Abstract
Nickel phosphide (Ni2P) is a promising material for the electrocatalytic generation of hydrogen from water. Here, we present a chemical picture of the fundamental mechanism of Volmer–Tafel steps in hydrogen evolution reaction (HER) activity under alkaline conditions at the (0001) and (100) surfaces of Ni2P using dispersion-corrected density functional theory calculations. Two terminations of each surface (Ni3P2- and Ni3P-terminated (0001); and Ni2P- and NiP-terminated (100)), which have been shown to coexist in Ni2P samples depending on the experimental conditions, were studied. Water adsorption on the different terminations of the Ni2P (0001) and (100) surfaces is shown to be exothermic (binding energy in the range of 0.33−0.68 eV) and characterized by negligible charge transfer to/from the catalyst surface (0.01−0.04 e−). High activation energy barriers (0.86−1.53 eV) were predicted for the dissociation of water on each termination of the Ni2P (0001) and (100) surfaces, indicating sluggish kinetics for the initial Volmer step in the hydrogen evolution reaction over a Ni2P catalyst. Based on the predicted Gibbs free energy of hydrogen adsorption (ΔGH*) at different surface sites, we found that the presence of Ni3-hollow sites on the (0001) surface and bridge Ni-Ni sites on the (100) surface bind the H atom too strongly. To achieve facile kinetics for both the Volmer and Heyrovsky–Tafel steps, modification of the surface structure and tuning of the electronic properties through transition metal doping is recommended as an important strategy.
1. Introduction
Rising global energy demands and the serious concerns of environmental contamination necessitate the development of renewable energy sources to alleviate our reliance on fossil fuels and simultaneously satisfy increasingly stringent environmental regulations. Molecular hydrogen (H2) is considered a promising energy carrier to meet future global energy demands owing to its high energy density and environmentally benign characteristics [1,2]. Among the several sources of H2 generation, the electrocatalytic hydrogen evolution reaction (HER) from water splitting is the most economical and effective route for a future hydrogen economy [3,4]. Although Pt-group metals are currently considered the best HER electrocatalysts owing to the optimum Gibb’s free energy of hydrogen adsorption (ΔGH* ≈ 0.09 eV) [5,6,7], the high cost and scarcity of these noble metal-based electrocatalysts severely limits their scalable applications [8]. Therefore, the development of cost-effective and earth-abundant catalysts as possible alternatives has received significant research interest recently.
Non-noble metal-based electrocatalysts—such as tungsten and molybdenum chalcogenides [9], carbides [10], nitrides [11], phosphides [12,13], and phosphosulfides [14,15] have received considerable attention. Among the reported catalysts, transition metal phosphides such as CoP [16], FeP [17], and Ni2P [18,19,20] have been regarded as very promising electrocatalysts for the HER owing to their low-cost, appropriate electronic structures, and high electrochemical stability. Nickel phosphide (Ni2P) in particular is an emerging catalyst for hydrogen evolution reactions [20,21,22,23], hydrodesulfurization (HDS) [24,25], hydrodenitrogenation (HDN) [26,27,28], hydrodeoxygenation (HDO) [29], hydrodechlorination (HDCl) [30], and water–gas shift reactions [31]. Ni2P is considered an attractive alternative to noble metal catalysts for HER [20] because both of its constituent elements nickel and phosphorous are cheap, abundant, and non-toxic, which makes Ni2P a promising cost-effective material for scalable renewable energy conversion systems.
Nickel phosphides are stable and durable in strong acid and alkali conditions, prolonging the turnover number (TON) and lifetime of the catalyst and hence can achieve enhanced HER efficiency [32,33]. The Ni2P (0001) surface is the most studied facet for HER activity owing to its comparable predicted hydrogen evolution activity to that of hydrogenase [22,34,35,36]. Earlier investigations have considered HER activity in acidic medium over Ni2P catalysts, whereby the Volmer–Tafel mechanism (H+(aq) → H*, 2H* → ↑H2) involves only characterizing the Gibb’s free energy hydrogen adsorption to the catalyst surface [22,23]. There exist limited studies of the HER activity of Ni2P under alkaline conditions and the underlying mechanism is still poorly understood [37,38]. The mechanism of the HER in alkaline media is slightly different to acidic media and can typically be treated as a combination of three elementary steps: the Volmer step—water dissociation and formation of a reactive intermediate (H2O + e− + cat → H*-cat + OH−)—followed by either the Heyrovsky step (H*-cat + H2O + e- → cat + OH− + ↑H2) or the Tafel recombination step (2H*-cat → ↑H2). Thus, the HER activity of an electrocatalyst in alkaline conditions is dominated by the prior Volmer step and subsequent Tafel step (i.e., Volmer reaction is the rate-determining step for the HER in alkaline electrolytes) [39].
The HER activity is often reported to be severely hindered by sluggish kinetics and high overpotential over Ni2P catalyst [12]. However, an atomic-level picture of the nature of the Ni2P catalyst surfaces and active sites that dictate the fundamental adsorption of water and the subsequent Volmer–Tafel steps in the hydrogen evolution reaction over Ni2P catalyst has not been investigated comprehensively. This information and insights are, however, vital in the quest to rationally design more active and stable Ni2P electrocatalysts. In this study, first-principles density functional theory (DFT) calculations were employed to investigate the mechanisms of water adsorption and dissociation (H2O → OH− + H+) over Ni2P. The investigation provides insight of thermodynamic stability, active sites of adsorption, and activation barriers on the Ni2P (0001) and (100) surfaces. The geometries for molecular water adsorption were first investigated, followed by climbing image nudged elastic band (CI-NEB) methodology to determine the activation energy barriers for HER on each surface. The Gibbs free energy of hydrogen adsorption ΔGH*, which quantifies the strength of H adsorption was calculated to provide insight into the HER activity of the different surfaces and terminations of the Ni2P electrocatalyst in an alkaline environment.
