Redox Isomerization of Allylic Alcohols Catalyzed by New Water-Soluble Rh(I)-N-Heterocyclic Carbene Complexes

 ,
,  ,
,  , ,
, ,  and
and
Abstract
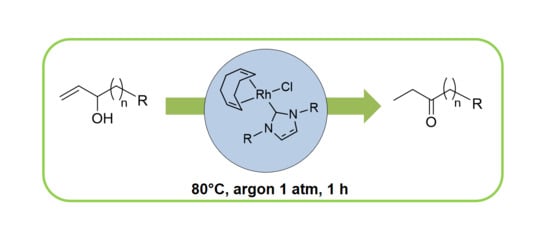
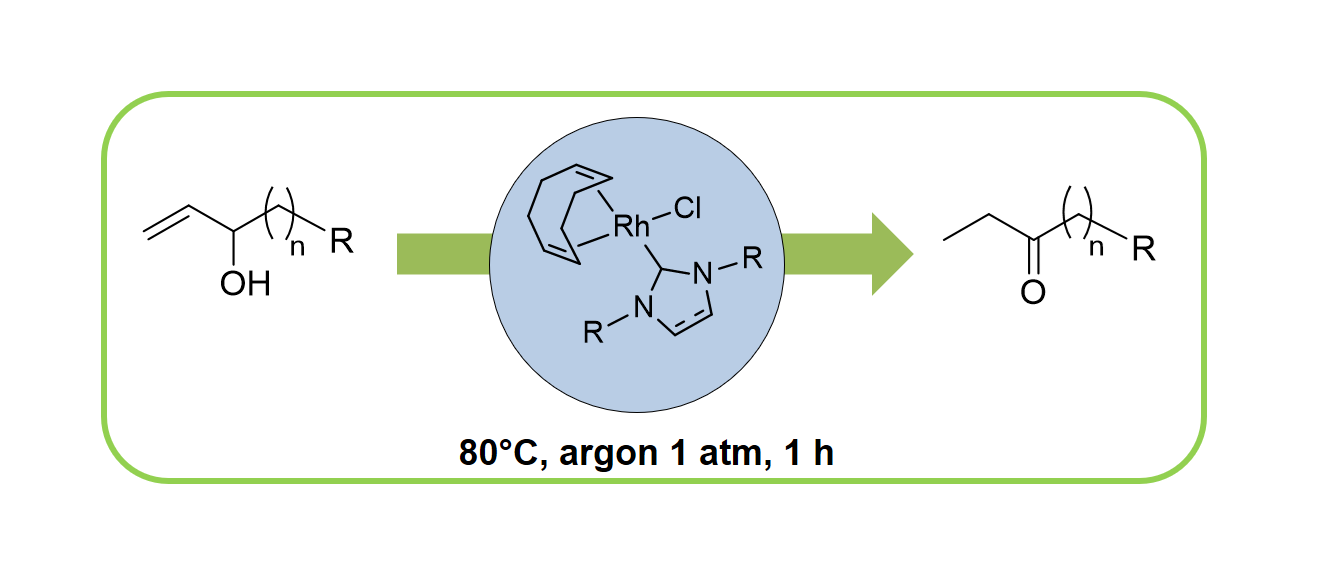
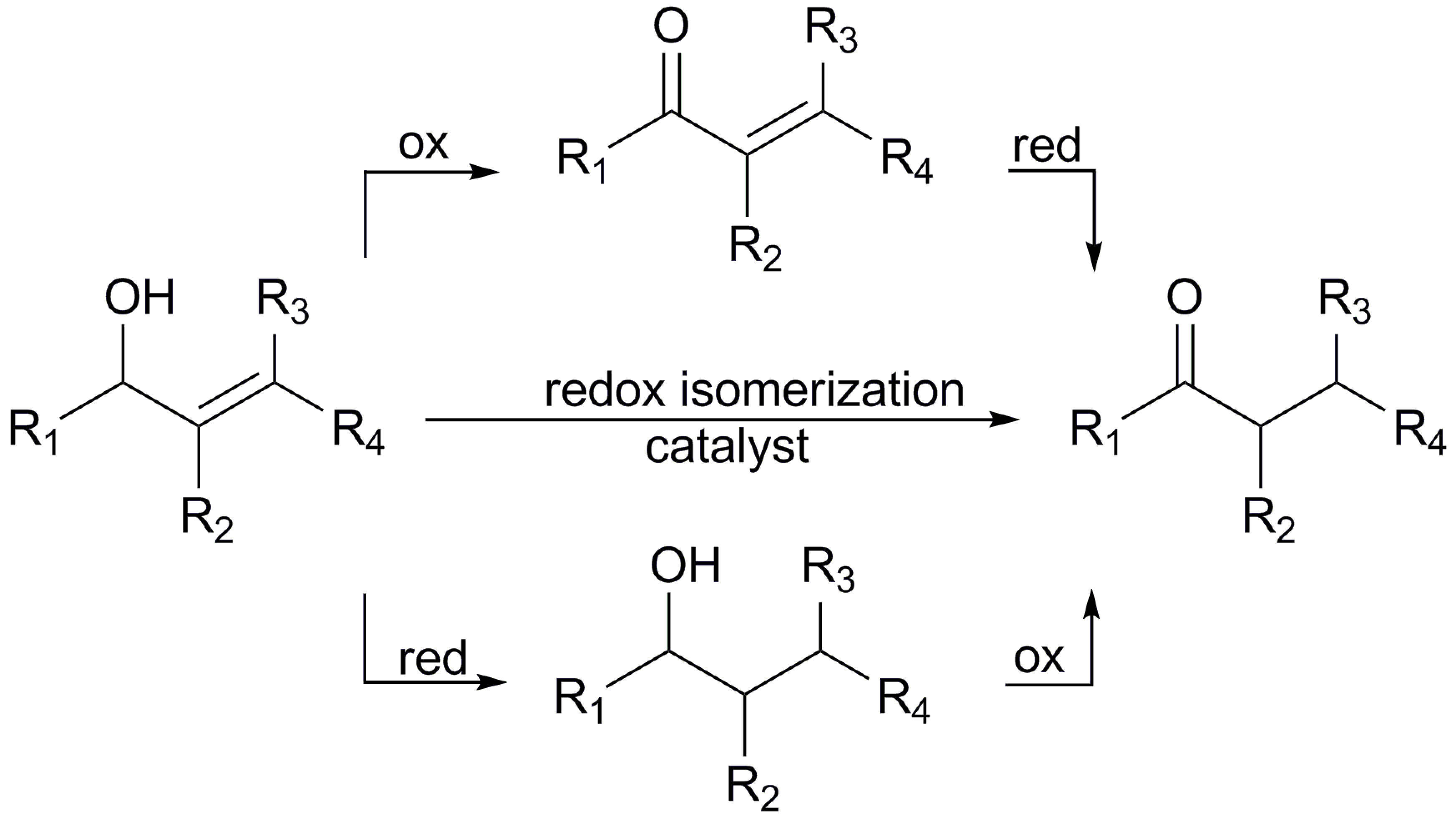
1. Introduction
2. Results and Discussion
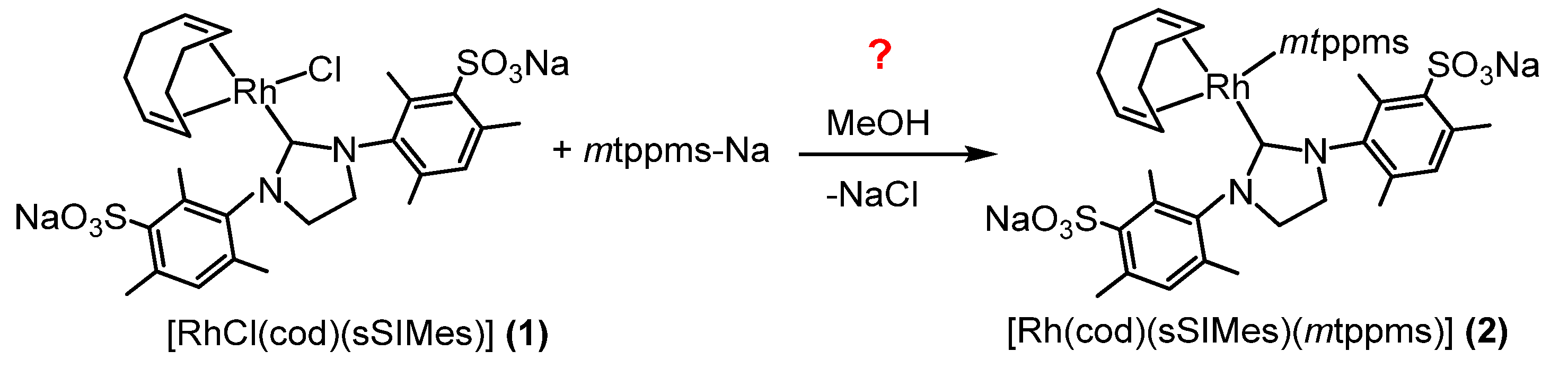
2.1. Synthetic Procedure and Characterization of Rh(I)-N-Heterocyclic Complexes 1–5
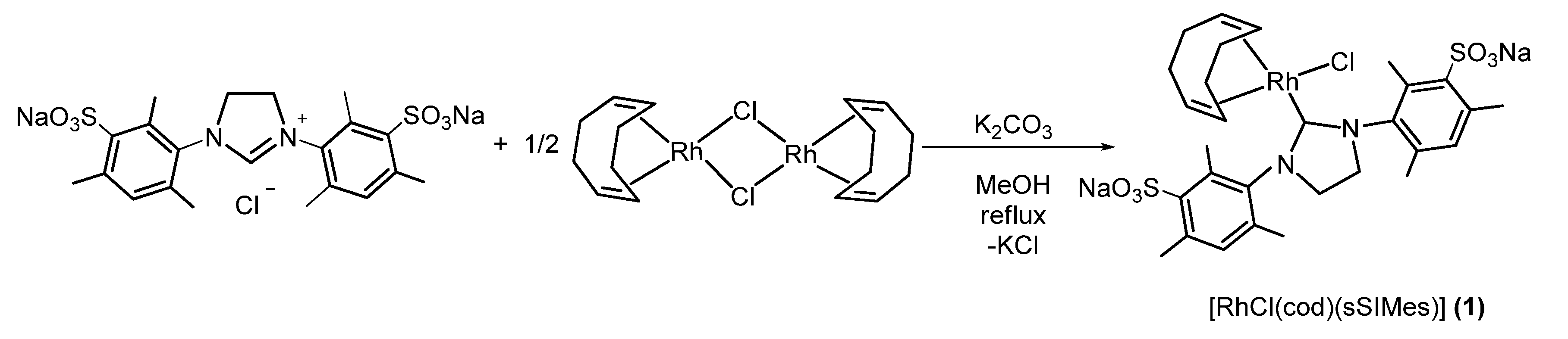
2.1.1. Synthesis of [RhCl(cod)(sSIMes)] (1) (Scheme 2).
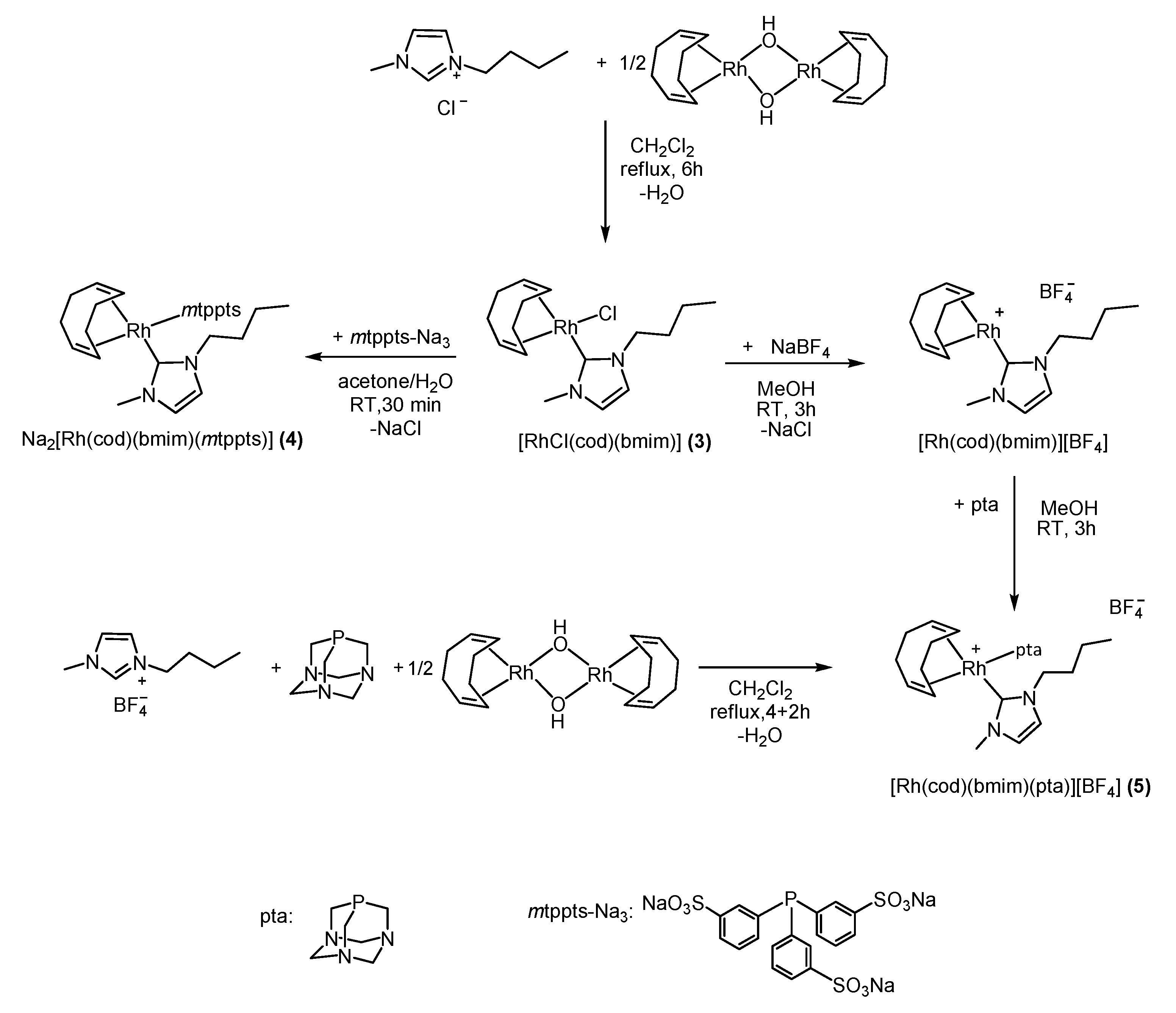
2.1.2. Synthesis of [RhCl(bmim)(cod)] (3)
2.1.3. Synthesis of Na2[Rh(bmim)(cod)(mtppts)] (4)
2.1.4. Synthesis of [Rh(bmim)(cod)(pta)]BF4 (5)
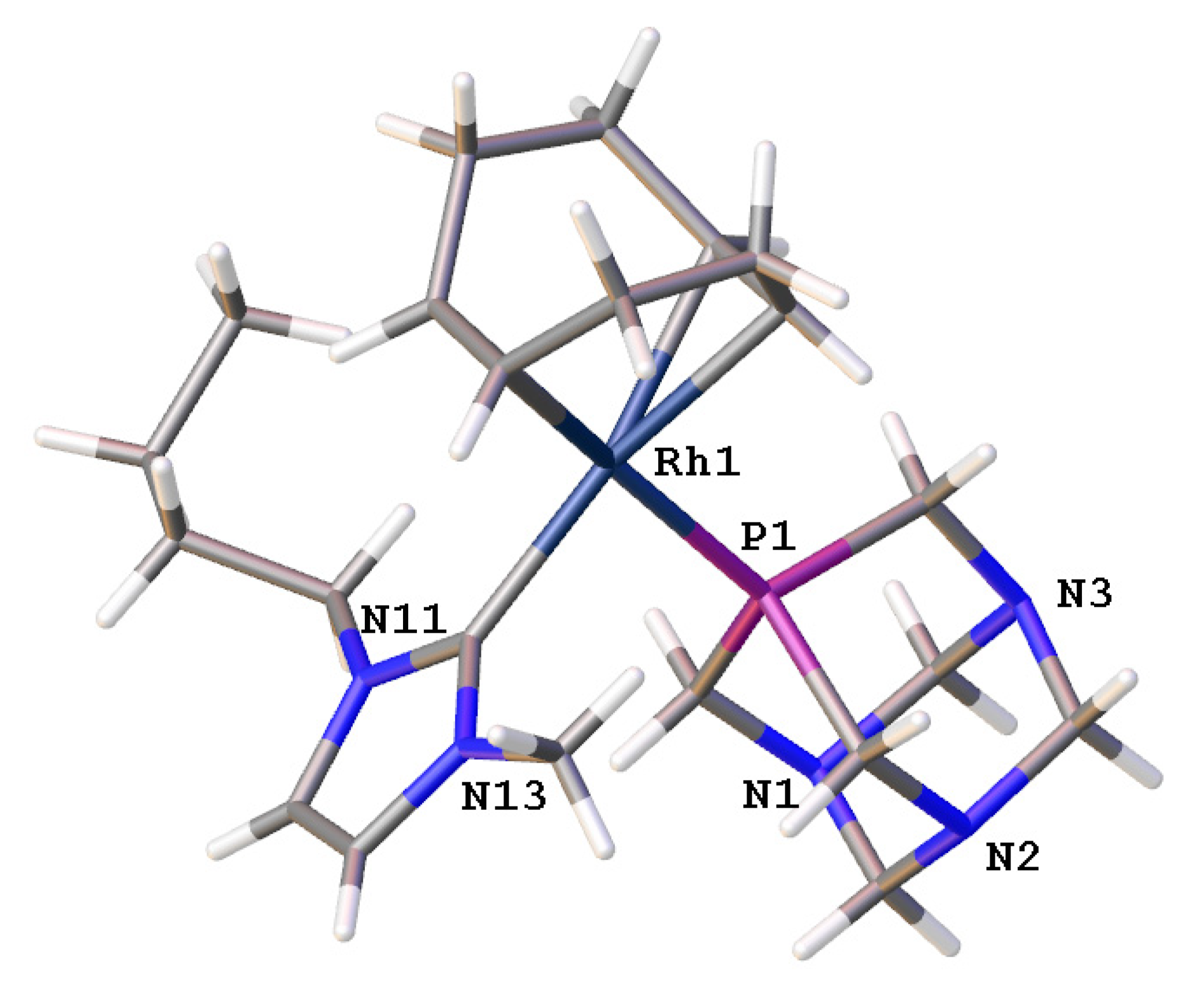
2.1.5. Single-crystal X-ray diffraction analysis of [Rh(bmim)(η4-cod)(pta)]BF4 (5)
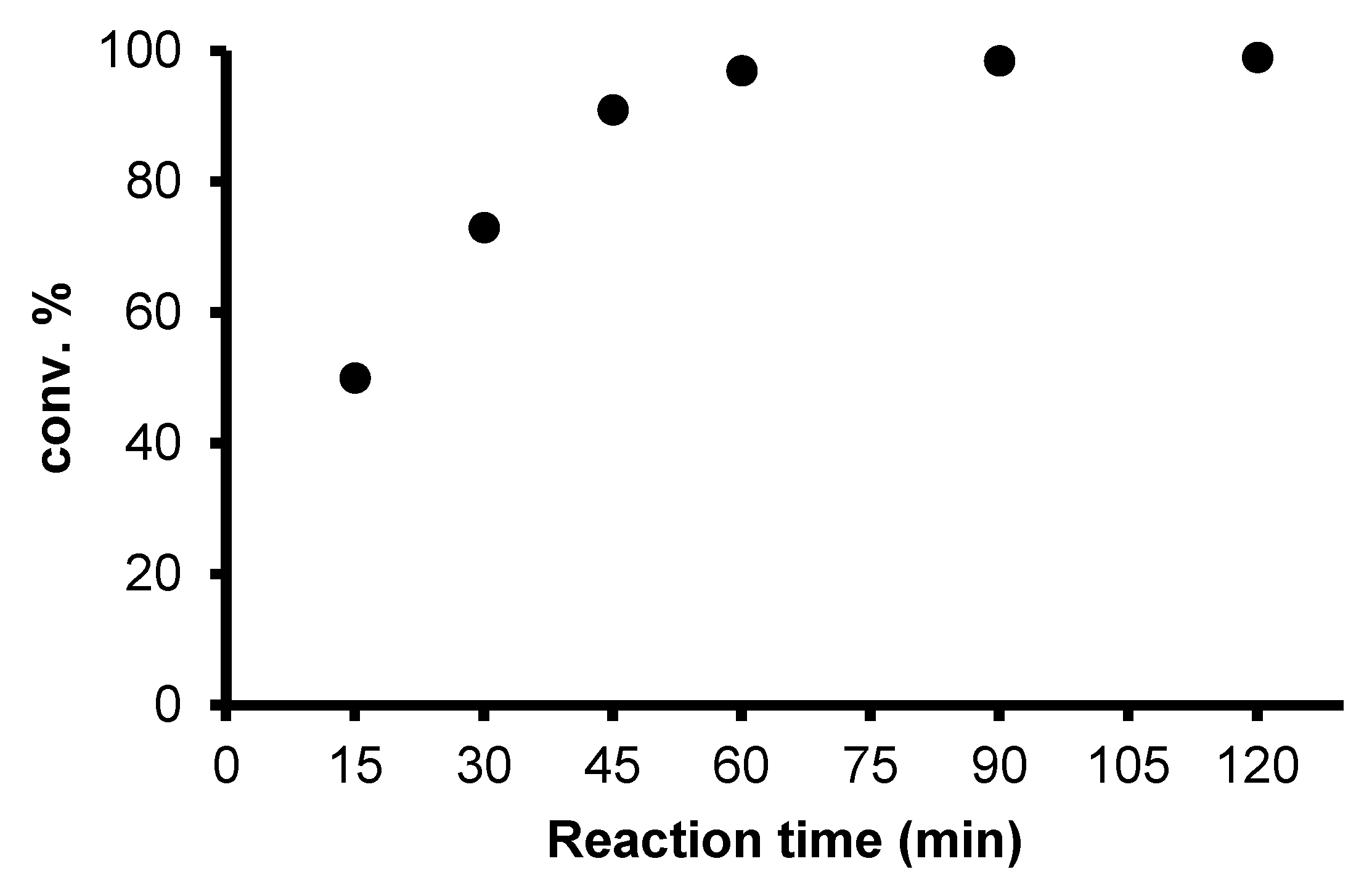
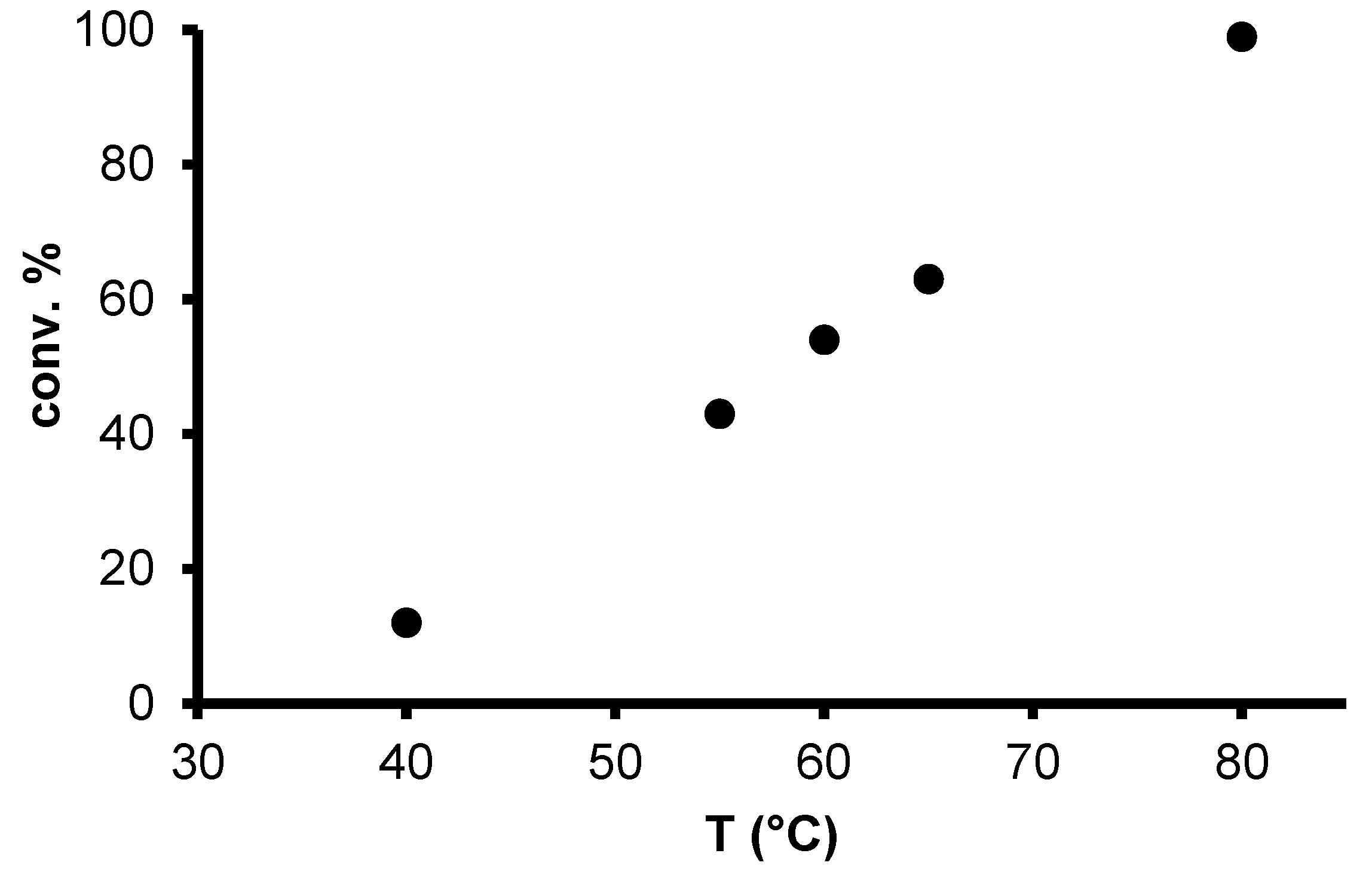
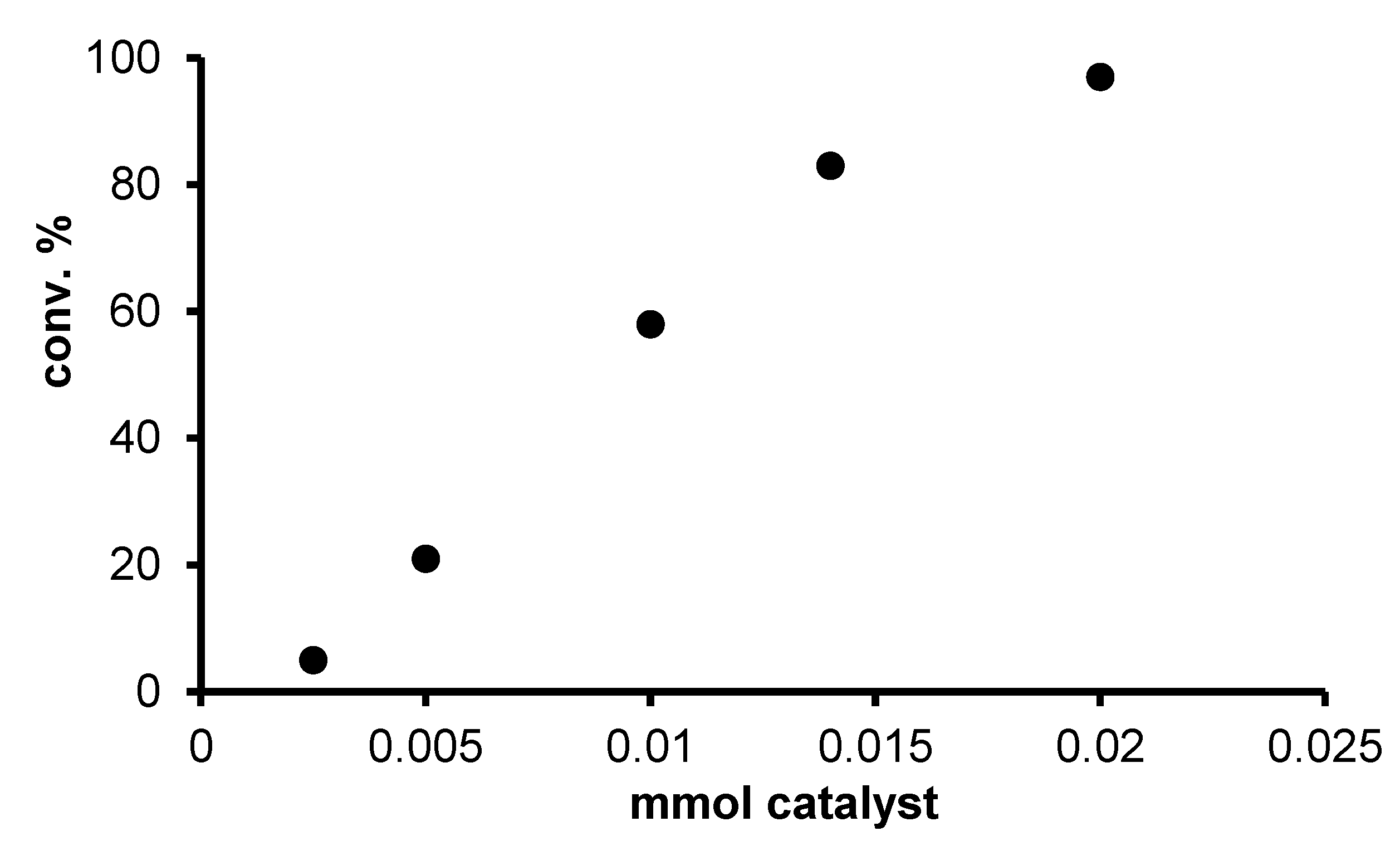
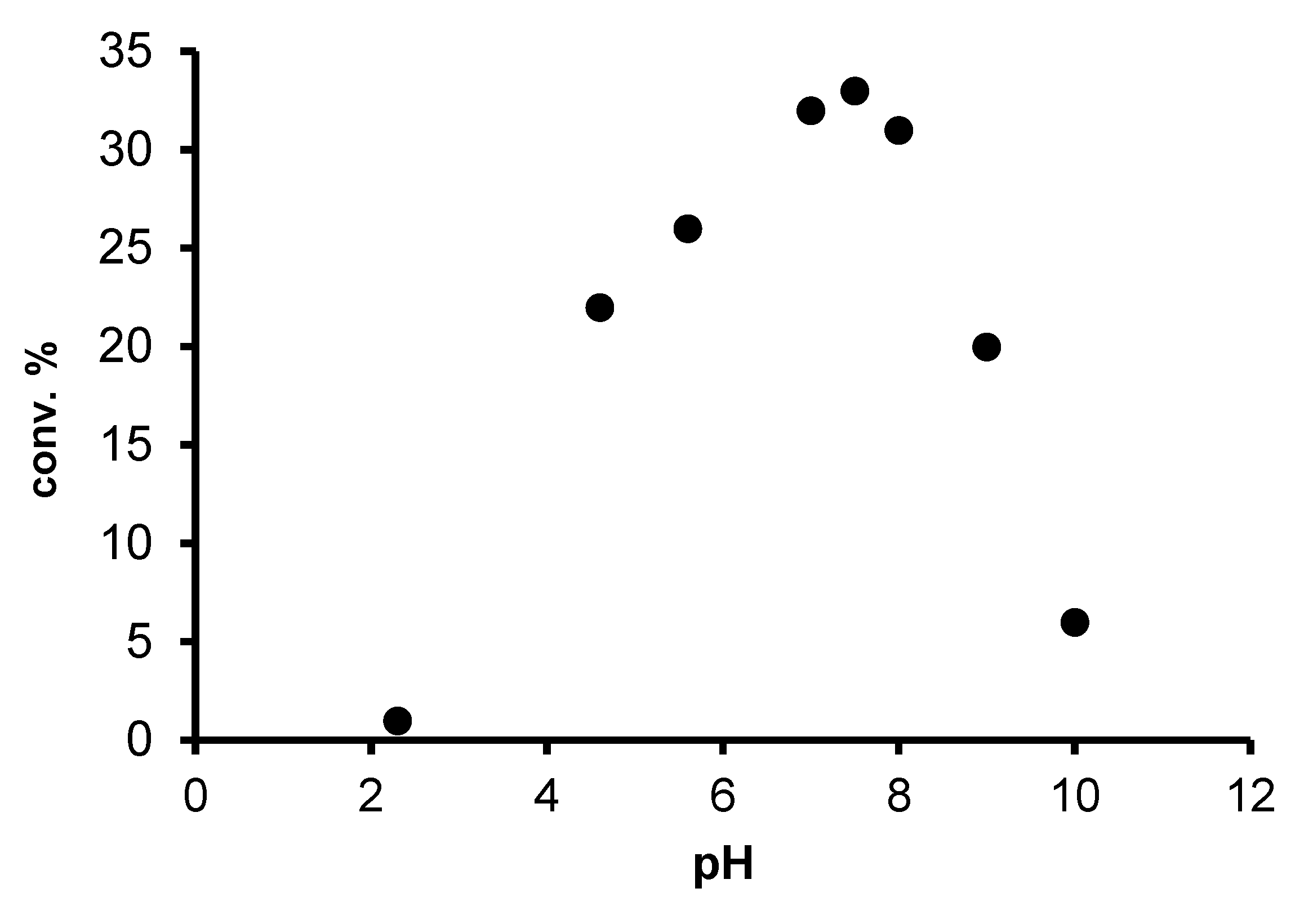
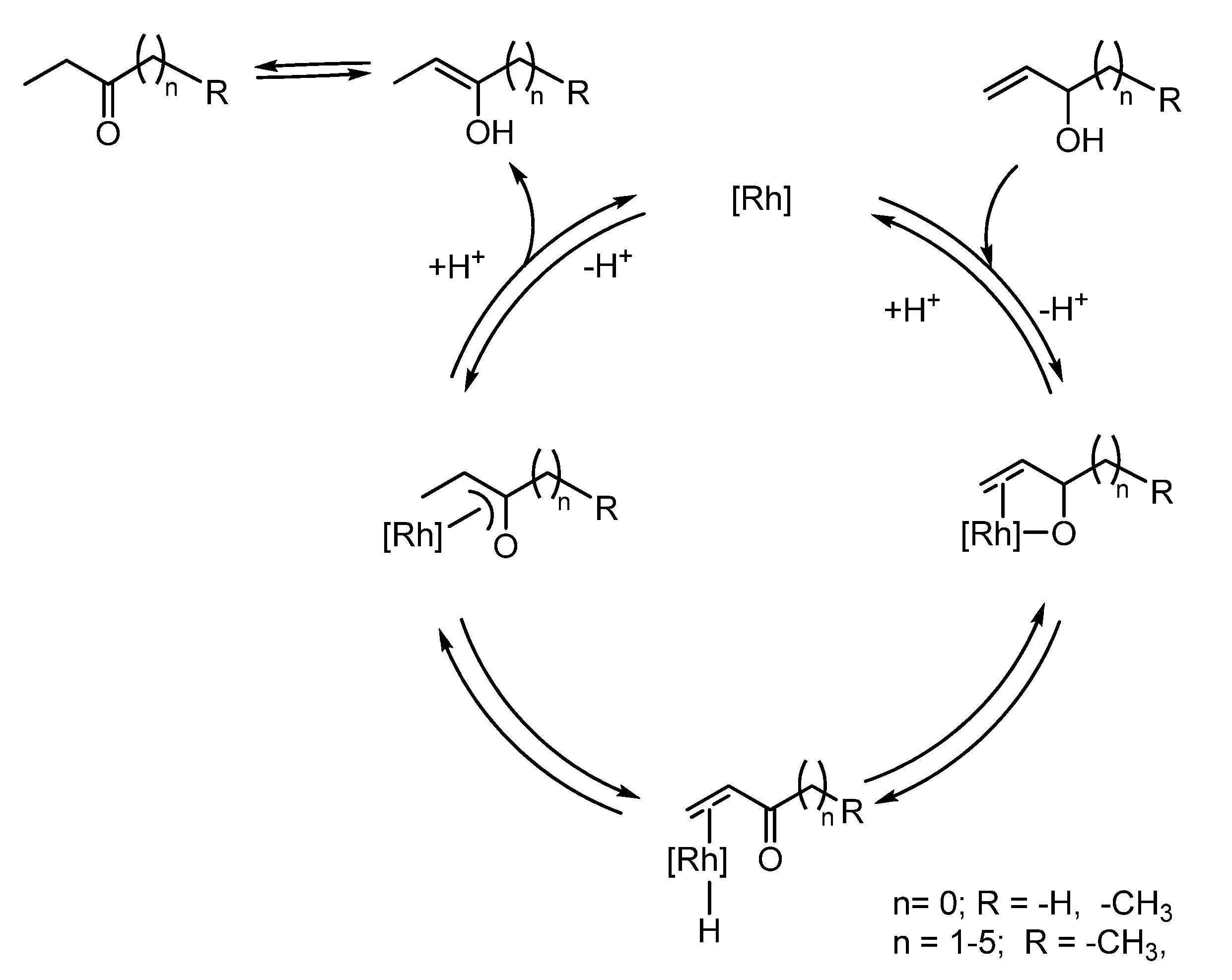
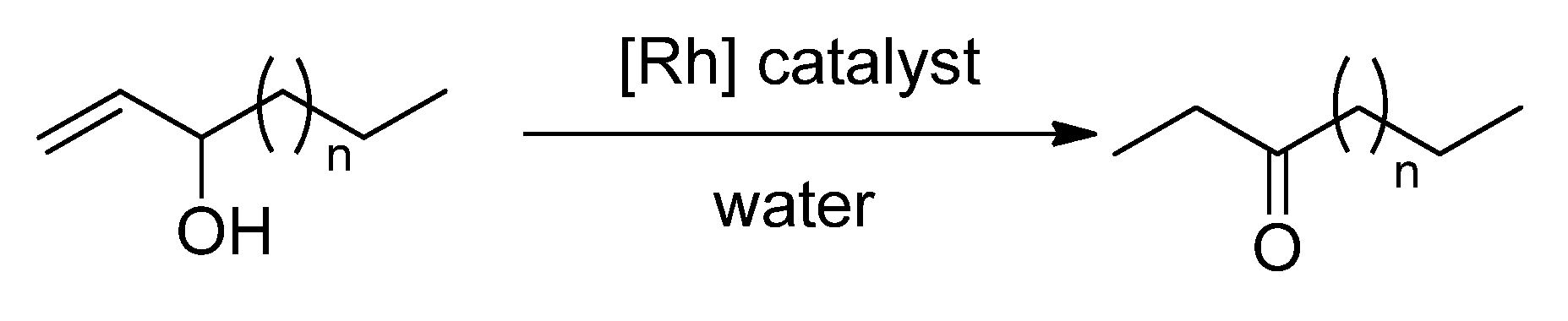
2.2. Redox isomerization of allylic alcohols with water soluble Rh(I)-NHC catalysts
3. Materials and Methods
3.1. Materials
3.2. General Methods
3.2.1. Preparation of [RhC(cod)(sSIMes)] (1)
3.2.2. Synthesis of [RhCl(bmim)(cod)] (3)
3.2.3. Synthesis of Na2[Rh(bmim)(cod)(mtppts)] (4)
3.2.4. Synthesis of [Rh(bmim)(cod)(pta)]BF4 (5)
3.2.5. General Procedure Redox Isomerization of Allylic Alcohols
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mantilli, L.; Mazet, C. Platinum Metals in the Catalytic Asymmetric Isomerization of Allylic Alcohols. Chem. Lett. 2011, 40, 341–344. [Google Scholar] [CrossRef]
- Van der Drift, R.C.; Bouwman, E.; Drent, E. Homogeneously catalysed isomerisation of allylic alcohols to carbonyl compounds. J. Organomet. Chem. 2002, 650, 1–24. [Google Scholar] [CrossRef]
- Fu, G.C. Recent Advances in Rhodium(I)-Catalyzed Asymmetric Olefin Isomerization and Hydroacylation Reactions. In Modern Rhodium-Catalyzed Organic Reactions; Evans, P.A., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 79–91. [Google Scholar]
