Influence of Surface Ligands on Charge-Carrier Trapping and Relaxation in Water-Soluble CdSe@CdS Nanorods


Abstract
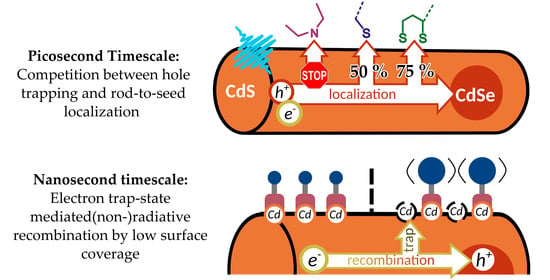
:
1. Introduction
2. Results
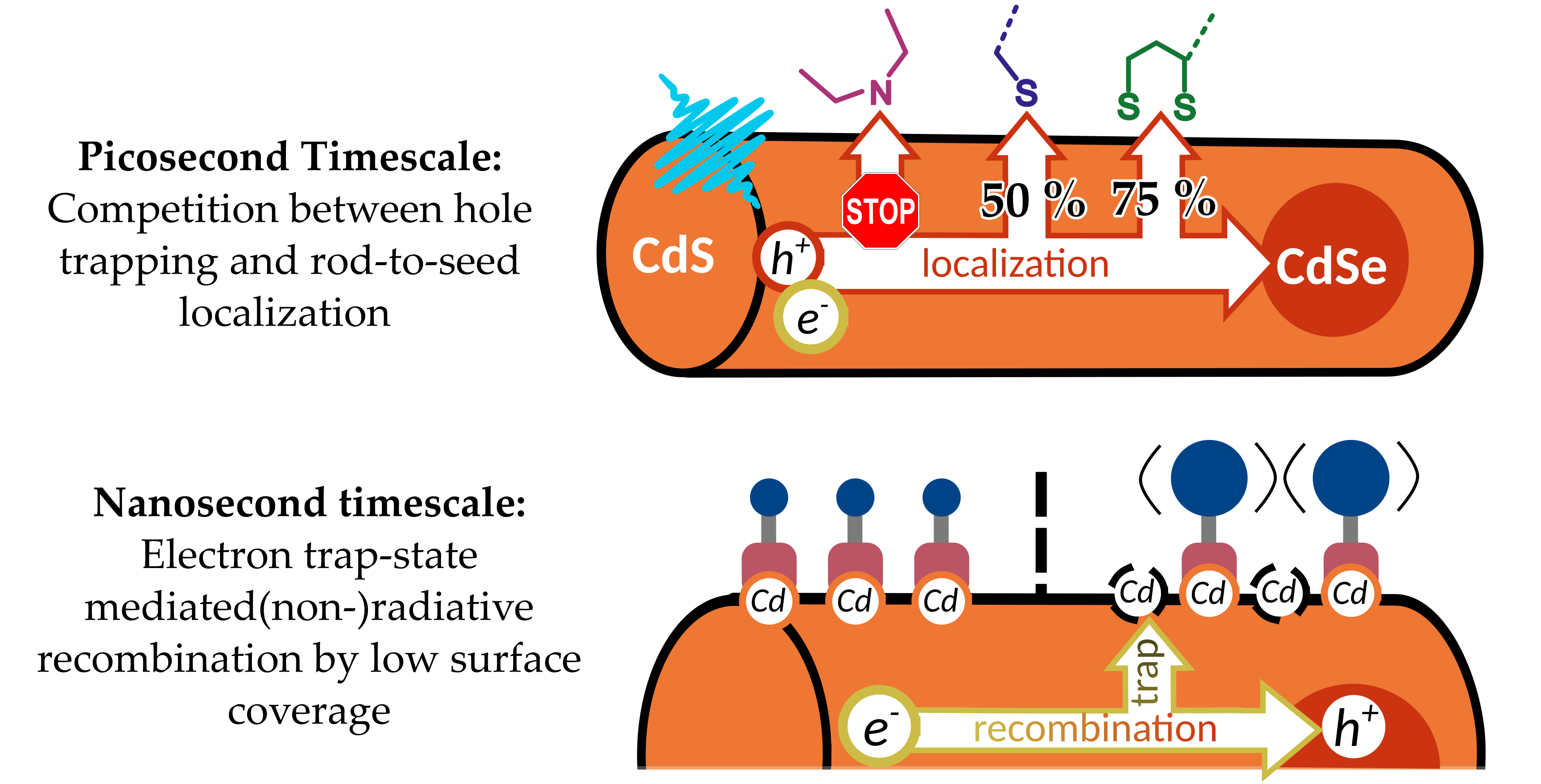
2.1. Particle Synthesis and Ligand Exchange
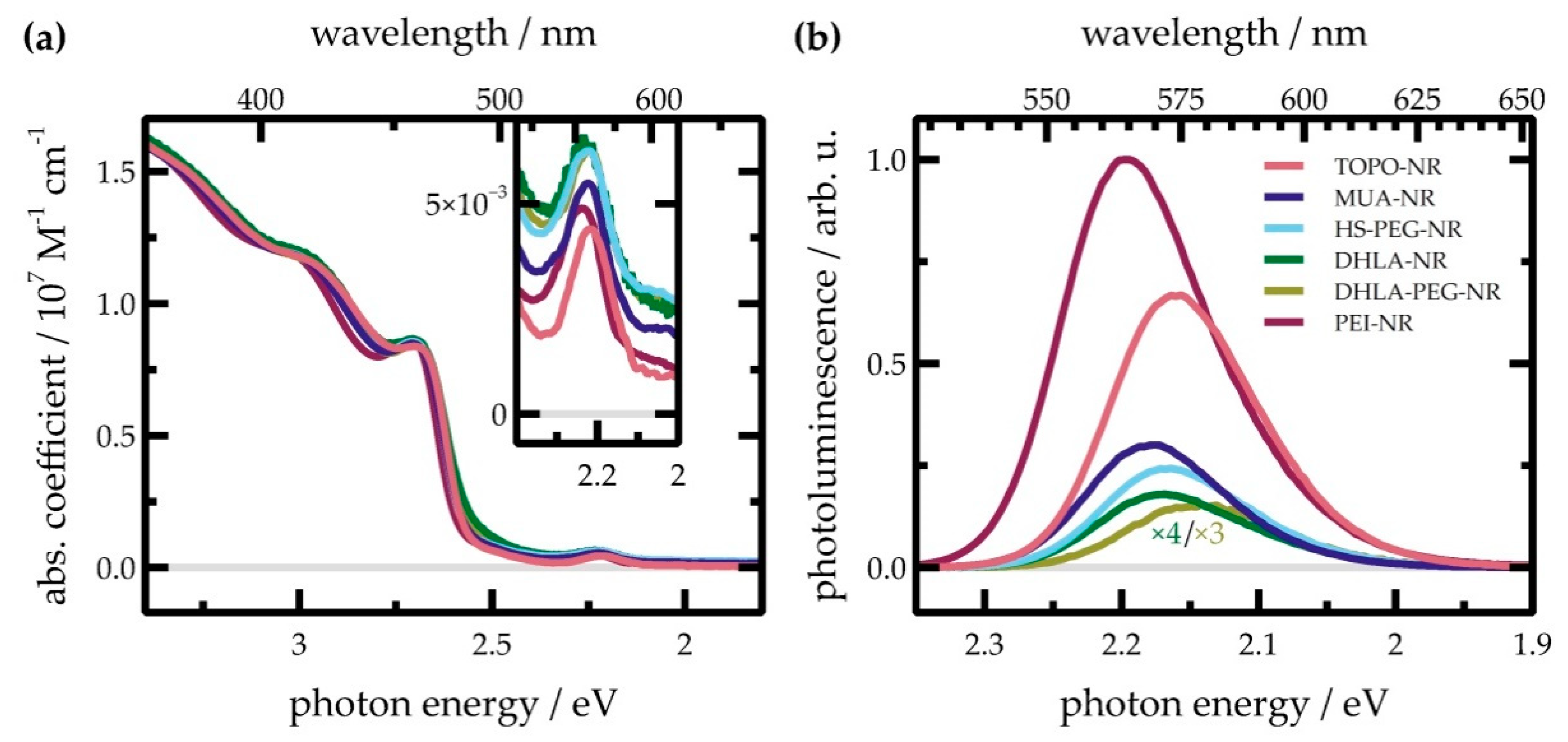
2.2. Steady-state UV/Vis Absorption Spectroscopy
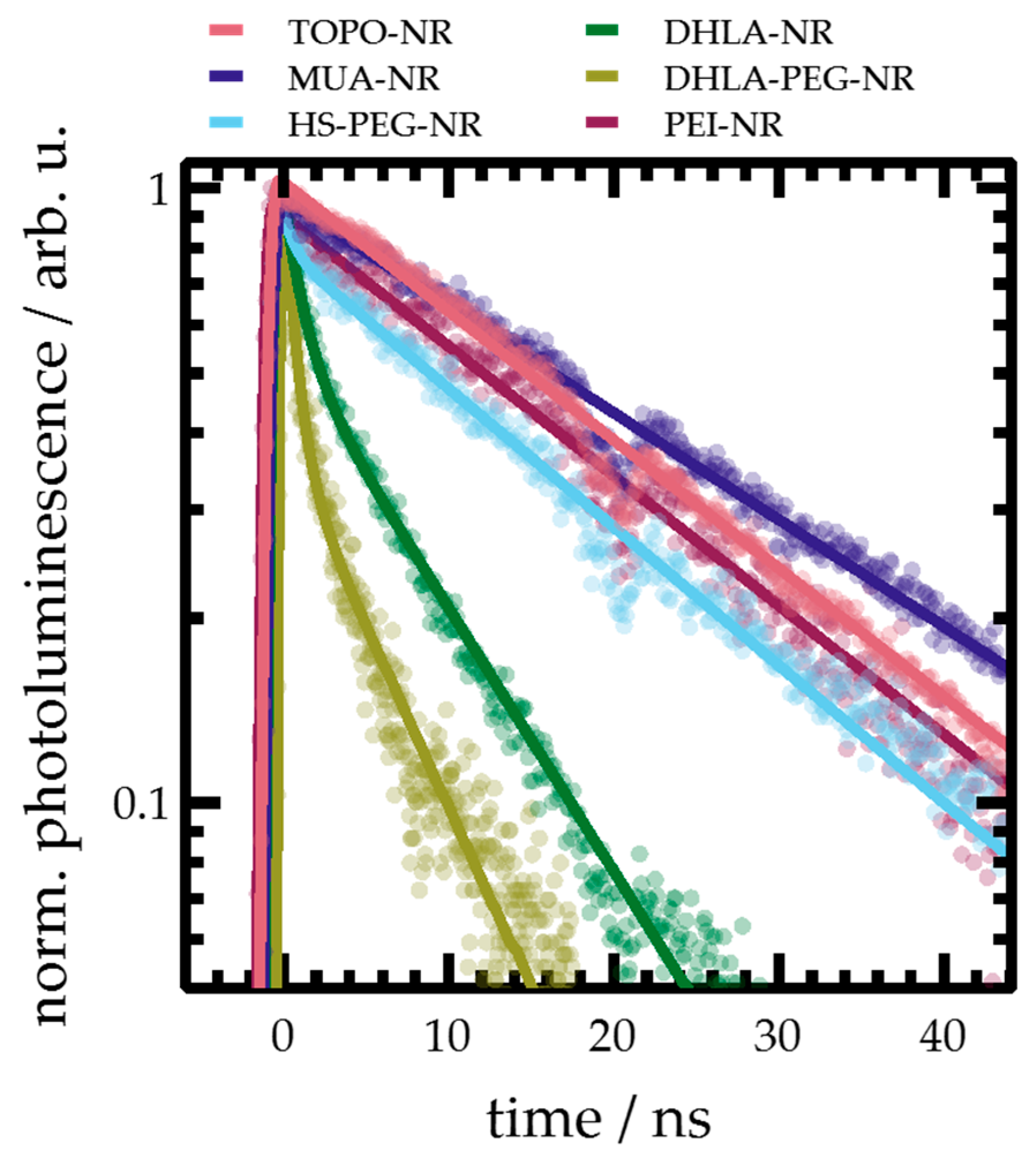
