High Surface Area VOx/TiO2/SBA-15 Model Catalysts for Ammonia SCR Prepared by Atomic Layer Deposition


Abstract
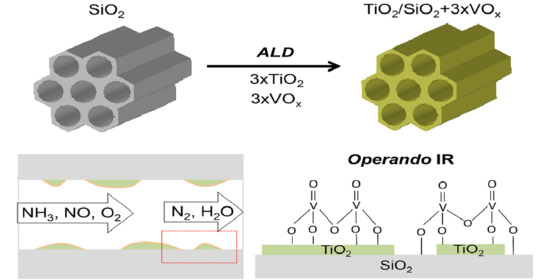
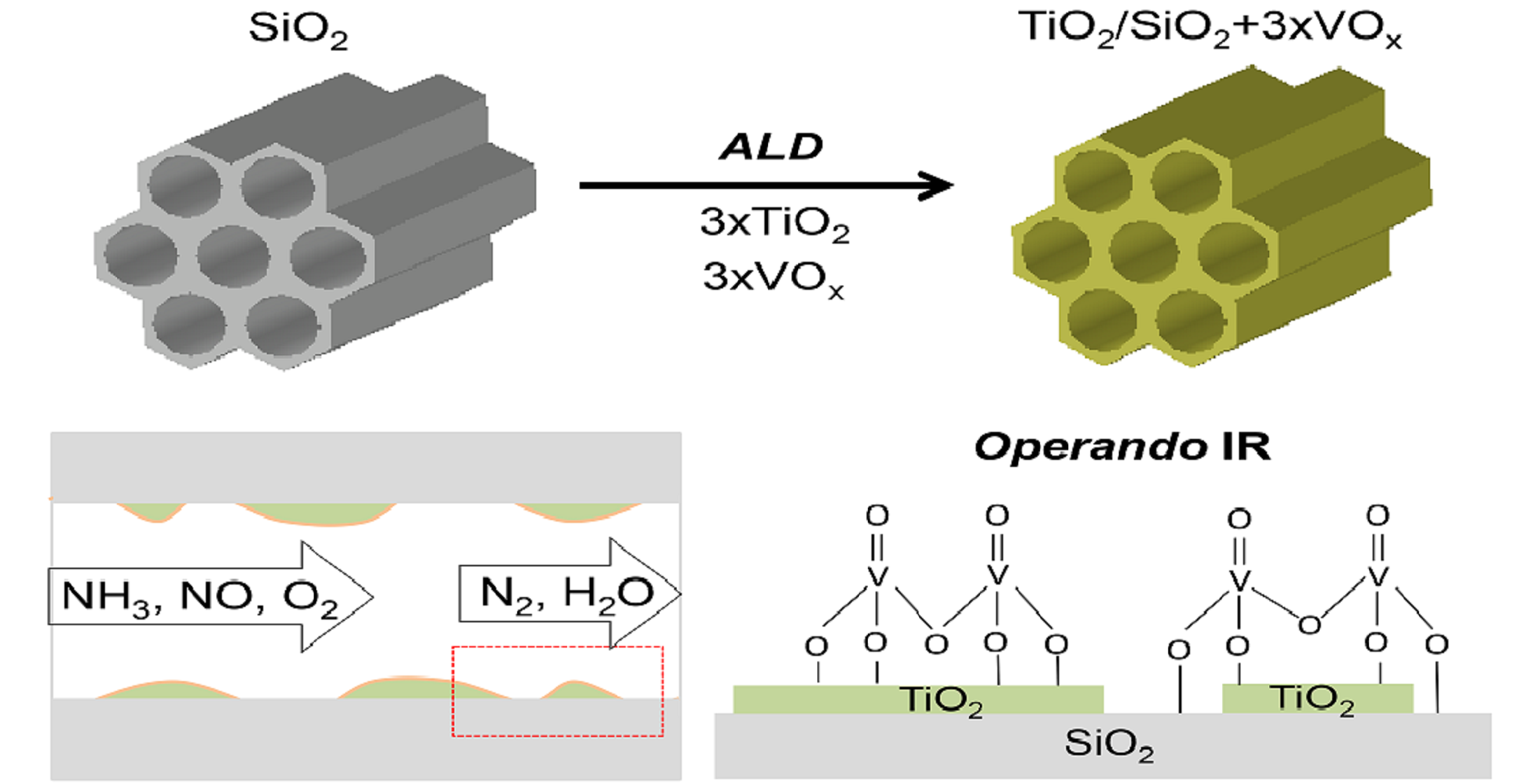
1. Introduction
2. Results
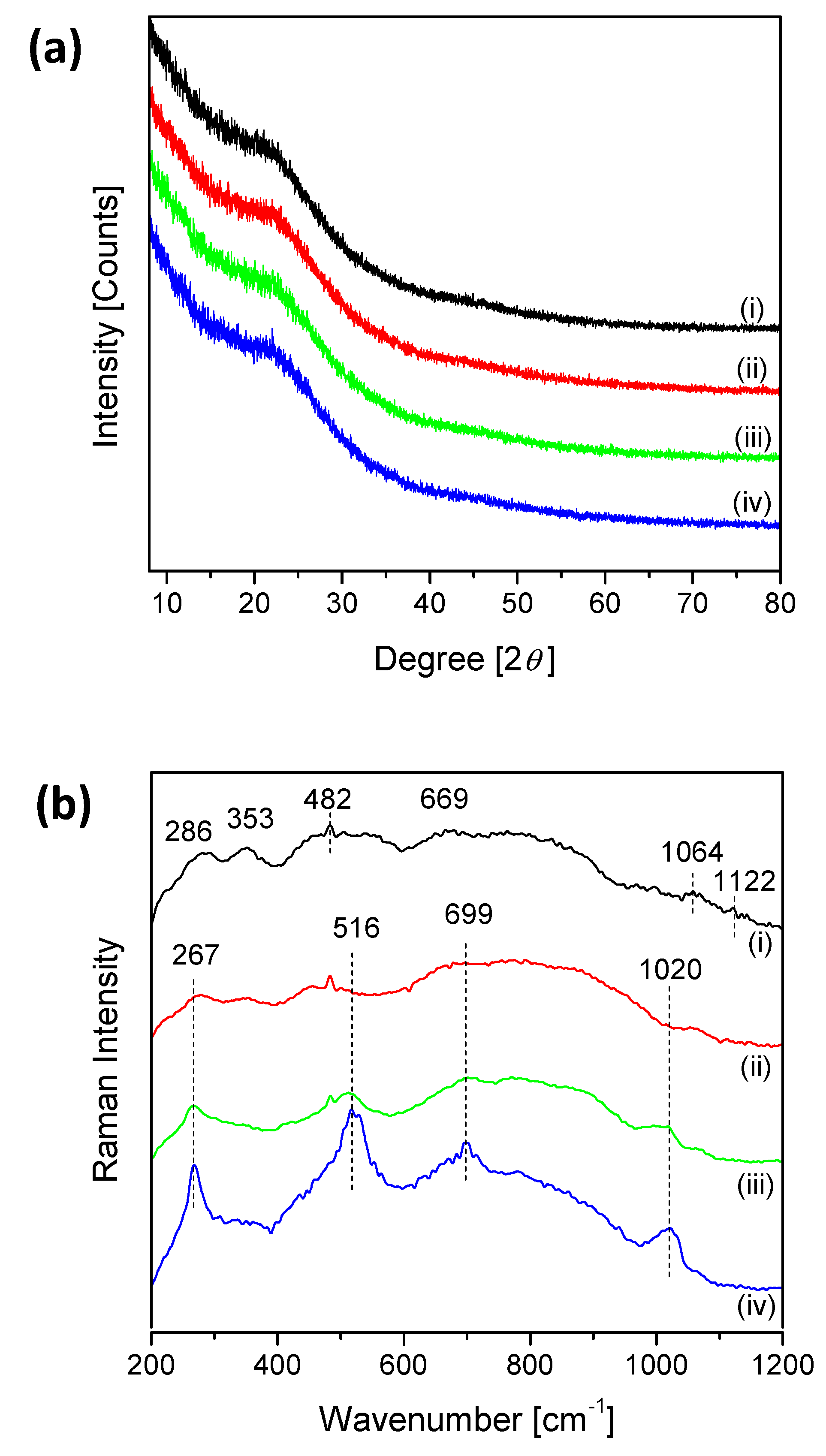
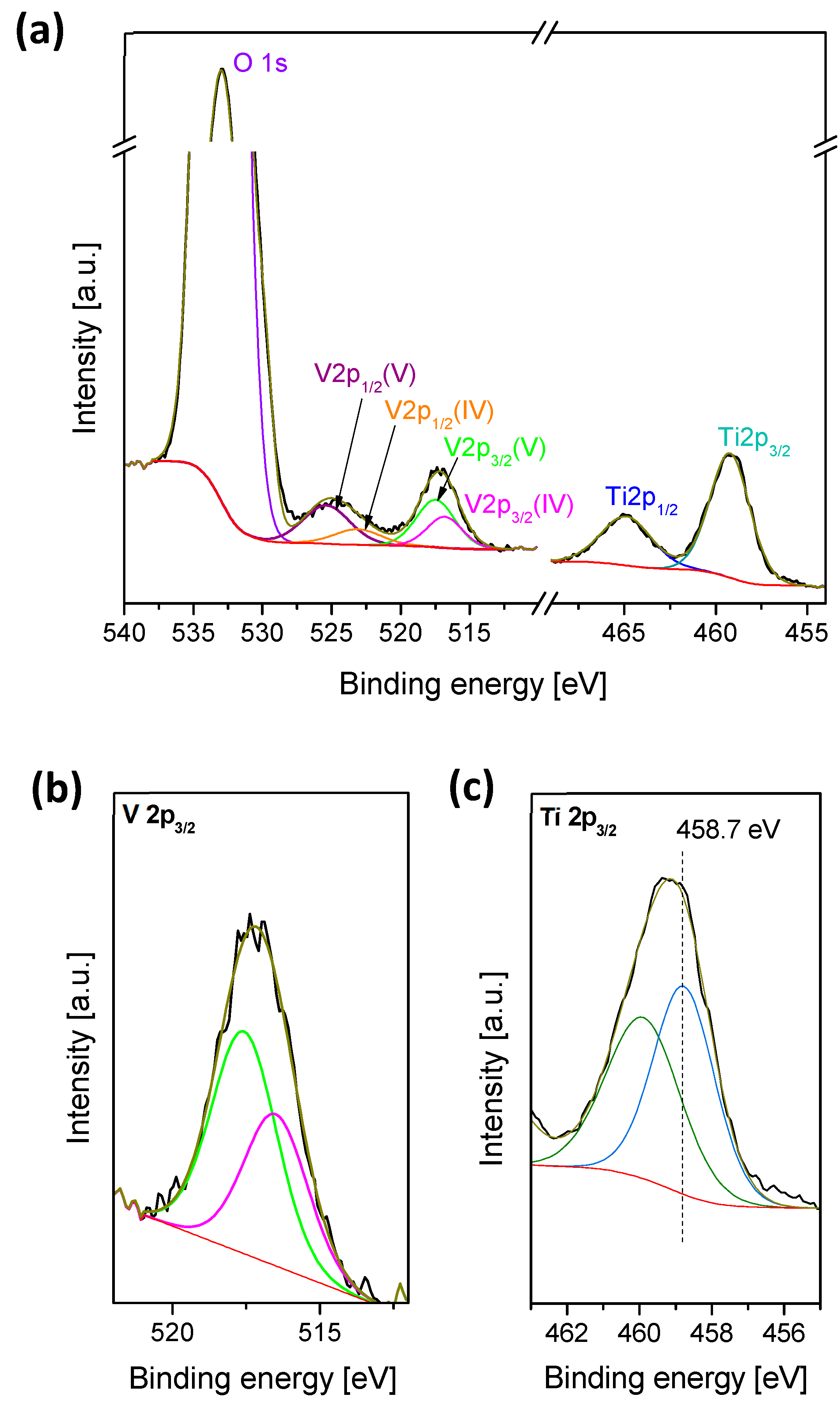
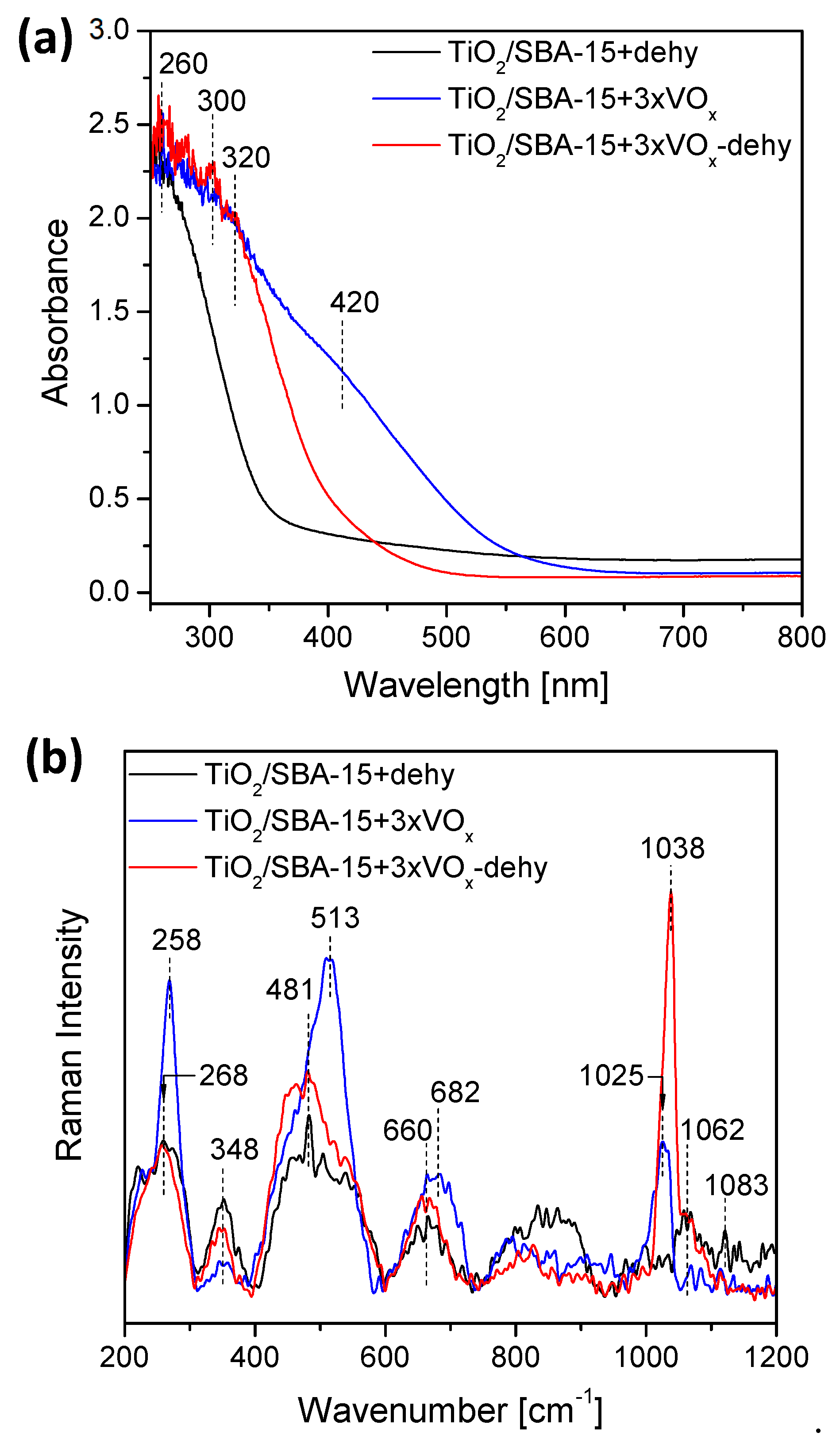
2.1. Catalyst Characterization
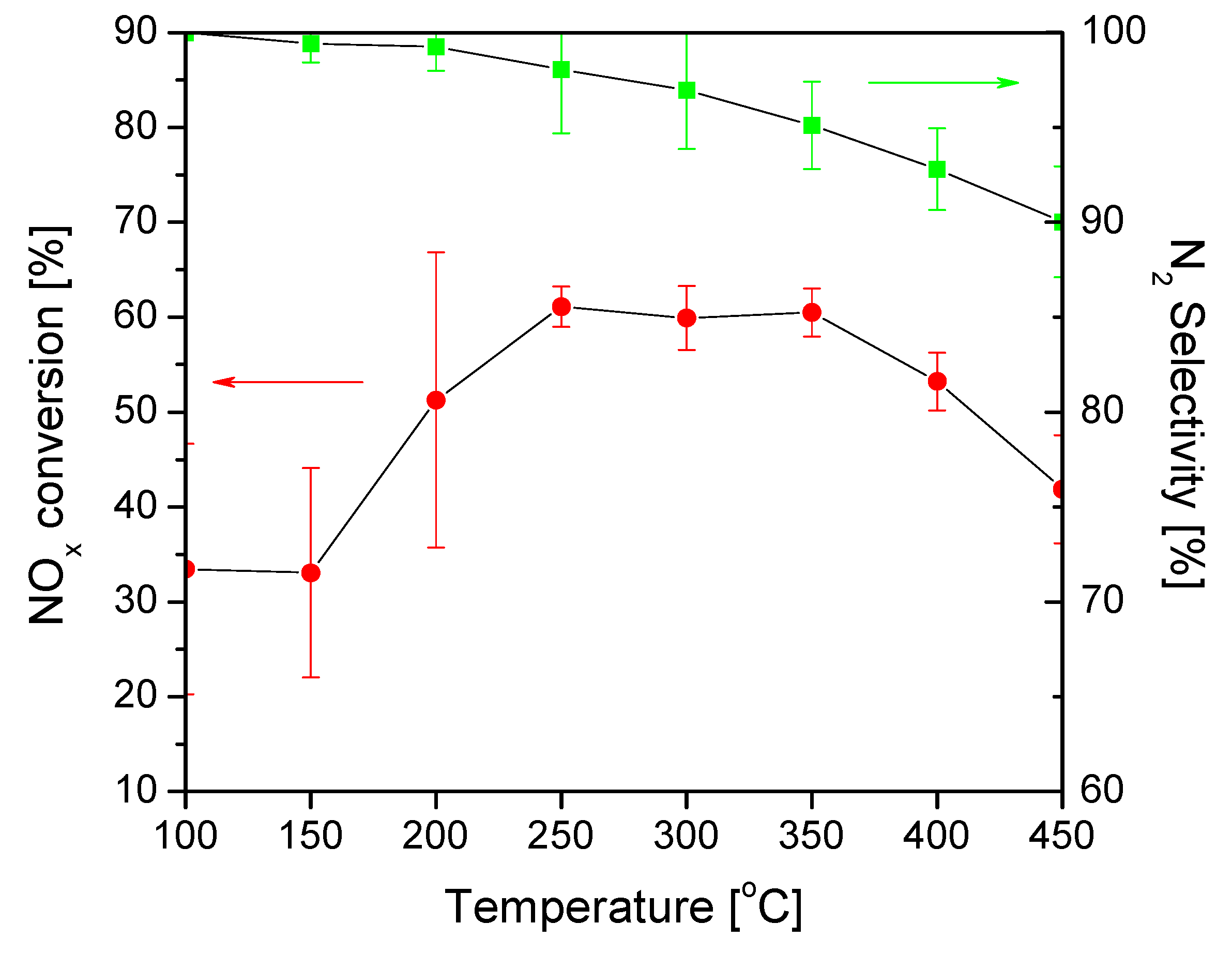
2.2. Catalytic Performance
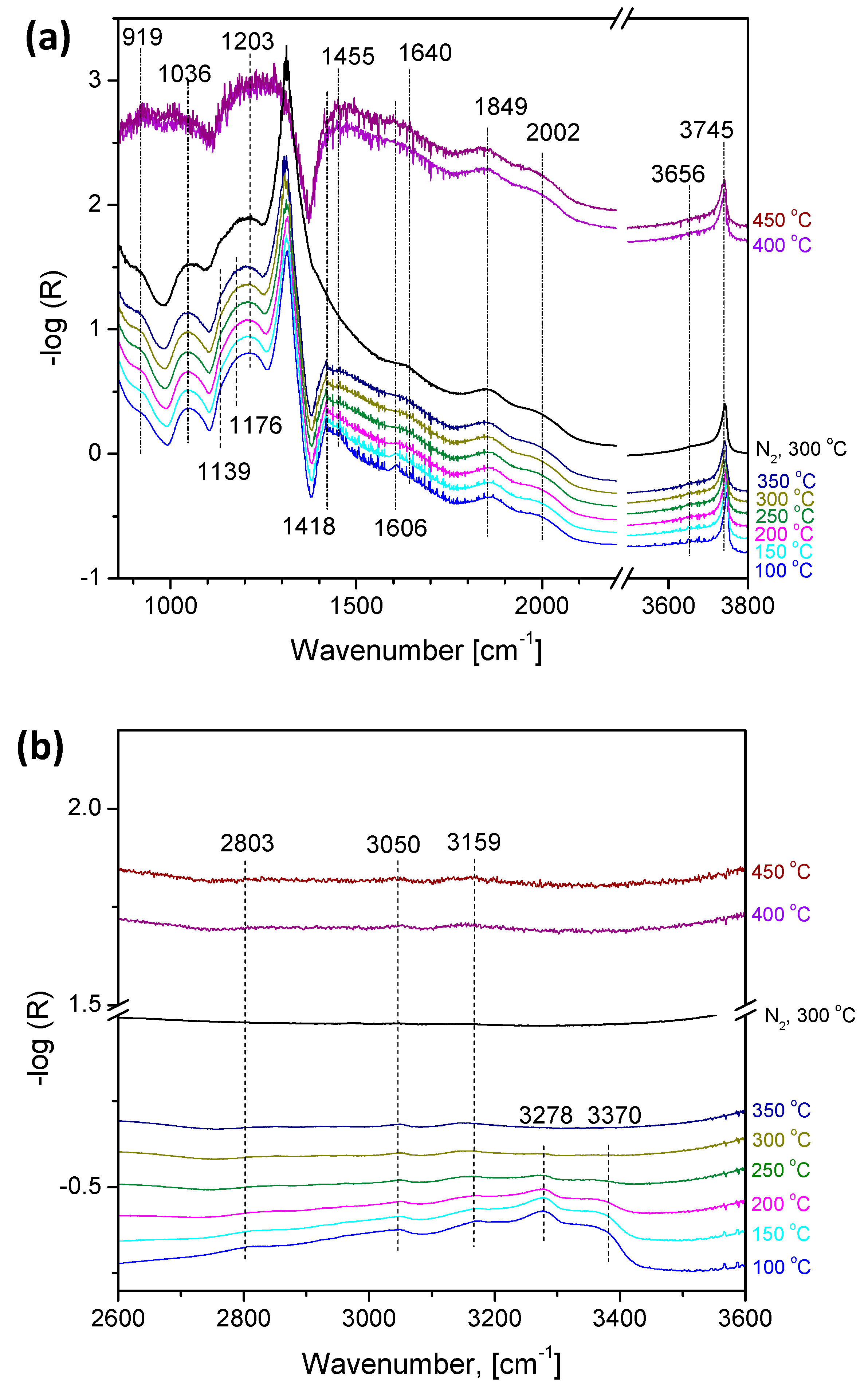
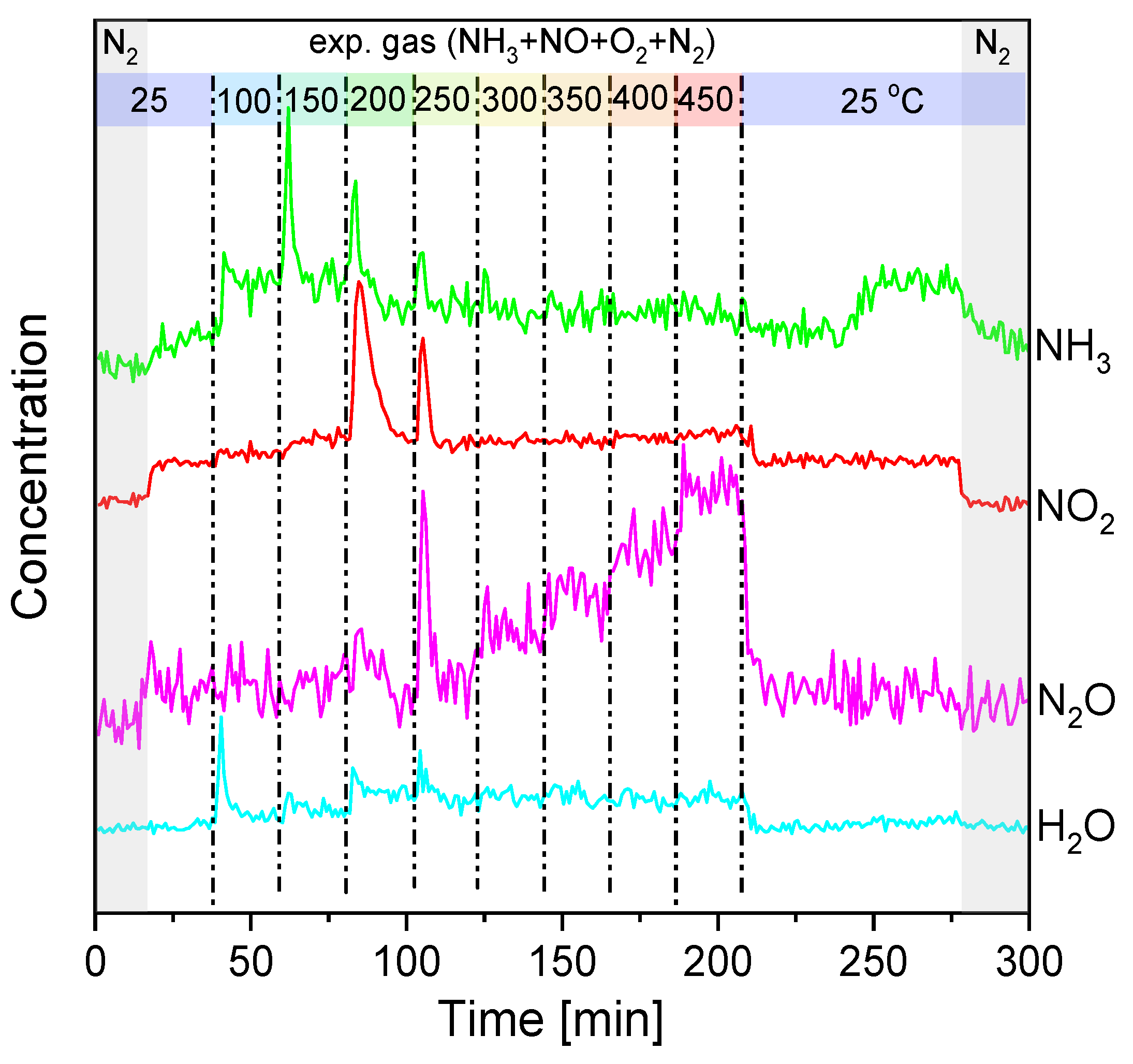
2.3. Operando Characterization of TiO2/SBA-15+3xVOx
3. Discussion
3.1. Surface Vanadia Structure
3.2. Rate-Determining Step
3.3. Structure-Activity Relationship
4. Experimental
4.1. Chemicals
4.2. Synthesis
4.3. Catalytic Performance
4.4. Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lai, J.K.; Wachs, I.E. A Perspective on the Selective Catalytic Reduction (SCR) of NO with NH3 by Supported V2O5–WO3/TiO2 Catalysts. ACS Catal. 2018, 8, 6537–6551. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Schill, L.; Godiksen, A.; Poreddy, R.; Mossin, S.; Jensen, A.D.; Fehrmann, R. Promoted V2O5/TiO2 catalysts for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B Environ. 2016, 183, 282–290. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, Q.; Wang, Y. Effect of rutile phase on V2O5 supported over TiO2 mixed phase for the selective catalytic reduction of NO with NH3. Appl. Surf. Sci. 2014, 314, 112–118. [Google Scholar] [CrossRef]
- Zhu, M.; Lai, J.K.; Tumuluri, U.; Ford, M.E.; Wu, Z.; Wachs, I.E. Reaction Pathways and Kinetics for Selective Catalytic Reduction (SCR) of Acidic NOx Emissions from Power Plants with NH3. ACS Catal. 2017, 7, 8358–8361. [Google Scholar] [CrossRef]
- Reiche, M. Vanadia grafted on TiO2–SiO2, TiO2 and SiO2 aerogels Structural properties and catalytic behaviour in selective reduction of NO by NH3. Appl. Catal. B Environ. 1999, 23, 187–203. [Google Scholar] [CrossRef]
- Engweiler, J.; Baiker, A. Vanadia supported on titania aerogels morphological properties and catalytic behaviour in the selective reduction of nitric oxide by ammonia. Appl. Catal. A Gen. 1994, 120, 187–205. [Google Scholar] [CrossRef]
- He, G.; Lian, Z.; Yu, Y.; Yang, Y.; Liu, K.; Shi, X.; Yan, Z.; Shan, W.; He, H. Polymeric vanadyl species determine the low-temperature activity of V-based catalysts for the SCR of NOx with NH3. Sci. Adv. 2018, 4, eaau4637. [Google Scholar] [CrossRef]
- Song, I.; Youn, S.; Lee, H.; Lee, S.G.; Cho, S.J.; Kim, D.H. Effects of microporous TiO2 support on the catalytic and structural properties of V2O5/microporous TiO2 for the selective catalytic reduction of NO by NH3. Appl. Catal. B Environ. 2017, 210, 421–431. [Google Scholar] [CrossRef]
- Ruff, P.; Schumacher, L.; Rogg, S.; Hess, C. Atomic Layer Deposition-Assisted Synthesis of Embedded Vanadia Catalysts. ACS Catal. 2019, 9, 6349–6361. [Google Scholar] [CrossRef]
- Hess, C. Direct correlation of the dispersion and structure in vanadium oxide supported on silica SBA-15. J. Catal. 2007, 248, 120–123. [Google Scholar] [CrossRef]
- Hess, C. Characterization of the synthesis and reactivity behavior of nanostructured vanadia model catalysts using XPS and vibrational spectroscopy. Surf. Sci. 2006, 600, 3695–3701. [Google Scholar] [CrossRef][Green Version]
- Hess, C.; Waleska, P.; Ratzka, M.; Janssens, T.V.W.; Rasmussen, S.B.; Beato, P. Hierarchical Vanadia Model Catalysts for Ammonia Selective Catalytic Reduction. Top. Catal. 2017, 60, 1631–1640. [Google Scholar] [CrossRef]
- Segura, Y.; Chmielarz, L.; Kuśtrowski, P.; Cool, P.; Dziembaj, R.; VanSant, E.F. Characterisation and reactivity of vanadia–titania supported SBA-15 in the SCR of NO with ammonia. Appl. Catal. B Environ. 2005, 61, 69–78. [Google Scholar] [CrossRef]
- Lu, J.L.; Elam, J.W.; Stair, P.C. Atomic layer deposition—Sequential self-limiting surface reactions for advanced catalyst “bottom-up” synthesis. Surf. Sci. Rep. 2016, 71, 410–472. [Google Scholar] [CrossRef]
