Synthesis and Investigation of Pinane-Based Chiral Tridentate Ligands in the Asymmetric Addition of Diethylzinc to Aldehydes



Abstract

1. Introduction
2. Results
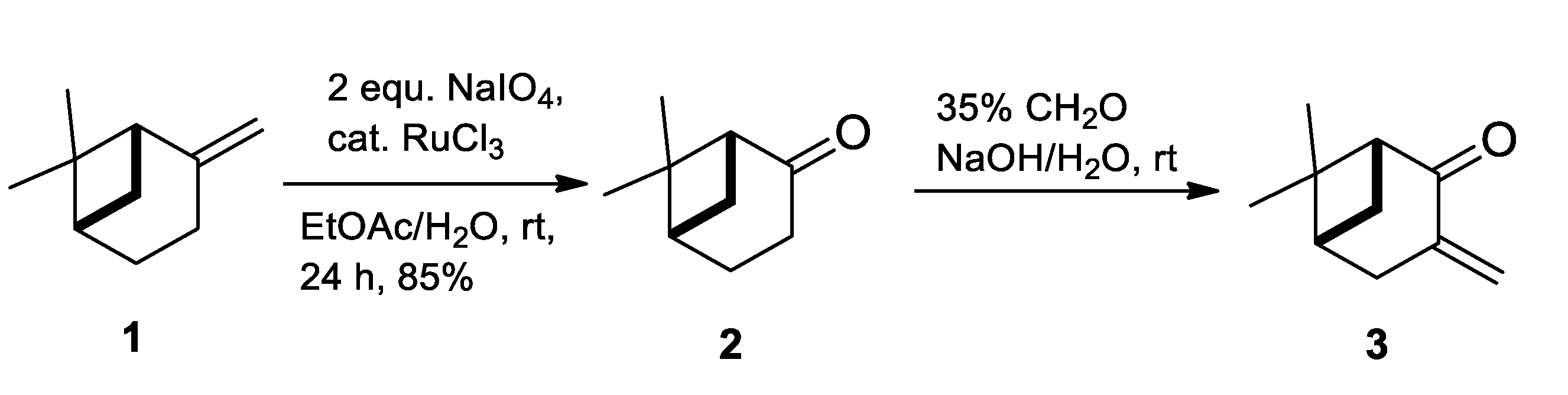
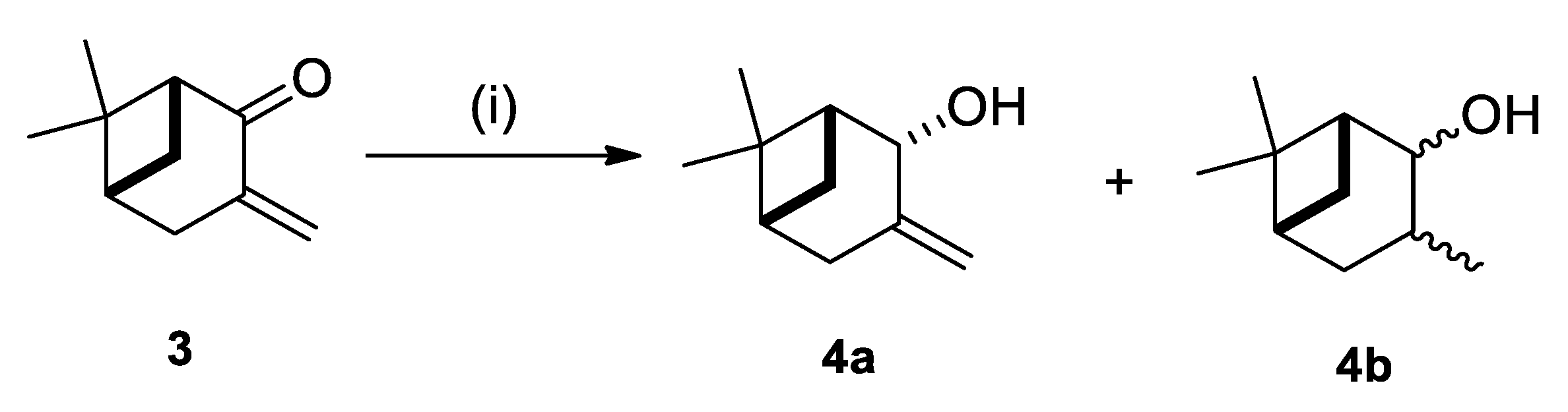
2.1. Synthesis of Allylic Alcohol 4a
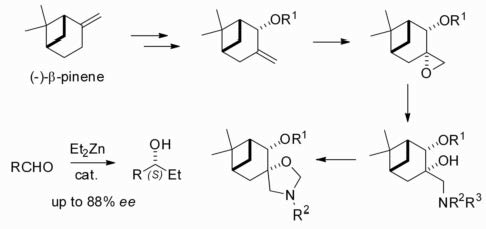
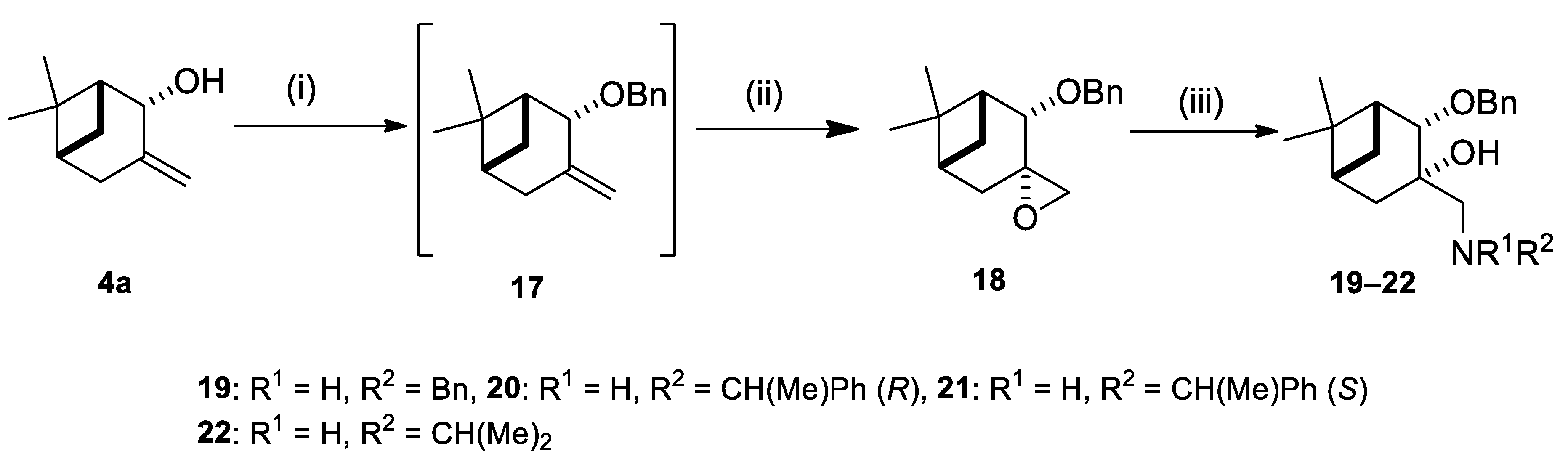
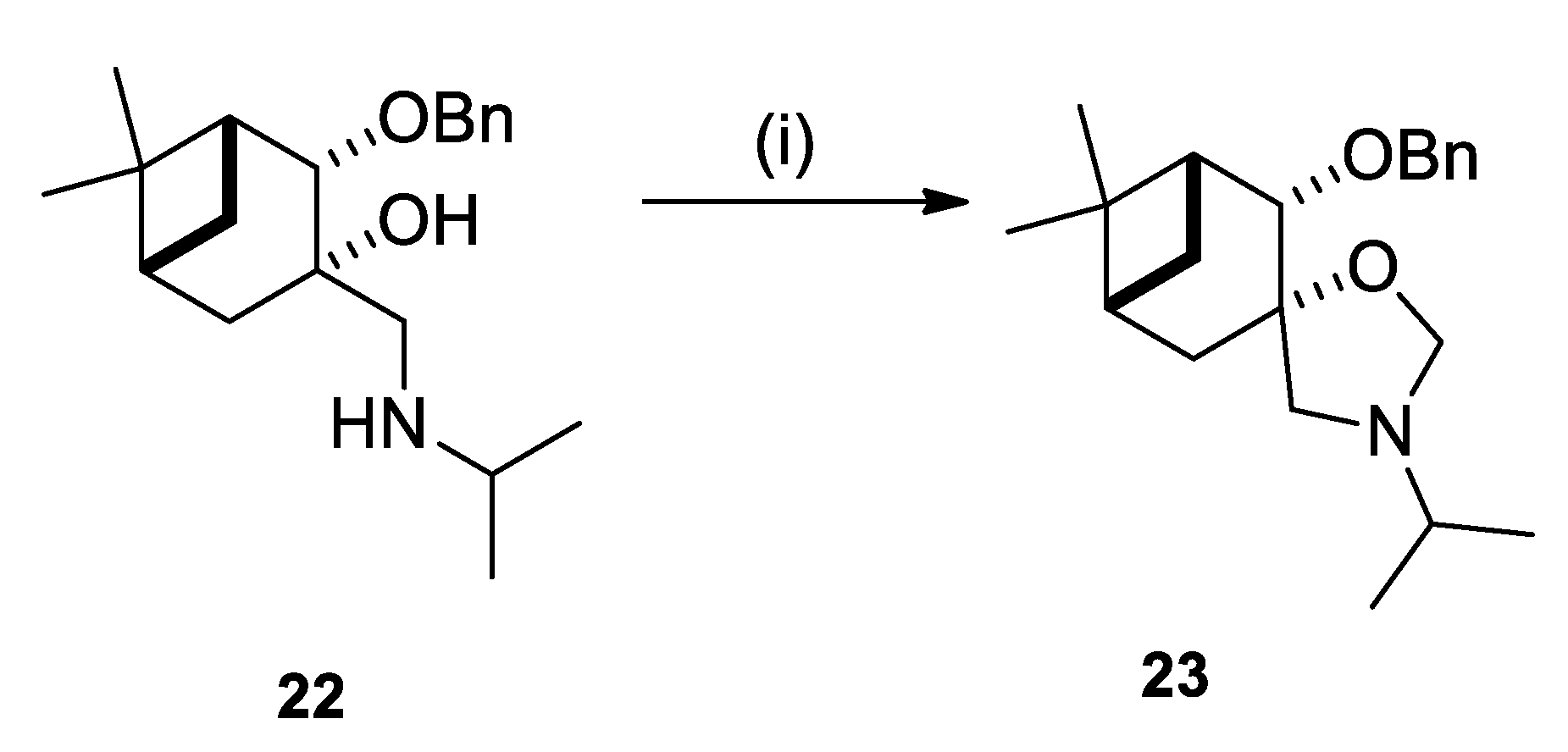
2.2. Synthesis of (–)-β-Pinene-Based Aminodiols
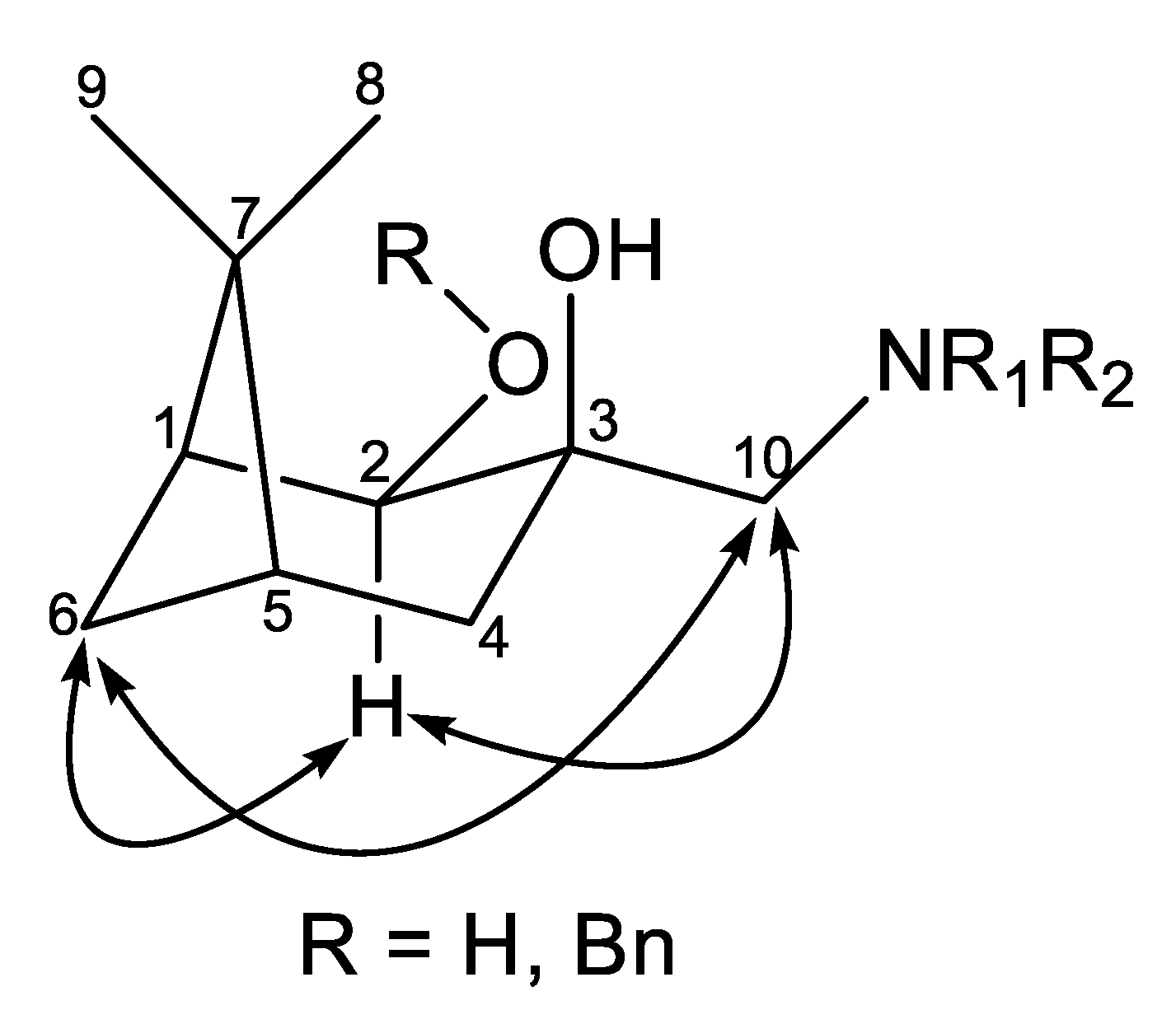
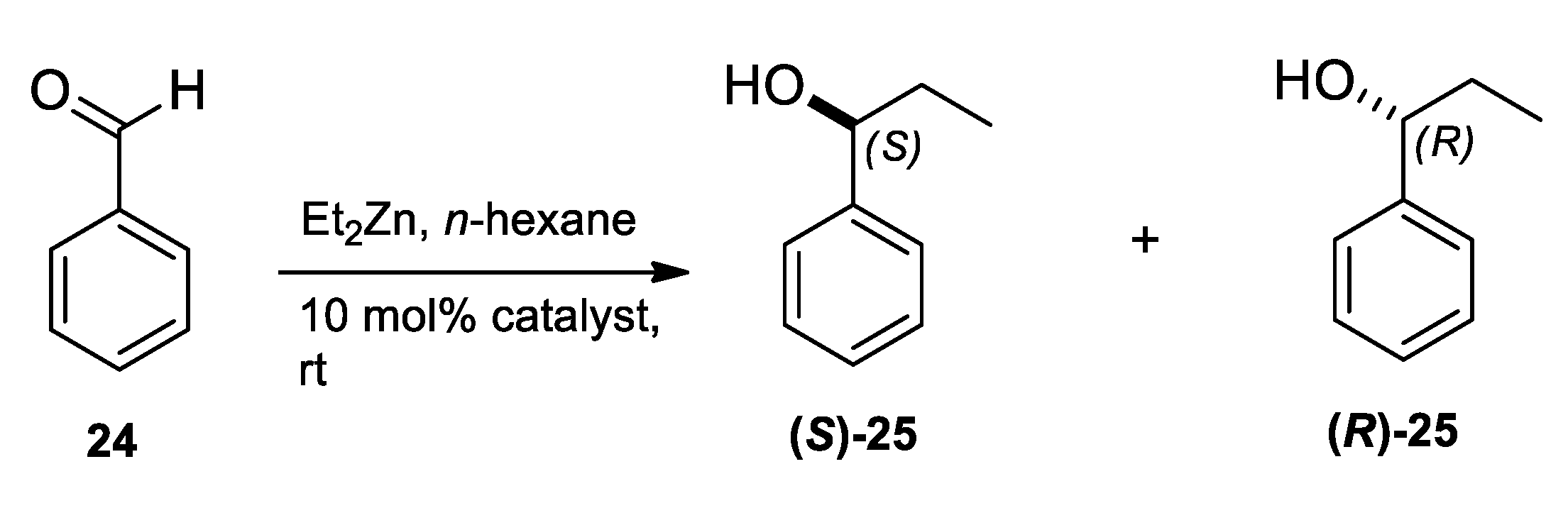
2.3. Application of Aminodiols as Chiral Ligands for Catalytic Addition of Diethylzinc to Aldehydes
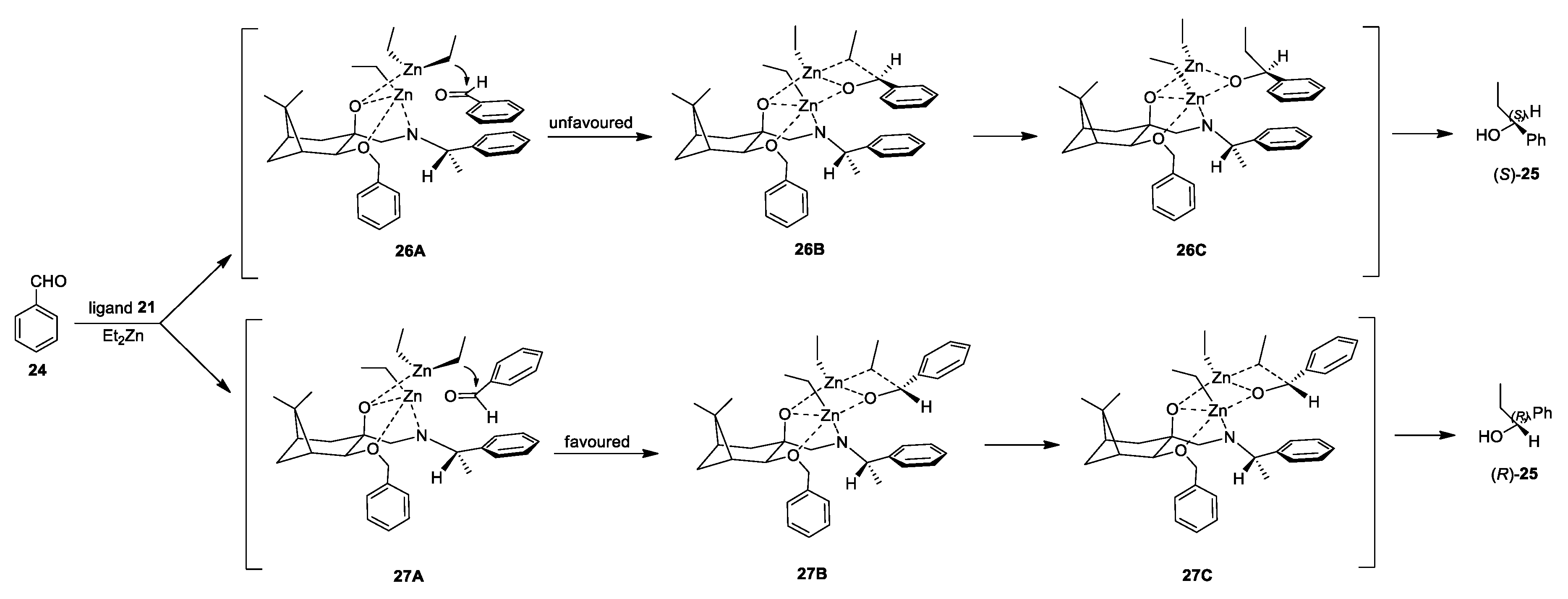
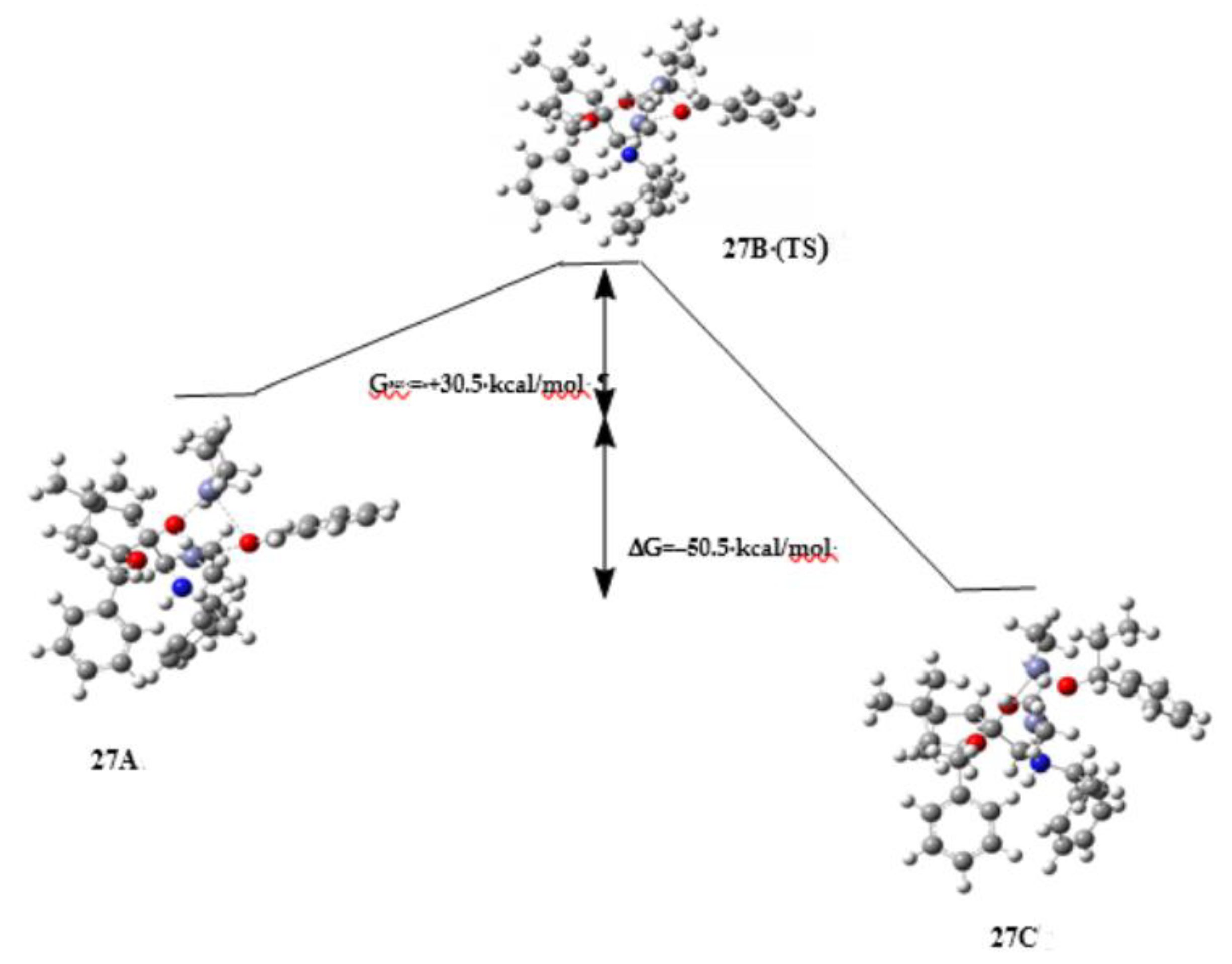
3. Discussion
4. Materials and Methods
4.1. Materials and General Methods
4.2. (1R,2R,5R)-6,6-Dimethyl-3-Methylenebicyclo [3.1.1]heptan-2-ol (4a)
4.3. (1R,2R,5R)-6,6-Dimethylspiro[bicyclo[3.1.1]heptane-3,2’-oxiran]-2-ol (5)
4.4. General Procedure for Ring Opening of Epoxide 5 with Primary and Secondary Amines
4.4.1. (1R,2R,3S,5R)-3-((Benzylamino)methyl)-6,6-Dimethylbicyclo [3.1.1]heptane-2,3-diol (6)
4.4.2. (1R,2S,3S,5R)-6,6-Dimethyl-3-((((R)-1-phenyl-ethyl)amino)methyl)bicyclo[3.1.1]heptane-2,3- diol (7)
4.4.3. (1R,2S,3S,5R)-6,6-Dimethyl-3-((((S)-1-phenyl-ethyl)amino)methyl)bicyclo[3.1.1]heptane-2,3- diol (8)
4.4.4. (1R,2R,3S,5R)-3-((Disopropylamino)methyl)-6,6-Dimethylbicyclo-[3.1.1]heptane-2,3-diol (9)
4.4.5. (1R,2S,3S,5R)-3-((Dibenzylamino)methyl)-6,6-Dimethylbicyclo[3.1.1]heptane-2,3-diol (10)
4.4.6. (1R,2S,3S,5R)-3-((Benzyl((R)-1-phenylethyl)amino)methyl)-6,6-dimethylbicyclo[3.1.1] heptane-2,3- diol (11)
4.4.7. (1R,2S,3S,5R)-3-((Benzyl((S)-1-phenylethyl)amino)methyl)-6,6-dimethylbicyclo[3.1.1] heptane-2,3- diol (12)
4.4.8. (1R,2S,3S,5R)-3-((4-Benzylpiperidin-1-yl)methyl)-6,6-dimethylbicyclo[3.1.1]heptane-2,3-diol (13)
4.4.9. (1R,2S,3S,5R)-3-((Benzylamino)methyl)-2-(benzyloxy)-6,6-dimethylbicyclo[3.1.1]heptan-3-ol (19)
4.4.10. (1R,2S,3S,5R)-2-(Benzyloxy)-6,6-dimethyl-3-((((R)-1-phenylethyl)amino)methyl)bicyclo[3.1.1] heptan-3-ol (20)
4.4.11. (1R,2S,3S,5R)-2-(Benzyloxy)-6,6-dimethyl-3-((((S)-1-phenylethyl)amino)methyl)bicyclo[3.1.1] heptan-3-ol (21)
4.4.12. (1R,2S,3S,5R)-2-(Benzyloxy)-3-((isopropylamino)methyl)-6,6-dimethylbicyclo[3.1.1]heptan-3- ol (22)
4.5. (1R,2R,3S,5R)-3-(Aminomethyl)-6,6-dimethylbicyclo-[3.1.1]heptane-2,3-diol (14)
4.6. General Procedure for Ring Closure of Aminodiol Derivatives with Formaldehyde
4.6.1. (1R,2S,3S,5R)-3’-Benzyl-6,6-dimethylspiro [bicyclo[3.1.1]heptane-3,5’-oxazolidin]-2-ol (15)
4.6.2. (1R,5R)-3’-Isopropyl-6,6-dimethylspiro[bicyclo[3.1.1]heptane-3,5’-oxazolidin]-2-ol (16)