2.3. Photoluminescence Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | τ1/ns | A1 | τ2/ns | A2 | krad/1011 s−1 |
|---|---|---|---|---|---|
| TOPO-NR | - | - | 19.7 ± 0.4 | - | 3.9 ± 0.2 |
| MUA-NR | - | - | 25.0 ± 0.3 | - | 1.9 ± 0.1 |
| HS-PEG-NR | 0.9 ± 0.2 | 0.31 ± 0.14 | 18.0 ± 2.0 | 0.69 ± 0.14 | 2.2 ± 0.4 |
| DHLA-NR | 0.8 ± 0.3 | 0.43 ± 0.03 | 8.4 ± 2.5 | 0.57 ± 0.03 | 1.8 ± 0.8 |
| DHLA-PEG-NR | 0.6 ± 0.2 | 0.64 ± 0.05 | 7.5 ± 1.1 | 0.36 ± 0.05 | 2.8 ± 0.7 |
| PEI-NR | - | - | 20.1 ± 0.4 | - | 4.3 ± 0.2 |
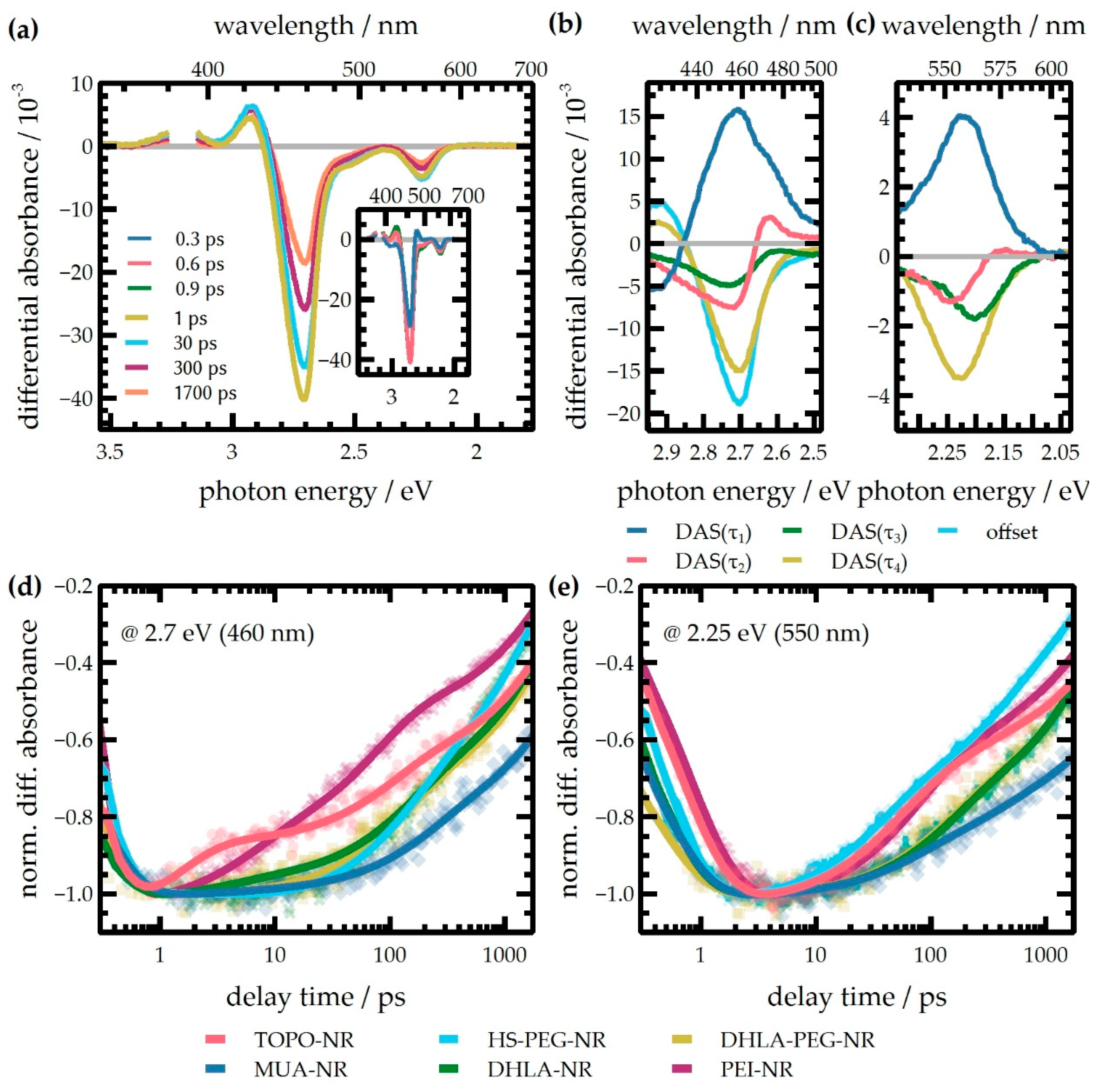
2.4. Transient Absorption Spectroscopy
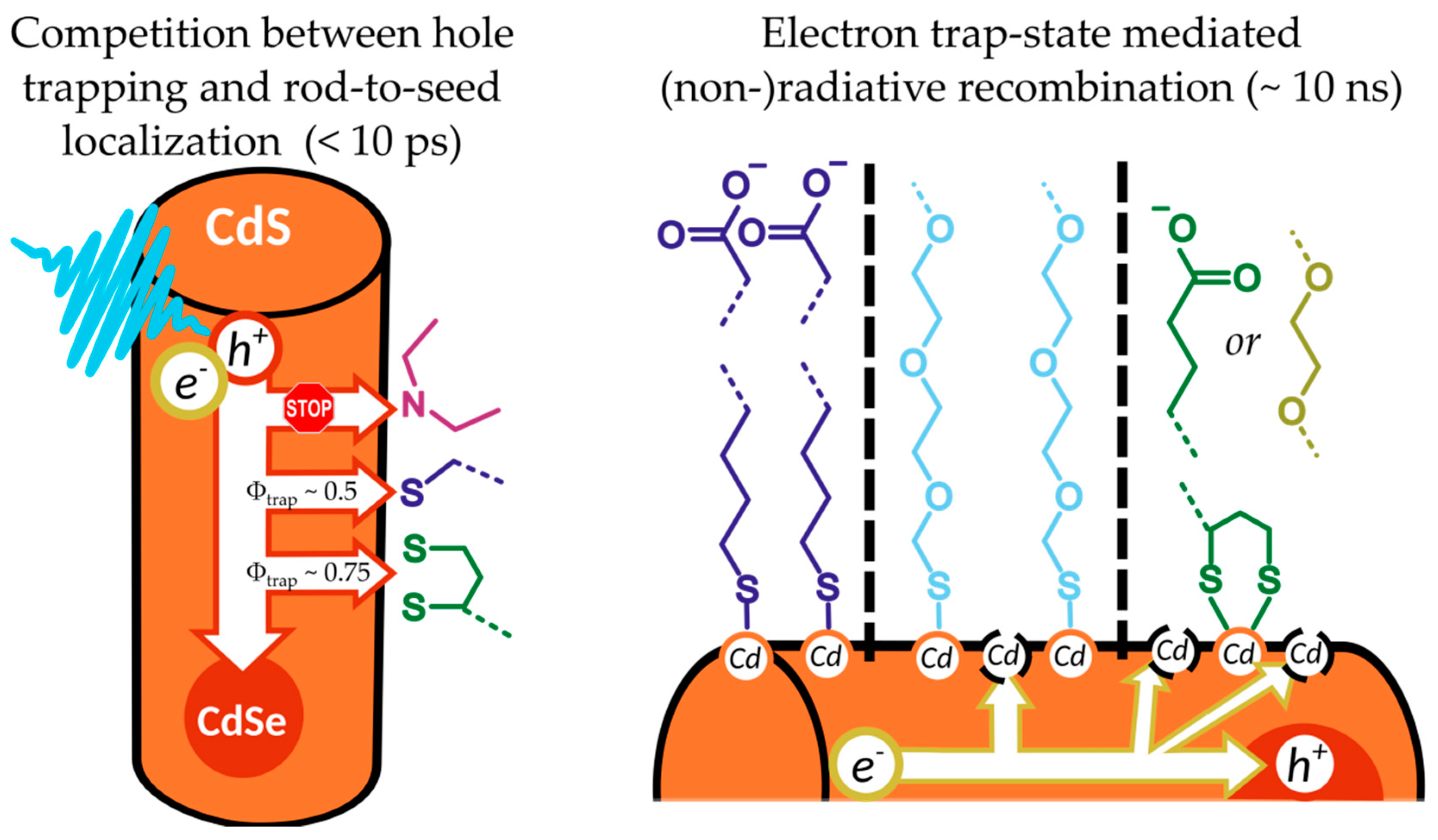
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalisman, P.; Houben, L.; Aronovitch, E.; Kauffmann, Y.; Bar-Sadan, M.; Amirav, L. The golden gate to photocatalytic hydrogen production. J. Mater. Chem. A 2015, 3, 19679–19682. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Zhu, H.; Lian, T. Ultrafast Exciton