Engineering of Bifunctional Enzymes with Uricase and Peroxidase Activities for Simple and Rapid Quantification of Uric Acid in Biological Samples

 , and
, and
Abstract
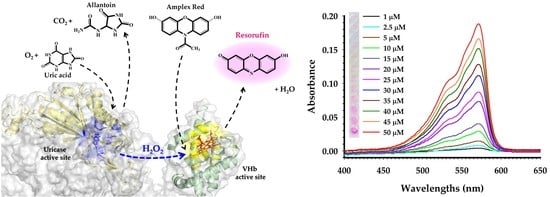
1. Introduction
2. Results and Discussion
2.1. Production of Native and Bifunctional Proteins with Uricase and Peroxidase Activities
2.2. Characterization of Enzymatic Activity and Kinetic Parameters
2.3. Optimization of Two-Enzymatic Cascade Reactions for Quantification of Uric Acid
2.4. Uric Acid Calibration Curve
2.5. Method Evaluation and Measurement of Uric Acid from Lyophilized Serum
3. Materials and Methods
3.1. Bacterial Strains, Plasmids and Chemicals
3.2. DNA Manipulations
3.3. Protein Expression and Purification
3.4. Molecular Weight Determination
3.5. Measurement of Enzymatic Activity and Kinetic Parameters
3.6. Investigation of Sequential Reactions Catalyzed by Uricase (CUOX) and Peroxidase (VHb)
3.7. Preparation of a UA Calibration Curve
3.8. Method Evaluation and Quantification of UA from Lyophilized Human Serum
3.9. Molecular Modeling
3.10. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dalbeth, N.; Choi, H.K.; Joosten, L.A.B.; Khanna, P.P.; Matsuo, H.; Perez-Ruiz, F.; Stamp, L.K. Gout. Nat. Rev. Dis. Primers 2019, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wen, X.; Kong, J. Recent progress on uric acid detection: A review. Crit. Rev. Anal. Chem. 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, X.; Lu, W.; Liao, H.; Liao, F. Uricase based methods for determination of uric acid in serum. Microchim. Acta 2009, 164, 1–6. [Google Scholar] [CrossRef]
- Domagk, G.F.; Schlicke, H.H. A colorimetric method using uricase and peroxidase for the determination of uric acid. Anal. Biochem. 1968, 22, 219–224. [Google Scholar] [CrossRef]
- Price, C.P.; James, D.R. Analytical reviews in clinical biochemistry: The measurement of urate. Ann. Clin. Biochem. 1988, 25, 484–498. [Google Scholar] [CrossRef]
- Gochman, N.; Schmitz, J.M. Automated determination of uric acid, with use of a uricase-peroxidase system. Clin. Chem. 1971, 17, 1154–1159. [Google Scholar] [CrossRef]
- Klose, S.; Stoltz, M.; Munz, E.; Portenhauser, R. Determination of uric acid on continuous-flow (AutoAnalyzer II and SMA) systems with a uricase/phenol/4-aminophenazone color test. Clin. Chem. 1978, 24, 250–255. [Google Scholar] [CrossRef]
- Trivedi, R.C.; Rebar, L.; Berta, E.; Stong, L. New enzymatic method for serum uric acid at 500 nm. Clin. Chem. 1978, 24, 1908–1911. [Google Scholar] [CrossRef]
- Fossati, P.; Prencipe, L.; Berti, G. Use of 3,5-dichloro-2-hydroxybenzenesulfonic acid/4-aminophenazone chromogenic system in direct enzymic assay of uric acid in serum and urine. Clin. Chem. 1980, 26, 227–231. [Google Scholar] [CrossRef]
- Majkić-Singh, N.; Stojanov, M.; Spasić, S.; Berkes, I. Spectrophotometric determination of serum uric acid by an enzymatic method with 2,2’-azino-di(3-ethylbenzthiazoline-6-sulfonate) (ABTS). Clin. Chim. Acta 1981, 116, 117–123. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Z.; Jiao, X.; Zhang, L.; Lv, Y. Uricase-Based Highly Sensitive and Selective Spectrophotometric Determination of Uric Acid Using BSA-Stabilized Au Nanoclusters as Artificial Enzyme. Spectrosc. Lett. 2012, 45, 511–519. [Google Scholar] [CrossRef]