- Uma, R.; Crévisy, C.; Grée, R. Transposition of Allylic Alcohols into Carbonyl Compounds Mediated by Transition Metal Complexes. Chem. Rev. 2003, 103, 27–52. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.; Bolaño, S.; Gonsalvi, L.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA) and derivatives. Part II. The quest for tailored ligands, complexes and related applications. Coord. Chem. Rev. 2010, 254, 555–607. [Google Scholar] [CrossRef]
- Lorenzo-Luis, P.; Romerosa, A.; Serrano-Ruiz, M. Catalytic Isomerization of Allylic Alcohols in Water. ACS Catal. 2012, 2, 1079–1086. [Google Scholar] [CrossRef]
- Garcia-Alvarez, J.; Garcia-Garrido, S.E.; Crochet, P.; Cadierno, V. Metal-Catalyzed Isomerization of Allylic and Propargylic Alcohols in Aqueous Media. Curr. Top. Catal. 2012, 10, 35–56. [Google Scholar] [CrossRef]
- Scalambra, F.; Lorenzo-Luis, P.; de los Rios, I.; Romerosa, A. Isomerization of allylic alcohols in water catalyzed by transition metal complexes. Coord. Chem. Rev. 2019, 393, 118–148. [Google Scholar] [CrossRef]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef]
- Annen, S.; Zweifel, T.; Ricatto, F.; Grützmacher, H. Catalytic Aerobic Dehydrogenative Coupling of Primary Alcohols and Water to Acids Promoted by a Rhodium(I) Amido N-Heterocyclic Carbene Complex. ChemCatChem 2010, 2, 1286–1295. [Google Scholar] [CrossRef]
- Levin, E.; Ivry, E.; Diesendruck, C.E.; Lemcoff, N.G. Water in N-Heterocyclic Carbene-Assisted Catalysis. Chem. Rev. 2015, 115, 4607–4692. [Google Scholar] [CrossRef]
- Pruchnik, F.P.; Smoleński, P.; Wajda-Hermanowicz, K. Rhodium(I) acetylacetonato complexes with functionalized phosphines. J. Organomet. Chem. 1998, 570, 63–69. [Google Scholar] [CrossRef]
- Bianchini, C.; Frediani, P.; Sernau, V. Zwitterionic Metal Complexes of the New Triphosphine NaO3S(C6H4)CH2C(CH2PPh2)3 in Liquid Biphasic Catalysis: An Alternative to Teflon “Ponytails” for Facile Catalyst Separation without Water. Organometallics 1995, 14, 5458–5459. [Google Scholar] [CrossRef]
- Corkum, E.G.; Kalapugama, S.; Hass, M.J.; Bergens, S.H. Solvent-free isomerization of allylic alcohols catalyzed by a rhodium catalyst-organic framework. RSC Adv. 2012, 2, 3473–3476. [Google Scholar] [CrossRef]
- Gómez, A.B.; Holmberg, P.; Bäckvall, J.-E.; Martín-Matute, B. Transition metal-catalyzed redox isomerization of codeine and morphine in water. RSC Adv. 2014, 4, 39519–39522. [Google Scholar] [CrossRef]