- Johnson, R.W.; Hultqvist, A.; Bent, S.F. A brief review of atomic layer deposition: From fundamentals to applications. Mater. Today 2014, 17, 236–246. [Google Scholar] [CrossRef]
- Miikkulainen, V.; Leskelä, M.; Ritala, M.; Puurunen, R.L. Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends. J. Appl. Phys. 2013, 113, 021301. [Google Scholar] [CrossRef]
- Li, S.; Xu, J.; Wang, L.; Yang, N.; Ye, X.; Yuan, X.; Xiang, H.; Liu, C.; Li, H. Effect of post-deposition annealing on atomic layer deposited SiO2 film for silicon surface passivation. Mater. Sci. Semicond. Process. 2020, 106. [Google Scholar] [CrossRef]
- Fares, C.; Islam, Z.; Haque, A.; Kneiß, M.; Von Wenckstern, H.; Grundmann, M.; Tadjer, M.; Ren, F.; Pearton, S.J. Effect of Annealing on the Band Alignment of ALD SiO2 on (AlxGa1−x)2O3 for x = 0.2–0.65. ECS J. Solid State Sci. Technol. 2019, 8, P751–P756. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, Y.; Ren, Y.M.; Orkoulas, G.; Christofides, P.D. Machine learning-based modeling and operation for ALD of SiO2 thin-films using data from a multiscale CFD simulation. Chem. Eng. Res. Des. 2019, 151, 131–145. [Google Scholar] [CrossRef]
- Fengler, S.; Kriegel, H.; Schieda, M.; Gutzmann, H.; Klassen, T.; Wollgarten, M.; Dittrich, T. Charge Transfer in c-Si(n++)/TiO2(ALD) at the Amorphous/Anatase Transition: A Transient Surface Photovoltage Spectroscopy Study. ACS Appl. Mater. Interfaces 2020, 12, 3140–3149. [Google Scholar] [CrossRef]
- Yang, Y.; Wei, W.; Wang, S.; Huang, T.; Yuan, M.; Zhang, R.; Yang, W.; Zhang, T.; Sun, Y.; Yuan, Y.J.; et al. Suppression of Photoinduced Surface Oxidation of Vanadium Dioxide Nanostructures by Blocking Oxygen Adsorption. ACS Omega 2019, 4, 17735–17740. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, C.; Jensen, N.S.; Boetker, J.; Madsen, A.; Kääriäinen, T.O.; Kaariainen, M.-L.; Hoppu, P.; George, S.M.; Murtomaa, M.; Sun, C.C.; et al. Improving Powder Characteristics by Surface Modification Using Atomic Layer Deposition. Org. Process. Res. Dev. 2019, 23, 2362–2368. [Google Scholar] [CrossRef]
- Kim, T.W.; Uchida, S.; Kondo, T.; Segawa, H. Optimization of TiO2 compact layer formed by atomic layer deposition for efficient perovskite solar cells. Appl. Phys. Lett. 2019, 115, 203902. [Google Scholar] [CrossRef]
- Jo, W.J.; Katsoukis, G.; Frei, H. Ultrathin Amorphous Silica Membrane Enhances Proton Transfer across Solid-to-Solid Interfaces of Stacked Metal Oxide Nanolayers while Blocking Oxygen. Adv. Funct. Mater. 2020, 30. [Google Scholar] [CrossRef]
- Yang, G.Q.; Wang, H.; Gong, T.; Song, Y.H.; Feng, H.; Ge, H.Q.; Ge, H.B.; Liu, Z.T.; Liu, Z.W. Understanding the active-site nature of vanadia-based catalysts for oxidative dehydrogenation of ethylbenzene with CO2 via atomic layer deposited VOx on γ-Al2O3. J. Catal. 2019, 380, 195–203. [Google Scholar] [CrossRef]
- Samek, I.A.; Bobbitt, N.S.; Snurr, R.Q.; Stair, P.C. Interactions of VOx Species with Amorphous TiO2 Domains on ALD-Derived Alumina-Supported Materials. J. Phys. Chem. C 2018, 123, 7988–7999. [Google Scholar] [CrossRef]
- Song, G.Y.; Oh, C.; Sinha, S.; Son, J.; Heo, J. Facile Phase Control of Multivalent Vanadium Oxide Thin Films (V2O5 and VO2) by Atomic Layer Deposition and Postdeposition Annealing. ACS Appl. Mater. Interfaces 2017, 9, 23909–23917. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Millot, Y.; Averseng, F.; Calers, C.; Valentin, L.; Dzwigaj, S. Influence of the Preparation Procedure of Vanadium-Containing SiBEA Zeolites on Their Catalytic Activity in Propene Epoxidation. J. Phys. Chem. C 2018, 122, 18570–18582. [Google Scholar] [CrossRef]
- Keränen, J.; Guimon, C.; Auroux, A.; Iiskola, E.I.; Niinistö, L. Gas-phase synthesis, structure and surface acid–base properties of highly dispersed vanadia/titania/silica catalysts. Phys. Chem. Chem. Phys. 2003, 5, 5333–5342. [Google Scholar] [CrossRef]
- Keränen, J.; Guimon, C.; Iiskola, E.; Auroux, A.; Niinistö, L. Atomic layer deposition and surface characterization of highly dispersed titania/silica-supported vanadia catalysts. Catal. Today 2003, 78, 149–157. [Google Scholar] [CrossRef]