2. Results and Discussion
2.1. Ni2P Bulk and Surface Characterization
Nickel phosphide (Ni2P) crystallizes in the hexagonal structure (Figure 1a) and belongs to space group P2m with a = b = 5.859 Å and c = 3.382 Å [40]. The fully optimized bulk Ni2P cell parameters (a = b = 5.821 Å and c = 3.320 Å) are within 2% of experimental values [40]. The calculated Ni−Ni and Ni−P bond lengths are 2.589 Å and 2.289 Å, which are both also within 2% of the experimental values [41]. The calculated partial density of states (PDOS) shown in Figure 1b reveals the metallic character of Ni2P, whereby the Ni d-states dominate the regions around the Fermi level, which is consistent with earlier DFT results [23,42].
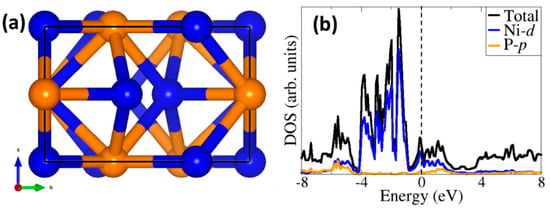
Figure 1.
(a) Hexagonal structure of bulk Ni2P and (b) projected density of states (PDOS) showing the metallic character of Ni2P. Colour code: Ni = blue, P = orange.
The Ni2P (0001) and (100) surfaces were created from the fully relaxed bulk structure in order to eliminate any residual forces which may be present during surface relaxation. All possible terminations that ensure symmetric slab models were investigated in order to determine the most stable termination. The symmetric slab model helps to avoid unphysical overall polarization of the slab and ensures a zero-dipole moment in the z-direction, perpendicular to the surface plane. The (0001) and (100) surfaces each have two symmetric slab terminations, both of which were investigated in order to determine the most stable termination. The (0001) can be Ni3P2- or Ni3P-terminated (Figure 2a,b). The Ni3P2 termination of the (0001) surface has relatively equivalent nickel sites—each with two neighbouring phosphorous and conjugating within a 3-Ni triangle (Ni3-hollow site), with average Ni−Ni bond length of 2.665 Å. At the Ni3P-terminated (0001) surface, each phosphorous is coordinated to three nickel atoms. The Ni3P2- and Ni3P-terminated (0001) surfaces possess similar surface energies calculated at 1.91 and 1.92 Jm−2, respectively, which suggests that both terminations could be formed (coexist), depending on the experimental conditions. In an earlier theoretical investigation [35], where the formation energy as a function of the chemical potential of the P atom (μP) for the Ni3P2-and Ni3P-terminated surfaces was derived, it was shown that both Ni3P2-and Ni3P-terminations have comparable range of thermodynamic stability, although over a wide range of μP (−10.10 eV < μP < −6.62 eV), there is preference for the Ni3P2 terminations. A scanning tunneling microscopy (STM) study of the (0001) surface validates the existence of both the Ni3P2- and Ni3P-terminations of the (0001) surface [43]. Similar to the (0001) surface, the (100) surface has two terminations: Ni2P- and NiP-terminated surfaces as shown in Figure 2c,d—whereby the Ni2P-termination has been observed experimentally, and the NiP is included due to the closeness in surface energies. The Ni2P-termination of the (100) surface shows there are two distinct nickel sites, differentiated by the number of neighbouring phosphorous atoms—having either one or two coordinated P atoms. The NiP-termination of the (100) surface exposes a single nickel and phosphorous atom at the topmost layer (Figure 2d). Both terminations are also found to have similar surface energies, calculated at 1.33 and 1.39 Jm−2 for the Ni2P- and NiP-terminations, respectively, which again suggests that both terminations could be formed depending on the experimental conditions. The (100) has been less extensively studied, and so the same amount of literature does not exist as for the (0001)-surface. Previous computational work has focused on the (100)-Ni2P termination [22]. However, in 2010, K. Edamoto et al. [44] did confirm two ordered surfaces exist on the (100) surface from their photoemission spectroscopy (PES) analysis. The predicted lower surface energy of the (100) compared to the (0001) surface, however, indicate that the (100) surface is thermodynamically more stable. The relative stabilities, geometries, and compositions of the Ni2P (0001) and (100) surfaces may dictate their chemical reactivity toward water adsorption and splitting. This has been investigated and discussed in the subsequent sections.
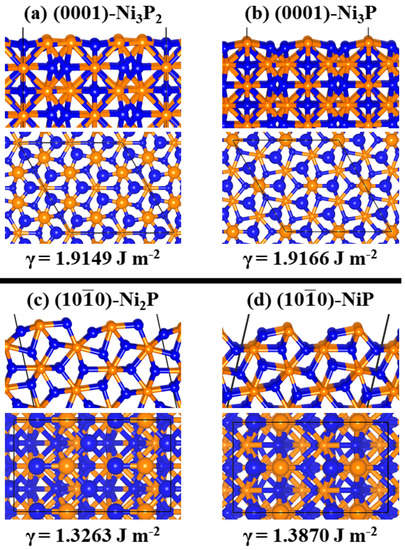
Figure 2.
Schematic illustration of the side (top row) and top (bottom row) views of the relaxed structures of the (a) Ni3P2- and (b) Ni3P-terminations of (0001) surface, (c) Ni2P- and (d) NiP-terminations of (100) surface. γ denotes the surface energy.
