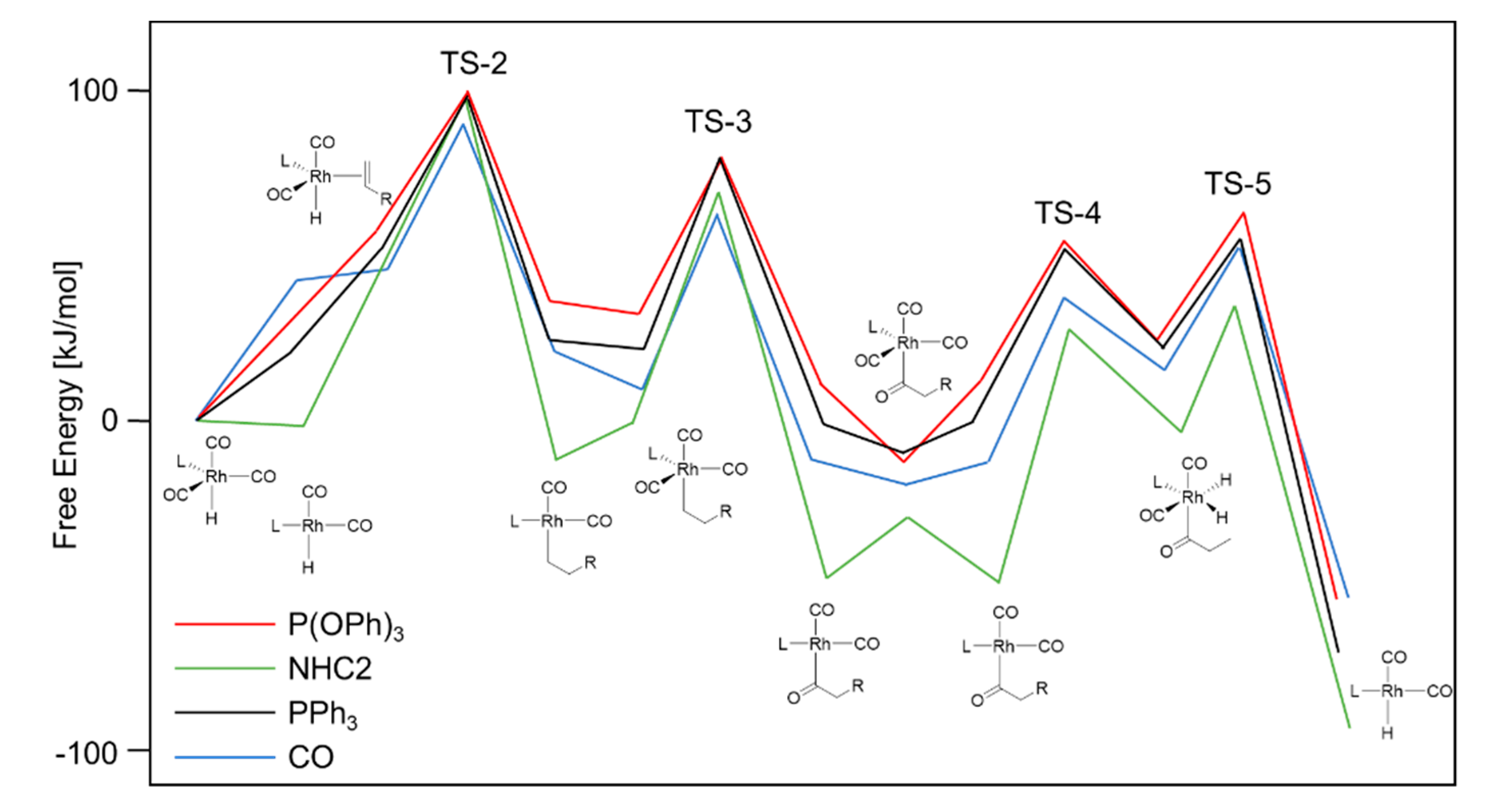
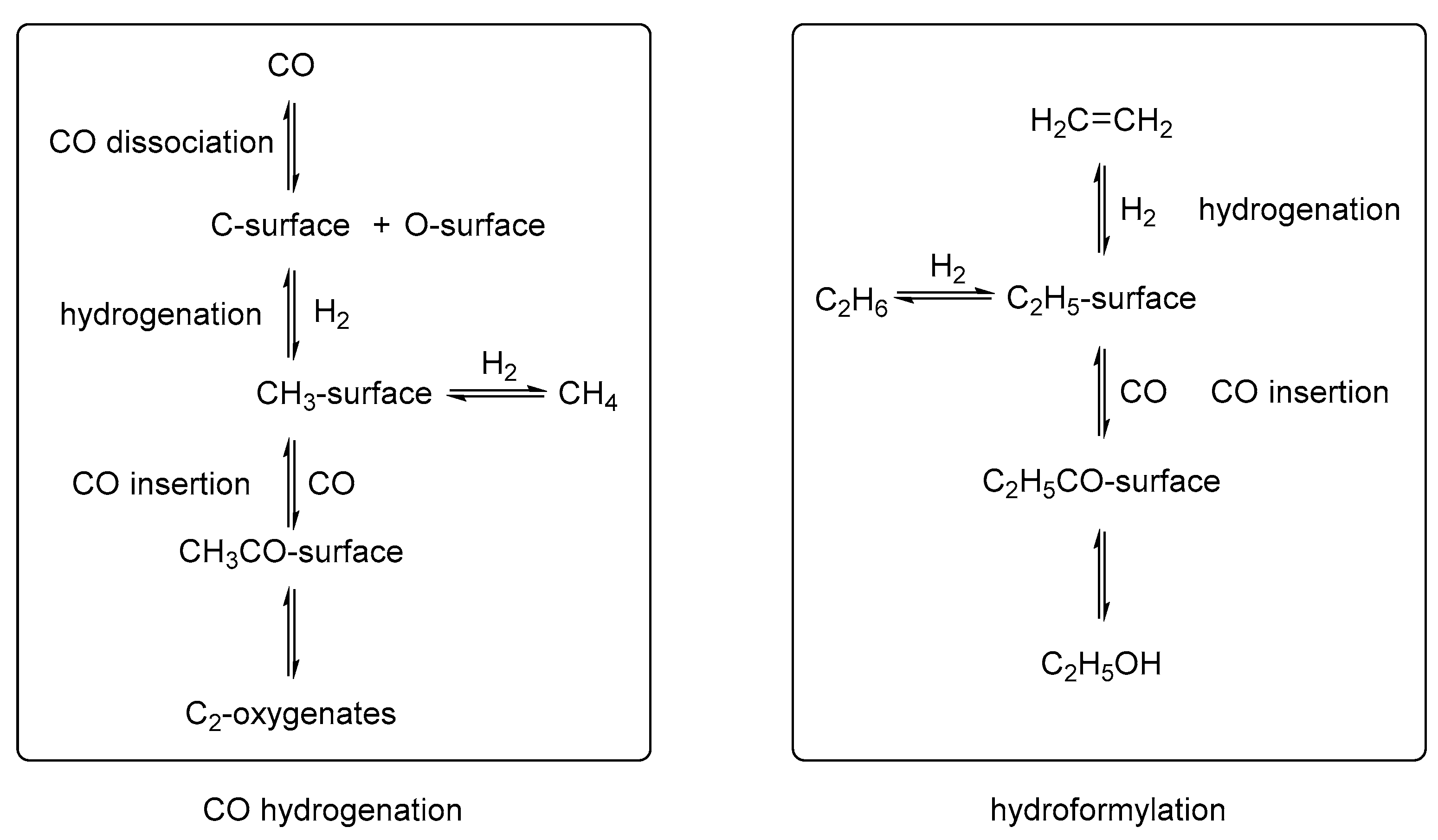
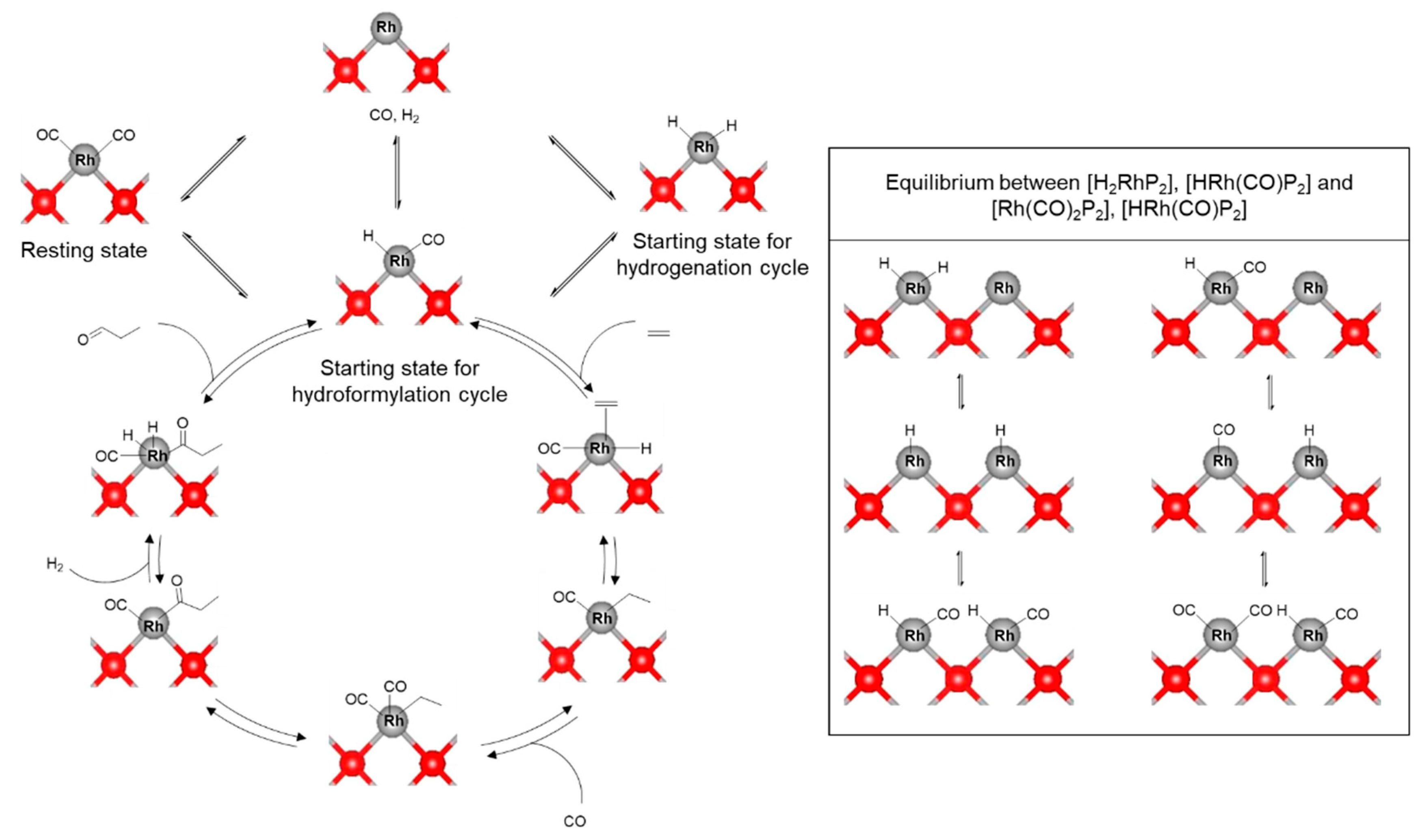
2. Mechanistic Considerations
- The catalytically active species is an in-situ formed unsaturated 16e- Rh hydride complex
- At least one CO ligand must be present within the catalytically active Rh complex
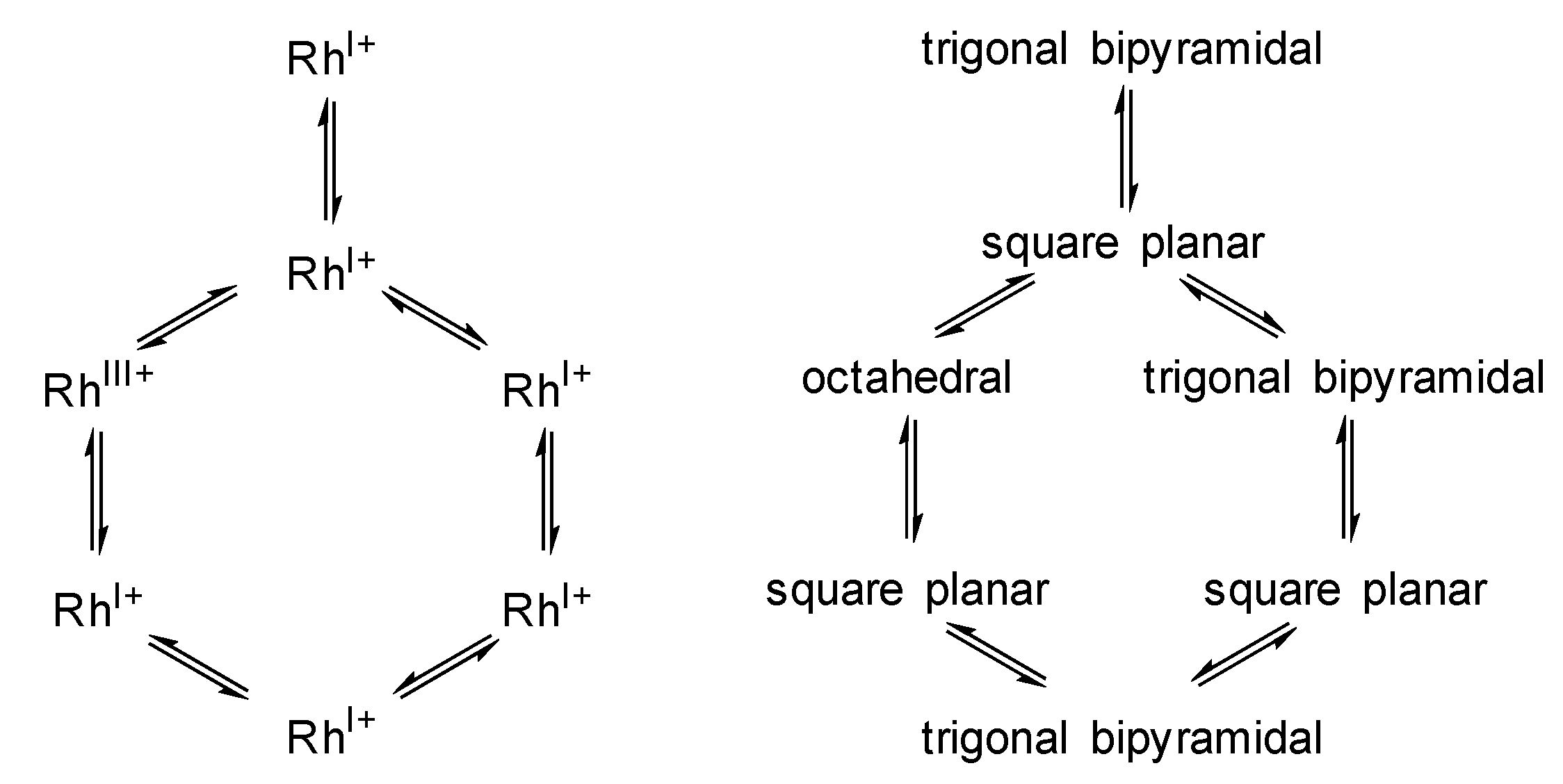
- The coordination number around the Rh center alters between 4 and 6 within the catalytic cycle
- A change in oxidation state of the Rh center from +I to +III (and back to +I) occurs during the catalytic cycle
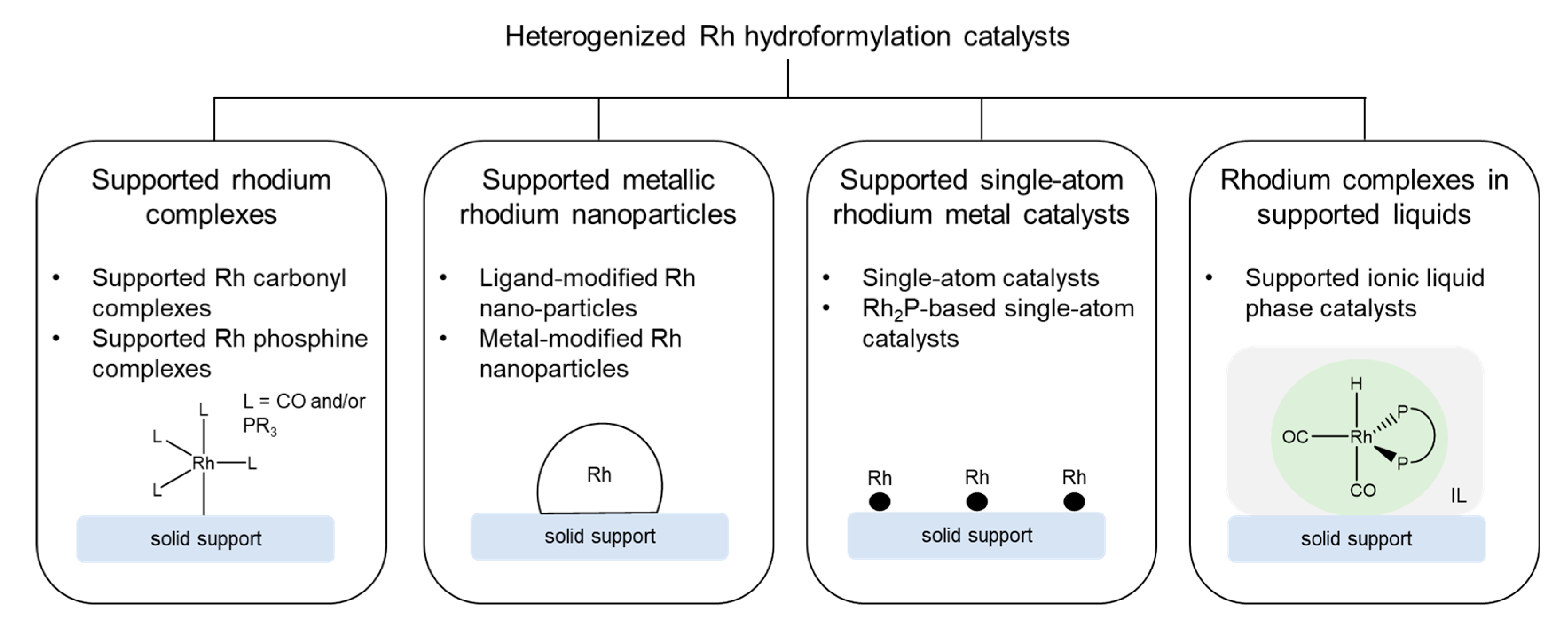
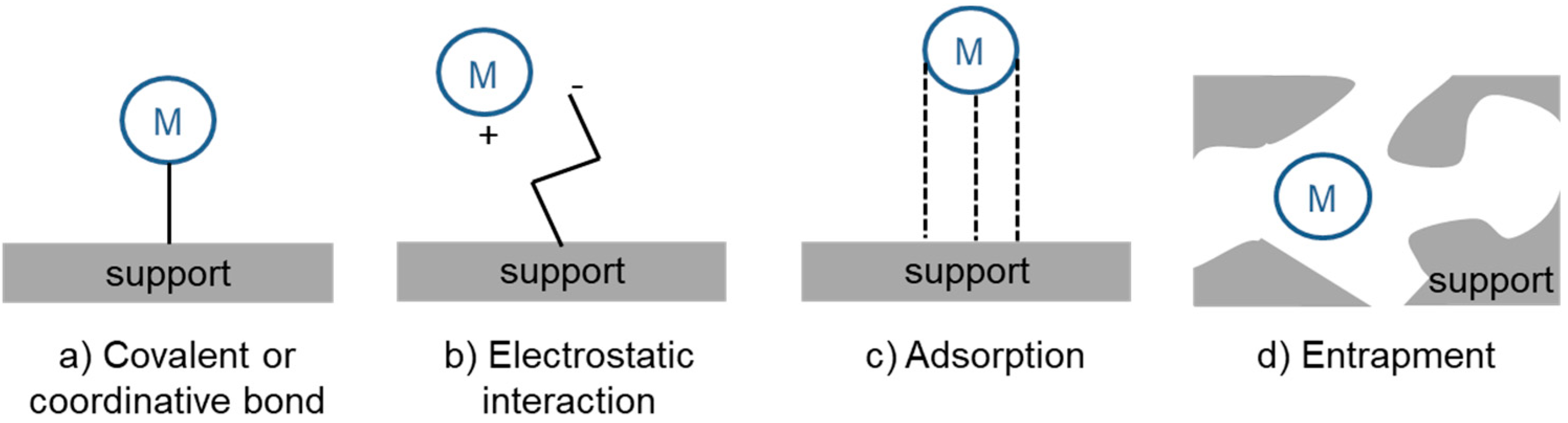
3. Heterogenization of Rh Hydroformylation Catalysts
3.1. Supported Rhodium Complexes
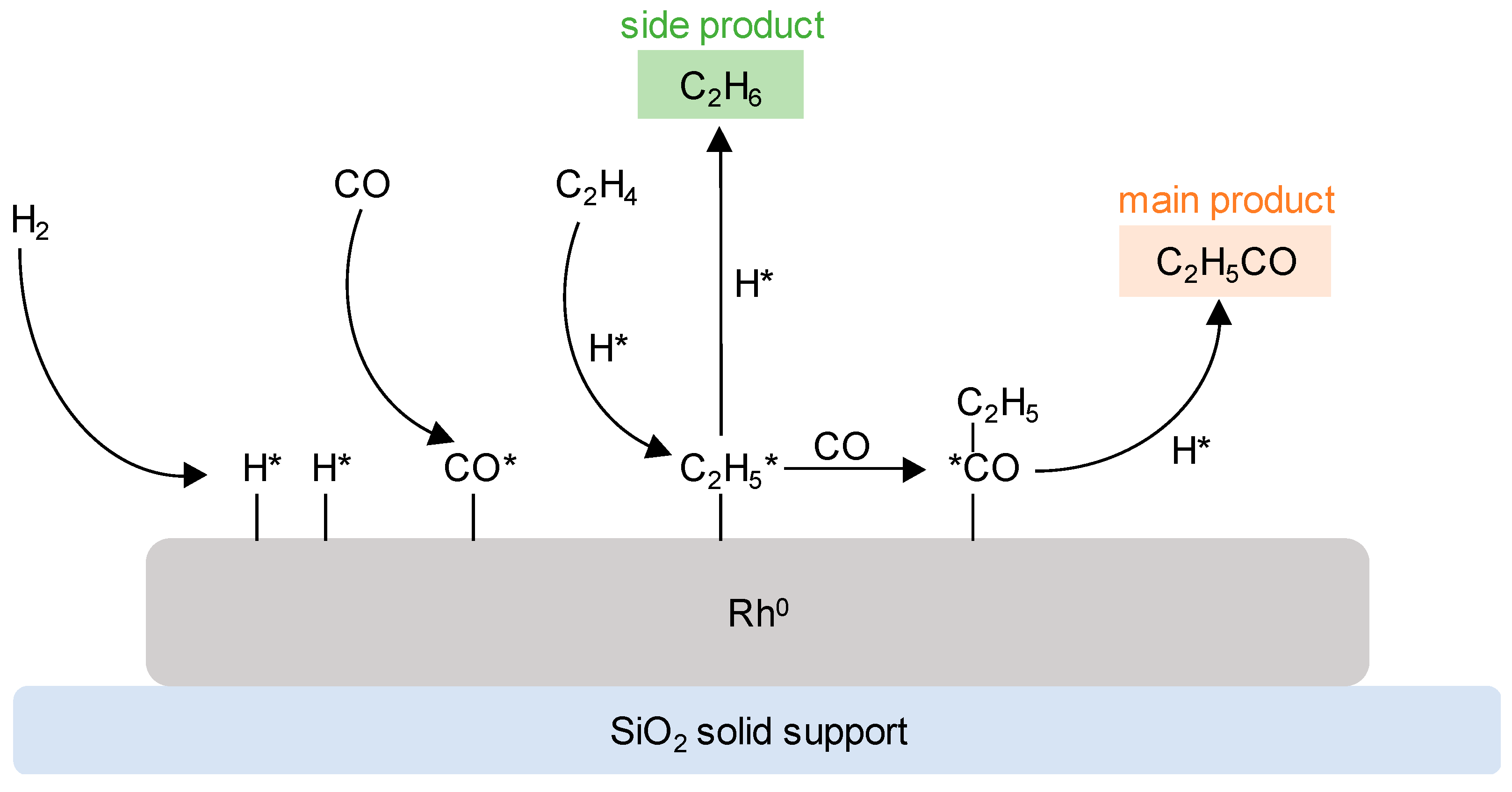
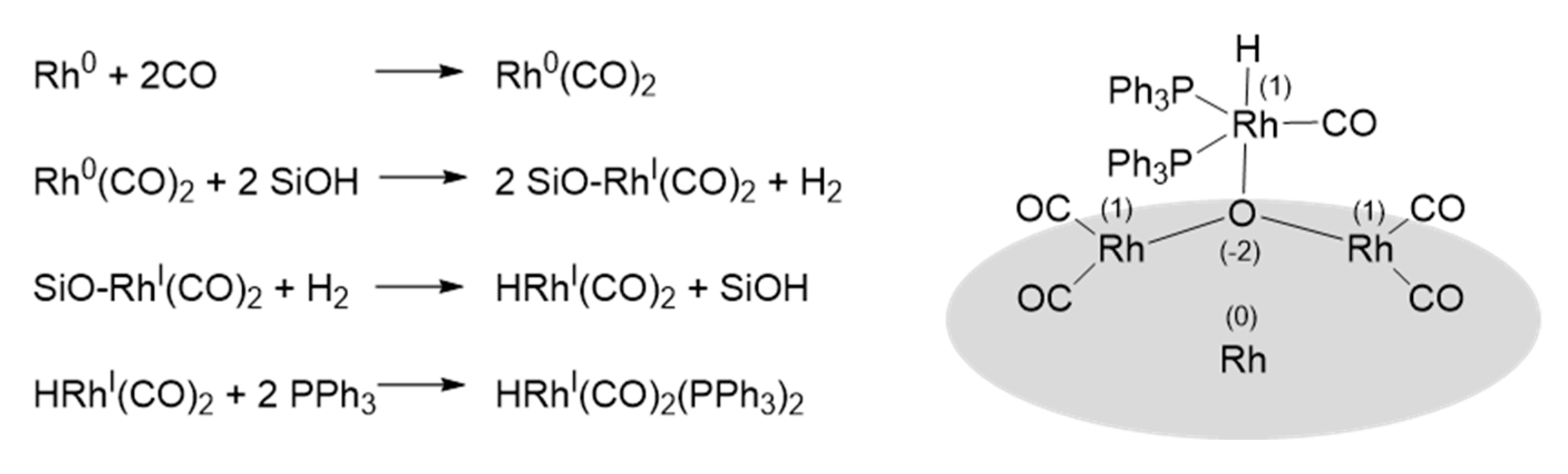
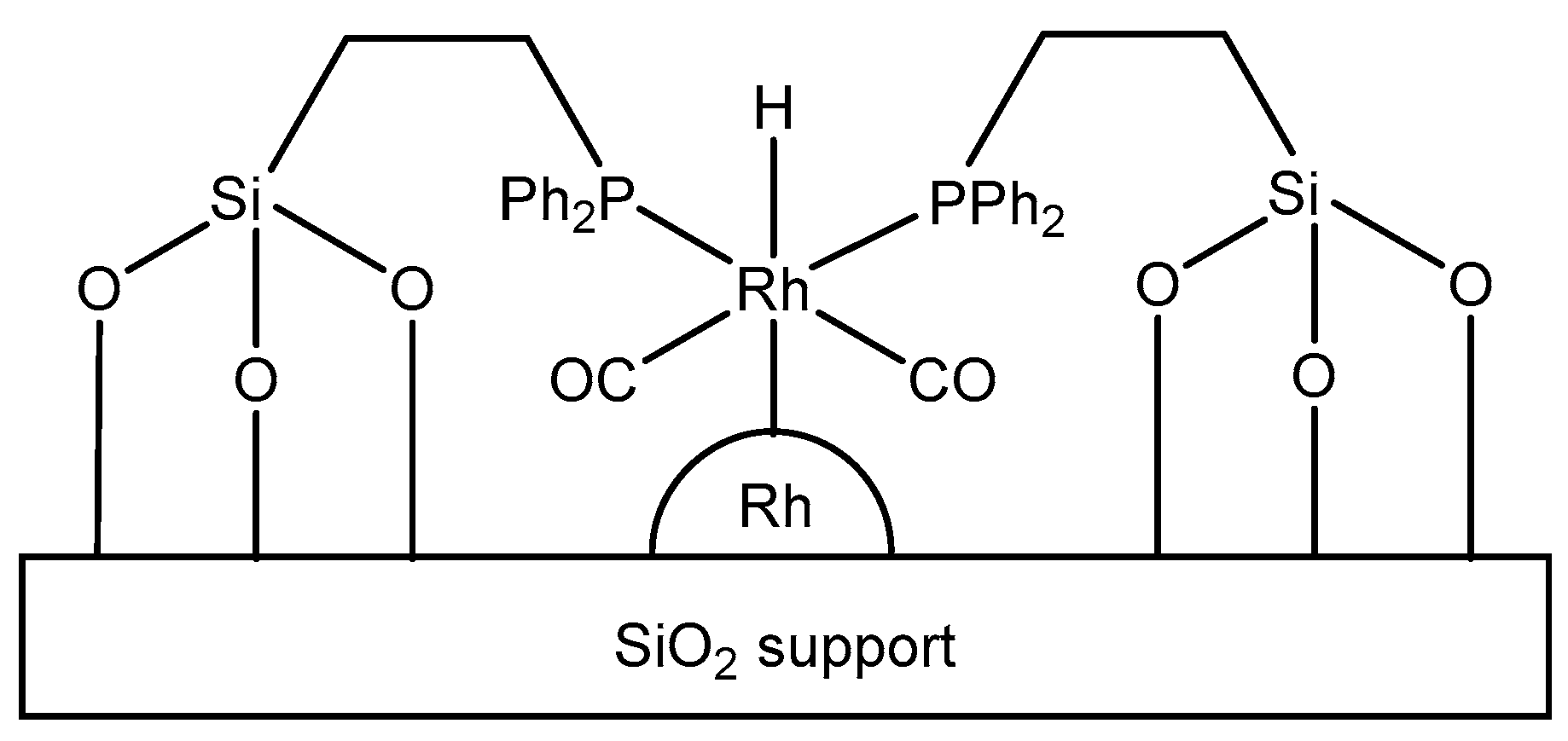
3.1.1. Supported Rhodium Carbonyl Complexes
3.1.2. Other Supported Rhodium Complexes
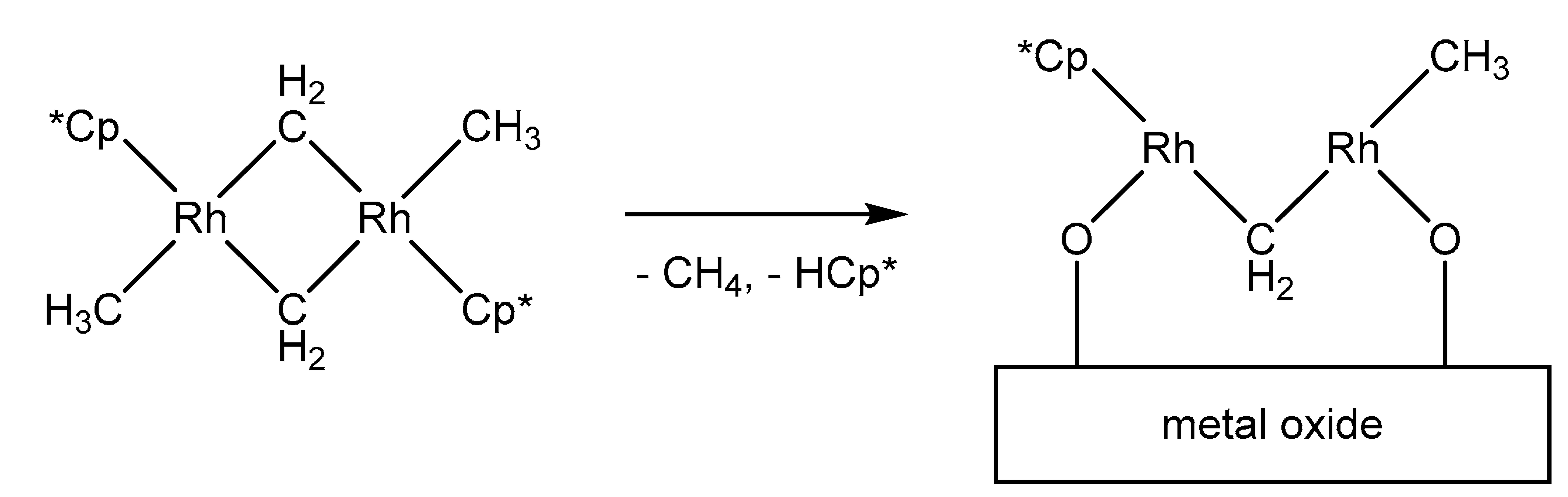
3.2. Supported Metallic Rhodium Nanoparticles
3.2.1. Supported Ligand-Modified Rhodium Nanoparticles
3.2.2. Supported Metal-Modified Rhodium Nanoparticles
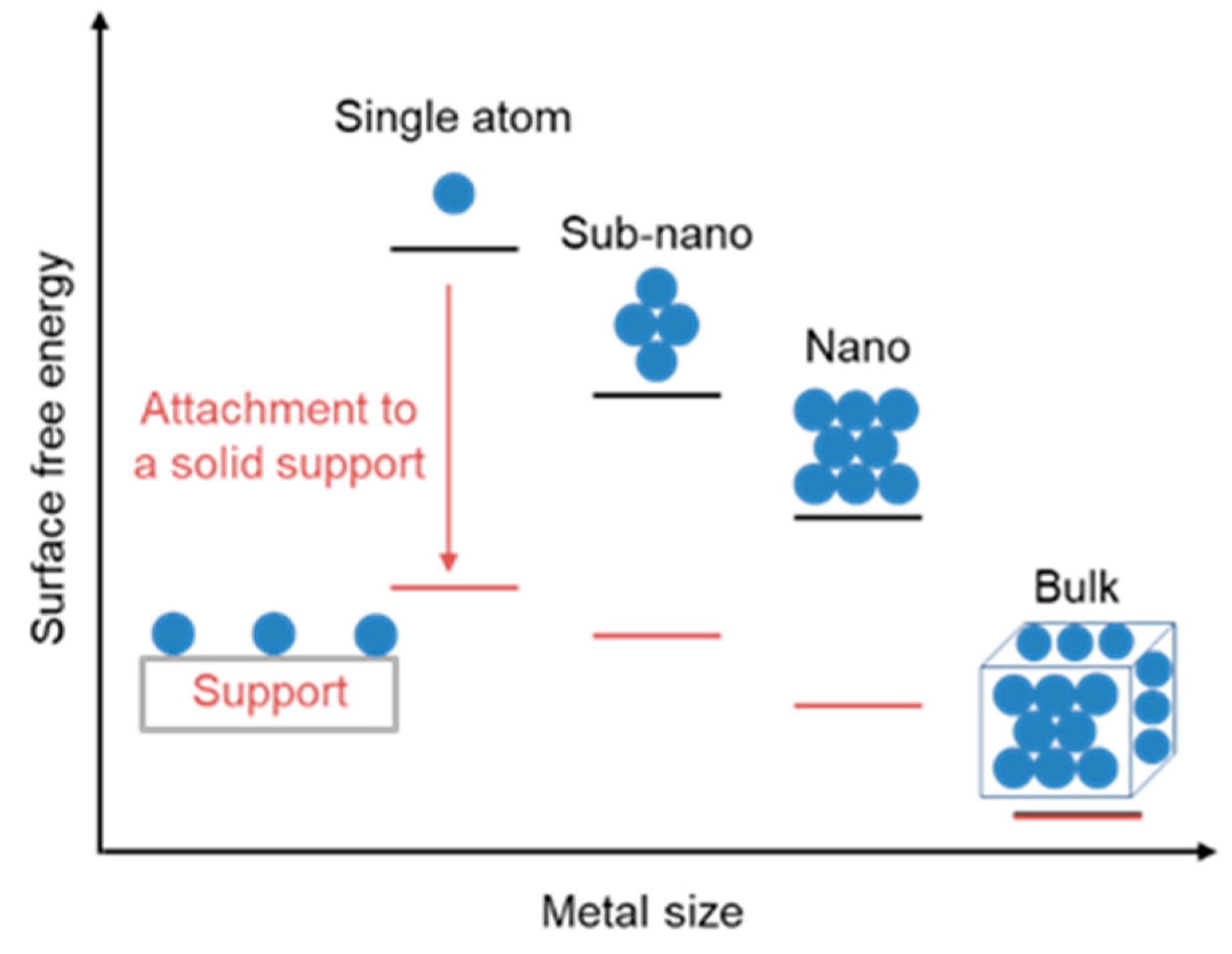
3.3. Supported Single-Atom Rhodium Metal Catalysts
3.3.1. Supported Single-Atom Catalysts
3.3.2. Supported Rh2P-Based Catalysts
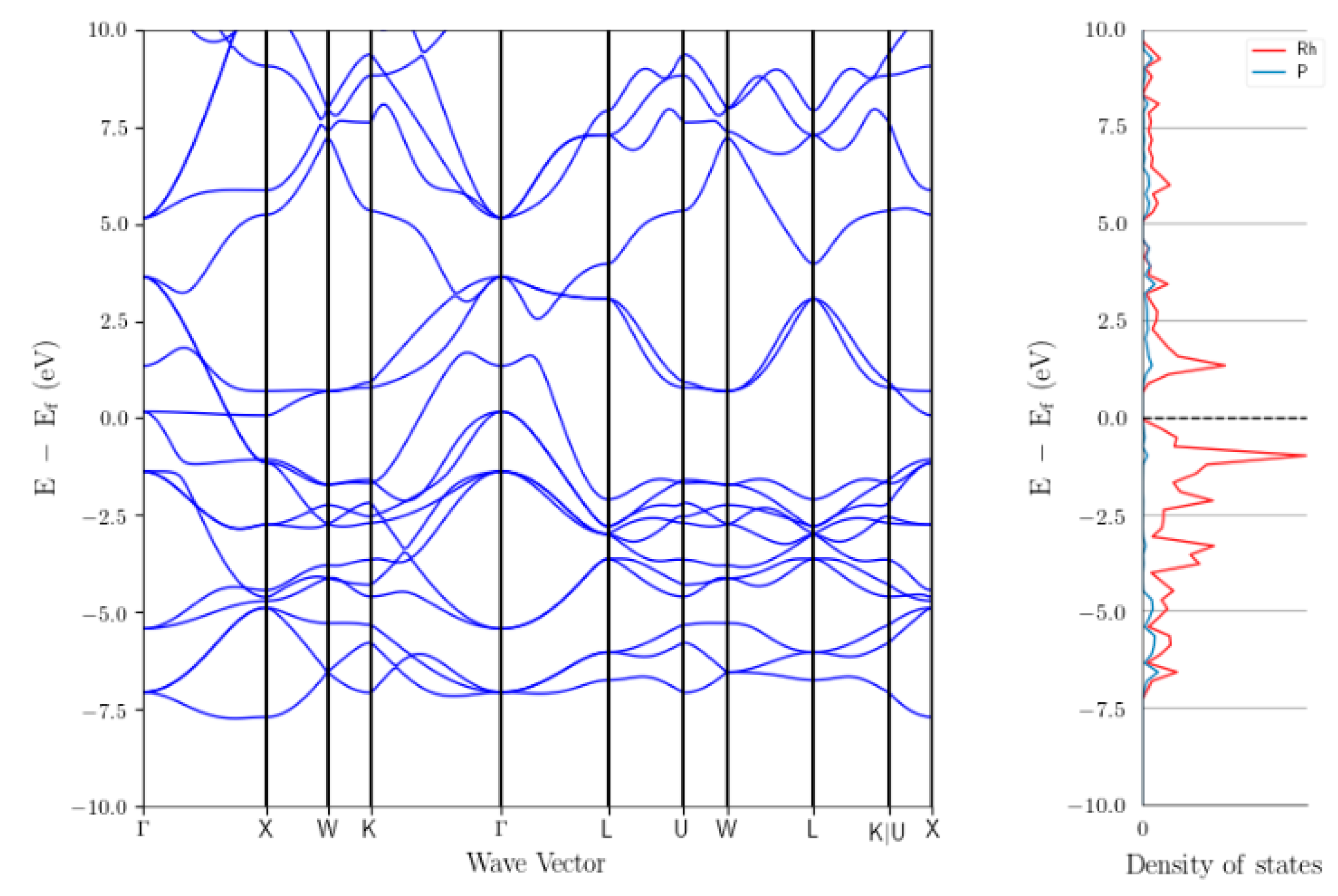
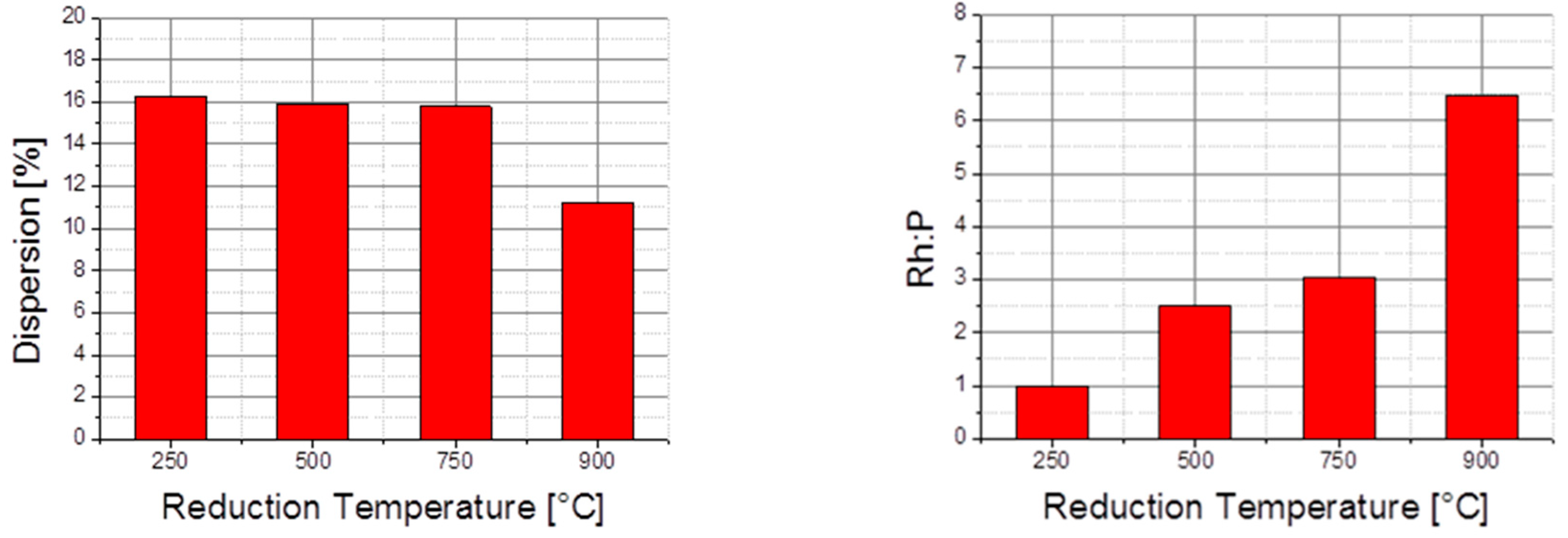
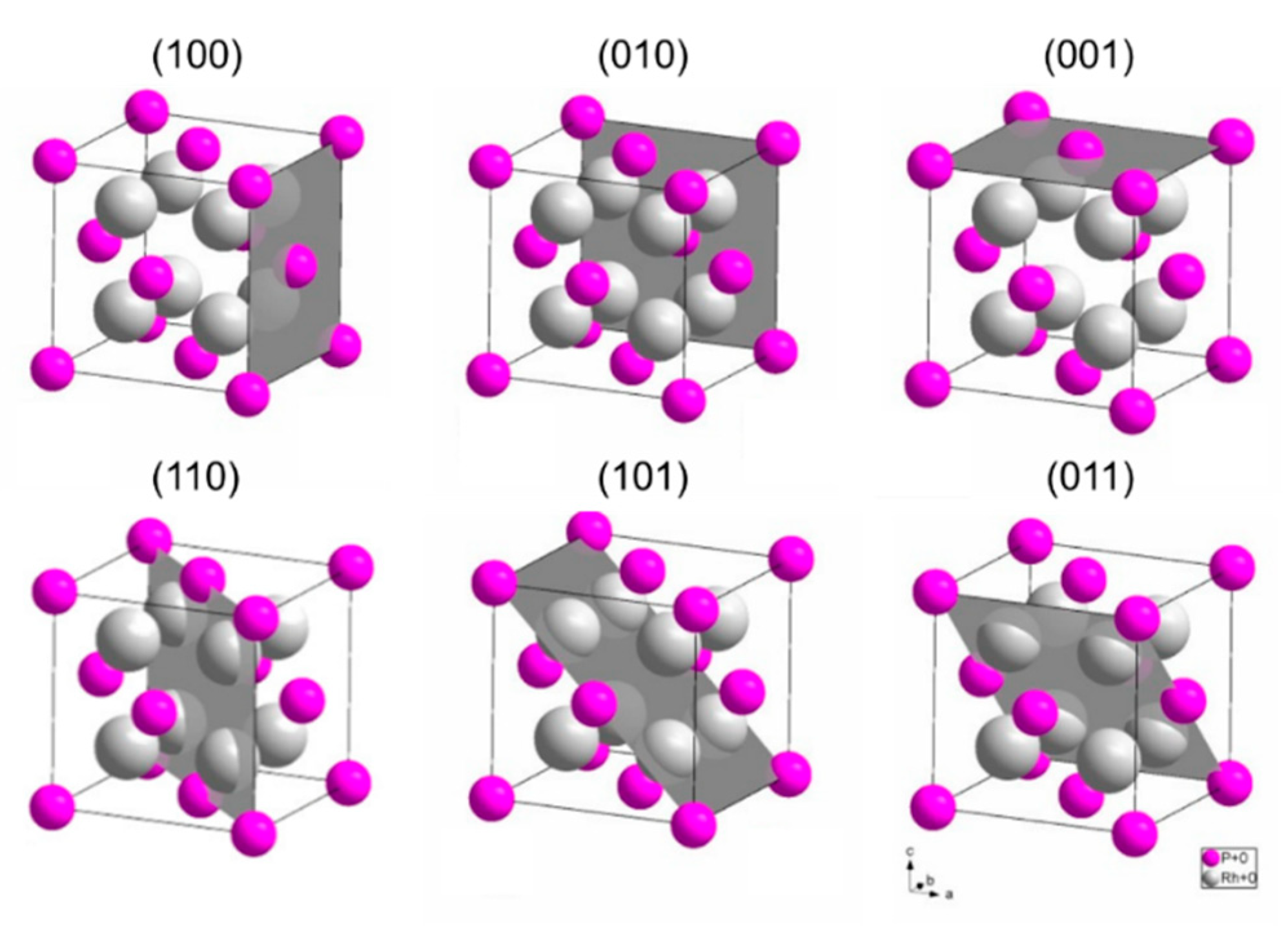
3.3.3. Investigation of The Surface Properties of Rh2P-Based Catalysts
- Based on the experimental and analytical data mentioned before, we extracted the following characteristic features of Rh2P catalysts, which are crucial for the hypothetic hydroformylation reaction mechanism shown in Figure 15.
- Surface of Rh2P is Rh terminated
- Site separation of Rh and P occurs
- Undercoordinated Rh sites are present, which are similar to Rh phosphine complexes
- Rh2P reduced at 900 °C coordinates CO in a linear fashion via Rh
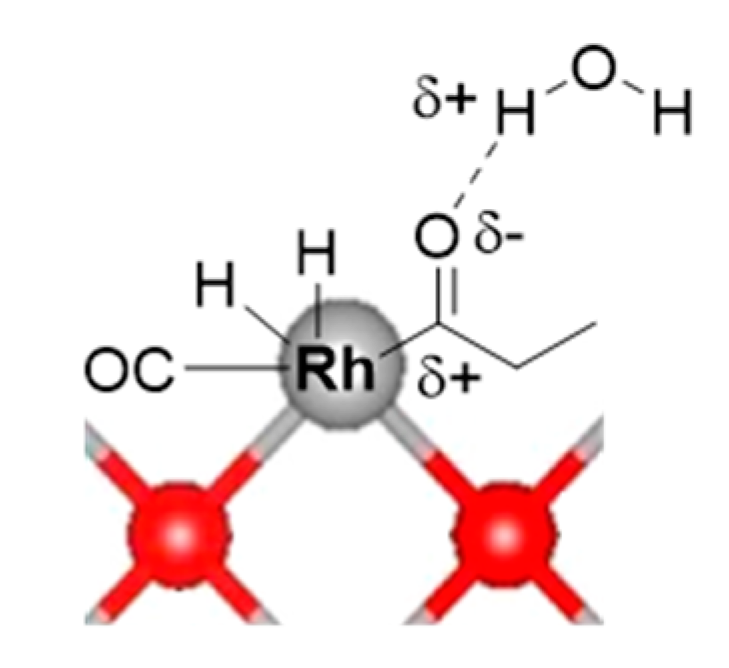
3.3.4. Potential Role of Water in The Reaction
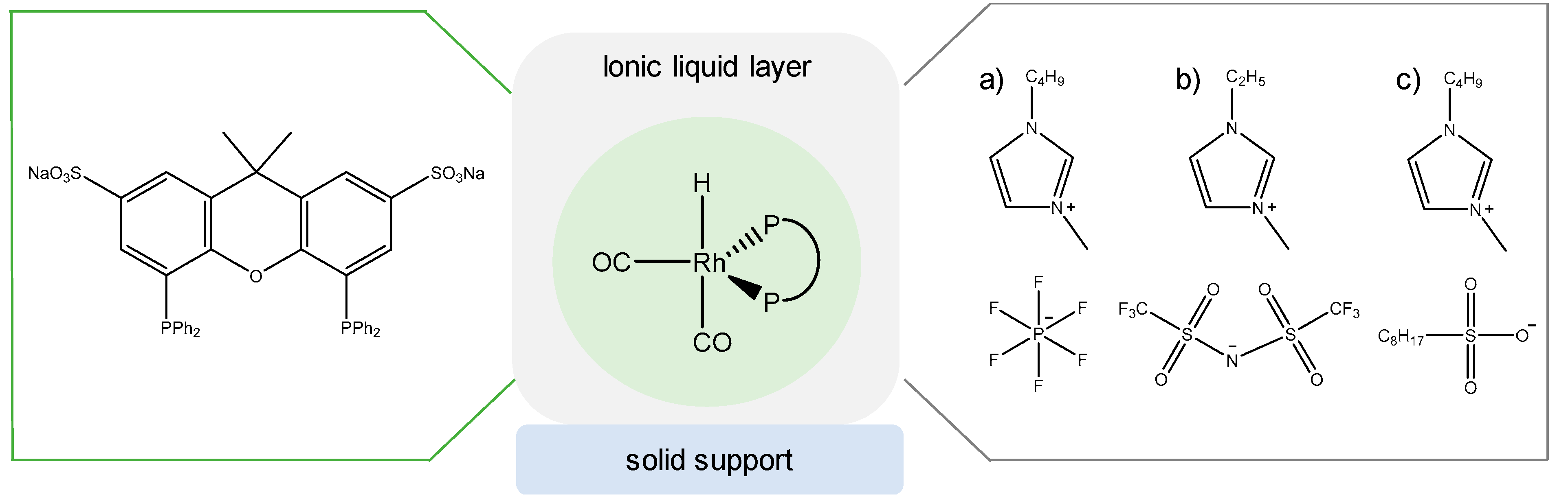
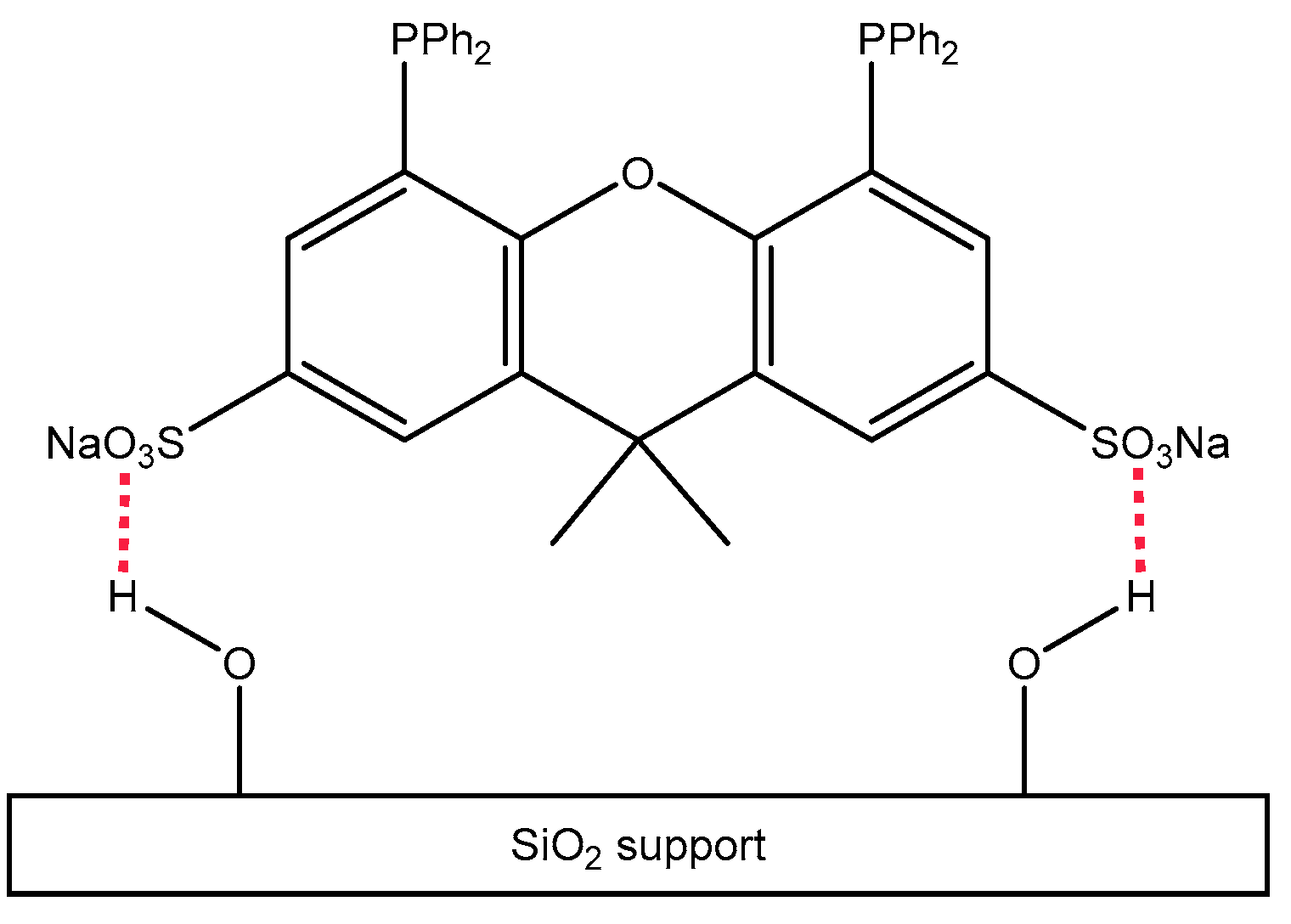
3.4. Rhodium Complexes in Supported Liquids
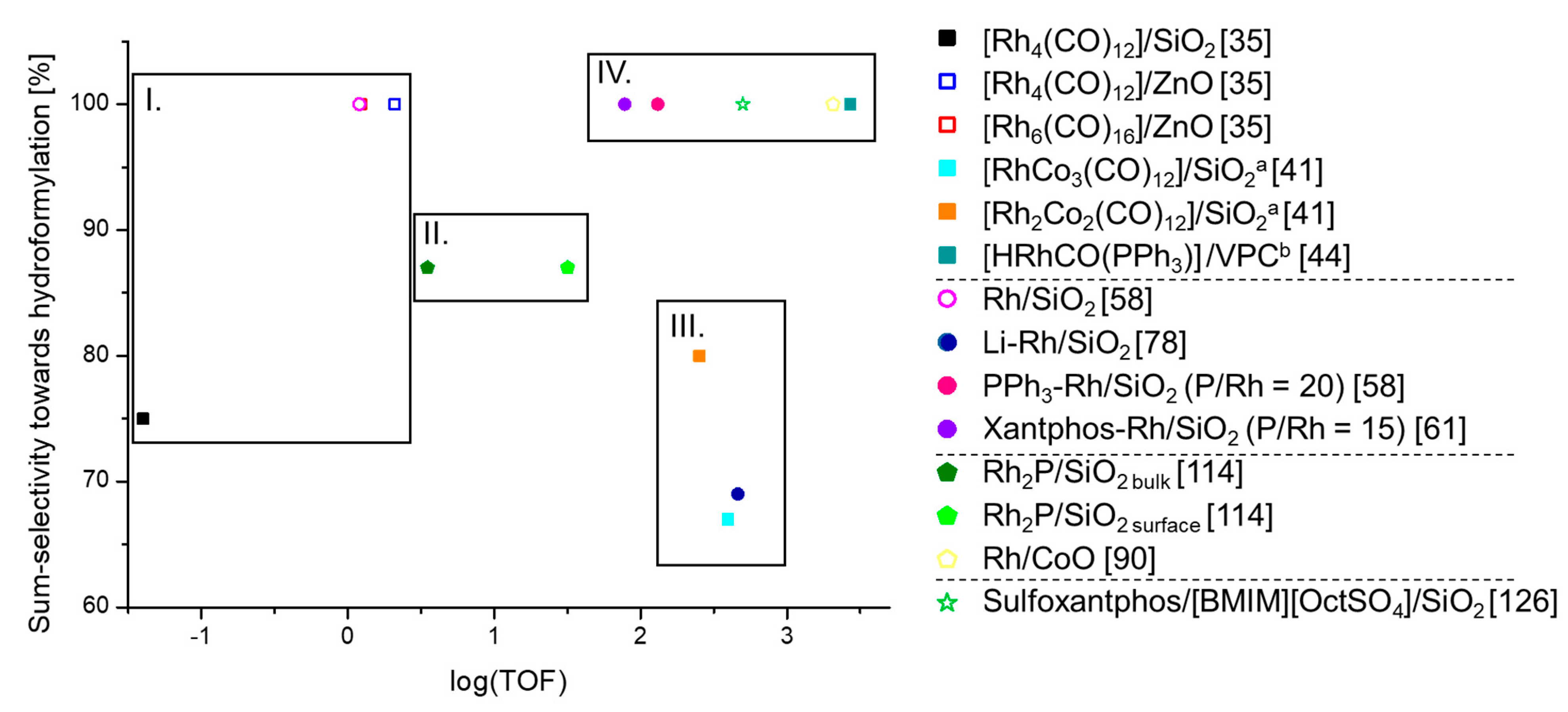
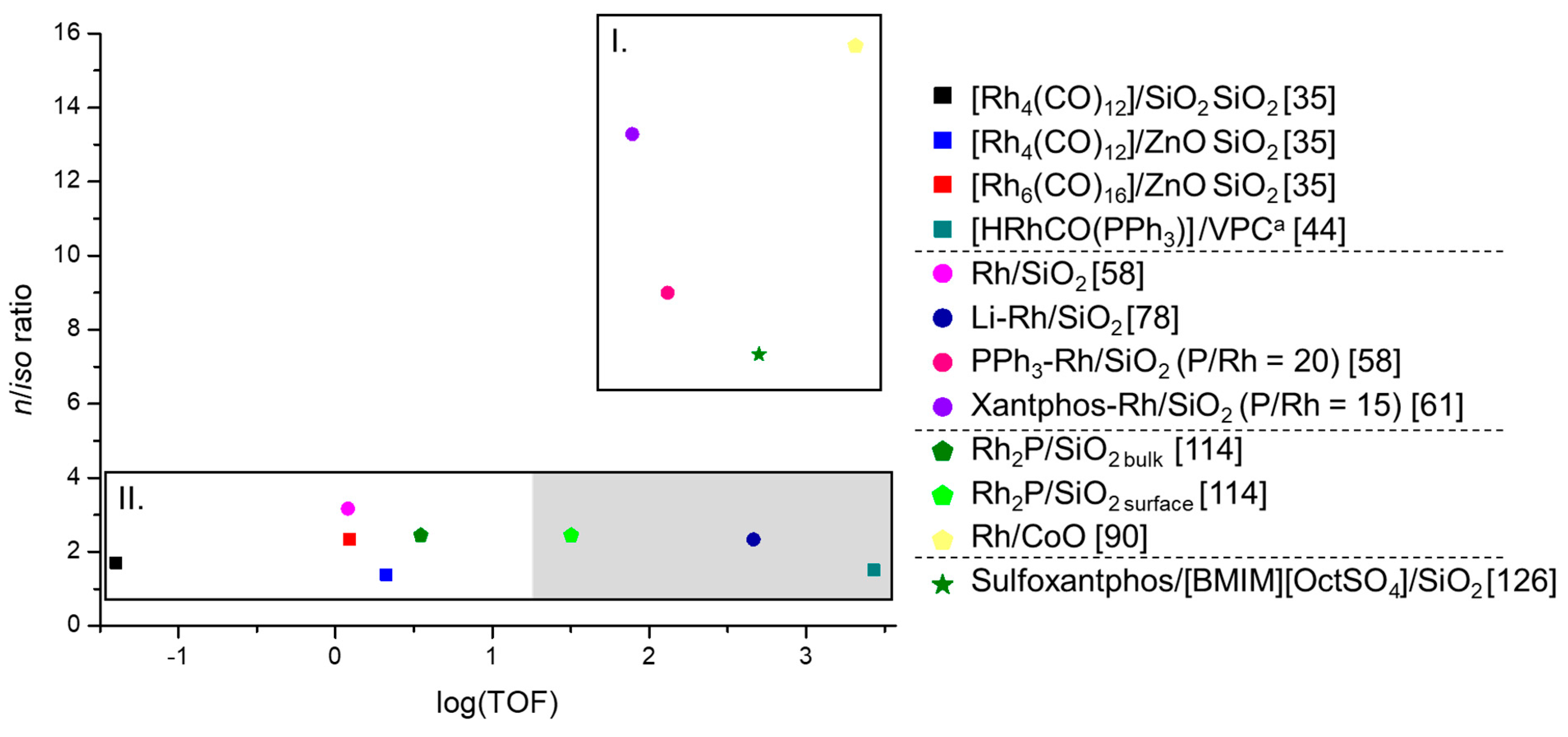
3.5. Comparison of The Different Heterogeneous Hydroformylation Rh Catalysts with Regard to Performance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
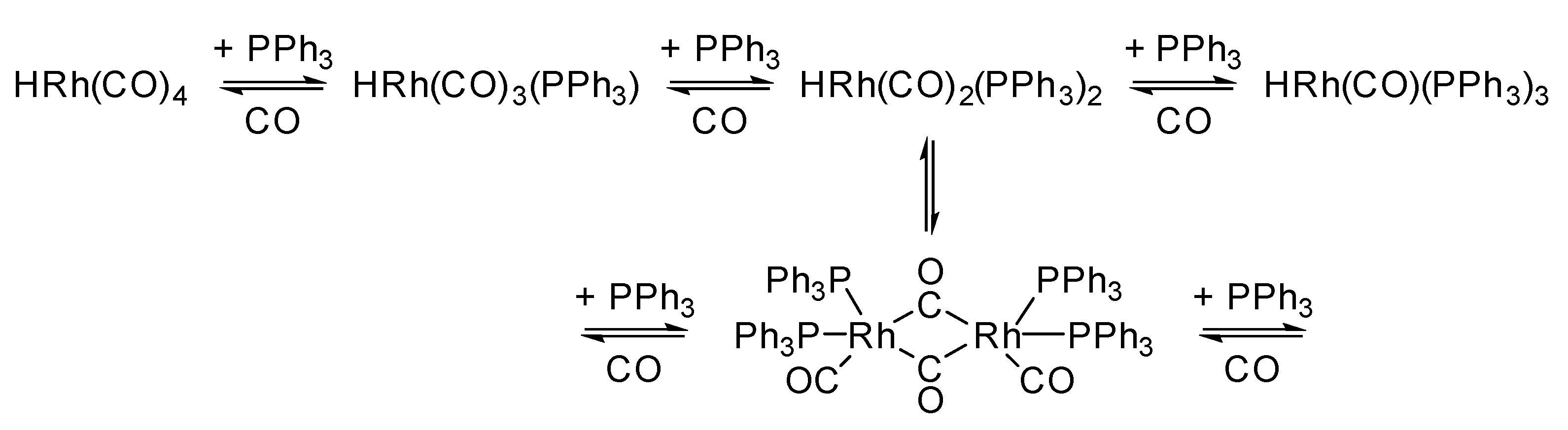{kind=link}
| Section | Section 3.1 | |||||
|---|---|---|---|---|---|---|
| Catalyst | [Rh4(CO)12] | [Rh4(CO)12] | [Rh6(CO)16] | [RhCo3(CO)12] | [Rh2Co2(CO)12] | [HRhCO(PPh3)]c |
| Rh [wt %] | 2.24 | 0.3 | 0.3 | - | - | - |
| Support | SiO2 | ZnO | ZnO | SiO2 | SiO2 | VPC |
| C3H6 [%] | 32 | 32 | 32 | 33.33 | 33.33 | 33.33 |
| CO [%] | 32 | 32 | 32 | 33.33 | 33.33 | 33.33 |
| H2 [%] | 36 | 36 | 36 | 33.33 | 33.33 | 33.33 |
| H2O [%] | - | - | - | - | - | - |
| Ar [%] | - | - | - | - | - | - |
| p [MPa] | 0.07 | 0.07 | 0.07 | 0.1 | 0.1 | 2.9 |
| T [K] | 428 | 423 | 423 | 443 | 443 | 393 |
| GHSV | - | - | - | - | - | - |
| Sum-sel.a [%] | 75 | - | - | 67b | 80b | 100 |
| Conv. [%] | - | - | - | 12.6 | 6.9 | - |
| n/iso ratio | 1.7 | 1.4 | 2.33 | - | - | 1.5 |
| TOFbulk [h−1] | 0.04 | 2.1 | 1.23 | 396b | 252b | 2700 |
| TOFsurf [h−1] | - | - | - | - | - | - |
| Ref. | [35] | [35] | [35] | [41] | [41] | [44] |
| Section | Section 3.2 | Section 3.3 | Section 3.4 | ||||
|---|---|---|---|---|---|---|---|
| Catalyst | Rh | Li-Rh | PPh3-Rh | Xantphos-Rh | Rh2Pa | Rh SAC | Rh SILPb |
| Rh [wt %] | 1.0 | 2 | 1.0 | 0.2 | 5.07 | 0.2 | 0.5 |
| Support | SiO2 | SiO2 | SiO2 | SiO2 | SiO2 | CoO | SiO2 |
| C3H6 [%] | 33.33 | 20 | 33.33 | 33.33 | 10 | 6 | 24 |
| CO [%] | 33.33 | 40 | 33.33 | 33.33 | 50 | 47 | 38 |
| H2 [%] | 33.33 | 40 | 33.33 | 33.33 | 10 | 47 | 38 |
| H2O [%] | - | - | - | - | 20 | - | - |
| Ar [%] | - | - | - | - | 10 | - | - |
| p [MPa] | 1 | 0.1 | 1.0 | 0.2 | 2.0 | 1.66 | 1.32 |
| T [K] | 393 | 413K | 393 | 398 | 503 | 373 | 413 |
| GHSV | 2000 | - | 2000 | - | 3000 | - | - |
| Sum-sel.a [%] | - | 59 | 100 | 100 | 87 | - | - |
| Conv. [%] | 0.12 | 0.85 | 27.1 | <1 | 7.3 | 94.6 | 28.1 |
| n/iso ratio | 3.17 | 2.33 | 9 | 13.29 | 2.45 | 15.67 | 7.13 |
| TOFbulk [h−1] | 1.2 | 462 | 131 | 78 | 3.5 | 2065 | 500 |
| TOFsurf [h−1] | - | - | - | - | 31.8 | - | - |
| Ref. | [58] | [78] | [58] | [61] | [114] | [90] | [126] |
References
- Roelen, O. Verfahren zur Herstellung von Sauerstoffhaltigen Verbindungen. DE Patent 849548, 20 September 1938. [Google Scholar]
- Roelen, O. Production of Oxygenated Carbon Compounds. U.S. Patent 2327066, 17 August 1943. [Google Scholar]