4.6.3. (1R,2S,3S,5R)-2-(Benzyloxy)-3’-isopropyl-6,6-dimethylspiro[bicyclo[3.1.1]heptane-3,5’- oxazolidine] (23)
4.7. (1R,2S,5R)-2-(Benzyloxy)-6,6-dimethyl-3-methylenebicyclo[3.1.1]heptane (17)
4.8. (1R,2S,3S,5R)-2-(Benzyloxy)-6,6-dimethylspiro[bicyclo[3.1.1]heptane-3,2’-oxirane] (18)
4.9. General Procedure for the Reaction of Diethylzinc with Aldehydes in the Presence of Chiral Catalysts
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caprio, V.; Williams, J.M.J. Catalysis in Asymmetric Synthesis, 2nd ed.; Wiley: Hoboken, NJ, USA, 2009; ISBN 978-1-4051-9091-6. [Google Scholar]
- Satyanarayana, T.; Kagan, H.B. The multi-substrate screening of asymmetric catalysts. Adv. Synth. Catal. 2005, 347, 737–748. [Google Scholar] [CrossRef]
- Gaunt, M.J.; Johansson, C.C.C.; McNally, A.; Vo, N.T. Enantioselective organocatalysis. Drug Discov. Today 2007, 12, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Kitamura, M. Enantioselective addition of organometallic reagents to carbonyl compounds: Chirality transfer, multiplication, and amplification. Angew. Chem. Int. Ed. Engl. 1991, 30, 49–69. [Google Scholar] [CrossRef]
- Soai, K.; Niwa, S. Enantioselective addition of organozinc reagents to aldehydes. Chem. Rev. 1992, 92, 833–856. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C. Nonlinear stereochemical effects in asymmetric reactions. Tetrahedron Asymmetry 1997, 8, 2997–3017. [Google Scholar] [CrossRef]
- Hanessian, S.; Soma, U.; Dorich, S.; Deschênes-Simard, B. Total synthesis of (+)-ent-Cyclizidine: Absolute configurational confirmation of antibiotic M146791. Org. Lett. 2011, 13, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Kang, S.H. Total synthesis of Azithromycin. Angew. Chem. Int. Ed. 2009, 48, 1827–1829. [Google Scholar] [CrossRef]
- Pastó, M.; Moyano, A.; Pericàs, M.A.; Riera, A. An enantioselective, stereodivergent approach to anti- and syn-α-hydroxy-β-amino acids from anti-3-amino-1,2-diols. Synthesis of the ready for coupling taxotere® side chain. Tetrahedron Asymmetry 1996, 7, 243–262. [Google Scholar] [CrossRef]
- Ohsaki, A.; Ishiyama, H.; Yoneda, K.; Kobayashi, J. Secu’amamine A, a novel indolizidine alkaloid from Securinega suffruticosa var. amamiensis. Tetrahedron Lett. 2003, 44, 3097–3099. [Google Scholar] [CrossRef]
- Azumi, M.; Ogawa, K.; Fujita, T.; Takeshita, M.; Yoshida, R.; Furumai, T.; Igarashi, Y. Bacilosarcins A and B, novel bioactive isocoumarins with unusual heterocyclic cores from the marine-derived bacterium Bacillus subtilis. Tetrahedron 2008, 64, 6420–6425. [Google Scholar] [CrossRef]