2.2. Molecular H2O Adsorption on Ni2P (0001) and (100) Surfaces
Seeing that the Volmer reaction (H2O + e− + cat → H*-cat + OH−) is the rate-determining step for the HER in alkaline electrolytes, the first interest is to characterize the adsorption and activation of water on the Ni2P (0001) and (100) surfaces. The most stable adsorption geometries for molecular water on the Ni3P2- and Ni3P-terminated (0001) surface are shown in Figure 3, whereas the calculated adsorption energies and optimized structural parameters are listed in Table 1. The adsorption of water on the Ni3P2- and Ni3P-terminated (0001) surface released adsorption energies of −0.55 and −0.59 eV, respectively. The similarity in the adsorption energies is consistent with the relative stability of the two terminations. The interacting Ni−O bond distance at the Ni3P2- and Ni3P-terminated (0001) surfaces is calculated at 2.219 Å and 2.165 Å, respectively. The O–H bond lengths calculated for water on each surface (Table 1) were slightly larger than for a free H2O molecule (0.972 Å) in vacuum. This indicates that the O–H bonds of the adsorbed species are slightly weakened when adsorbed on the Ni2P (0001) surface—which was confirmed via O–H bond stretching vibrational frequencies (Table 1). Due to the fact that vibrational modes and bond strengths may be related to charge transfer processes and electron density rearrangement between the surface species and the adsorbing water molecule (Figure 3c,d), Bader population and differential charge density analyses were performed to quantify any charge transfer and electron density redistribution within the Ni2P−H2O systems. Overall, it was found that the adsorption of water on both the Ni3P2- and Ni3P-terminated (0001) surface is characterized by only small charge transfer (< 0.03 e−), suggesting a small degree of water oxidation (Table 1). The calculated electron density difference isosurface plots (Figure 3c,d) reveal accumulation of electron density within the bonding region of Ni−O, which is consistent with chemisorption. Evidence of electron density redistribution between hydrogen and surface species can be also seen, suggestive of hydrogen-bonded interactions that we believe contributes to the stabilization of adsorbed water on the catalyst surface.
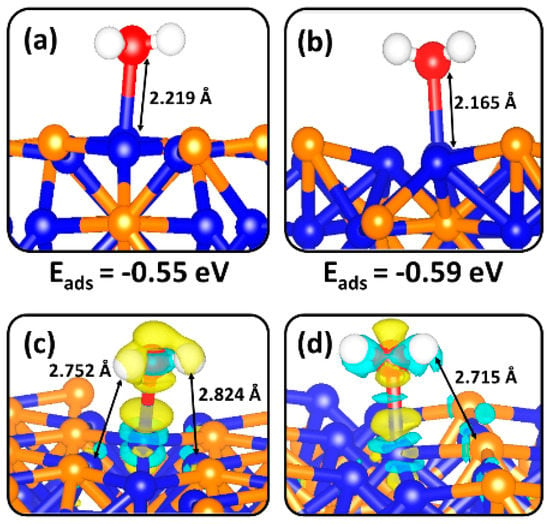
Figure 3.
The optimized structures of the most favourable adsorption configurations of molecular H2O on the (a) Ni3P2- and (b) Ni3P-terminations of (0001) surface. The corresponding isosurfaces contours of the charge density difference for adsorbed H2O molecule are shown in (c) and (d). The cyan and yellow contours indicate electron density accumulation and depletion by ±0.003 e/Å3, respectively.

Table 1.
Molecular adsorption energies (Eads), bond lengths (d), bond angles (α), Bader charge of adsorbed water (q(H2O)), and vibrational frequencies (υ) for water adsorption on Ni2P (0001) and (100) surfaces. The gas phase O–H stretching modes are predicted at 3988 and 3866 cm−1.
The lowest-energy molecular H2O geometries at the two terminations of the (100) surface are shown in Figure 4. Two stable adsorption water geometries are obtained on the Ni2P-terminated (100) surface, which released adsorption energies of −0.68 eV when adsorbed at edge Ni-site (Figure 4a) and −0.33 eV when adsorbed at step Ni-site (Figure 4b). The stronger binding at the edge Ni-site can be attributed to its lower coordination number (N = 4) compared to that at a step Ni-site (N =6). At the NiP-terminated (100) surface, the lowest-energy water adsorption geometry (Figure 4c) released adsorption energy −0.58 eV. The interacting Ni−O bond distances at the Ni2P-terninated (100) surface are calculated at 2.152 Å and 2.298 Å for the lowest-energy structure (Figure 4a) and next stable structure (Figure 4b). At the NiP-terminated (100) surface (Figure 4c), the Ni–O bond distance is 2.156 Å. We observe small elongation in the O–H bonds of the adsorbed water molecule compared to the gas phase molecule, indicating that the O–H bonds are somewhat activated. Bader charge analysis reveals only small charge transfer between the surface-Ni and O of the adsorbed water molecule (Table 1). The calculated differential charge density isosurface (Figure 4d–f)) reveals electron density depletion from the Ni sites and accumulation in the Ni–O bonding regions which is consistent with chemisorption. Evidence of hydrogen-bonded interrelations is also seen in the accumulated electron densities in regions between the hydrogen and the adjacent surface P-sites.
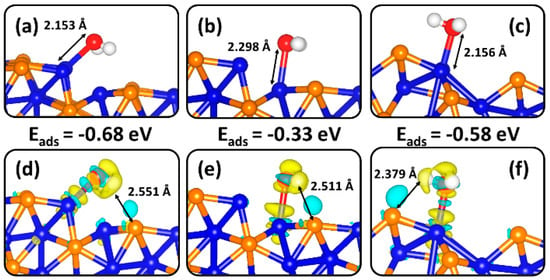
Figure 4.
The most favourable, fully relaxed binding conformations of molecular H2O on the (a,b) Ni2P- and (c) NiP-terminations of the (100) surface. The corresponding isosurfaces contours of the difference in charge density for an adsorbed H2O molecule are shown in (d–f). The cyan and yellow contours indicate electron density accumulation and depletion by ±0.003 e/Å3, respectively.