- Franke, R.; Selent, D.; Börner, A. Applied hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Pospech, J.; Fleischer, I.; Franke, R.; Buchholz, S.; Beller, M. Alternative metals for homogeneous catalyzed hydroformylation reactions. Angew. Chem. Int. Ed. 2013, 52, 2852–2872. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.; Osborn, J.A.; Wilkinson, G. Hydroformylation of alkenes by use of rhodium complex catalysts. J. Chem. Soc. A 1968, 3, 3133–3142. [Google Scholar] [CrossRef]
- Brown, C.K.; Wilkinson, G. Homogeneous hydroformylation of alkenes with hydridocarbonyltris- (triphenylphosphine)rhodium(I) as catalyst. J. Chem. Soc. A 1970, 2753–2764. [Google Scholar] [CrossRef]
- Tolman, C.A. Phosphorus ligand exchange equilibriums on zerovalent nickel. Dominant role for steric effects. J. Am. Chem. Soc. 1970, 92, 2953–2956. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Durand, D.J.; Fey, N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561–6594. [Google Scholar] [CrossRef] [PubMed]
- Wiese, K.-D.; Obst, D. Catalytic Carbonylation Reactions; Beller, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Jiao, Y.; Torne, M.S.; Gracia, J.; Niemantsverdriet, J.H.; van Leeuwen, P.W. Ligand effects in rhodium-catalyzed hydroformylation with bisphosphines: Steric or electronic. Catal. Sci. Technol. 2017, 7, 1404–1414. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamashita, M.; Nozaki, K. Tandem Hydroformylation/Hydrogenation of Alkenes to Normal Alcohols Using Rh/Ru Dual Catalyst or Ru Single Component Catalyst. J. Am. Chem. Soc. 2012, 134, 18746–18757. [Google Scholar] [CrossRef]
- Fleischer, I.; Dyballa, K.M.; Jennerjahn, R.; Jackstell, R.; Franke, R.; Spannenberg, A.; Beller, M. From olefins to alcohols: Efficient and regioselective ruthenium-catalyzed domino hydroformylation/ reduction sequence. Angew. Chem. Int. Ed. 2013, 52, 2949–2953. [Google Scholar] [CrossRef]
- Agbossou, F.; Carpentier, J.; Mortreux, F. Asymmetric hydroformylation. Chem. Rev. 1995, 95, 2485–2506. [Google Scholar] [CrossRef]
- Shaharun, M.S.; Mukhtar, H.; Dutta, B.K. Selectivity of Rhodium-catalyzed Hydroformylation of 1-Octene in a Thermomorphic Solvent System. J. Appl. Sci. 2011, 11, 1157–1163. [Google Scholar] [CrossRef]
- Caiazzoa, A.; Settambolob, R.; Pontornoa, L.; Lazzaron, R. Chemoselectivity in the rhodium-catalyzed hydroformylation of 4-vinylpyridine: Crucial role of phosphine ligand in promoting carbonylation instead of hydrogenation. J. Organomet. Chem. 2000, 599, 298–303. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Crocker, L.S.; Morgan, M.K. Thermochemical studies of carbonyl compounds. 5. Enthalpies of reduction of carbonyl groups. J. Am. Chem. Soc. 1991, 113, 3447–3450. [Google Scholar]
- Gorbunov, D.N.; Volkov, A.V.; Kardasheva, Y.S.; Maksimov, A.L.; Karakhanov, E.A. Hydroformylation in petroleum chemistry and organic synthesis: Implementation of the process and solving the problem of recycling homogeneous catalysts. Pet. Chem. 2015, 55, 587–603. [Google Scholar] [CrossRef]
- Evans, D.; Yagupsky, G.; Wilkinson, G. The reaction of hydridocarbonyltris(triphenylphosphine)rhodium with carbon monoxide, and of the reaction products, hydridodicarbonylbis(triphenylphosphine)rhodium and dimeric species, with hydrogen. J. Chem. Soc. A 1968, 2660–2665. [Google Scholar] [CrossRef]
- Kiss, G.; Mozeleski, E.J.; Nadler, K.C.; Van Driessche, E.; DeRoover, C. Hydroformylation of ethene with triphenylphosphine modified rhodium catalyst: Kinetic and mechanistic studies. J. Mol. Catal. A 1999, 138, 155–176. [Google Scholar] [CrossRef]
- Kégl, T. Computational aspects of hydroformylation. RSC Adv. 2015, 5, 4304–4327. [Google Scholar] [CrossRef]
- Lazzaroni, R.; Settambolo, R.; Alagona, G.; Ghio, C. Investigation of alkyl metal intermediate formation in the rhodium-catalyzed hydroformylation: Experimental and theoretical approaches. Coord. Chem. Rev. 2010, 254, 696–706. [Google Scholar] [CrossRef]
- Rush, L.E.; Pringle, P.G.; Harvey, J.N. Computational kinetics of cobalt-catalyzed alkene hydroformylation. Angew. Chem. Int. Ed. 2014, 53, 8672–8676. [Google Scholar] [CrossRef]
- Sparta, M.; Børve, K.J.; Jensen, V.R. Activity of Rhodium-Catalyzed Hydroformylation: Added Insight and Predictions from Theory. J. Am. Chem. Soc. 2007, 129, 8487–8499. [Google Scholar] [CrossRef] [PubMed]
- Gellrich, U.; Koslowski, T.; Breit, B. Full kinetic analysis of a rhodium-catalyzed hydroformylation: Beyond the rate-limiting step picture. Catal. Sci. Technol. 2015, 5, 129–133. [Google Scholar] [CrossRef]
- Gellrich, U.; Seiche, W.; Keller, M.; Breit, B. Mechanistic Insights into a Supramolecular Self-Assembling Catalyst System: Evidence for Hydrogen Bonding during Rhodium-Catalyzed Hydroformylation. Angew. Chem. Int. Ed. 2012, 51, 11033–11038. [Google Scholar] [CrossRef] [PubMed]