- Faigl, F.; Erdélyi, Z.; Deák, S.; Nyerges, M.; Mátravölgyi, B. A new pyrrolidine-derived atropisomeric amino alcohol as a highly efficient chiral ligand for the asymmetric addition of diethylzinc to aldehydes. Tetrahedron Lett. 2014, 55, 6891–6894. [Google Scholar] [CrossRef]
- Pu, L.; Yu, H.-B. Catalytic asymmetric organozinc additions to carbonyl compounds. Chem. Rev. 2001, 101, 757–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-C.; Li, G.-W.; Hu, W.-B.; Hua, Y.-Z.; Song, X.; Lu, H.-J. A mathematical expression for the enantioselectivity and thermodynamic factors in the conformational equilibrium of catalysts in the addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2014, 25, 1360–1365. [Google Scholar] [CrossRef]
- Wiseman, J.M.; McDonald, F.E.; Liotta, D.C. 1-Deoxy-5-hydroxysphingolipids as new anticancer Principles: An efficient procedure for stereoselective syntheses of 2-amino-3,5-diols. Org. Lett. 2005, 7, 3155–3157. [Google Scholar] [CrossRef] [PubMed]
- Woltering, T.J.; Wostl, W.; Hilpert, H.; Rogers-Evans, M.; Pinard, E.; Mayweg, A.; Göbel, M.; Banner, D.W.; Benz, J.; Travagli, M.; et al. BACE1 inhibitors: A head group scan on a series of amides. Bioorg. Med. Chem. Lett. 2013, 23, 4239–4243. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Morioka, Y.; Tomida, M.; Takahashi, T.; Hattori, M.; Hanasaki, K.; Koike, K.; Chiba, H.; Shinohara, S.; Kanemasa, T.; et al. 2-Arylimino-5,6-dihydro-4H-1,3-thiazines as a new class of cannabinoid receptor agonists. Part 2: Orally bioavailable compounds. Bioorg. Med. Chem. Lett. 2007, 17, 3925–3929. [Google Scholar] [CrossRef]
- Kai, H.; Morioka, Y.; Murashi, T.; Morita, K.; Shinonome, S.; Nakazato, H.; Kawamoto, K.; Hanasaki, K.; Takahashi, F.; Mihara, S.; et al. 2-Arylimino-5,6-dihydro-4H-1,3-thiazines as a new class of cannabinoid receptor agonists. Part 1: Discovery of CB2 receptor selective compounds. Bioorg. Med. Chem. Lett. 2007, 17, 4030–4034. [Google Scholar] [CrossRef]
- Kai, H.; Morioka, Y.; Koriyama, Y.; Okamoto, K.; Hasegawa, Y.; Hattori, M.; Koike, K.; Chiba, H.; Shinohara, S.; Iwamoto, Y.; et al. 2-Arylimino-5,6-dihydro-4H-1,3-thiazines as a new class of cannabinoid receptor agonists. Part 3: Synthesis and activity of isosteric analogs. Bioorg. Med. Chem. Lett. 2008, 18, 6444–6447. [Google Scholar] [CrossRef]
- Xu, X.; Qian, X.; Li, Z.; Song, G.; Chen, W. Synthesis and fungicidal activity of fluorine-containing phenylimino-thiazolidines derivatives. J. Fluor. Chem. 2005, 126, 297–300. [Google Scholar] [CrossRef]
- Cherng, Y.-J.; Fang, J.-M.; Lu, T.-J. Pinane-type tridentate reagents for enantioselective reactions: Reduction of ketones and addition of diethylzinc to aldehydes. J. Org. Chem. 1999, 64, 3207–3212. [Google Scholar] [CrossRef]