2.3. Dissociative H2O Adsorption on Ni2P (0001) and (100) Surfaces
To gain atomic-level insights into the fundamental mechanism of HER activity on Ni2P surfaces, the thermodynamics and kinetic energy barrier of water dissociation (i.e., the Volmer step) were systematically studied. The stretched O–H bonds observed for H2O adsorption suggest that these molecular adsorbed conformations are likely precursors for initiating the Volmer step of HER. Ni2P surface structures will dictate their water splitting activity, hence this is the current focus of our investigation. Different co-adsorption structures for OH and H species were considered on the Ni2P (0001) and (100) surfaces in order to determine lowest-energy configurations. Shown in Figure 5i–l and Table 2 are the co-adsorption energies and structural parameters for the lowest-energy configurations. The structures and energetics of the less-stable explored co-adsorption geometries are shown in Supplementary Information Figures S1 and S2, and Table S1. The most preferred co-adsorption geometry on the (0001)-Ni3P2 termination is found for the configuration in which the OH species form bidentate Ni–O bonds with the H atom binding at the 3-fold Ni (Ni3-hollow) site (Figure 5i), releasing a co-adsorption energy of −1.07 eV. Compared to the (0001)-Ni3P2-terminated surface, the adsorption energy for dissociative water adsorption has been found to be endothermic at all explored sites on the Ni3P-terminated (0001) surface, with the least endothermic energetics (0.45 eV, Figure 5j) predicted for the geometry in which the OH binds at top-Ni site and the H atom bridges Ni-P site. At the (100)-Ni2P terminated surface, the most favourable dissociative adsorption structure is found for the configuration in which both OH and H species preferentially bind at adjacent bridge Ni–Ni sites (Figure 5k) releasing a co-adsorption energy of −0.69 eV. Compared to the Ni2P-terminated (100) surface, unfavourable (endothermic) dissociative adsorption is obtained at all binding sites on the NiP-terminated (100) surface with the least endothermic energy (0.05 eV) predicted for the configuration in which OH and H bind at adjacent top-Ni and top-P sites, respectively, as shown in Figure 5k.
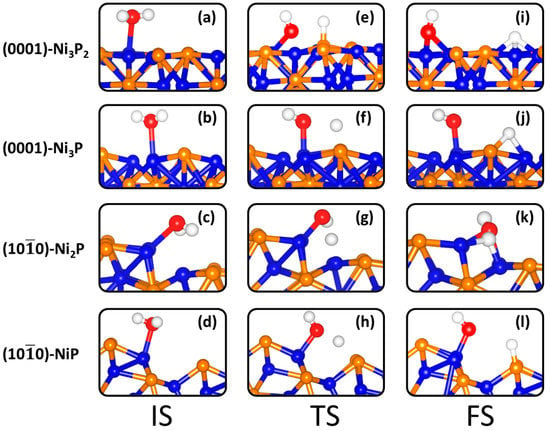
Figure 5.
The initial (IS) (a–d), transition (TS) (e–h), and final (FS) (i–l) states of the most favourable path for water dissociation on the (0001) and (100) surfaces of Ni2P.
Table 2.
Dissociative adsorption energies (Eads) and bond lengths (d) for water on Ni2P (0001) and (100) surfaces. H-P bond distances are denoted as d(P).
To ascertain whether the dissociation or desorption of water will take place on the Ni2P catalyst surfaces, we have calculated the activation energy barriers for water dissociation on the (0001) and (100) surfaces (Figure 6) and compared it with the molecular water adsorption energies on the corresponding surfaces (Table 1). The schematic representations of the initial (IS), transition (TS), and final (FS) states of the dissociation of water on the Ni2P(0001) and Ni2P(100) surfaces are shown in Figure 5. The adsorption energy of the water molecule on each termination of the Ni2P(0001) and Ni2P(100) surfaces are obviously smaller, in absolute value, than the energy required for water dissociation (1.53 and 1.12 eV on the Ni3P2- and Ni3P-terminated (0001) surfaces; 0.96 and 0.82 on the Ni2P- and NiP-terminated (100) surfaces, Figure 6), suggesting sluggish kinetics for the initial Volmer step in the hydrogen evolution reaction over Ni2P catalyst, and thus requiring a high overpotential for the dissociation to happen [45].
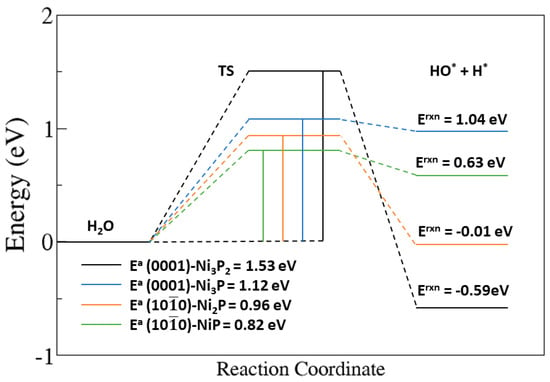
Figure 6.
Activation barriers (Ea) and reaction energies (Erxn) for dissociative water adsorption to the (0001) and (100) surfaces of Ni2P.
2.4. H Adsorption on Ni2P (0001) and (100) Surfaces
After the splitting of water (H2O → H + OH) i.e., the Volmer step, the subsequent combination of the adsorbed H* into molecular hydrogen (Tafel step) is vital in HER. The Gibbs free energy of hydrogen adsorption (ΔGH*), which quantifies the strength of hydrogen adsorption on the catalyst surfaces is known to be the best descriptor for hydrogen evolution activity [4,7]. For an active HER catalyst, the value of |ΔGH*| must be close to zero, indicating that the free energy of adsorbed H is close to that of the reactant or product. The ΔGH* value was calculated using the relation , where ∆EH is the hydrogen adsorption energy calculated as ∆EZPE is the difference in zero-point energy between the adsorbed hydrogen and hydrogen in the gas phase, and ∆SH is the entropy difference between adsorbed state and gas phase. The vibrational and configurational entropies of the adsorbed H*-intermediate are assumed to be negligible, and thus the entropy difference is simply meV K−1, where is the entropy of molecule hydrogen in gas phase. Under standard conditions, is about 0.24 eV, therefore [4,7,46].