- De Bellefon, C.; Caravieilhes, S.; Kuntz, É.G. Efficient catalytic isomerization of allylic alcohols to carbonyl compounds with water soluble rhodium complexes. Comptes Rendus Académie Sci. Ser. IIC Chem. 2000, 3, 607–614. [Google Scholar] [CrossRef]
- Knight, D.A.; Schull, T.L. Rhodium Catalyzed Allylic Isomerization in Water. Synth. Commun. 2003, 33, 827–831. [Google Scholar] [CrossRef]
- Smoleński, P.; Kirillova, M.V.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Isomerisation and controlled condensation in an aqueous medium of allyl alcohol catalysed by new water-soluble rhodium complexes with 1,3,5-triaza-7-phosphaadamantane (PTA). Dalton Trans. 2013, 42, 10867–10874. [Google Scholar] [CrossRef]
- Wilson, M.E.; Whitesides, G.M. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J. Am. Chem. Soc. 1978, 100, 306–307. [Google Scholar] [CrossRef]
- Ward, T.R. Artificial Metalloenzymes Based on the Biotin−Avidin Technology: Enantioselective Catalysis and Beyond. Acc. Chem. Res. 2011, 44, 47–57. [Google Scholar] [CrossRef]
- Velazquez, H.D.; Verpoort, F. N-heterocyclic carbene transition metal complexes for catalysis in aqueous media. Chem. Soc. Rev. 2012, 41, 7032–7060. [Google Scholar] [CrossRef]
- Wang, W.; Cui, L.; Sun, P.; Shi, L.; Yue, C.; Li, F. Reusable N-Heterocyclic Carbene Complex Catalysts and Beyond: A Perspective on Recycling Strategies. Chem. Rev. 2018, 118, 9843–9929. [Google Scholar] [CrossRef] [PubMed]
- Schaper, L.-A.; Hock, S.J.; Herrmann, W.A.; Kühn, F.E. Synthesis and Application of Water-Soluble NHC Transition-Metal Complexes. Angew. Chem. Int. Ed. 2013, 52, 270–289. [Google Scholar] [CrossRef]
- Almássy, A.; Nagy, C.E.; Bényei, A.C.; Joó, F. Novel Sulfonated N-Heterocyclic Carbene Gold(I) Complexes: Homogeneous Gold Catalysis for the Hydration of Terminal Alkynes in Aqueous Media. Organometallics 2010, 29, 2484–2490. [Google Scholar] [CrossRef]
- Czégéni, C.E.; Papp, G.; Kathó, Á.; Joó, F. Water-soluble gold(I)–NHC complexes of sulfonated IMes and SIMes and their catalytic activity in hydration of alkynes. J. Mol. Catal. Chem. 2011, 340, 1–8. [Google Scholar] [CrossRef]