- Bergmeier, S.C. The synthesis of vicinal amino alcohols. Tetrahedron 2000, 56, 2561–2576. [Google Scholar] [CrossRef]
- Braga, A.L.; Rubim, R.M.; Schrekker, H.S.; Wessjohann, L.A.; De Bolster, M.W.G.; Zeni, G.; Sehnem, J.A. The facile synthesis of chiral oxazoline catalysts for the diethylzinc addition to aldehydes. Tetrahedron Asymmetry 2003, 14, 3291–3295. [Google Scholar] [CrossRef]
- Matsuda, K.; Yamamoto, S.; Irie, M. Diastereoselective cyclization of a diarylethene having a chiral N-phenylethylamide substituent in crystals. Tetrahedron Lett. 2001, 42, 7291–7293. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Duque-Soladana, J.P.; Rosón, C.D. Regio- and stereoselective 6-exo-trig radical cyclisations onto chiral perhydro-1,3-benzoxazines: Synthesis of enantiopure 3-alkylpiperidines. Tetrahedron Asymmetry 2000, 11, 2809–2821. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Nieto, J.; Del Pozo, S. Synthesis of enantiopure 3-azabicyclo[3.2.0]heptanes by diastereoselective intramolecular [2 + 2] photocycloaddition reactions on chiral perhydro-1,3-benzoxazines. J. Org. Chem. 2003, 68, 4923–4931. [Google Scholar] [CrossRef]
- Alberola, A.; Andrés, C.; Pedrosa, R. Diastereoselective ring opening of 2-substituted N-Benzyl-4,4, 7α-trimethyl-trans-octahydro-1,3-benzoxazines by Grignard reagents. Highly enantioselective synthesis of primary amines. Synlett 1990, 1990, 763–765. [Google Scholar] [CrossRef]
- Andrés, C.; Nieto, J.; Pedrosa, R.; Villamañán, N. Synthesis of enantiopure primary amines by stereoselective ring opening of chiral octahydro-1,3-benzoxazines by Grignard and Organoaluminum reagents. J. Org. Chem. 1996, 61, 4130–4135. [Google Scholar] [CrossRef]
- Philipova, I.; Dimitrov, V.; Simova, S. Synthesis of new enantiopure aminodiols and their use as ligands for the addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 1999, 10, 1381–1391. [Google Scholar] [CrossRef]
- Zhang, A.-L.; Yang, L.-W.; Yang, N.-F.; Liu, D.-C. Investigation of catalytic activity and catalytic mechanism of chiral amino diol tridentate ligands in the asymmetric addition of aldehydes in the present of methyllithium reagent. J. Organomet. Chem. 2014, 768, 50–55. [Google Scholar] [CrossRef]
- Stoyanova, M.P.; Shivachev, B.L.; Nikolova, R.P.; Dimitrov, V. Highly efficient synthesis of chiral aminoalcohols and aminodiols with camphane skeleton. Tetrahedron Asymmetry 2013, 24, 1426–1434. [Google Scholar] [CrossRef]
- Schwindt, M.A.; Belmont, D.T.; Carlson, M.; Franklin, L.C.; Hendrickson, V.S.; Karrick, G.L.; Poe, R.W.; Sobieray, D.M.; Van De Vusse, J. Unique and efficient synthesis of [2S-(2 R*,3S*,4R*)]-2-amino-1-cyclohexyl-6-methyl-3,4-heptanediol, a popular C-terminal component of many renin inhibitors. J. Org. Chem. 1996, 61, 9564–9568. [Google Scholar] [CrossRef]