- Lei, Z.; Long, A.; Wen, C.; Zhang, J.; Chen, B. Experimental and Kinetic Study of Low Temperature Selective Catalytic Reduction of NO with NH3 over the V2O5/AC Catalyst. Ind. Eng. Chem. Res. 2011, 50, 5360–5368. [Google Scholar] [CrossRef]
- Jiang, Y.; Gao, X.; Zhang, Y.; Wu, W.; Luo, Z.; Cen, K. PbCl2-poisoning kinetics of V2O5/TiO2 catalysts for the selective catalytic reduction of NO with NH3. Environ. Prog. Sustain. Energy 2015, 34, 1085–1091. [Google Scholar] [CrossRef]
- Yang, S.; Wang, C.; Ma, L.; Peng, Y.; Qu, Z.; Yan, N.; Chen, J.; Chang, H.; Li, J. Substitution of WO3in V2O5/WO3–TiO2 by Fe2O3 for selective catalytic reduction of NO with NH3. Catal. Sci. Technol. 2013, 3, 161–168. [Google Scholar] [CrossRef]
- Topsøe, N.Y.; Anstrom, M.; Dumesic, J. Raman, FTIR and Theoretical Evidence for Dynamic Structural Rearrangements of Vanadia/Titania DeNOx Catalysts. Catal. Lett. 2001, 76, 11–20. [Google Scholar] [CrossRef]
- Damma, D.; Ettireddy, P.R.; Reddy, B.M.; Smirniotis, P.G. A review of low temperature NH3-SCR for removal of NOx. Catalysts 2019, 9, 349. [Google Scholar] [CrossRef]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.Y.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef]
- Topsoe, N. Characterization of the nature of surface sites on vanadia-titania catalysts by FTIR. J. Catal. 1991, 128, 499–511. [Google Scholar] [CrossRef]
- Topsoe, N.Y.; Dumesic, J.A.; Topsoe, H. Vanadia-titania catalysts for selective catalytic reduction of nitric-oxide by ammonia: II Studies of active sites and formulation of catalytic cycles. J. Catal. 1995, 151, 241–252. [Google Scholar] [CrossRef]
- Topsøe, N.Y.; Slabiak, T.; Clausen, B.S.; Srnak, T.Z.; Dumesic, J.A. Influence of water on the reactivity of vanadia/titania for catalytic reduction of NOx. J. Catal. 1992, 134, 742–746. [Google Scholar] [CrossRef]
- Topsøe, N.Y.; Topsøe, H. Combined in-situ FTIR and on-line activity studies: Applications to vanadia-titania DeNOx catalyst. Catal. Today 1991, 9, 77–82. [Google Scholar] [CrossRef]
- Ramis, G.; Yi, L.; Busca, G. Ammonia activation over catalysts for the selective catalytic reduction of NOx and the selective catalytic oxidation of NH3. An FT-IR study. Catal. Today 1996, 28, 373–380. [Google Scholar] [CrossRef]
- Marberger, A.; Ferri, D.; Elsener, M.; Kröcher, O. The Significance of Lewis Acid Sites for the Selective Catalytic Reduction of Nitric Oxide on Vanadium-Based Catalysts. Angew. Chem. Int. Ed. 2016, 55, 11989–11994. [Google Scholar] [CrossRef] [PubMed]
- Marberger, A.; Ferri, D.; Elsener, M.; Sagar, A.; Artner, C.; Schermanz, K.; Kröcher, O. Relationship between structures and activities of supported metal vanadates for the selective catalytic reduction of NO by NH3. Appl. Catal. B Environ. 2017, 218, 731–742. [Google Scholar] [CrossRef]
- Zhu, M.; Lai, J.K.; Tumuluri, U.; Wu, Z.; Wachs, I.E. Nature of Active Sites and Surface Intermediates during SCR of NO with NH3 by Supported V2O5–WO3/TiO2 Catalysts. J. Am. Chem. Soc. 2017, 139, 15624–15627. [Google Scholar] [CrossRef]
- Risse, T.; Hollmann, D.; Brückner, A. Chapter 1. In situ electron paramagnetic resonance (EPR)—A unique tool for analysing structure and reaction behaviour of paramagnetic sites in model and real catalysts. Catalysists 2015, 27, 1–32. [Google Scholar] [CrossRef]
- Silversmit, G.; Poelman, H.; Sack, I.; Buyle, G.; Marin, G.B.; De Gryse, R. An in-situ Reduction/Oxidation XAS Study on the EL10V8 VOx/TiO2(Anatase) Powder Catalyst. Catal. Lett. 2006, 107, 61–71. [Google Scholar] [CrossRef]
- Silversmit, G.; Van Bokhoven, J.A.; Poelman, H.; Van Dereerden, A.; Marin, G.B.; Reyniers, M.; De Gryse, R. The Structure of a VOxTiO2anatase Powder Catalyst under Reduction and Oxidation at 623K. Phys. Scr. 2005, 115, 798. [Google Scholar] [CrossRef]
- Silversmit, G.; Van Bokhoven, J.A.; Poelman, H.; Van Der Eerden, A.M.; Marin, G.B.; Reyniers, M.F.; De Gryse, R. The structure of supported and unsupported vanadium oxide under calcination, reduction and oxidation determined with XAS. Appl. Catal. A Gen. 2005, 285, 151–162. [Google Scholar] [CrossRef]