Different binding sites were explored for H adsorption on the Ni2P (0001) and (100) surfaces in order to determine the optimum ΔGH* value. The optimized adsorption structures with the predicted ΔGH* values at the Ni2P (0001) and (100) surfaces are shown in Figure 7 and Figure 8, respectively. The calculated optimum |ΔGH*| values (Figure 9) at the Ni3P2- and Ni3P-terminated (0001) surfaces were calculated at 0.13 eV and 0.20 eV, respectively, predicted at the top-Ni (Figure 7a) and bridge Ni-P (Figure 7e) sites. The small |ΔGH*| values indicate a facile Tafel step on both terminations, but the smaller ΔGH* value for the Ni3P2-termination (0.13 eV) suggest a superior HER activity. Compared to the top-Ni and bridge Ni-P sites on the Ni3P2-terminated (0001) surface, the Ni3-hollow site binds H too strongly (ΔGH* = −0.42 eV, Figure 7d) similar to the reported value of −0.45 eV by Wexler et al. [47]. The stronger binding of H at the Ni3-hollow site points to sluggish Heyrovsky–Tafel step as the diffusion of the adsorbed H* to combine is restricted. At the Ni2P- and NiP-terminated (100) surfaces, the optimum |ΔGH*| value is calculated to be 0.34 eV (Figure 8a) and 0.01 eV (Figure 8d), suggesting the NiP-terminated (100) surface provides a nearly thermoneutral H adsorption and consequently higher catalytic activity towards HER. Similar to the Ni3-hollow site at the Ni3P2-terminated (0001) surface, we found that the Ni−Ni (2.797 Å) bridge site at the Ni2P-terminated (100) surface binds H too strongly (ΔGH* = −0.44, Figure 8b).
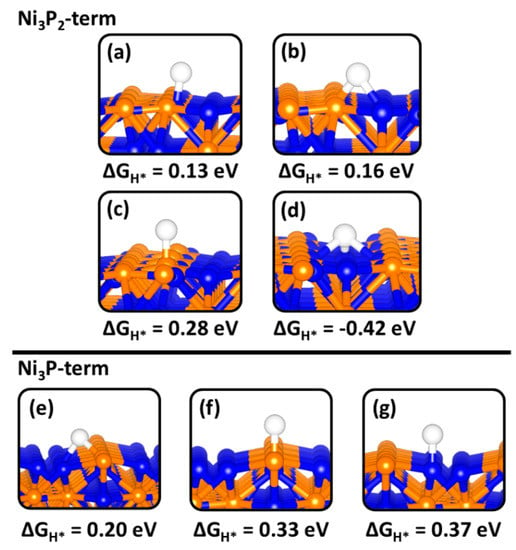
Figure 7.
Gibbs free of hydrogen adsorption (ΔGH*) at different binding sites on the (0001) surface of Ni2P. Whereby (a–d) represent Ni-top, Ni-P-bridge, P-top and 3-Ni-bridge, respectively on the Ni3P2-termination. Along with (e–g) showing a Ni-P-bridge, P-top and Ni-top respectively on the Ni3P-termination.
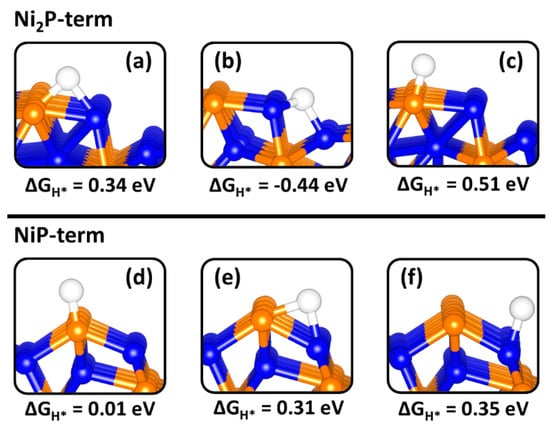
Figure 8.
Gibbs free of hydrogen adsorption (ΔGH*) at different binding sites on the (100) surface of Ni2P. Whereby (a–c) show a Ni-P-bridge, Ni-Ni-bridge and P-top respectively on the (100)-Ni2P termination and (d–f) are the P-top, Ni-P-bridge and Ni-top respectively on the (100)-NiP termination.
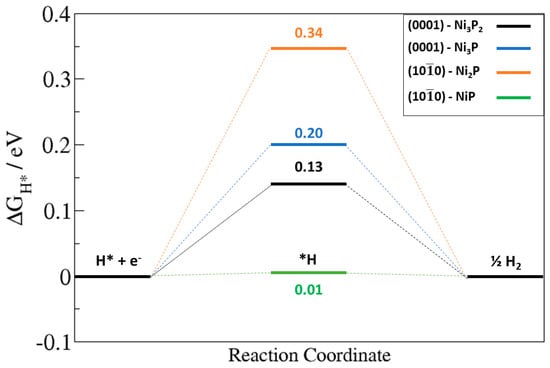
Figure 9.
The optimum Gibbs free of hydrogen adsorption (|ΔGH*|) on (0001) and (100) surfaces of Ni2P.
3. Summary and Conclusions
The adsorption and dissociation reaction mechanisms of water over the Ni2P (0001) and (100) surfaces have been investigated by means of first-principles DFT-D3 calculations. Results regarding the thermodynamic stability, active sites, and activation barriers of both the Volmer and Tafel steps have been systematically characterized. The dissociative adsorption of water is demonstrated to be thermodynamically favourable (exothermic) at both the Ni2P (0001) and (100) surfaces, with activation energy barrier of 1.12 and 0.82 eV, respectively. The calculated dissociative adsorption energetics and high activation energy barriers for the dissociation, however, suggest sluggish kinetics for the initial Volmer step in the hydrogen evolution reaction over a Ni2P catalyst. This indicates that a high overpotential is required to drive the HER activity on pristine Ni2P catalyst. Based on predicted ΔGH* values at different surface sites, we predict that the Ni3-hollow sites on the (0001) and the Ni-Ni bridge sites on the (100) surfaces bind H too strongly, hence the modification of the surface structure and tuning of the electronic properties through transition metal doping could be an important strategy to achieve facile kinetics for both the Volmer and Heyrovsky–Tafel steps. Future investigations will therefore expand the results presented here to detail transition metal doped Ni2P catalysts. This will provide full atomic-level insights into the geometric and electronic properties that dictate their improved HER activity. The insights derived from the present study on the pristine Ni2P (0001) and (100) surfaces in terms of surface stability, active sites, adsorption energies and activation barriers for water dissociation will be useful in investigating the ligand (changes in the electronic structure) and ensemble (structure sensitivity) effects of transition metal doped Ni2P electrocatalysts towards achieving improved HER activity.