- Rigoli, J.W.; Guzei, I.A.; Schomaker, J.M. Aminodiols via stereocontrolled oxidation of methyleneaziridines. Org. Lett. 2014, 16, 1696–1699. [Google Scholar] [CrossRef] [PubMed]
- Panev, S.; Linden, A.; Dimitrov, V. Chiral aminoalcohols with a menthane skeleton as catalysts for the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2001, 12, 1313–1321. [Google Scholar] [CrossRef]
- Cherng, Y.J.; Fang, J.M.; Lu, T.J. A new pinane-type tridentate modifier for asymmetric reduction of ketones with lithium aluminum hydride. Tetrahedron Asymmetry 1995, 6, 89–92. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Hetényi, A.; Fulop, F. Synthesis of enantiomeric spirooxazolines and spirooxazolidines by the regioselective ring closure of (–)-α-pinene-based aminodiols. Arkivoc 2007, 2008, 33. [Google Scholar]
- Szakonyi, Z.; Csillag, K.; Fülöp, F. Stereoselective synthesis of carane-based aminodiols as chiral ligands for the catalytic addition of diethylzinc to aldehydes. Tetrahedron Asymmetry 2011, 22, 1021–1027. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Csőr, Á.; Csámpai, A.; Fülöp, F. Stereoselective synthesis and modelling-driven optimisation of Carane-based aminodiols and 1,3-oxazines as catalysts for the enantioselective addition of diethylzinc to benzaldehyde. Chem. Eur. J. 2016, 22, 7163–7173. [Google Scholar] [CrossRef]
- Lee, D.-S.; Chang, S.-M.; Ho, C.-Y.; Lu, T.-J. Enantioselective addition of diethylzinc to aldehydes catalyzed by chiral O, N, O-tridentate phenol ligands derived from Camphor. Chirality 2016, 28, 65–71. [Google Scholar] [CrossRef]
- Tashenov, Y.; Daniels, M.; Robeyns, K.; Van Meervelt, L.; Dehaen, W.; Suleimen, Y.; Szakonyi, Z. Stereoselective syntheses and application of chiral bi- and tridentate ligands derived from (+)-Sabinol. Molecules 2018, 23, 771. [Google Scholar] [CrossRef]
- Csillag, K.; Németh, L.; Martinek, T.A.; Szakonyi, Z.; Fülöp, F. Stereoselective synthesis of pinane-type tridentate aminodiols and their application in the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2012, 23, 144–150. [Google Scholar] [CrossRef]
- Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Stereoselective synthesis and application of tridentate aminodiols derived from (+)-pulegone. Tetrahedron Asymmetry 2016, 27, 480–486. [Google Scholar] [CrossRef]
- Le, T.M.; Szilasi, T.; Volford, B.; Szekeres, A.; Fülöp, F.; Szakonyi, Z. Stereoselective synthesis and investigation of Isopulegol-based chiral ligands. Int. J. Mol. Sci. 2019, 20, 4050. [Google Scholar] [CrossRef] [PubMed]