- Feng, H.; Elam, J.W.; Libera, J.A.; Pellin, M.J.; Stair, P.C. Oxidative dehydrogenation of cyclohexane over alumina-supported vanadium oxide nanoliths. J. Catal. 2010, 269, 421–431. [Google Scholar] [CrossRef]
- Feng, Z.; Lu, J.; Feng, H.; Stair, P.C.; Elam, J.W.; Bedzyk, M.J. Catalysts Transform While Molecules React: An Atomic-Scale View. J. Phys. Chem. Lett. 2013, 4, 285–291. [Google Scholar] [CrossRef]
- Feng, Z.; Cheng, L.; Kim, C.Y.; Elam, J.W.; Zhang, Z.; Curtiss, L.A.; Zapol, P.; Bedzyk, M.J. Atomic-Scale Study of Ambient-Pressure Redox-Induced Changes for an Oxide-Supported Submonolayer Catalyst: VOx/α-TiO2(110). J. Phys. Chem. Lett. 2012, 3, 2845–2850. [Google Scholar] [CrossRef]
- Arnarson, L.; Rasmussen, S.B.; Falsig, H.; Lauritsen, J.V.; Moses, P.G. Coexistence of Square Pyramidal Structures of Oxo Vanadium (+5) and (+4) Species Over Low-Coverage VOX/TiO2 (101) and (001) Anatase Catalysts. J. Phys. Chem. C 2015, 119, 23445–23452. [Google Scholar] [CrossRef]
- Avdeev, V.I.; Bedilo, A.F. Molecular mechanism of propane oxidative dehydrogenation on surface oxygen radical sites of VOx/TiO2 catalysts. Res. Chem. Intermed. 2015, 42, 5237–5252. [Google Scholar] [CrossRef]
- Avdeev, V.I.; Bedilo, A.F. Molecular Mechanism of Oxygen Isotopic Exchange over Supported Vanadium Oxide Catalyst VOx/TiO2. J. Phys. Chem. C 2013, 117, 2879–2887. [Google Scholar] [CrossRef]
- Avdeev, V.I.; Tapilin, V.M. Water Effect on the Electronic Structure of Active Sites of Supported Vanadium Oxide Catalyst VOx/TiO2(001). J. Phys. Chem. C 2010, 114, 3609–3613. [Google Scholar] [CrossRef]
- Jehng, J.-M.; Deo, G.; Weckhuysen, B.M.; Wachs, I.E. Effect of water vapor on the molecular structures of supported vanadium oxide catalysts at elevated temperatures. J. Mol. Catal. A Chem. 1996, 110, 41–54. [Google Scholar] [CrossRef]
- Ruff, P.; Lauterbach, S.; Kleebe, H.J.; Hess, C. Surface structuring of mesoporous materials by controlled synthesis of nanocavities. Microporous Mesoporous Mater. 2016, 235, 160–169. [Google Scholar] [CrossRef]
- Thielemann, J.P.; Girgsdies, F.; Schlögl, R.; Hess, C. Pore structure and surface area of silica SBA-15: Influence of washing and scale-up. Beilstein J. Nanotechnol. 2011, 2, 110–118. [Google Scholar] [CrossRef]
- Huang, Y.; Gao, D.; Tong, Z.; Zhang, J.; Luo, H. Oxidation of NO over cobalt oxide supported on mesoporous silica. J. Nat. Gas Chem. 2009, 18, 421–428. [Google Scholar] [CrossRef]
- Lubas, M.; Jasinski, J.; Sitarz, M.; Kurpaska, L.; Podsiad, P. Raman spectroscopy of TiO2 thin films formed by hybrid treatment for biomedical applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 133, 867–871. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, L.; Shao, M.; Huang, J.; Ding, M.; Deng, X.; Wei, X.; Xu, X. Anodic Oxidation Synthesis of One-Dimensional TiO2Nanostructures for Photocatalytic and Field Emission Properties. J. Nanomater. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Waleska, P.S.; Rupp, S.; Hess, C. Operando Multiwavelength and Time-Resolved Raman Spectroscopy: Structural Dynamics of a Supported Vanadia Catalyst at Work. J. Phys. Chem. C 2018, 122, 3386–3400. [Google Scholar] [CrossRef]
- Tallant, D.R.; Bunker, B.C.; Balfe, C.A.; Brinker, C.J. Raman Spectra of Rings in Silicate Materials. MRS Proc. 1986, 73, 261. [Google Scholar] [CrossRef]
- Chligui, M.; Guimbretiere, G.; Canizares, A.; Matzen, G.; Vaills, Y.; Simon, P. New Features in the Raman Spectrum of Silica: Key-Points in the Improvement on Structure Knowledge. Available online: https://hal.archives-ouvertes.fr/hal-00520823/document (accessed on 27 November 2020).
- Huang, T.; Hu, Z.; Xu, G.S.; Zhang, X.L.; Zhang, J.Z.; Chu, J.H. Inherent optical behavior and structural variation in Na0.5Bi0.5TiO3-6%BaTiO3 revealed by temperature dependent Raman scattering and ultraviolet-visible transmittance. Appl. Phys. Lett. 2014, 104, 111908. [Google Scholar] [CrossRef]