- De, S.; Udvardy, A.; Czégéni, C.E.; Joó, F. Poly-N-heterocyclic carbene complexes with applications in aqueous media. Coord. Chem. Rev. 2019, 400, 213038. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Gooβen, L.J.; Spiegler, M. Functionalized imidazoline-2-ylidene complexes of rhodium and palladium. J. Organomet. Chem. 1997, 547, 357–366. [Google Scholar] [CrossRef]
- Syska, H.; Herrmann, W.A.; Kühn, F.E. Water-soluble carbene complexes as catalysts for the hydrogenation of acetophenone under hydrogen pressure. J. Organomet. Chem. 2012, 703, 56–62. [Google Scholar] [CrossRef]
- Virboul, M.A.N.; Lutz, M.; Siegler, M.A.; Spek, A.L.; van Koten, G.; Klein Gebbink, R.J.M. One-Pot Synthesis and Immobilisation of Sulfonate-Tethered N-Heterocyclic Carbene Complexes on Polycationic Dendrimers. Chem. Eur. J. 2009, 15, 9981–9986. [Google Scholar] [CrossRef]
- Basauri-Molina, M.; Riemersma, C.F.; Würdemann, M.; Klein Gebbink, R.J.M. Lipase active site covalent anchoring of Rh(NHC) catalysts: Towards chemoselective artificial metalloenzymes. Chem. Commun. 2015, 51, 6792–6795. [Google Scholar] [CrossRef]
- Sauer, D.F.; Qu, Y.; Mertens, M.A.S.; Schiffels, J.; Polen, T.; Schwaneberg, U.; Okuda, J. Biohybrid catalysts for sequential one-pot reactions based on an engineered transmembrane protein. Catal. Sci. Technol. 2019, 9, 942–946. [Google Scholar] [CrossRef]
- Strohmeier, W.; Weigelt, L. Homogene katalytische umlagerung von isobutenol zu butyraldehyd mit RhHCO(PPh3)3 und metallhalogeniden. J. Organomet. Chem. 1975, 86, C17–C19. [Google Scholar] [CrossRef]
- Alper, H.; Hachem, K. Rhodium(I)-catalyzed biphasic isomerization of allylic alcohols. J. Org. Chem. 1980, 45, 2269–2270. [Google Scholar] [CrossRef]
- Sasson, Y.; Zoran, A.; Blum, J. Reversible ion-pair extraction in a biphasic system. application in transition metal-catalyzed isomerization of allylic compounds. J. Mol. Catal. 1981, 11, 293–300. [Google Scholar] [CrossRef]
- De Bellefon, C.; Abdallah, R.; Ireland, T. Discovering New Water Soluble Catalysts for the Liquid/Liquid Isomerization of Allylic Alcohols Using Microdevices. Chem. Ing. Tech. 2004, 76, 633–637. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W. Isomerization of allylic alcohols to carbonyl compounds by aqueous-biphase rhodium catalysis. New J. Chem. 2001, 25, 11–12. [Google Scholar] [CrossRef]