- Gleich, D.; Hutter, J. Computational Approaches to Activity in Rhodium-Catalysed Hydroformylation. Chem. Eur. J. 2004, 10, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, P.W.M.N.; Casey, C.P.; Whiteker, G.T. Rhodium Catalyzed Hydroformylation; van Leeuwen, P.W.M.N., Ed.; Academic Publishers: Dordrecht, The Netherlands, 2000. [Google Scholar]
- Lenarda, M.; Storaro, L.; Ganzerla, R. Hydroformylation of simple olefins catalyzed by metals and clusters supported on unfunctionalized inorganic carriers. J. Mol. Catal. A 1996, 111, 203–237. [Google Scholar] [CrossRef]
- Sun, Q.; Dai, Z.; Liu, X.; Sheng, N.; Deng, F.; Meng, X.; Xiao, F.-S. Highly Efficient Heterogeneous Hydroformylation over Rh-Metalated Porous Organic Polymers: Synergistic Effect of High Ligand Concentration and Flexible Framework. J. Am. Chem. Soc. 2015, 137, 5204–5209. [Google Scholar] [CrossRef]
- Sun, Q.; Jiang, M.; Shen, Z.; Jin, Y.; Pan, S.; Wang, L.; Meng, X.; Chen, W.; Ding, Y.; Lib, J.; et al. Porous organic ligands (POLs) for synthesizing highly efficient heterogeneous catalysts. Chem. Commun. 2014, 50, 11844–11847. [Google Scholar] [CrossRef]
- Neves, Â.C.B.; Calvete, M.J.F.; Pinho e Melo, T.M.V.; Pereira, D.M.M. Immobilized Catalysts for Hydroformylation Reactions: A Versatile Tool for Aldehyde Synthesis. Eur. J. Org. Chem. 2012, 2012, 6309–6320. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Yan, L.; Ding, Y. A mini review on strategies for heterogenization of rhodium-based hydroformylation catalysts. Front. Chem. Sci. Eng. 2018, 12, 113–123. [Google Scholar] [CrossRef]
- Wang, Z.; Ding, K.; Uozumi, Y. Handbook of Asymmetric Heterogenous Catalysis; Ding, K., Uozumi, Y., Eds.; WILEY-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Ichikawa, M. Catalytic Hydroformylation of Olefins Over the Rhodium, Bimetallic Rh-Co, and Cobalt Carbonyl Clusters Supported with Some Metal-Oxides. J. Catal. 1979, 59, 67–78. [Google Scholar] [CrossRef]
- Fukuoka, A.; Rao, L.-F.; Kosugi, N.; Kuroda, H.; Ichikawa, M. Selective hydroformylation of ethene and propene catalysed on nay zeolite-entrapped Rh6 and bimetallic RhFe clusters and their structural characterization by extended x-ray absorption fine structure and fourier transform infrared spectroscopy. Appl. Catal. 1989, 50, 295–301. [Google Scholar] [CrossRef]
- Ichikawa, M.; Rao, L.-F.; Kimura, T.; Fukuoka, A. Heterogenized bimetallic clusters: Their structures and bifunctional catalysis. J. Mol. Catal. 1990, 62, 1–35. [Google Scholar] [CrossRef]
- Kosmulski, M. Isoelectric points and points of zero charge of metal (hydr)oxides: 50 years after Parks’ review. Adv. Colloid Interfac. Sci. 2016, 238, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, Y.; Guo, W.; Liu, A.; Li, D.; Guo, X. Study on catalysis by carbonyl cluster-derived SiO2-supported rhodium for ethene hydroformylation. Catal. Lett. 1995, 32, 61–81. [Google Scholar] [CrossRef]
- Basu, P.; Panayotov, D.; Yates, J.T. Rhodium-carbon monoxide surface chemistry: The involvement of surface hydroxyl groups on alumina and silica supports. J. Am. Chem. Soc. 1988, 110, 2074–2081. [Google Scholar] [CrossRef]
- Huang, L.; Xu, Y.; Piao, G.; Liu, A.; Zhang, W. Enhancement of olefin hydroformylation related to supported bimetallic Rh-Co clusters. Catal. Lett. 1994, 23, 87–95. [Google Scholar] [CrossRef]
- Huang, L. Formation of bimetallic RhCo3 clusters from monometallic carbonyl clusters on SiO2 as probed by hydroformylation. J. Mol. Catal. A 1997, 125, 47–52. [Google Scholar] [CrossRef]
- de Munk, N.A.; Verbruggen, M.W.; de Leur, J.E.; Scholten, J.J.F. Gas Phase Hydroformylation of Propene with Porous Resin Anchored Rhodium Complexes. Part II. The Catalytic Performance. J. Mol. Catal. 1981, 11, 331–342. [Google Scholar] [CrossRef]
- Yoneda, N.; Nakagawa, Y.; Mimami, T. Hydroformylation catalyzed by immobilized rhodium complex to polymer support. Catal. Today 1997, 36, 357–364. [Google Scholar] [CrossRef]
- Bando, K.K.; Asakura, K.; Arakawa, H.; Isobe, K.; Iwasawa, Y. Surface Structures and Catalytic Hydroformylation Activities of Rh Dimers Attached on Various Inorganic Oxide Supports. J. Phys. Chem. 1996, 100, 13636–13645. [Google Scholar] [CrossRef]
- Alvila, L.; Pakkanen, T.A.; Pakkanen, T.T.; Krause, O. Catalytic hydroformylation of 1-hexene over Co2Rh2(CO)12 supported on inorganic carrier materials. J. Mol. Catal. 1992, 75, 333–345. [Google Scholar] [CrossRef]
- Van Vu, T.; Kosslick, H.; Schulz, A.; Harloff, J.; Paetzold, E.; Schneider, M.; Radnik, J.; Steinfeldt, N.; Fulda, G.; Kragl, U. Selective hydroformylation of olefins over the rhodium supported large porous metal–organic framework MIL-101. Appl. Catal. A 2013, 468, 410–417. [Google Scholar] [CrossRef]