4. Computational Details
All the calculations were performed using the Vienna Ab Initio Simulation Package (VASP) [48]. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) was used for the calculation of the electronic exchange–correlation potential [49]. The projector augmented wave (PAW) method was used to describe the electron-ion interactions [50]. Wave functions were expanded in a plane wave basis with a high energy cut-off of 600 eV. The convergence criterion was set to 10−6 eV between two ionic steps for the self-consistency process, with Hellmann–Feynman forces on each ion reached 0.01 eV Å−1. We have accounted for Van der Waals dispersion forces through the Grimme DFT-D3 scheme [51], which adds a semi-empirical dispersion correction to the conventional Kohn–Sham DFT energy as implemented in the VASP code. The coefficients of the R−6 term in the DFT-D3 scheme is influenced by the neighborhood of each atom and they are they continuously change along with environment changes during geometry optimization. The Brillouin zone was sampled using a Monkhorst-Pack [52] k-point grid of 5 × 5 × 11 for the bulk and 3 × 3 × 1 for the surface calculations.
The METADISE code [53,54] was employed to create the Ni2P (0001) and (100) surfaces (Figure 1), which are known to be active for HER [19,22] from the fully relaxed bulk material. The slab thickness of the (0001) and (100) surfaces which ensured convergence of the surface energy within 0.001 eV is found to be 13.28 Å and 10.08 Å, respectively. A vacuum size of 15 Å was added in the z-direction of the slab to avoid interactions between periodic slabs. The surface energy (γ) of each surface termination was calculated using the relation
where Esurface and Ebulk are the energies of fully surface and bulk structures, respectively, n is the number of repeat unit cells in the z-direction, and A is the surface area.
A (2 × 2) supercells of the Ni2P (0001) and (100) surfaces (Figure 2) was employed for the characterization of the adsorption structures and properties of water. The area of these surfaces is large enough to reduce or avoid lateral interactions between water and its dissociated products in periodic cells. To obtain the lowest-energy adsorption geometries, atoms of the topmost three surface layers and the adsorbate species were allowed to relax without constraints until the residual forces reached 0.01 eV Å−1. The strength of water adsorption on the different Ni2P surfaces was characterized by calculating the adsorption energy (Eads) using the equation
where Esurface + adsorbate is the energy of the slab with adsorbed molecule, Esurface is the energy of the naked surface, and Eadsorbate is the energy of adsorbates in the gas phase. Based on this definition, a negative or positive value of Eads indicates an exothermic or endothermic adsorption process. Bader charge analysis [55,56] was used to quantify any charge transfers between the Ni2P surface and adsorbate molecules. Transition state structures (confirmed via frequency calculations) and the corresponding activation energy barriers for water dissociation was determined using the climbing image nudged elastic band (cNEB) method [57].
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/3/307/s1, Figure S1: The optimized structures of dissociated water configurations (C) on the Ni3P2- and Ni3P-terminations of Ni2P(0001) surface; Figure S2: The optimized structures of dissociated water configurations (C) on the Ni2P- and NiP-terminations of the Ni2P (100) surface, and Table S1: Dissociative adsorption energies (Eads) and bond lengths (d) for water on Ni2P (0001) and (100) surfaces. H-P bond distances are denoted as d(P). Sites Ni, P, Ni-Ni and Ni-P denote top-Ni, top-P, bridge-Ni-Ni and bridge-Ni-P.
Author Contributions
R.W.C. performed the DFT simulations and data analysis and wrote the paper. N.Y.D. contributed to the study design and scientific discussion of the results. Both co-authors contributed to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the UK’s Engineering and Physical Sciences Research Council (EPSRC), grant number EP/S001395/1.