- Schumann, H.; Ravindar, V.; Meltser, L.; Baidossi, W.; Sasson, Y.; Blum, J. Effect of the CO2H groups of carboxylated triarylphosphines on (COD)RhCl(PAr3)-catalyzed isomerization of 1-octen-3-ol under phase transfer conditions. J. Mol. Catal. Chem. 1997, 118, 55–61. [Google Scholar] [CrossRef]
- Leung, D.H.; Bergman, R.G.; Raymond, K.N. Highly Selective Supramolecular Catalyzed Allylic Alcohol Isomerization. J. Am. Chem. Soc. 2007, 129, 2746–2747. [Google Scholar] [CrossRef]
- Ahlsten, N.; Lundberg, H.; Martín-Matute, B. Rhodium-catalysed isomerisation of allylic alcohols in water at ambient temperature. Green Chem. 2010, 12, 1628–1633. [Google Scholar] [CrossRef]
- Sahoo, S.; Lundberg, H.; Edén, M.; Ahlsten, N.; Wan, W.; Zou, X.; Martín-Matute, B. Single Site Supported Cationic Rhodium(I) Complexes for the Selective Redox Isomerization of Allylic Alcohols. ChemCatChem 2012, 4, 243–250. [Google Scholar] [CrossRef]
- Azua, A.; Sanz, S.; Peris, E. Sulfonate-Functionalized NHC-Based Ruthenium Catalysts for the Isomerization of Allylic Alcohols in Water. Recyclability Studies. Organometallics 2010, 29, 3661–3664. [Google Scholar] [CrossRef]
- Bruneau, C.; Achard, M. Allylic ruthenium(IV) complexes in catalysis. Coord. Chem. Rev. 2012, 256, 525–536. [Google Scholar] [CrossRef]
- Crochet, P.; Cadierno, V. Arene-ruthenium(II) complexes with hydrophilic P-donor ligands: Versatile catalysts in aqueous media. Dalton Trans. 2014, 43, 12447–12462. [Google Scholar] [CrossRef] [PubMed]
- Horváth, H.; Kathó, Á.; Udvardy, A.; Papp, G.; Szikszai, D.; Joó, F. New Water-Soluble Iridium(I)–N-Heterocyclic Carbene–Tertiary Phosphine Mixed-Ligand Complexes as Catalysts of Hydrogenation and Redox Isomerization. Organometallics 2014, 33, 6330–6340. [Google Scholar] [CrossRef]
- Bittermann, A.; Baskakov, D.; Herrmann, W.A. Carbene Complexes Made Easily: Decomposition of Reissert Compounds and Further Synthetic Approaches. Organometallics 2009, 28, 5107–5111. [Google Scholar] [CrossRef]
- Czégéni, C.E.; De, S.; Udvardy, A.; Derzsi, N.J.; Papp, G.; Papp, G.; Joó, F. Selective Hydration of Nitriles to Corresponding Amides in Air with Rh(I)-N-Heterocyclic Complex Catalysts. Catalysts 2020, 10, 125. [Google Scholar] [CrossRef]