- Van Vu, T.; Kosslick, H.; Schulz, A.; Harloff, J.; Paetzold, E.; Lund, H.; Kragl, U.; Schneider, M.; Fulda, G. Influence of the textural properties of Rh/MOF-5 on the catalytic properties in the hydroformylation of olefins. Microporous Mesoporous Mater. 2012, 154, 100–106. [Google Scholar]
- Sharma, S.K.; Parikh, P.A.; Jasra, R.V. Hydroformylation of alkenes using heterogeneous catalyst prepared by intercalation of HRh(CO)(TPPTS)3 complex in hydrotalcite. J. Mol. Catal. A. 2010, 316, 153–162. [Google Scholar] [CrossRef]
- Sudheesh, N.; Sharma, S.K.; Shukla, R.S.; Jasra, R.V. HRh(CO)(PPh3)3 encapsulated mesopores of hexagonal mesoporous silica (HMS) acting as nanophase reactors for effective catalytic hydroformylation of olefins. J. Mol. Catal. A 2008, 296, 61–70. [Google Scholar] [CrossRef]
- Sudheesh, N.; Sharma, S.K.; Shukla, R.S.; Jasra, R.V. Investigations on the kinetics of hydroformylation of 1-hexene using HRh(CO)(PPh3)3 encapsulated hexagonal mesoporous silica as a heterogeneous catalyst. J. Mol. Catal. A 2010, 316, 23–29. [Google Scholar] [CrossRef]
- Duanmu, C.; Wu, L.; Gu, J.; Xu, X.; Feng, L.; Gu, X. Magnetic nanoparticle supported triphenylphosphine ligand for the Rh-catalyzed hydroformylation reaction. Catal. Commun. 2014, 48, 45–49. [Google Scholar] [CrossRef]
- Gorbunov, D.; Safronova, D.; Kardasheva, Y.; Maximov, A.; Rosenberg, E.; Karakhanov, E. New Heterogeneous Rh-Containing Catalysts Immobilized on a Hybrid Organic–Inorganic Surface for Hydroformylation of Unsaturated Compounds. ACS Appl. Mater. Interfaces 2018, 10, 26566–26575. [Google Scholar] [CrossRef]
- Chuang, S.S.C.; Pien, S.-I. Infrared spectroscopic studies of ethene hydroformylation on Rh/SiO2: An investigation of the relationships between homogeneous and heterogeneous hydroformylation. J. Mol. Catal. 1989, 55, 12–22. [Google Scholar] [CrossRef]
- Chuang, S.C.; Pien, S.-I. Infrared Study of the CO Insertion Reaction on Reduced, Oxidized, and Sulfided Rh/SiO2 Catalysts. J. Catal. 1992, 135, 618–634. [Google Scholar] [CrossRef]
- McClure, S.M.; Lundwall, M.J.; Goodman, D.W. Planar oxide supported rhodium nanoparticles as model catalysts. Proc. Natl. Acad. Sci. USA 2011, 108, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Tan, C.D.; Baker, R.T.K. Ethene hydroformylation on graphite nanofiber supported rhodium catalysts. Catal. Today 2001, 65, 19–29. [Google Scholar] [CrossRef]
- Yan, L.; Ding, Y.J.; Zhu, H.J.; Xiong, J.M.; Wang, T.; Pan, Z.D.; Lin, L.W. Ligand modified real heterogeneous catalysts for fixed-bed hydroformylation of propene. J. Mol. Catal. 2005, 234, 1–7. [Google Scholar] [CrossRef]
- Li, Y.; Yunjie, D.; Jia, L.; Hejun, Z.; Liwu, L. Influence of Phosphine Concentration on Propene Hydroformylation over the PPh3-Rh/SiO2 Catalyst. Chin. J. Catal. 2011, 32, 31–35. [Google Scholar]
- Kim, T.; Celik, F.E.; Hanna, D.G.; Shylesh, S.; Werner, S.; Bell, A.T. Gas-Phase Hydroformylation of Propene over Silica-Supported PPh3-Modified Rhodium Catalysts. Top. Catal. 2011, 54, 299–307. [Google Scholar] [CrossRef]
- Shylesh, S.; Hanna, D.; Molinar, A.; Koong, X.-Q.; Reimer, J.A.; Alexis, T. Bell, In Situ Formation of Wilkinson-Type Hydroformylation Catalysts: Insights into the Structure, Stability, and Kinetics of Triphenylphosphine- and Xantphos-Modified Rh/SiO2. ACS Catalysis 2013, 3, 348–357. [Google Scholar] [CrossRef]
- Li, X.; Ding, Y.; Jiao, G.; Li, J.; Lin, R.; Gong, L.; Yan, L.; Zhu, H. A new concept of tethered ligand-modified Rh/SiO2 catalyst for hydroformylation with high stability. Appl. Catal. A 2009, 353, 266–270. [Google Scholar] [CrossRef]
- Liu, J.; Yan, L.; Jiang, M.; Li, C.; Ding, Y. Effect of lengthening alkyl spacer on hydroformylation performance of tethered-phosphine modified Rh/SiO2 catalyst. Chin. J. Catal. 2016, 37, 268–272. [Google Scholar] [CrossRef]
- Liu, J.; Yan, L.; Ding, Y.; Jiang, M.; Dong, W.; Song, X.; Zhu, H. Promoting effect of Al on tethered ligand-modified Rh/SiO2 catalysts for ethene hydroformylation. Appl. Catal. A 2015, 492, 127–132. [Google Scholar] [CrossRef]
- Tana, M.; Ishikuro, Y.; Hosoi, Y.; Yamane, N.; Ai, P.; Zhang, P.; Yanga, G.; Wuc, M.; Yange, R.; Tsubaki, N. PPh3 functionalized Rh/rGO catalyst for heterogeneous hydroformylation: Bifunctional reduction of graphene oxide by organic ligand. Chem. Eng. J. 2017, 330, 863–869. [Google Scholar] [CrossRef]
- Trunschke, A.; Bigttcher, H.-C.; Fukuoka, A.; Ichikawa, M.; Miessner, H. Olefin hydroformylation and Selective Hydrogenation of Acetaldehyde on Mo-Promoted Rh/SiO2 Catalysts Derived from Metal Salt and Heteronuclear Cluster Precursors. Catal. Lett. 1991, 8, 221–228. [Google Scholar] [CrossRef]