- Kreisel, J.; Glazer, A.M.; Jones, G.; Thomas, P.A.; Abello, L.; Lucazeau, G. An x-ray diffraction and Raman spectroscopy investigation of A-site substituted perovskite compounds: The (Na1−xKx)0.5Bi0.5TiO3(0lexle1) solid solution. J. Phys. Condens. Matter 2000, 12, 3267–3280. [Google Scholar] [CrossRef]
- Walrafen, G.E.; Chu, Y.C.; Hokmabadi, M.S. Raman spectroscopic investigation of irreversibly compacted vitreous silica. J. Chem. Phys. 1990, 92, 6987–7002. [Google Scholar] [CrossRef]
- Li, C.; Xiong, G.; Liu, J.; Ying, P.; Xin, A.Q.; Feng, Z. Identifying Framework Titanium in TS-1 Zeolite by UV Resonance Raman Spectroscopy. J. Phys. Chem. B 2001, 105, 2993–2997. [Google Scholar] [CrossRef]
- Hess, C.; Tzolova-Müller, G.; Herbert, R. The Influence of Water on the Dispersion of Vanadia Supported on Silica SBA-15: A Combined XPS and Raman Study. J. Phys. Chem. C 2007, 111, 9471–9479. [Google Scholar] [CrossRef]
- Eberhardt, M.A.; Proctor, A.; Houalla, M.; Hercules, D.M. Investigation of V Oxidation States in Reduced V/Al2O3Catalysts by XPS. J. Catal. 1996, 160, 27–34. [Google Scholar] [CrossRef]
- Sawatzky, G.A.; Post, D. X-ray photoelectron and Auger spectroscopy study of some vanadium oxides. Phys. Rev. B 1979, 20, 1546–1555. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, W.; Zhong, Q.; Cai, W.; Li, H. Promotional effect of Si-doped V2O5/TiO2 for selective catalytic reduction of NOx by NH3. J. Environ. Sci. 2013, 25, 1703–1711. [Google Scholar] [CrossRef]
- Gao, X.; Bare, S.R.; Fierro, J.L.G.; Wachs, I.E. Structural Characteristics and Reactivity/Reducibility Properties of Dispersed and Bilayered V2O5/TiO2/SiO2 Catalysts. J. Phys. Chem. B 1999, 103, 618–629. [Google Scholar] [CrossRef]
- Nitsche, D.; Hess, C. Structure of Isolated Vanadia and Titania: A Deep UV Raman, UV–Vis, and IR Spectroscopic Study. J. Phys. Chem. C 2016, 120, 1025–1037. [Google Scholar] [CrossRef]
- Waleska, P.; Hess, C. Structural Dynamics of Dispersed Titania During Dehydration and Oxidative Dehydrogenation Studied by In Situ UV Raman Spectroscopy. Catal. Lett. 2018, 148, 2537–2547. [Google Scholar] [CrossRef]
- Gao, X.; Bare, S.R.; Fierro, J.L.G.; Banares, M.A.; Wachs, I.E. Preparation and in-Situ Spectroscopic Characterization of Molecularly Dispersed Titanium Oxide on Silica. J. Phys. Chem. B 1998, 102, 5653–5666. [Google Scholar] [CrossRef]
- Beck, B.; Harth, M.; Hamilton, N.G.; Carrero, C.A.; Uhlrich, J.J.; Trunschkec, A.; Shaikhutdinov, S.; Schubert, H.; Freund, H.; Schlögl, R.; et al. Partial oxidation of ethanol on vanadia catalysts on supporting oxides with different redox properties compared to propane. J. Catal. 2012, 296, 120–131. [Google Scholar] [CrossRef]
- Schraml-Marth, M.; Pohl, M.; Krauss, H.-L.; Wokaun, A. Spectroscopic investigation of the structure of silica-supported vanadium oxide catalysts at submonolayer coverages. J. Chem. Soc. Faraday Trans. 1991, 87, 2635–2646. [Google Scholar] [CrossRef]
- Arena, F.; Frusteri, F.; Martra, G.; Coluccia, S.; Parmaliana, A. Surface structures, reduction pattern and oxygen chemisorption of V2O5/SiO2 catalysts. J. Chem. Soc. Faraday Trans. 1997, 93, 3849–3854. [Google Scholar] [CrossRef]
- Kwon, D.W.; Park, K.H.; Hong, S.C. Influence of VOx surface density and vanadyl species on the selective catalytic reduction of NO by NH3 over VOx/TiO2 for superior catalytic activity. Appl. Catal. A Gen. 2015, 499, 1–12. [Google Scholar] [CrossRef]
- Burcham, L.J.; Deo, G.; Gao, X.; Wachs, I.E. In situ IR, Raman, and UV-Vis DRS spectroscopy of supported vanadium oxide catalysts during methanol oxidation. Top. Catal. 2000, 11, 85–100. [Google Scholar] [CrossRef]
- Gao, X.; Wachs, I.E. Investigation of Surface Structures of Supported Vanadium Oxide Catalysts by UV−vis−NIR Diffuse Reflectance Spectroscopy. J. Phys. Chem. B 2000, 104, 1261–1268. [Google Scholar] [CrossRef]
- Srinivas, D.; Hölderich, W.; Kujath, S.; Valkenberg, M.; Raja, T.; Saikia, L.; Hinze, R.; Ramaswamy, V. Active sites in vanadia/titania catalysts for selective aerial oxidation of β-picoline to nicotinic acid. J. Catal. 2008, 259, 165–173. [Google Scholar] [CrossRef]