- Legoux, R.; Delpech, B.; Dumont, X.; Guillemot, J.C.; Ramond, P.; Shire, D.; Caput, D.; Ferrara, P.; Loison, G. Cloning and expression in Escherichia coli of the gene encoding Aspergillus flavus urate oxidase. J. Biol. Chem. 1992, 267, 8565–8570. [Google Scholar] [PubMed]
- Itaya, K.; Yamamoto, T.; Fukumoto, J. Studies on yeast uricase. Agric. Biol. Chem. 1967, 31, 1256–1264. [Google Scholar]
- Zhao, Y.; Zhao, L.; Yang, G.; Tao, J.; Bu, Y.; Liao, F. Characterization of a uricase from Bacillus fastidious A.T.C.C. 26904 and its application to serum uric acid assay by a patented kinetic uricase method. Biotechnol. Appl. Biochem. 2006, 45, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sakasegawa, S.-I.; Misaki, H.; Sugiyama, M. Molecular cloning and expression of uricase gene from Arthrobacter globiformis in Escherichia coli and characterization of the gene product. J. Biosci. Bioeng. 2004, 98, 153–158. [Google Scholar] [CrossRef]
- Bomalaski, J.S.; Holtsberg, F.W.; Ensor, C.M.; Clark, M.A. Uricase formulated with polyethylene glycol (uricase-PEG 20): Biochemical rationale and preclinical studies. J. Rheumatol. 2002, 29, 1942–1949. [Google Scholar]
- Saud Al-Bagmi, M.; Shahnawaz Khan, M.; Alhasan Ismael, M.; Al-Senaidy, A.M.; Ben Bacha, A.; Mabood Husain, F.; Alamery, S.F. An efficient methodology for the purification of date palm peroxidase: Stability comparison with horseradish peroxidase (HRP). Saudi J. Biol. Sci. 2019, 26, 301–307. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, Y.; Yang, X.; Zhao, H.; Hu, X.; Pu, J.; Liao, J.; Long, G.; Liao, F. Optimization of pH values to formulate the bireagent kit for serum uric acid assay. Biotechnol. Appl. Biochem. 2015, 62, 137–144. [Google Scholar] [CrossRef]
- Lavery, C.B.; Macinnis, M.C.; Macdonald, M.J.; Williams, J.B.; Spencer, C.A.; Burke, A.A.; Irwin, D.J.G.; D’Cunha, G.B. Purification of peroxidase from Horseradish (Armoracia rusticana) roots. J. Agric. Food Chem. 2010, 58, 8471–8476. [Google Scholar] [CrossRef] [PubMed]
- Spadiut, O.; Herwig, C. Production and purification of the multifunctional enzyme horseradish peroxidase. Pharm. Bioprocess. 2013, 1, 283–295. [Google Scholar] [CrossRef]
- Morawski, B.; Lin, Z.; Cirino, P.; Joo, H.; Bandara, G.; Arnold, F.H. Functional expression of horseradish peroxidase in Saccharomyces cerevisiae and Pichia pastoris. Protein Eng. 2000, 13, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Krainer, F.W.; Dietzsch, C.; Hajek, T.; Herwig, C.; Spadiut, O.; Glieder, A. Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway. Microb. Cell Fact. 2012, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Santama, N.; Dacey, S.; Edwards, M.; Bray, R.C.; Thorneley, R.N.; Burke, J.F. Expression of a synthetic gene for horseradish peroxidase C in Escherichia coli and folding and activation of the recombinant enzyme with Ca2+ and heme. J. Biol. Chem. 1990, 265, 13335–13343. [Google Scholar] [PubMed]
- Grigorenko, V.; Chubar, T.; Kapeliuch, Y.; Börchers, T.; Spener, F.; Egorova, A. New approaches for functional expression of recombinant horseradish peroxidase C in Escherichia coli. Biocatal. Biotransform. 1999, 17, 359–379. [Google Scholar] [CrossRef]