- Lenarda, M.; Ganzerla, R.; Storaro, L.; Trovarelli, A.; Zanoni, R.; Kaspar, J. Vapour phase hydroformylation of ethene and propene catalyzed by a rhodium-containing aluminum pillared smectite clay. J. Mol. Catal. 1992, 72, 75–84. [Google Scholar] [CrossRef]
- Lenarda, M.; Ganzerla, R.; Enzo, S.; Storaro, L.; Zanoni, R. Vapour phase propene hydroformylation catalyzed by the Rh/Al system on silica. J. Mol. Catal. 1993, 80, 105–116. [Google Scholar] [CrossRef]
- Lenarda, M.; Ganzerla, R.; Storaro, L.; Zanoni, R. Catalysis by the Rh/B system Part 1. Vapour-phase hydroformylation of ethene at atmospheric pressure on Rh/B on silica. J. Mol. Catal. 1993, 78, 339–350. [Google Scholar] [CrossRef]
- Storaro, L.; Ganzerla, R.; Lenarda, M.; Zanoni, R.; Righini, G. Highly selective vapor phase propene hydroformylation catalyzed by Rh/B and Rh-Co/B systems on silica. J. Mol. Catal. A 1996, 112, 43–54. [Google Scholar] [CrossRef]
- Navidi, N.; Thybaut, J.W.; Marin, G.B. Experimental investigation of ethene hydroformylation to propanal on Rh and Co based catalysts. Appl. Catal. A 2014, 469, 357–366. [Google Scholar] [CrossRef]
- Hanaoka, T.; Arakawa, H.; Matsuzaki, T.; Sugi, Y.; Kanno, K.; Abe, Y. Ethene Hydroformylation and Carbon Monoxide Hydrogenation over Modified and Unmodified Silica Supported Rhodium Catalysts. Catal. Today 2000, 58, 271–280. [Google Scholar] [CrossRef]
- Ni, B.; Wang, X. Face the Edges: Catalytic Active Sites of Nanomaterials. Adv. Sci. 2015, 2, 1–22. [Google Scholar] [CrossRef]
- Freund, H.-J.; Libuda, J.; Bäumer, M.; Risse, T.; Carlsson, A. Cluster, Facets, and Edges: Site-Dependent Selective Chemistry on Model Catalysts. Chem. Rec. 2003, 3, 181–200. [Google Scholar] [CrossRef]
- Hejral, U.; Franz, D.; Volkov, S.; Francoual, S.; Strempfer, J.; Stierle, A. Identification of a Catalytically Highly Active Surface Phase for CO Oxidation over PtRh Nanoparticles under Operando Reaction Conditions. Phys. Rev. Lett. 2018, 120, 126101–126106. [Google Scholar] [CrossRef]
- Murzin, D.Y. Thermodynamic analysis of nanoparticle size effect on catalytic kinetics. Chem. Eng. Sci. 2009, 64, 1046–1052. [Google Scholar] [CrossRef]
- Van Santen, R. Complementary Structure Sensitive and Insensitive Catalytic Relationships. Accounts Chem. Res. 2009, 42, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Sordelli, L.; Guidotti, M.; Andreatta, D.; Vlaic, G.; Psaro, R. Characterization and catalytic performances of alkali-metal promoted Rh/SiO2 catalysts for propene hydroformylation. J. Mol. Catal. A 2003, 204–205, 509–518. [Google Scholar] [CrossRef]
- Qiao, B.T.; Wang, A.Q.; Yang, X.F.; Allard, L.F.; Jiang, Z.; Cui, Y.T.; Liu, J.Y.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nature Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Liang, J.; Yu, Q.; Yang, X.; Zhang, T.; Li, J. A systematic theoretical study on FeOx-supported single-atom catalysts: M1/FeOx for CO oxidation. Nano Res. 2018, 11, 1599–1611. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M.; Gates, B.C. Atomically Dispersed Supported Metal Catalysts. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 545–574. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, X.; Zhang, L.; Lang, R.; Qiao, B. Single-atom catalysis: Bridging the homo- and heterogeneous catalysis. Chin. J. Catal. 2018, 39, 893–898. [Google Scholar] [CrossRef]
- Liu, J.Y. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Bayram, E.; Lu, J.; Aydin, C.; Browning, N.D.; Özkar, S.; Finney, E.; Gates, B.C.; Finke, R.G. Agglomerative Sintering of an Atomically Dispersed Ir1/Zeolite Y Catalyst: Compelling Evidence Against Ostwald Ripening but for Bimolecular and Autocatalytic Agglomeration Catalyst Sintering Steps. ACS Catal. 2015, 5, 3514–3527. [Google Scholar] [CrossRef]
- Qin, R.; Liu, P.; Fu, G.; Zheng, N. Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications. Small Methods 2018, 2, 1–21. [Google Scholar]
- Choi, C.H.; Kim, M.; Kwon, H.C.; Cho, S.J.; Yun, S.; Kim, H.-T.; Mayrhofer, K.J.J.; Kim, H.; Choi, M. Tuning selectivity of electrochemical reactions by atomically dispersed platinum catalyst. Nature Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhao, M.; Ding, X.; Ma, K.; Wu, C.; Gates, I.D.; Gao, Z. The effect of coordination environment on the kinetic and thermodynamic stability of single-atom iron catalysts. Phys. Chem. Chem. Phys. 2020, 22, 3983–3989. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Li, T.; Matsumura, D.; Miao, S.; Ren, Y.; Cui, Y.-T.; Tan, Y.; Qiao, B.; Li, L.; Wang, A.; et al. Hydroformylation of Olefins by a Rhodium Single-Atom Catalyst with Activity Comparable to RhCl(PPh3)3. Angew. Chem. Int. Ed. 2016, 55, 16054–16058. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, W.; Wang, S.; Gao, Z.; Luo, Z.; Wang, X.; Zeng, R.; Li, A.; Li, H.; Wang, M.; et al. Atomic-level insights in optimizing reaction paths for hydroformylation reaction over Rh/CoO single-atom catalyst. Nat. Commun. 2016, 7, 14036. [Google Scholar] [CrossRef]