Acknowledgments
R.W.C. acknowledges the College of Physical Sciences and Engineering, Cardiff University for studentship. We also acknowledge the use of computational facilities of the Advanced Research Computing at Cardiff (ARCCA) Division, Cardiff University, and HPC Wales. Information on the data that underpins the results presented here, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2020.0102983129.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dunn, S. Hydrogen futures: Toward a sustainable energy system. Int. J. Hydrogen Energy 2002, 27, 235–264. [Google Scholar] [CrossRef]
- Turner, J.; Sverdrup, G.; Mann, M.K.; Maness, P.C.; Kroposki, B.; Ghirardi, M.; Evans, J.R.; Blake, D. Renewable hydrogen production. Int. J. Energy Res. 2008, 32, 379–407. [Google Scholar] [CrossRef]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Ojha, K.; Saha, S.; Dagar, P.; Ganguli, A.K. Nanocatalysts for hydrogen evolution reactions. Phys. Chem. Chem. Phys. 2018, 20, 6777–6799. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef]
- Noerskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U.; Nørskov, J.K.; Bligaard, T.; Logadottir, A.; et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Markovic, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241. [Google Scholar] [CrossRef]
- Kong, D.; Cha, J.J.; Wang, H.; Lee, H.R.; Cui, Y. First-row transition metal dichalcogenide catalysts for hydrogen evolution reaction. Energy Environ. Sci. 2013, 6, 3553–3558. [Google Scholar] [CrossRef]
- Wan, C.; Regmi, Y.N.; Leonard, B.M. Multiple phases of molybdenum carbide as electrocatalysts for the hydrogen evolution reaction. Angew. Chem. Int. Ed. 2014, 53, 6407–6410. [Google Scholar] [CrossRef]
- Chen, W.F.; Sasaki, K.; Ma, C.; Frenkel, A.I.; Marinkovic, N.; Muckerman, J.T.; Zhu, Y.; Adzic, R.R. Hydrogen-evolution catalysts based on non-noble metal nickel–molybdenum nitride nanosheets. Angew. Chem. Int. Ed. 2012, 51, 6131–6135. [Google Scholar] [CrossRef]
- Stern, L.A.; Feng, L.; Song, F.; Hu, X. Ni2P as a Janus catalyst for water splitting: The oxygen evolution activity of Ni2P nanoparticles. Energy Environ. Sci. 2015, 8, 2347–2351. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, Y.; Chen, Y.; Liu, Y.; Liu, C. Cobalt phosphide-based electrocatalysts: Synthesis and phase catalytic activity comparison for hydrogen evolution. J. Mater. Chem. A 2016, 4, 4745–4754. [Google Scholar] [CrossRef]
- Cabán-Acevedo, M.; Stone, M.L.; Schmidt, J.R.; Thomas, J.G.; Ding, Q.; Chang, H.C.; Tsai, M.L.; He, J.-H.; Jin, S. Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide. Nat. Mater. 2015, 14, 1245. [Google Scholar] [CrossRef]
- Mukherjee, D.; Austeria, P.M.; Sampath, S. Two-Dimensional, Few-Layer Phosphochalcogenide, FePS3: A New Catalyst for Electrochemical Hydrogen Evolution over Wide pH Range. ACS Energy Lett. 2016, 1, 367–372. [Google Scholar] [CrossRef]
- Callejas, J.F.; Read, C.G.; Popczun, E.J.; McEnaney, J.M.; Schaak, R.E. Nanostructured Co2P Electrocatalyst for the Hydrogen Evolution Reaction and Direct Comparison with Morphologically Equivalent CoP. Chem. Mater. 2015, 27, 3769–3774. [Google Scholar] [CrossRef]
- Schipper, D.E.; Zhao, Z.; Thirumalai, H.; Leitner, A.P.; Donaldson, S.L.; Kumar, A.; Qin, F.; Wang, Z.; Grabow, L.C.; Bao, J.; et al. Effects of Catalyst Phase on the Hydrogen Evolution Reaction of Water Splitting: Preparation of Phase-Pure Films of FeP, Fe2P, and Fe3P and Their Relative Catalytic Activities. Chem. Mater. 2018, 30, 3588–3598. [Google Scholar] [CrossRef]
- Chu, S.; Chen, W.; Chen, G.; Huang, J.; Zhang, R.; Song, C.; Wang, X.; Li, C.; Ostrikov, K. In situ engineering bi-metallic phospho-nitride bi-functional electrocatalysts for overall water splitting. Appl. Catal. B Environ. 2019, 243, 537–545. [Google Scholar] [CrossRef]
- Morales-García, Á.; He, J.; Lyu, P.; Nachtigall, P. Exploring the stability and reactivity of Ni2P and Mo2C catalysts using ab initio atomistic thermodynamics and conceptual DFT approaches. Biomass Convers. Biorefin. 2017, 7, 377–383. [Google Scholar] [CrossRef]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef]
- Pei, Y.; Cheng, Y.; Chen, J.; Smith, W.; Dong, P.; Ajayan, P.M.; Ye, M.; Shen, J. Recent developments of transition metal phosphides as catalysts in the energy conversion field. J. Mater. Chem. A 2018, 6, 23220–23243. [Google Scholar] [CrossRef]
- Hansen, M.H.; Stern, L.A.; Feng, L.; Rossmeisl, J.; Hu, X. Widely available active sites on Ni2P for electrochemical hydrogen evolution–insights from first principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 10823–10829. [Google Scholar] [CrossRef]
- Moon, J.S.; Jang, J.H.; Kim, E.G.; Chung, Y.H.; Yoo, S.J.; Lee, Y.K. The nature of active sites of Ni2P electrocatalyst for hydrogen evolution reaction. J. Catal. 2015, 326, 92–99. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Kim, J.Y.; Hanson, J.C.; Sawhill, S.J.; Bussell, M.E. Physical and Chemical Properties of MoP, Ni2P, and MoNiP Hydrodesulfurization Catalysts: Time-Resolved X-ray Diffraction. J. Phys. Chem. B 2003, 107, 6276–6285. [Google Scholar] [CrossRef]
- Kawai, T.; Bando, K.K.; Lee, Y.K.; Oyama, S.T.; Chun, W.-J.; Asakura, K. EXAFS measurements of a working catalyst in the liquid phase: An in situ study of a Ni2P hydrodesulfurization catalyst. J. Catal. 2006, 241, 20–24. [Google Scholar] [CrossRef]