- Outouch, R.; Oubaassine, S.; Ait Ali, M.; El Firdoussi, L.; Spannenberg, A. Crystal structure of (1S,2R,4S)-1-[(morpholin-4-yl)methyl]-4-(prop-1-en-2-yl)cyclohexane-1,2-diol. Acta Crystallogr. Sect. E Crystallogr. Commun. 2015, 71, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.H.; Spina, K.P.; Woods, K.W.; Polakowski, J.; Martin, D.L.; Yao, Z.; Stein, H.H.; Cohen, J.; Barlow, J.L. Studies directed toward the design of orally active renin inhibitors. 1. Some factors influencing the absorption of small peptides. J. Med. Chem. 1993, 36, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P.L.; Gillard, J.; Bailey, M.; Beaulieu, C.; Duceppe, J.-S.; Lavallée, P.; Wernic, D. Practical synthesis of BILA 2157 BS, a potent and orally active renin inhibitor: Use of an enzyme-catalyzed hydrolysis for the preparation of homochiral succinic acid derivatives. J. Org. Chem. 1999, 64, 6622–6634. [Google Scholar] [CrossRef]
- Kawai, A.; Hara, O.; Hamada, Y.; Shioiri, T. Stereoselective synthesis of the hydroxy amino acid moiety of Al-77-B, a gastroprotective substance from Bacillus pumilus Al-77. Tetrahedron Lett. 1988, 29, 6331–6334. [Google Scholar] [CrossRef]
- Wang, G.T.; Li, S.; Wideburg, N.; Krafft, G.A.; Kempf, D.J. Synthetic chemical diversity: Solid phase synthesis of libraries of C2 symmetric inhibitors of HIV protease containing diamino diol and diamino alcohol cores. J. Med. Chem. 1995, 38, 2995–3002. [Google Scholar] [CrossRef]
- Grajewska, A.; Rozwadowska, M.D. Stereoselective synthesis of cytoxazone and its analogues. Tetrahedron Asymmetry 2007, 18, 803–813. [Google Scholar] [CrossRef]
- Mishra, R.K.; Coates, C.M.; Revell, K.D.; Turos, E. Synthesis of 2-Oxazolidinones from β-Lactams: Stereospecific Total Synthesis of (−)-Cytoxazone and All of Its Stereoisomers. Org. Lett. 2007, 9, 575–578. [Google Scholar] [CrossRef]
- Allepuz, A.C.; Badorrey, R.; Díaz-de-Villegas, M.D.; Gálvez, J.A. Diastereoselective reduction of ketimines derived from (R)-3,4-dihydroxybutan-2-one: An alternative route to key intermediates for the synthesis of anticancer agent ES-285. Tetrahedron Asymmetry 2010, 21, 503–506. [Google Scholar] [CrossRef]
- Malkov, A.V.; Baxendale, I.R.; Bella, M.; Langer, V.; Fawcett, J.; Russell, D.R.; Mansfield, D.J.; Valko, M.; Kočovský, P. Synthesis of new chiral 2,2′-bipyridyl-type ligands, their coordination to Molybdenum(0), Copper(II), and Palladium(II), and application in asymmetric allylic Substitution, allylic oxidation, and cyclopropanation. Organometallics 2001, 20, 673–690. [Google Scholar] [CrossRef]
- Chelucci, G.; Loriga, G.; Murineddu, G.; Pinna, G.A. Synthesis of chiral C2-symmetric 1,10-Phenanthrolines from naturally occurring monoterpenes. Synthesis 2003, 0073–0078. [Google Scholar] [CrossRef]