- Oyama, S.T. Novel catalysts for advanced hydroprocessing: Transition metal phosphides. J. Catal. 2003, 216, 343–352. [Google Scholar] [CrossRef]
- Oyama, S.T.; Gott, T.; Zhao, H.; Lee, Y.-K. Transition metal phosphide hydroprocessing catalysts: A review. Catal. Today 2009, 143, 94–107. [Google Scholar] [CrossRef]
- Kanda, Y.; Uemichi, Y. Noble Metal Phosphides as New Hydrotreating Catalysts: Highly Active Rhodium Phosphide Catalyst. J. Jpn. Pet. Inst. 2015, 58, 20–32. [Google Scholar] [CrossRef][Green Version]
- Prins, R.; Bussell, M. Metal Phosphides: Preparation, Characterization and Catalytic Reactivity. Catal. Lett. 2012, 142, 1413–1436. [Google Scholar] [CrossRef]
- Wang, X.; Clark, P.; Oyama, S.T. Synthesis, Characterization, and Hydrotreating Activity of Several Iron Group Transition Metal Phosphides. J. Catal. 2002, 208, 321–331. [Google Scholar] [CrossRef]
- Kanda, Y.; Temma, C.; Nakata, K.; Kobayashi, T.; Sugioka, M.; Uemichi, Y. Preparation and performance of noble metal phosphides supported on silica as new hydrodesulfurization catalysts. Appl. Catal. A 2010, 386, 171–178. [Google Scholar] [CrossRef]
- Kanda, Y.; Matsukura, Y.; Sawada, A.; Sugioka, M.; Uemichi, Y. Low-temperature synthesis of rhodium phosphide on alumina and investigation of its catalytic activity toward the hydrodesulfurization of thiophene. Appl. Catal. A 2016, 515, 25–31. [Google Scholar] [CrossRef][Green Version]
- Bowker, R.H.; Smith, M.C.; Carrillo, B.A.; Bussell, M.E. Synthesis and Hydrodesulfurization properties of Noble metal phosphides: Ruthenium and palladium. Top. Catal. 2012, 55, 999–1009. [Google Scholar] [CrossRef]
- Hayes, J.R.; Bowker, R.H.; Gaudette, A.F.; Smith, M.C.; Moak, C.E.; Nam, C.Y.; Pratum, T.K.; Bussell, M. Hydrodesulfurization properties of rhodium phosphide: Comparison with rhodium metal and sulfide catalysts. J. Catal. 2010, 276, 249–258. [Google Scholar] [CrossRef]
- Habas, S.; Baddour, F.; Ruddy, D.; Nash, C.P.; Wang, J.; Pan, M.; Hensley, J.E.; A Schaidle, J. A Facile Molecular Precursor Route to Metal Phosphide Nanoparticles and Their Evaluation as Hydrodeoxygenation Catalysts. Chem. Mat. 2015, 27.22, 7580–7592. [Google Scholar] [CrossRef]
- Stinner, C.; Prins, R.; Weber, T. Binary and Ternary Transition-Metal Phosphides as HDN Catalysts. J. Catal. 2001, 202, 187–194. [Google Scholar] [CrossRef]
- Rupflin, L.A.; Boscagli, C.; Schunk, S.A. Platinum Group Metal Phosphides as Efficient Catalysts in Hydroprocessing and Syngas-Related Catalysis. Catalysts 2018, 8, 122. [Google Scholar] [CrossRef]
- Rupflin, L.A.; Mormul, J.; Lejkowski, M.; Titlbach, S.; Papp, R.; Gläser, R.; Dimitrakopoulou, M.; Huang, X.; Trunschke, A.; Willinger, M.G.; et al. Platinum Group Metal Phosphides as Heterogeneous Catalysts for the Gas-Phase Hydroformylation of Small Olefins. ACS Catal. 2017, 7, 3584–3590. [Google Scholar] [CrossRef]
- Schunk, S.A.; Alvarado Rupflin, L.; Mormul, J.; Lejkowski, M.; Titlbach, S.; Papp, R.; Gläser, R.; Dimitrakopoulou, M.; Huang, X.; Trunschke, A.; et al. New horizons for heterogeneously catalyzed CO insertion reactions: From molecular sieves to extended complex solids. DGMK Tagungsbericht 2007, 125–128. [Google Scholar]
- Duan, H.; Li, D.; Tang, Y.; He, Y.; Ji, S.; Wang, R.; Lv, H.; Lopes, P.P.; Paulikas, A.P.; Li, H.; et al. High Performance Rh2P Electrocatalyst for Efficient Water Splitting. J. Am. Chem. Soc. 2017, 139, 5494–5502. [Google Scholar] [CrossRef]
- Zumbusch, M. Über die Strukturen des Uransubsulfids und der Subphosphide des Iridiums und Rhodiums. Zeitschrift Für Anorganische Und Allgemeine Chemie 1940, 243, 322–329. [Google Scholar] [CrossRef]
- El Ghadraoui, E.; Guerin, R.; Sergent, M. Diphosphure de Trirhodium Rh3P2: Premier Exemple d’une Structure Lacunaire Ordonnee de Type anti-PbFCl. Acta Cryst. C 1983, 39, 1493–1494. [Google Scholar] [CrossRef]
- Rundqvist, S.; Hede, A.; Varde, E.; Westin, G. X-Ray Investigation on Rhodium Phosphides. The Crystal Structure of Rh4P3. Acta Chem. Scand. 1960, 14, 893–902. [Google Scholar] [CrossRef]
- Kjekshus, A.; Nolander, B.; Klæboe, P.; Cyvin, S.J.; Lagerlund, I.; Ehrenberg, L. On the Properties of Binary Compounds with the CoSb2 Type Crystal Structure. Acta Chem. Scan. 1971, 25, 411–422. [Google Scholar] [CrossRef]
- Rundqvist, S.; Ersson, N. Structure and bonding in skutterudite-type phosphides. Arkiv foer Kemi 1969, 30, 103. [Google Scholar]