- Asad, S.; Dabirmanesh, B.; Ghaemi, N.; Etezad, S.M.; Khajeh, K. Studies on the refolding process of recombinant horseradish peroxidase. Mol. Biotechnol. 2013, 54, 484–492. [Google Scholar] [CrossRef]
- Gundinger, T.; Spadiut, O. A comparative approach to recombinantly produce the plant enzyme horseradish peroxidase in Escherichia coli. J. Biotechnol. 2017, 248, 15–24. [Google Scholar] [CrossRef]
- Humer, D.; Spadiut, O. Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis. Int. J. Mol. Sci. 2019, 20, 916. [Google Scholar] [CrossRef]
- Kvist, M.; Ryabova, E.S.; Nordlander, E.; Bülow, L. An investigation of the peroxidase activity of Vitreoscilla hemoglobin. J. Biol. Inorg. Chem. 2007, 12, 324–334. [Google Scholar] [CrossRef]
- Isarankura-Na-Ayudhya, C.; Yainoy, S.; Tantimongcolwat, T.; Bülow, L.; Prachayasittikul, V. Engineering of a novel chimera of superoxide dismutase and Vitreoscilla hemoglobin for rapid detoxification of reactive oxygen species. J. Biosci. Bioeng. 2010, 110, 633–637. [Google Scholar] [CrossRef]
- Isarankura-Na-Ayudhya, C.; Tansila, N.; Worachartcheewan, A.; Bülow, L.; Prachayasittikul, V. Biochemical and cellular investigation of Vitreoscilla hemoglobin (VHb) variants possessing efficient peroxidase activity. J. Microbiol. Biotechnol. 2010, 20, 532–541. [Google Scholar]
- Li, W.; Zhang, Y.; Xu, H.; Wu, L.; Cao, Y.; Zhao, H.; Li, Z. pH-induced quaternary assembly of Vitreoscilla hemoglobin: The monomer exhibits better peroxidase activity. Biochim. Biophys. Acta 2013, 1834, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Giangiacomo, L.; Mattu, M.; Arcovito, A.; Bellenchi, G.; Bolognesi, M.; Ascenzi, P.; Boffi, A. Monomer-dimer equilibrium and oxygen binding properties of ferrous Vitreoscilla hemoglobin. Biochemistry 2001, 40, 9311–9316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Yainoy, S.; Phuadraksa, T.; Wichit, S.; Sompoppokakul, M.; Songtawee, N.; Prachayasittikul, V.; Isarankura-Na-Ayudhya, C. Production and Characterization of Recombinant Wild Type Uricase from Indonesian Coelacanth (L. menadoensis) and Improvement of Its Thermostability by In Silico Rational Design and Disulphide Bridges Engineering. Int. J. Mol. Sci. 2019, 20, 1269. [Google Scholar] [CrossRef]
- Liu, C.Y.; Webster, D.A. Spectral characteristics and interconversions of the reduced oxidized, and oxygenated forms of purified cytochrome o. J. Biol. Chem. 1974, 249, 4261–4266. [Google Scholar]
- Dröse, S.; Galkin, A.; Brandt, U. Chapter 26 Measurement of superoxide formation by mitochondrial complex I of Yarrowia lipolytica. Meth. Enzymol. 2009, 456, 475–490. [Google Scholar]
- Ovádi, J.; Tompa, P.; Vértessy, B.; Orosz, F.; Keleti, T.; Welch, G.R. Transient-time analysis of substrate-channelling in interacting enzyme systems. Biochem. J. 1989, 257, 187–190. [Google Scholar] [CrossRef]
- Spivey, H.O.; Ovádi, J. Substrate channeling. Methods 1999, 19, 306–321. [Google Scholar] [CrossRef]
- Liu, M.; He, Y.; Zhou, J.; Ge, Y.; Zhou, J.; Song, G. A “naked-eye” colorimetric and ratiometric fluorescence probe for uric acid based on Ti3C2 MXene quantum dots. Anal. Chim. Acta 2020, 1103, 134–142. [Google Scholar] [CrossRef]