- Ruiz-Varilla, A.M.; Baquero, E.A.; Silbestri, G.F.; Gonzalez-Arellano, C.; de Jesús, E.; Flores, J.C. Synthesis and behavior of novel sulfonated water-soluble N-heterocyclic carbene (η4-diene) platinum(0) complexes. Dalton Trans. 2015, 44, 18360–18369. [Google Scholar] [CrossRef]
- Horváth, H.; Papp, G.; Kovács, H.; Kathó, Á.; Joó, F. Iridium(I)NHC-phosphine complex-catalyzed hydrogen generation and storage in aqueous formate/bicarbonate solutions using a flow reactor - Effective response to changes in hydrogen demand. Int. J. Hydrogen Energy 2019, 44, 28527–28532. [Google Scholar] [CrossRef]
- Park, K.H.; Kim, S.Y.; Son, S.U.; Chung, Y.K. Use of Rhodium N-Heterocyclic Carbene Complexes in Catalytic Cyclization and Hydrosilylation of 1,6-Enynes to 2-Methyl-1-silylmethylidene-2-cyclopentane. Eur. J. Org. Chem. 2003, 2003, 4341–4345. [Google Scholar] [CrossRef]
- Joó, F. Aqueous Organometallic Catalysis. In Catalysis by Metal Complexes Series; Springer: Dordrecht, The Netherlands, 2001; p. 39. [Google Scholar]
- Gil, W.; Lis, T.; Trzeciak, A.M.; Ziółkowski, J.J. Structure and catalytic activity of rhodium(I) carbene complexes in polymerization of phenylacetylene. Inorg. Chim. Acta 2006, 359, 2835–2841. [Google Scholar] [CrossRef]
- Gil, W.; Trzeciak, A.M.; Ziółkowski, J.J. Rhodium(I) N-Heterocyclic Carbene Complexes as Highly Selective Catalysts for 1-Hexene Hydroformylation. Organometallics 2008, 27, 4131–4138. [Google Scholar] [CrossRef]
- Nichol, G.S.; Kneebone, J.; Anna, L.J.; Rajaseelan, E. CCDC 785398: Experimental Crystal Structure Determination. 2014. [Google Scholar] [CrossRef]
- Siegler, M.A.; Spek, A.L.; Rubio, M.; Reek, J.N.H. CCDC 890216: Experimental Crystal Structure Determination. 2014. [Google Scholar] [CrossRef]
- Joó, F.; Kovács, J.; Bényei, A.C.; Nádasdi, L.; Laurenczy, G. The Effect of pH on the Reactions of Catalytically Important Rh(I) Complexes in Aqueous Solution: Reaction of [RhCl(tppms)3] and trans-[RhCl(CO)(tppms)2] with Hydrogen (TPPMS=mono-sulfonated triphenylphosphine). Chem. Eur. J. 2001, 7, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Fekete, M.; Joó, F. Redox isomerization of allylic alcohols in aqueous–organic biphasic systems catalyzed by water-soluble Ru(II)-N-heterocyclic carbene complexes. Catal. Commun. 2006, 7, 783–786. [Google Scholar] [CrossRef]