- Okamoto, H. The P-Rh (phosphorus-rhodium) system. Bull. Alloy Phase Diagr. 1990, 11, 415–417. [Google Scholar] [CrossRef]
- Persson, K. Materials Data on PRh2 (SG:225) by Materials Project. Data Explor. 2014. [Google Scholar] [CrossRef]
- Muetterties, E.L.; Sauer, J.C. Catalytic Properties of Metal Phosphides. I. Qualitative Assay of Catalytic Properties. J. Am. Chem. Soc. 1974, 96, 3410–3415. [Google Scholar] [CrossRef]
- Alvarado Rupflin, L. Rhodiumphosphid Rh2P als neues Katalysatormaterial für die Aktivierung von Kohlenstoffmonoxid. Ph.D. Thesis, Universität Leipzig, Leipzig, Germany, 2016. [Google Scholar]
- Dyson, P.J.; Jessop, P.G. Solvent effects in catalysis: Rational improvements of catalysts via manipulation of solvent interactions. Catal. Sci. Technol. 2016, 6, 3302–3316. [Google Scholar] [CrossRef]
- Kulyaev, P.O.; Pidko, E.A. Operando Modeling of Multicomponent Reactive Solutions in Homogeneous Catalysis: From Non-standard Free Energies to Reaction Network Control. ChemCatChem 2020, 12, 795–802. [Google Scholar] [CrossRef]
- Bazhenov, A.S.; Lefferts, L.; Honkala, K. Adsorption and Activation of Water on Cuboctahedral Rhodium and Platinum Nanoparticles. J. Phys. Chem. C 2017, 121, 4324–4331. [Google Scholar] [CrossRef]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Riisager, R.; Fehrmann, M.; Haumann, P. Wasserscheid, Supported ionic liquids: Versatile reaction and separation media. Top. Catal. 2006, 40, 91–102. [Google Scholar] [CrossRef]
- Riisager, A.; Fehrmann, R.; Haumann, M.; Wasserscheid, P. Supported Ionic Liquid Phase (SILP) Catalysis: An Innovative Concept for Homogeneous Catalysis in Continuous Fixed-Bed Reactors. Eur. J. Inorg. Chem. 2006, 695–706. [Google Scholar] [CrossRef]
- Haumann, M.; Riisager, A. Hydroformylation in Room Temperature Ionic Liquids (RTILs): Catalyst and Process Developments. Chem. Rev. 2008, 108, 1474–1497. [Google Scholar] [CrossRef]
- Van Doorslaer, C.; Wahlen, J.; Mertens, P.; Binnemans, K.; De Vos, D. Immobilization of molecular catalysts in supported ionic liquid phases. Dalton Trans. 2010, 39, 8377–8390. [Google Scholar] [CrossRef]
- Steinrück, H.-P.; Libuda, J.; Wasserscheid, P.; Cremer, T.; Kolbeck, C.; Laurin, M.; Maier, F.; Sobota, M.; Schulz, P.; Stark, M. Surface Science and Model Catalysis with Ionic Liquid-Modified Materials. Adv. Mater. 2011, 23, 2571–2587. [Google Scholar] [CrossRef]
- Riisager, A.; Wasserscheid, P.; Van Hal, R.; Fehrmann, R. Continuous fixed-bed gas-phase hydroformylation using supported ionic liquid-phase (SILP) Rh catalysts. J. Catal. 2003, 219, 452–455. [Google Scholar] [CrossRef]
- Riisager, A.; Fehrmann, R.; Flicker, S.; Van Hal, R.; Haumann, M.; Wasserscheid, P. Very Stable and Highly Regioselective Supported Ionic-Liquid-Phase (SILP) Catalysis: Continuous-Flow Fixed-Bed Hydroformylation of Propene. Angew. Chem. Int. Ed. 2005, 44, 815–819. [Google Scholar] [CrossRef]
- Riisager, A.; Fehrmann, R.; Haumann, M.; Gorle, B.S.K.; Wasserscheid, P. Stability and Kinetic Studies of Supported Ionic Liquid Phase Catalysts for Hydroformylation of Propene. Ind. Eng. Chem. Res. 2005, 44, 9853–9859. [Google Scholar] [CrossRef]
- Hanna, D.G.; Shylesh, S.; Werner, S.; Bell, A.T. The kinetics of gas-phase propene hydroformylation over a supported ionic liquid-phase (SILP) rhodium catalyst. J. Catal. 2012, 292, 166–172. [Google Scholar] [CrossRef]
- Shylesh, S.; Hanna, D.; Werner, S.; Bell, A.T. Factors Influencing the Activity, Selectivity, and Stability of Rh-Based Supported Ionic Liquid Phase (SILP) Catalysts for Hydroformylation of Propene. ACS Catal. 2012, 2, 487–493. [Google Scholar] [CrossRef]
- Haumann, M.; Dentler, K.; Joni, J.; Riisager, A.; Wasserscheid, P. Continuous Gas-Phase Hydroformylation of 1-Butene using Supported Ionic Liquid Phase (SILP) Catalysts. Adv. Synth. Catal. 2007, 349, 425–431. [Google Scholar] [CrossRef]
- Haumann, M.; Jakuttis, M.; Werner, S.; Wasserscheid, P. Supported ionic liquid phase (SILP) catalyzed hydroformylation of 1-butene in a gradient-free loop reactor. J. Catal. 2009, 263, 321–327. [Google Scholar] [CrossRef]
- Jakuttis, M.; Schönweiz, A.; Werner, S.; Franke, R.; Wiese, K.-D.; Haumann, M.; Wasserscheid, P. Rhodium-Phosphite SILP Catalysis for the Highly Selective Hydroformylation of Mixed C4 Feedstocks. Angew. Chem. Int. Ed. 2011, 50, 4492–4495. [Google Scholar] [CrossRef]
- Weiß, A.; Giese, M.; Lijewski, M.; Franke, R.; Wasserscheid, P.; Haumann, M. Modification of nitrogen doped carbon for SILP catalyzed hydroformylation of ethylene. Catal. Sci. Technol. 2017, 7, 5562–5571. [Google Scholar] [CrossRef]