- Due-Hansen, J.; Rasmussen, S.B.; Mikolajska, E.; Bañares, M.A.; Avila, P.C.; Fehrmann, R. Redox behaviour of vanadium during hydrogen–oxygen exposure of the V2O5-WO3/TiO2 SCR catalyst at 250 °C. Appl. Catal. B Environ. 2011, 107, 340–346. [Google Scholar] [CrossRef]
- Davis, E.A.; Mott, N.F. Conduction in non-crystalline systems V. Conductivity, optical absorption and photoconductivity in amorphous semiconductors. Philos. Mag. 1970, 22, 0903–0922. [Google Scholar] [CrossRef]
- Ghosh, A.; Bhattacharya, S. Optical and other physical properties of semiconducting cadmium vanadate glasses. J. Appl. Phys. 2007, 101, 83511. [Google Scholar] [CrossRef]
- Tauc, J. Absorption edge and internal electric fields in amorphous semiconductors. Mater. Res. Bull. 1970, 5, 721–729. [Google Scholar] [CrossRef]
- Waleska, P.S.; Hess, C. Oligomerization of Supported Vanadia: Structural Insight Using Surface-Science Models with Chemical Complexity. J. Phys. Chem. C 2016, 120, 18510–18519. [Google Scholar] [CrossRef]
- Borovkov, V.Y.; Mikheeva, E.P.; Zhidomirov, G.M.; Lapina, O.B. Theoretical and Experimental Studies of the Nature of the Catalytic Activity of VOx/TiO2 Systems. Kinet. Catal. 2003, 44, 710–717. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Reshetnikov, S.I.; Kiwi-Minsker, L.; Renken, A. Deactivation kinetics of V/Ti-oxide in toluene partial oxidation. Appl. Catal. A Gen. 2001, 220, 31–39. [Google Scholar] [CrossRef]
- Kamata, H. The role of K2O in the selective reduction of NO with NH3 over a V2O5(WO3)/TiO2 commercial selective catalytic reduction catalyst. J. Mol. Catal. A Chem. 1999, 139, 189–198. [Google Scholar] [CrossRef]
- Lu, J.; Kosuda, K.M.; Van Duyne, R.P.; Stair, P.C. Surface Acidity and Properties of TiO2/SiO2 Catalysts Prepared by Atomic Layer Deposition: UV−visible Diffuse Reflectance, DRIFTS, and Visible Raman Spectroscopy Studies. J. Phys. Chem. C 2009, 113, 12412–12418. [Google Scholar] [CrossRef][Green Version]
- Mu, J.C.; Li, X.Y.; Sun, W.B.; Fan, S.Y.; Wang, X.Y.; Wang, L.; Qin, M.C.; Gan, G.Q.; Yin, Z.F.; Zhang, D.K. Inductive Effect Boosting Catalytic Performance of Advanced Fe1−xVxO delta Catalysts in Low-Temperature NH3 Selective Catalytic Reduction: Insight into the Structure, Interaction, and Mechanisms. ACS Catal. 2018, 8, 6760–6774. [Google Scholar] [CrossRef]
- Cheng, K.; Liu, J.; Zhao, Z.; Wei, Y.; Jiang, G.; Duan, A. Direct synthesis of V–W–Ti nanoparticle catalysts for selective catalytic reduction of NO with NH3. RSC Adv. 2015, 5, 45172–45183. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, Z.; Yang, H.; Wang, C. Effect of MoO3 on vanadium based catalysts for the selective catalytic reduction of NOx with NH3 at low temperature. J. Environ. Sci. 2017, 56, 169–179. [Google Scholar] [CrossRef]
- Topsoe, N.; Topsoe, H.; Dumesic, J.A. Vanadia/Titania Catalysts for Selective Catalytic Reduction (SCR) of Nitric-Oxide by Ammonia. J. Catal. 1995, 151, 226–240. [Google Scholar] [CrossRef]
- Anstrom, M. Density functional theory studies of mechanistic aspects of the SCR reaction on vanadium oxide catalysts. J. Catal. 2003, 213, 115–125. [Google Scholar] [CrossRef]
- Farber, M.; Harris, S.P. Kinetics of ammonia-nitric oxide reactions on vanadium oxide catalysts. J. Phys. Chem. 1984, 88, 680–682. [Google Scholar] [CrossRef]
- Bredow, T.; Homann, T.; Jug, K. Adsorption of NO, NH3 and H2O on V2O5/TiO2 catalysts. Res. Chem. Intermed. 2004, 30, 65–73. [Google Scholar] [CrossRef]
- Yao, H.; Chen, Y.; Zhao, Z.; Wei, Y.; Liu, Z.; Zhai, D.; Liu, B.; Xu, C. Periodic DFT study on mechanism of selective catalytic reduction of NO via NH3 and O2 over the V2O5 (001) surface: Competitive sites and pathways. J. Catal. 2013, 305, 67–75. [Google Scholar] [CrossRef]
- Nickl, J.; Dutoit, D.; Baiker, A.; Scharf, U.; Wokaun, A. Vanadia catalysts for the selective catalytic reduction of NO by NH3 prepared by vapour deposition of vanadyl alkoxide onto various supports. Ber. Bunsenges. Phys. Chem. 1993, 97, 217–228. [Google Scholar]