- González, B.; Lorenzo-Luis, P.; Serrano-Ruiz, M.; Papp, É.; Fekete, M.; Csépke, K.; Ősz, K.; Kathó, Á.; Joó, F.; Romerosa, A. Catalysis of redox isomerization of allylic alcohols by [RuClCp(mPTA)2](OSO2CF3)2 and [RuCp(mPTA)2(OH2-κO)](OSO2CF3)3·(H2O)(C4H10O)0.5. Unusual influence of the pH and interaction of phosphate with catalyst on the reaction rate. J. Mol. Catal. Chem. 2010, 326, 15–20. [Google Scholar] [CrossRef]
- Daigle, D.J.; Decuir, T.J.; Robertson, J.B.; Darensbourg, D.J. 1,3,5-Triaz-7-Phosphatricyclo[3.3.1.13,7]Decane and Derivatives. In Inorganic Syntheses, 1st ed.; Darensbourg, M.Y., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1998; Volume 32, pp. 40–45. [Google Scholar]
- Herrmann, W.A.; Kohlpaintner, C.W.; Hanson, B.E.; Kang, X. Syntheses of Water-Soluble Phosphines and their Transition Metal Complexes. In Inorganic Syntheses, 1st ed.; Darensbourg, M.Y., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1998; Volume 32, pp. 8–25. [Google Scholar]
- Joó, F.; Kovács, J.; Kathó, Á.; Bényei, A.C.; Decuir, T.; Darensbourg, D.J.; Miedaner, A.; Dubois, D.L. (Meta- Sulfonatophenyl) Diphenylphosphine, Sodium Salt and its Complexes with Rhodium(I), Ruthenium(II), Iridium(I). In Inorganic Syntheses, 1st ed.; Darensbourg, M.Y., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1998; Volume 32, pp. 1–8. [Google Scholar]
- Giordano, G.; Crabtree, R.H.; Heintz, R.M.; Forster, D.; Morris, D.E. Di-μ-Chloro-Bis(η4-1,5-Cyclooctadiene)-Dirhodium(I). In Inorganic Syntheses, 1st ed.; Angelici, R.J., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1990; Volume 28, pp. 1–8. [Google Scholar]
- Fandos, R.; Hernández, C.; Otero, A.; Rodríguez, A.; Ruiz, M.J.; García Fierro, J.L.; Terreros, P. Rhodium and Iridium Hydroxide Complexes [M(μ-OH)(COD)]2 (M = Rh, Ir) as Versatile Precursors of Homo and Early−Late Heterobimetallic Compounds. X-ray Crystal Structures of Cp*Ta(μ3-O)4[Rh(COD)]4 (Cp* = η5-C5Me5) and [Ir(2-O-3-CN-4,6-Me2-C5HN)(COD)]2. Organometallics 1999, 18, 2718–2723. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.a.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. Sect. E 2020, 76, 1–11. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | [S]/[Rh] | Added PR3 Phosphine | [P]/[Rh] | T (°C) | Octan-3-on (%) a | Octan-3-ol (%) a |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 50 | - | - | 55 | 15 | 0 |