- Wang, W.; Li, X.; Sun, Z.; Wang, A.; Liu, Y.; Chen, Y.; Duan, X. Influences of calcination and reduction methods on the preparation of Ni2P/SiO2 and its hydrodenitrogenation performance. Appl. Catal. A Gen. 2016, 509, 45–51. [Google Scholar] [CrossRef]
- Sun, Z.; Li, X.; Wang, A.; Wang, Y.; Chen, Y. The Effect of CeO2 on the Hydrodenitrogenation Performance of Bulk Ni2P. Top. Catal. 2012, 55, 1010–1021. [Google Scholar] [CrossRef]
- Krishnan, P.S.; Ramya, R.; Umasankar, S.; Shanthi, K. Promotional effect of Ni2P on mixed and separated phase MoS2/Al-SBA-15 (10) catalyst for hydrodenitrogenation of ortho-Propylaniline. Microporous Mesoporous Mater. 2017, 242, 208–220. [Google Scholar] [CrossRef]
- Oyama, S.T. Novel catalysts for advanced hydroprocessing: Transition metal phosphides. J. Catal. 2003, 216, 343–352. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Yang, Q.; Li, K.; Yao, C. An approach to preparing highly dispersed Ni2P/SiO2 catalyst. Catal. Commun. 2010, 11, 571–575. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Takahashi, Y.; Nakamura, K. Water-gas-shift reaction on a Ni2P (001) catalyst: Formation of oxy-phosphides and highly active reaction sites. J. Catal. 2009, 262, 294–303. [Google Scholar] [CrossRef]
- Mishra, I.K.; Zhou, H.; Sun, J.; Qin, F.; Dahal, K.; Bao, J.; Chen, S.; Ren, Z. Hierarchical CoP/Ni5P4/CoP microsheet arrays as a robust pH-universal electrocatalyst for efficient hydrogen generation. Energy Environ. Sci. 2018, 11, 2246–2252. [Google Scholar] [CrossRef]
- Han, A.; Jin, S.; Chen, H.; Ji, H.; Sun, Z.; Du, P. A robust hydrogen evolution catalyst based on crystalline nickel phosphide nanoflakes on three-dimensional graphene/nickel foam: High performance for electrocatalytic hydrogen production from pH 0–14. J. Mater. Chem. A 2015, 3, 1941–1946. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Catalysts for Hydrogen Evolution from the [NiFe] Hydrogenase to the Ni2P(001) Surface: The Importance of Ensemble Effect. J. Am. Chem. Soc. 2005, 127, 14871–14878. [Google Scholar] [CrossRef]
- Li, Q.; Hu, X. First-principles study of Ni2P(0001) surfaces. Phys. Rev. B 2006, 74, 35414. [Google Scholar] [CrossRef]
- Hernandez, A.B.; Ariga, H.; Takakusagi, S.; Kinoshita, K.; Suzuki, S.; Otani, S.; Oyama, S.T.; Asakura, K. Dynamical LEED analysis of Ni2P(0001)-1× 1: Evidence for P-covered surface structure. Chem. Phys. Lett. 2011, 513, 48–52. [Google Scholar] [CrossRef][Green Version]
- Zhang, L.; Ren, X.; Guo, X.; Liu, Z.; Asiri, A.M.; Li, B.; Chen, L.; Sun, X. Efficient Hydrogen Evolution Electrocatalysis at Alkaline pH by Interface Engineering of Ni2P–CeO2. Inorg. Chem. 2018, 57, 548–552. [Google Scholar] [CrossRef]
- Yu, L.; Mishra, I.K.; Xie, Y.; Zhou, H.; Sun, J.; Zhou, J.; Ni, Y.; Luo, D.; Yu, F.; Yu, Y.; et al. Ternary Ni2(1-x)Mo2xP nanowire arrays toward efficient and stable hydrogen evolution electrocatalysis under large-current-density. Nano Energy 2018, 53, 492–500. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3 d M (Ni, Co, Fe, Mn) hydr (oxy) oxide catalysts. Nat. Mater. 2012, 11, 550. [Google Scholar] [CrossRef]
- Rundqvist, S. X-ray investigations of Mn3P, Mn2P and Ni2P. Acta Chem. Scand. 1962, 16, 992–998. [Google Scholar] [CrossRef]
- Lee, Y.K.; Oyama, S.T. Bifunctional nature of a SiO2-supported Ni2P catalyst for hydrotreating: EXAFS and FTIR studies. J. Catal. 2006, 239, 376–389. [Google Scholar] [CrossRef]
- He, J.; Morales-García, Á.; Bludský, O.; Nachtigall, P. The surface stability and equilibrium crystal morphology of Ni2P nanoparticles and nanowires from an ab-initio atomistic thermodynamic approach. CrystEngComm 2016, 18, 3808–3818. [Google Scholar] [CrossRef]
- Moula, M.G.; Suzuki, S.; Chun, W.J.; Otani, S.; Oyama, S.T.; Asakura, K. Surface structures of Ni2P (0001)—Scanning tunneling microscopy (STM) and low-energy electron diffraction (LEED) characterizations. Surf. Interface Anal. 2006, 38, 1611–1614. [Google Scholar] [CrossRef]
- Edamoto, K.; Inomata, H.; Ozawa, K.; Nakagawa, Y.; Asakura, K.; Otani, S. Electronic structure of the Ni2P(100) surface: Angle-resolved photoemission study. Solid State Commun. 2010, 150, 1120–1123. [Google Scholar] [CrossRef]
- Feng, J.X.; Xu, H.; Dong, Y.T.; Lu, X.F.; Tong, Y.-X.; Li, G.R. Efficient hydrogen evolution electrocatalysis using cobalt nanotubes decorated with titanium dioxide nanodots. Angew. Chem. Int. Ed. 2017, 56, 2960–2964. [Google Scholar] [CrossRef]
- Zhang, F.S.; Wang, J.W.; Luo, J.; Liu, R.R.; Zhang, Z.M.; He, C.-T.; Lu, T.B. Extraction of nickel from NiFe-LDH into Ni2P@NiFe hydroxide as a bifunctional electrocatalyst for efficient overall water splitting. Chem. Sci. 2018, 9, 1375–1384. [Google Scholar] [CrossRef]
- Wexler, R.B.; Martirez, M.P.J.; Rappe, A.M. Active role of phosphorus in the hydrogen evolving activity of nickel phosphide (0001) surfaces. ACS Catal. 2017, 7, 7718–7725. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab-initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Watson, G.W.; Kelsey, E.T.; de Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic simulation of dislocations, surfaces and interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433–438. [Google Scholar] [CrossRef]
- Tasker, P.W. The stability of ionic crystal surfaces. J. Phys. C Solid State Phys. 1979, 12, 4977–4984. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 84204. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).