- Wang, X.; Yao, Q.; Tang, X.; Zhong, H.; Qiu, P.; Wang, X. A highly selective and sensitive colorimetric detection of uric acid in human serum based on MoS2-catalyzed oxidation TMB. Anal. Bioanal. Chem. 2019, 411, 943–952. [Google Scholar] [CrossRef]
- Lu, H.-F.; Li, J.-Y.; Zhang, M.-M.; Wu, D.; Zhang, Q.-L. A highly selective and sensitive colorimetric uric acid biosensor based on Cu(II)-catalyzed oxidation of 3,3′,5,5′-tetramethylbenzidine. Sens. Actuators B Chem. 2017, 244, 77–83. [Google Scholar] [CrossRef]
- Zhuang, Q.-Q.; Lin, Z.-H.; Jiang, Y.-C.; Deng, H.-H.; He, S.-B.; Su, L.-T.; Shi, X.-Q.; Chen, W. Peroxidase-like activity of nanocrystalline cobalt selenide and its application for uric acid detection. Int. J. Nanomed. 2017, 12, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xiong, Y.; Liao, C.; Ye, F. Colorimetric detection of uric acid in human urine and serum based on peroxidase mimetic activity of MIL-53(Fe). Anal. Methods 2015, 7, 9894–9899. [Google Scholar] [CrossRef]
- Suárez, A.S.G.; Stefan, A.; Lemma, S.; Conte, E.; Hochkoeppler, A. Continuous enzyme-coupled assay of phosphate- or pyrophosphate-releasing enzymes. BioTechniques 2012, 53, 99–103. [Google Scholar] [CrossRef] [PubMed]
- George, P. The chemical nature of the second hydrogen peroxide compound formed by cytochrome c peroxidase and horseradish peroxidase. I. Titration with reducing agents. Biochem. J. 1953, 54, 267–276. [Google Scholar] [CrossRef]
- Roskoski, R. Enzyme Assays☆. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger PyMOL: The PyMOL Molecular Graphics System; Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Molecular Weight (kDa) | Uricase Activity | Peroxidase Activity | ||||||
|---|---|---|---|---|---|---|---|---|---|
| U/mg | U/μmol | U/mg (%) | U/μmol (%) | U/mg | U/μmol | U/mg (%) | U/μmol (%) | ||
| CUOX | 143 | 9.7 ± 0.2 | 1387.1 ± 28.6 | 100 | 100 | - | - | - | - |
| VHb | 35 | - | - | - | - | 20.0 ± 0.5 | 700.00 ± 17.5 | 100 | 100 |
| CV | 207 | 6.7 ± 0.3 * | 1386.9 ± 71.7 | 69.07 | 99.98 | 0.68 ± 0.1 ** | 140.76 ± 22.4 | 3.40 | 20.10 |
| VC | 207 | 5.9 ± 0.1 * | 1221.3 ± 31.1 | 60.82 | 88.04 | 0.54 ± 0.1 ** | 111.78 ± 16.4 | 2.70 | 15.96 |
| Protein | Uricase | Peroxidase | ||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (mM−1s−1) | (mM) | kcat (s−1) | (mM−1s−1) | |
| CUOX | 0.079 | 29.08 | 368.04 | - | - | - |
| VHb | - | - | - | 1.84 | 4.72 | 2.56 |
| CV | 0.052 | 38.77 | 735.01 | 2.12 | 2.15 | 1.02 |
| VC | 0.046 | 46.93 | 1003.83 | 4.04 | 2.02 | 0.50 |
| Analytical Method | Uricase/rxn | Peroxidase or Peroxidase-Like/ rxn | Reaction Phase with Optimal Condition (pH/Temp./Time) | Total Reaction Time | Total Volume (μL) | Linear Range (μM) | LOD (μM) | Reference |
|---|---|---|---|---|---|---|---|---|
| 1. Uricase/HRP/ oxPOD and GSH-MQDs | 200 μg * | HRP 20 μg * | 1. pH NR **, 37 °C, 30 min 2. pH 7.2–7.4, 37 °C, time NR ** | >30 min | 2000 | 1.2–100 | 0.2 | [39] |
| 2. Uricase/ MoS2/TMB | 2.5 μg * | MoS2 0.18 μg | 1. pH 8.5, 35 °C, 15 min 2. pH 4, 50 °C, 60 min | 75 min | 2000 | 0.5–100 | 0.3 | [40] |
| 3. Uricase/Cu2+/ TMB | 2.5 μg * | Cu2+ 8 μmol | 1. pH 8.5, 35 °C, 15 min 2. pH 4, 45 °C, 60 min | 75 min | 2000 | 1–100 | 0.64 | [41] |
| 4. Uricase/ Co-Se-nano- crystalline/ 4-AAP/TOPS | 1 U | Co-Se- nanocrystal 260 μg | 1. pH 8.5, 40 °C, 10 min 2. pH 8.5, 40 °C, 30 min | 40 min | 4000 | 2–40 | 0.5 | [42] |