- Ha, H.N.T.; Duc, D.T.; Dao, T.V.; Le, T.M.; Riisager, A.; Fehrmann, R. Characterization and parametrical study of Rh-TPPTS supported ionic liquid phase (SILP) catalysts for ethylene hydroformylation. Catal. Commun. 2012, 25, 136–141. [Google Scholar] [CrossRef]

























| Catalyst | [Co (CO)4H] | [Co(PBu3)(CO)3H] | [Rh(PPh3)3(CO)H] | [Rh(TPPTS)3(CO)H] |
|---|---|---|---|---|
| T [K] | 383–457 | 433–473 | 358–388 | 323–423 |
| p [MPa] | 20–35 | 3–10 | 1.5–2 | 1–10 |
| n:iso | 4 | 7.3 | 11.5 | 19 |
| Olefins | C2–C20 | Long-chain olefins | Small-chain olefins | Small-chain olefins |
| Process | Ruhrchemie BASF Kuhlmann | Shell | Low Pressure Oxo | Ruhrchemie/Rhone Polenc |
| Catalyst Precursor | Support | Substrate | TOFbulk [h−1] | Sum-sel.a [%] | n-Regio-sel. [%] | Ref. |
|---|---|---|---|---|---|---|
| [Rh6(CO)16] | MgO | propylene | 0.720 | 78 | 32 | [35] |
| TiO2 | propylene | 0.154 | 86 | 62 | [35] | |
| La2O3 | propylene | 0.038 | 50 | 75 | [35] | |
| ZnO | ethylene propylene | 3.60 1.23 | - - | - 70 | [35] [35] | |
| NaY | ethylene propylene | 1.32b 0.23c | 96 92 | - 57 | [36] [36] | |
| [Rh4(CO)12] | TiO2 | propylene | 0.41 | 70 | 56 | [35] |
| La2O3 | propylene | 0.77 | 45 | 68 | [35] | |
| SiO2 | ethylene propylene | 78b 0.04 | 66d 75 | - 63 | [41] [35,37,42] | |
| Al2O3 ZnO | propylene ethylene propylene | 0 9.79 2.05 | - - - | - - 58 | [35] [35] [35] | |
| [Fe3Rh2(CO)14C] | SiO2 | propylene | 5.04c | - | - | [37] |
| [RhCo3(CO)12] | SiO2 ZnO | ethylene propylene propylene | 1152b 396c 0.92 | 80d 67e - | - - 80 | [41,42] [41] [35] |
| [Rh2Co2(CO)12] | SiO2 ZnO | ethylene propylene propylene - | 504b 252c 1.34 | 74d 80e - | - - 70 | [41] [41] [35] |
| [(RhCp*CH3)2(CH2)2] | SiO2 | ethylene | 0.192 | 89 | - | [45] |
| [HRhCO(PPh3)]f | VPC | propylene | 2700 | 100 | 60 | [44] |
| Catalyst Precursor | Support | Ligand | Substrate | Ref. |
|---|---|---|---|---|
| [Rh2Co2(CO)12] | SiO2 | Amines | 1-hexene | [47] |
| Al2O3 | Amines | 1-hexene | [47] | |
| MgOx SiO2 | - | 1-hexene | [47] | |
| zeolites | - | 1-hexene | [47] | |
| [Rh(acac)(cod)] | MOF | - | C6–C16 olefins | [48,49] |
| [HRh(CO)(PPh3)3] | Hydrotalcite | TPPTS | 1-hexene | [50,51,52] |
| VPC | PPh3 | octene | [44] | |
| SPION | PPh3 | styrene | [53] |
| Phase | Space Group | Bravais Lattice | Structure | Ref. | Unit Cell of Rh2P |
|---|---|---|---|---|---|
| Rh2P | Fmm | Cubic | Anti-CaF2 | [106] |  |
| Rh3P | Pm2 | Cubic | Anti-PbFCl | [107] | |
| Rh4P3 | Pnma | Orthorhombic | Rn4P3 | [108] | |
| RhP2 | P21/c | Monoclinic | CoSb2 | [109] | |
| RhP3 | Im | Cubic | CoAs3 | [110] |
| Support | Ionic Liquid | Ligand | Substrate | TOF (h−1) | Sum-sel.a (%) | n-Regio-sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| SiO2 | [BMIM][OctSO4] | TPPTS-Cs | ethylene | 800 | 90 | - | [133] |
| SiO2 | [BMIM][OctSO4] | Sulpho- xantphos | propylene butylene | 60–500 564 | - - | 88–96 91 | [124,125,126,127,128] |
| SiO2 | [EMIM][NTf2] | Sulpho- xantphos Diphos- phite | C4-mixture | 3600 | - | 99 | [130] |
| N-doped carbon | [EMIM][NTf2] | Xantphos | ethylene | 650 | 95 | - | [131] |
| Section | Section 3.1 | Section 3.2 | Section 3.2 | Section 3.3 | Section 3.4 |
|---|---|---|---|---|---|
| Catalyst | [Rh4(CO)12] | Rh | PPh3-Rh | Rh2Pb | Rh SILPc |
| Rh [wt %] | 2.24 | 1.0 | 1.0 | 5.07 | 0.2 |
| C3H6 [%] | 32 | 33.33 | 33.33 | 10 | 32 |
| CO [%] | 32 | 33.33 | 33.33 | 50 | 34 |
| H2 [%] | 36 | 33.33 | 33.33 | 10 | 34 |
| H2O [%] | - | - | - | 20 | - |
| Ar [%] | - | - | - | 10 | - |
| p [MPa] | 0.07 | 1 | 1.0 | 2.0 | 1.0 |
| T [K] | 428 | 393 | 393 | 503 | 413 |
| GHSV | - | 2000 | 2000 | 3000 | 462 |
| Sum-sel.a [%] | 75 | - | 100 | 87 | 88 |
| Conv. [%] | - | 0.12 | 27.1 | 7.3 | 28.1 |
| TOFbulk [h−1] | 0.04 | 1.2 | 131 | 3.5 | 500 |
| TOFsurf [h−1] | - | - | - | 31.8 | - |
| Ref. | [35] | [58] | [58] | [114] | [126] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanf, S.; Alvarado Rupflin, L.; Gläser, R.; Schunk, S.A. Current State of the Art of the Solid Rh-Based Catalyzed Hydroformylation of Short-Chain Olefins. Catalysts 2020, 10, 510. https://doi.org/10.3390/catal10050510
Hanf S, Alvarado Rupflin L, Gläser R, Schunk SA. Current State of the Art of the Solid Rh-Based Catalyzed Hydroformylation of Short-Chain Olefins. Catalysts. 2020; 10(5):510. https://doi.org/10.3390/catal10050510
Chicago/Turabian StyleHanf, Schirin, Luis Alvarado Rupflin, Roger Gläser, and Stephan Andreas Schunk. 2020. "Current State of the Art of the Solid Rh-Based Catalyzed Hydroformylation of Short-Chain Olefins" Catalysts 10, no. 5: 510. https://doi.org/10.3390/catal10050510
APA StyleHanf, S., Alvarado Rupflin, L., Gläser, R., & Schunk, S. A. (2020). Current State of the Art of the Solid Rh-Based Catalyzed Hydroformylation of Short-Chain Olefins. Catalysts, 10(5), 510. https://doi.org/10.3390/catal10050510