- Pinaeva, L.; Suknev, A.; Budneva, A.; Paukshtis, E.; Bal’Zhinimaev, B. On the role of oxygen in the reaction of NO reduction by NH3 over monolayer V2O5 TiO2 catalyst. J. Mol. Catal. A Chem. 1996, 112, 115–124. [Google Scholar] [CrossRef]
- Mihai, O.; Creaser, D.; Olsson, L. Adsorption and Oxidation Investigations over Pt/Al2O3 Catalyst: A Microcalorimetric Study. Catalists 2016, 6, 73. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, B.; Liu, B.; Sun, S. A review of Mn-containing oxide catalysts for low temperature selective catalytic reduction of NOx with NH3: Reaction mechanism and catalyst deactivation. RSC Adv. 2017, 7, 26226–26242. [Google Scholar] [CrossRef]
- Klyushina, A.; Pacultová, K.; Karásková, K.; Jirátová, K.; Ritz, M.; Fridrichová, D.; Volodarskaja, A.; Obalová, L. Effect of preparation method on catalytic properties of Co-Mn-Al mixed oxides for N2O decomposition. J. Mol. Catal. A Chem. 2016, 425, 237–247. [Google Scholar] [CrossRef]
- Hamilton, N.; Wolfram, T.; Müller, G.T.; Hävecker, M.; Kröhnert, J.; Carrero, C.; Schomäcker, R.; Trunschkec, A.; Schlögl, R. Topology of silica supported vanadium–titanium oxide catalysts for oxidative dehydrogenation of propane. Catal. Sci. Technol. 2012, 2, 1346–1359. [Google Scholar] [CrossRef]
- Deo, G.; Wachs, I. Effect of Additives on the Structure and Reactivity of the Surface Vanadium Oxide Phase in V2O5/TiO2 Catalysts. J. Catal. 1994, 146, 335–345. [Google Scholar] [CrossRef]
- Jehng, J.M.; Wachs, I.E. The molecular structures and reactivity of V2O5/TiO2/SiO2 catalysts. Catal. Lett. 1992, 13, 9–19. [Google Scholar] [CrossRef]
- Rouquerol, F.O.; Rouquerol, J.; Sing, K.S.W.; Llewellyn, P.L.; Maurin, G. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, 2nd ed.; Elsevier/AP: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Haber, J.; Machej, T.; Czeppe, T. The phenomenon of wetting at solid/solid interface. Surf. Sci. 1985, 151, 301–310. [Google Scholar] [CrossRef]
- Wang, C.B.; Cai, Y.; Wachs, I.E. Reaction-Induced Spreading of Metal Oxides onto Surfaces of Oxide Supports during Alcohol Oxidation: Phenomenon, Nature, and Mechanisms. Langmuir 1999, 15, 1223–1235. [Google Scholar] [CrossRef]
- Zhu, H.; Ould-Chikh, S.; Dong, H.; Llorens, I.; Saih, Y.; Anjum, D.H.; Hazemann, J.L.; Basset, J.M. VOx/SiO2Catalyst Prepared by Grafting VOCl3 on Silica for Oxidative Dehydrogenation of Propane. Chem. Cat. Chem. 2015, 7, 3332–3339. [Google Scholar] [CrossRef]
- Moisii, C.; Curran, M.D.; Van De Burgt, L.J.; Stiegman, A.E. Raman spectroscopy of discrete silica supported vanadium oxide: Assignment of fundamental stretching modes. J. Mater. Chem. 2005, 15, 3519–3524. [Google Scholar] [CrossRef]
- Nuguid, R.J.G.; Ferri, D.; Marberger, A.; Nachtegaal, M.; Kröcher, O. Modulated Excitation Raman Spectroscopy of V2O5/TiO2: Mechanistic Insights into the Selective Catalytic Reduction of NO with NH3. ACS Catal. 2019, 9, 6814–6820. [Google Scholar] [CrossRef]
- Lietti, L.; Alemany, J.L.; Forzatti, P.; Busca, G.; Ramis, G.; Giamello, E.; Bregani, F. Reactivity of V2O5-WO3/TiO2 catalysts in the selective catalytic reduction of nitric oxide by ammonia. Catal. Today 1996, 29, 143–148. [Google Scholar] [CrossRef]
- Arnarson, L.; Falsig, H.; Rasmussen, S.B.; Lauritsen, J.V.; Moses, P.G. A complete reaction mechanism for standard and fast selective catalytic reduction of nitrogen oxides on low coverage VO /TiO2(0 0 1) catalysts. J. Catal. 2017, 346, 188–197. [Google Scholar] [CrossRef]
- Giakoumelou, I.; Fountzoula, C.; Kordulis, C.; Boghosian, S. Molecular structure and catalytic activity of V2O5/TiO2 catalysts for the SCR of NO by NH3: In situ Raman spectra in the presence of O2, NH3, NO, H2, H2O, and SO2. J. Catal. 2006, 239, 1–12. [Google Scholar] [CrossRef]
- Dong, L.; Sun, C.; Tang, C.; Zhang, B.; Zhu, J.; Liu, B.; Gao, F.; Hu, Y.; Dong, L.; Chen, Y. Investigations of surface VOx species and their contributions to activities of VOx/Ti0.5Sn0.5O2 catalysts toward selective catalytic reduction of NO by NH3. Appl. Catal. A Gen. 2012, 431, 126–136. [Google Scholar] [CrossRef]