| 5. Uricase/ MIL53(Fe)/ TMB | 0.1 U | MIL53(Fe) 20 μg | 1. pH 9.0, 37 °C, 15 min 2. pH 4, 55 °C, 40 min | 55 min | 1000 | 4.5–60 | 1.3 | [43] |
| 6. Uricase/HRP/ 4-AAP/TOOS | 0.4 U | HRP 1.2 U | 1. pH 6.5, 25 °C, 5 min 2. pH 7.2–7.6, 25°C, 20 min | 25 min | 1020 | Up to 34 | 12 | [18] |
| 7. UOX/VHb/ Amplex Red | 0.05 U | VHb 0.005 U | 1. pH 8.0, 30 °C, 10 min | 10 min | 100 | 2.5–50 | 1 | This study |
| Detected (μM) | Added (μM) | Found (μM) | Recovery (%) | RSD (%, n = 5) |
|---|---|---|---|---|
| 327 ± 5.2 | 50 | 374.4 ± 6.1 | 94.8 | 1.63 |
| 75 | 399.4 ± 7.4 | 96.5 | 1.85 | |
| 100 | 424.0 ± 8.0 | 97.0 | 1.90 | |
| 125 | 452.6 ± 10.0 | 100.5 | 2.22 | |
| 150 | 477.2 ± 10.4 | 100.1 | 2.19 |
| Sample | Developed Assay | Automated Analyzer | Relative Error (%) | Average Relative Error (%) |
|---|---|---|---|---|
| Serum 1 | 285 | 298 | 4.5 | 3.06 |
| Serum 2 | 349 | 360 | 3.1 | |
| Serum 3 | 466 | 453 | 2.7 | |
| Serum 4 | 539 | 525 | 2.5 | |
| Serum 5 | 672 | 655 | 2.5 |
| Primers | Description | DNA sequence (5′ → 3′) |
|---|---|---|
| CUOX_FP | for construction of pETDuetCUOX | CGGGATCCCATGTCAACAACGCTCTCATC |
| CUOX_RP | CCCAAGCTTTTACAACTTGGTCTTCTCC | |
| Remove XhoI CUOX_FP | for removal of XhoI site from CUOX gene | CCACCTTTGCTCTTGAGAACTCTCCATCTG |
| Remove XhoI CUOX_RP | CAGATGGAGAGTTCTCAAGAGCAAAGGTGG | |
| Remove stop CUOX_FP | for removal of stop codon from CUOX gene | GAGAAGACCAAGTTGAAGCTTGCGGCCGC |
| Remove stop CUOX_RP | GCGGCCGCAAGCTTCAACTTGGTCTTCTC | |
| Insert HindIII VHb_FP | for insertion of HindIII site to VHb gene | CATCACGTGGATGACGACAAGCTTATGTTAGACCAGCAAACC |
| Insert HindIII VHb_RP | GGTTTGCTGGTCTAACATAAGCTTGTCGTCATCCACGTGATG | |
| Insert BamHI VHb_FP | for insertion of BamHI site to VHb gene | GCTCAAGCGGTTGAAGGATCCACCGGGCTTCTCCTC |
| Insert BamHI VHb_RP | GAGGAGAAGCCCGGTGGATCCTTCAACCGCTTGAGC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phuadraksa, T.; Chittrakanwong, J.; Tullayaprayouch, K.; Onsirisakul, N.; Wichit, S.; Yainoy, S. Engineering of Bifunctional Enzymes with Uricase and Peroxidase Activities for Simple and Rapid Quantification of Uric Acid in Biological Samples. Catalysts 2020, 10, 428. https://doi.org/10.3390/catal10040428
Phuadraksa T, Chittrakanwong J, Tullayaprayouch K, Onsirisakul N, Wichit S, Yainoy S. Engineering of Bifunctional Enzymes with Uricase and Peroxidase Activities for Simple and Rapid Quantification of Uric Acid in Biological Samples. Catalysts. 2020; 10(4):428. https://doi.org/10.3390/catal10040428
Chicago/Turabian StylePhuadraksa, Thanawat, Jurairat Chittrakanwong, Kittitouch Tullayaprayouch, Naruthai Onsirisakul, Sineewanlaya Wichit, and Sakda Yainoy. 2020. "Engineering of Bifunctional Enzymes with Uricase and Peroxidase Activities for Simple and Rapid Quantification of Uric Acid in Biological Samples" Catalysts 10, no. 4: 428. https://doi.org/10.3390/catal10040428
APA StylePhuadraksa, T., Chittrakanwong, J., Tullayaprayouch, K., Onsirisakul, N., Wichit, S., & Yainoy, S. (2020). Engineering of Bifunctional Enzymes with Uricase and Peroxidase Activities for Simple and Rapid Quantification of Uric Acid in Biological Samples. Catalysts, 10(4), 428. https://doi.org/10.3390/catal10040428