- Badjah-Hadj-Ahmed, A.Y.; Meklati, B.Y.; Waton, H.; Pham, Q.T. Structural studies in the bicyclo[3.1.1]heptane series by 1H and 13C NMR. Magn. Reson. Chem. 1992, 30, 807–816. [Google Scholar] [CrossRef]
- Molander, G.A.; Cavalcanti, L.N. Oxidation of Organotrifluoroborates via Oxone. J. Org. Chem. 2011, 76, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Gemal, A.L.; Luche, J.L. Lanthanoids in organic synthesis. 6. Reduction of alpha.-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J. Am. Chem. Soc. 1981, 103, 5454–5459. [Google Scholar] [CrossRef]
- Friedrich, D.; Bohlmann, F. Total synthesis of various elemanolides. Tetrahedron 1988, 44, 1369–1392. [Google Scholar] [CrossRef]
- Rodríguez-Berríos, R.R.; Torres, G.; Prieto, J.A. Stereoselective VO(acac)2 catalyzed epoxidation of acyclic homoallylic diols. Complementary preparation of C2-syn-3,4-epoxy alcohols. Tetrahedron 2011, 67, 830–836. [Google Scholar] [CrossRef]
- Shivani Pujala, B.; Chakraborti, A.K. Zinc(II) perchlorate hexahydrate Catalyzed opening of epoxide ring by amines: Applications to synthesis of (RS)/(R)-Propranolols and (RS)/(R)/(S)-Naftopidils. J. Org. Chem. 2007, 72, 3713–3722. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Hetényi, A.; Fülöp, F. Synthesis and application of monoterpene-based chiral aminodiols. Tetrahedron 2008, 64, 1034–1039. [Google Scholar] [CrossRef]
- Jia, Y.X.; Wu, B.; Li, X.; Ren, S.K.; Tu, Y.Q.; Chan, A.S.C.; Kitching, W. Synthetic studies of the HIV-1 protease inhibitive didemnaketals: Stereocontrolled synthetic approach to the key mother spiroketals. Org. Lett. 2001, 3, 847–849. [Google Scholar] [CrossRef]
- Kim, J.H.; Lim, H.J.; Cheon, S.H. Synthesis of (+)-hernandulcin and (+)-epihernandulcin. Tetrahedron Lett. 2002, 43, 4721–4722. [Google Scholar] [CrossRef]
- Waddell, T.G.; Ross, P.A. Chemistry of 3,4-epoxy alcohols. Fragmentation reactions. J. Org. Chem. 1987, 52, 4802–4804. [Google Scholar] [CrossRef]
- Kim, J.H.; Lim, H.J.; Cheon, S.H. A facile synthesis of (6S,1′S)-(+)-hernandulcin and (6S,1′R)-(+)-epihernandulcin. Tetrahedron 2003, 59, 7501–7507. [Google Scholar] [CrossRef]
- Tanaka, T.; Yasuda, Y.; Hayashi, M. New chiral Schiff base as a tridentate ligand for catalytic enantioselective addition of diethylzinc to aldehydes. J. Org. Chem. 2006, 71, 7091–7093. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, C.; Pastó, M.; Riera, A.; Pericàs, M.A. Modular amino alcohol ligands containing bulky alkyl groups as chiral controllers for Et2Zn addition to aldehydes: Illustration of a design principle. J. Org. Chem. 2003, 68, 3130–3138. [Google Scholar] [CrossRef] [PubMed]