| 2 | 1 + H2 (1 bar) | 50 | - | - | 55 | 18 | 2 |
| 3 | 1 | 50 | mtppms | 1 | 55 | 43 | 0 |
| 4 | 1 | 50 | mtppms | 2 | 55 | 24 | 0 |
| 5 | 1 | 50 | mtppms | 3 | 55 | 13 | 0 |
| 6 | 1 | 50 | mtppms | 1 | 80 | 98 | 0 |
| 7 b | 1 | 100 | mtppms | 1 | 80 | 58 | 0 |
| 8 b | 1 | 100 | mtppts | 1 | 80 | 44 | 0 |
| 9 b | 1 | 100 | pta | 1 | 80 | 85 | 0 |
| 10 b | 1 | 100 | PPh3 | 1 | 80 | 98 | 0 |
| 11 | - | 50 | mtppms | - | 80 | 0 | 0 |
| 12 | [RhCl(cod)]2 | 50 | - | - | 80 | 3 | 0 |
| Entry | Run | Octan-3-on (%) a | TOF (h−1) |
|---|---|---|---|
| 1 | 1 | 82 | 27 |
| 2 | 2 | 68 | 23 |
| 3 | 3 | 64 | 21 |
| Substrate | 1/mtppms-Na | 5 a | ||
|---|---|---|---|---|
| Conversion (%) | TOF(h−1) | Conversion (%) | TOF(h−1) | |
| oct-1-en-3-ol | 73 b | 146 | 18 | 13 |
| hept-1-en-3-ol | 76 | 152 | 53 | 38 |
| hex-1-en-3-ol | 75 | 150 | 83 | 59 |
| pent-1-en-3-ol | 70 c | 140 | 94 | 67 |
| but-1-en-3-ol | 45 c | 90 | 75 | 51 |
| prop-1-en-3-ol | 44 c | 88 | 73 | 52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czégéni, C.E.; Fekete, M.; Tóbiás, E.; Udvardy, A.; Horváth, H.; Papp, G.; Joó, F. Redox Isomerization of Allylic Alcohols Catalyzed by New Water-Soluble Rh(I)-N-Heterocyclic Carbene Complexes. Catalysts 2020, 10, 1361. https://doi.org/10.3390/catal10111361
Czégéni CE, Fekete M, Tóbiás E, Udvardy A, Horváth H, Papp G, Joó F. Redox Isomerization of Allylic Alcohols Catalyzed by New Water-Soluble Rh(I)-N-Heterocyclic Carbene Complexes. Catalysts. 2020; 10(11):1361. https://doi.org/10.3390/catal10111361
Chicago/Turabian StyleCzégéni, Csilla Enikő, Marianna Fekete, Eszter Tóbiás, Antal Udvardy, Henrietta Horváth, Gábor Papp, and Ferenc Joó. 2020. "Redox Isomerization of Allylic Alcohols Catalyzed by New Water-Soluble Rh(I)-N-Heterocyclic Carbene Complexes" Catalysts 10, no. 11: 1361. https://doi.org/10.3390/catal10111361
APA StyleCzégéni, C. E., Fekete, M., Tóbiás, E., Udvardy, A., Horváth, H., Papp, G., & Joó, F. (2020). Redox Isomerization of Allylic Alcohols Catalyzed by New Water-Soluble Rh(I)-N-Heterocyclic Carbene Complexes. Catalysts, 10(11), 1361. https://doi.org/10.3390/catal10111361