- Zhang, Q.M.; Song, C.; Lyu, G.; Bin, F.; Pang, H.T.; Song, J. Effect of metal oxide partial substitution of V2O5 in V2O5–WO3/TiO2 on selective catalytic reduction of NO with NH3. J. Ind. Eng. Chem. 2015, 24, 79–86. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, Q. Promotional effect of WO3 on O2− over V2O5/TiO2 catalyst for selective catalytic reduction of NO with NH3. J. Mol. Catal. A Chem. 2013, 373, 108–113. [Google Scholar] [CrossRef]
- Wang, P.; Guo, R.T. The promotion effect of copper doping on the potassium resistance of V/TiO2 catalyst for selective catalytic reduction of NO with NH3. Chem. Pap. 2017, 71, 2253–2259. [Google Scholar] [CrossRef]
- Handy, B.E.; Baiker, A.; Schraml-Marth, M.; Wokaun, A. Vanadia supported on TiO2-SiO2 mixed oxide gels: Structure of the dispersed phase and activity for the selective catalytic reduction of NO with NH3. J. Catal. 1992, 133, 1–20. [Google Scholar] [CrossRef]
- Handy, B.E.; Maciejewski, M.; Baiker, A. Vanadia, vanadia-titania, and vanadia-titania-silica gels: Structural genesis and catalytic behavior in the reduction of nitric oxide with ammonia. J. Catal. 1992, 134, 75–86. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kuma, R.; Masaki, S.; Sugishima, N. TiO2-SiO2 and V2O5/TiO2-SiO2 catalyst: Physico-chemical characteristics and catalytic behavior in selective catalytic reduction of NO by NH3. Appl. Catal. B Environ. 2005, 60, 173–179. [Google Scholar] [CrossRef]
- Won, J.M.; Kim, M.S.; Hong, S.C. Effect of vanadium surface density and structure in VOx/TiO2 on selective catalytic reduction by NH3. Korean J. Chem. Eng. 2018, 35, 2365–2378. [Google Scholar] [CrossRef]
- Dutoit, D.C.M.; Reiche, M.A.; Baiker, A. Vanadia-silica aerogels—Structure and catalytic properties in selective reduction of NO by NH3. Appl. Catal. B-Environ. 1997, 13, 275–288. [Google Scholar] [CrossRef]
- Soyer, S.; Uzun, A.; Senkan, S.; Onal, I. A quantum chemical study of nitric oxide reduction by ammonia (SCR reaction) on V2O5 catalyst surface. Catal. Today 2006, 118, 268–278. [Google Scholar] [CrossRef]
- Dai, H.; Bell, A.T.; Iglesia, E. Effects of molybdena on the catalytic properties of vanadia domains supported on alumina for oxidative dehydrogenation of propane. J. Catal. 2004, 221, 491–499. [Google Scholar] [CrossRef]
- Zhao, D.; Sun, J.; Li, Q.; Stucky, G.D. Morphological Control of Highly Ordered Mesoporous Silica SBA-15. Chem. Mater. 2000, 12, 275–279. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Stotal [m2/g] | Dp [nm] | Vtotal [cm3/g] | LV [V/nm2] | V:Ti (wt%) a |
|---|---|---|---|---|---|
| SBA-15 | 952 | 6.96 | 0.82 | -- | -- |
| TiO2/SBA-15+1xVOx | 410 | 6.33 | 0.49 | 0.4 | 1.4:5.8 |
| TiO2/SBA-15+3xVOx | 366 | 6.33 | 0.43 | 1.6 | 5.1:5.9 |
| TiO2/SBA-15+5xVOx | 355 | 6.33 | 0.43 | 2.0 | 6.0:5.6 |
| Temperature, °C | 100 | 150 | 200 | 250 | 300 | 350 | 400 | 450 |
|---|---|---|---|---|---|---|---|---|
| NOx conversion, % | 33.4 | 33.1 | 51.3 | 61.1 | 59.9 | 60.5 | 53.2 | 41.9 |
| N2 selectivity, % | 100 | 99.4 | 99.3 | 98.1 | 97.0 | 95.1 | 92.8 | 90.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Hess, C. High Surface Area VOx/TiO2/SBA-15 Model Catalysts for Ammonia SCR Prepared by Atomic Layer Deposition. Catalysts 2020, 10, 1386. https://doi.org/10.3390/catal10121386
Shen J, Hess C. High Surface Area VOx/TiO2/SBA-15 Model Catalysts for Ammonia SCR Prepared by Atomic Layer Deposition. Catalysts. 2020; 10(12):1386. https://doi.org/10.3390/catal10121386
Chicago/Turabian StyleShen, Jun, and Christian Hess. 2020. "High Surface Area VOx/TiO2/SBA-15 Model Catalysts for Ammonia SCR Prepared by Atomic Layer Deposition" Catalysts 10, no. 12: 1386. https://doi.org/10.3390/catal10121386
APA StyleShen, J., & Hess, C. (2020). High Surface Area VOx/TiO2/SBA-15 Model Catalysts for Ammonia SCR Prepared by Atomic Layer Deposition. Catalysts, 10(12), 1386. https://doi.org/10.3390/catal10121386