- Roothaan, C.C.J. Self-consistent field theory for open shells of electronic systems. Rev. Mod. Phys. 1960, 32, 179–185. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 (Revision A.02); Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reductant | Additive | Solvent | T (°C) | t (h) | Ratio 4a/4b [a] | Yield [b] (%) |
|---|---|---|---|---|---|---|---|
| 1 | NaBH4 | - | MeOH | −20 | 6 | 3:1 | 86 |
| 2 | NaBH4 | - | MeOH | 0 | 1 | 1:1 | 86 |
| 3 | NaBH4 | CeCl3 | MeOH | 0 | 0.5 | 100:0 | 87 |
| 5 | NaBH4 | - | Et2O | 0 | 3 | 3:1 | 76 |
| 4 | NaBH4 | - | EtOH | 0 | 48 | 1:3 | 84 |
| Entry | Ligand a | Yield b (%) | eec (%) | Configuration d |
|---|---|---|---|---|
| 1 | 6 | 83 | 5 | (R) |
| 2 | 7 | 92 | 23 | (R) |
| 3 | 8 | 80 | 31 | (R) |
| 4 | 9 | 85 | 4 | (R) |
| 5 | 10 | 85 | 80 | (R) |
| 6 | 11 | 90 | 3 | (R) |
| 7 | 12 | 95 | 16 | (R) |
| 8 | 13 | 80 | 60 | (R) |
| 9 | 14 | 83 | - | - |
| 10 | 15 | 83 | 26 | (R) |
| 11 | 16 | 85 | 35 | (R) |
| 12 | 19 | 95 | 73 | (R) |
| 13 | 20 | 87 | 74 | (S) |
| 14 | 21 | 83 | 80 | (R) |
| 15 | 22 | 80 | 40 | (R) |
| 16 | 23 | 93 | 10 | (R) |
| Entry | Catalyst | Products | R | Yield a (%) | eeb (%) | Configuration c |
|---|---|---|---|---|---|---|
| 1 | 20 | 29a | (4-MeO)C6H4 | 80 | 92 | (S) |
| 2 | 20 | 29b | (3-MeO)C6H4 | 78 | 84 | (S) |
| 3 | 20 | 29c | (3-Me)C6H4 | 75 | 78 | (S) |
| 4 | 21 | 29a | (4-MeO)C6H4 | 83 | 85 | (R) |
| 5 | 21 | 29b | (3-MeO)C6H4 | 92 | 87 | (R) |
| 6 | 21 | 29c | (3-Me)C6H4 | 80 | 84 | (R) |
| 7 | 21 | 29d | cyclohexyl | 85 | 48 | (R) |
| 8 | 21 | 29e | n-butyl | 80 | 45 | (R) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raji, M.; Le, T.M.; Fülöp, F.; Szakonyi, Z. Synthesis and Investigation of Pinane-Based Chiral Tridentate Ligands in the Asymmetric Addition of Diethylzinc to Aldehydes. Catalysts 2020, 10, 474. https://doi.org/10.3390/catal10050474
Raji M, Le TM, Fülöp F, Szakonyi Z. Synthesis and Investigation of Pinane-Based Chiral Tridentate Ligands in the Asymmetric Addition of Diethylzinc to Aldehydes. Catalysts. 2020; 10(5):474. https://doi.org/10.3390/catal10050474
Chicago/Turabian StyleRaji, Mounir, Tam Minh Le, Ferenc Fülöp, and Zsolt Szakonyi. 2020. "Synthesis and Investigation of Pinane-Based Chiral Tridentate Ligands in the Asymmetric Addition of Diethylzinc to Aldehydes" Catalysts 10, no. 5: 474. https://doi.org/10.3390/catal10050474
APA StyleRaji, M., Le, T. M., Fülöp, F., & Szakonyi, Z. (2020). Synthesis and Investigation of Pinane-Based Chiral Tridentate Ligands in the Asymmetric Addition of Diethylzinc to Aldehydes. Catalysts, 10(5), 474. https://doi.org/10.3390/catal10050474