Dynamics and Light-Driven H2 Evolution in Colloidal Semiconductor Nanorods and Pt-Tipped Nanorods. Acc. Chem. Res. 2015, 48, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Han, Z.; Peterson, J.J.; Odoi, M.Y.; Sowers, K.L.; Krauss, T.D. Photocatalytic Hydrogen Generation by CdSe/CdS Nanoparticles. Nano Lett. 2016, 16, 5347–5352. [Google Scholar] [CrossRef] [PubMed]
- Amirav, L.; Alivisatos, P. Photocatalytic Hydrogen Production with Tunable Nanorod Heterostructures. J. Phys. Chem. Lett. 2010, 1, 1051–1054. [Google Scholar] [CrossRef]
- Wu, K.; Hill, L.J.; Chen, J.; McBride, J.R.; Pavlopolous, N.G.; Richey, N.E.; Pyun, J.; Lian, T. Universal Length Dependence of Rod-to-Seed Exciton Localization Efficiency in Type I and Quasi-Type II CdSe@CdS Nanorods. ACS Nano 2015, 9, 4591–4599. [Google Scholar] [CrossRef] [PubMed]
- Eshet, H.; Grünwald, M.; Rabani, E. The Electronic Structure of CdSe/CdS Core/Shell Seeded Nanorods: Type-I or Quasi-Type-II? Nano Lett. 2013, 13, 5880–5885. [Google Scholar] [CrossRef] [Green Version]
- Sitt, A.; Della Sala, F.; Menagen, G.; Banin, U. Multiexciton Engineering in Seeded Core/Shell Nanorods: Transfer from Type-I to Quasi-type-II Regimes. Nano Lett. 2009, 9, 3470–3476. [Google Scholar] [CrossRef]
- Wu, K.; Lian, T. Quantum confined colloidal nanorod heterostructures for solar-to-fuel conversion. Chem. Soc. Rev. 2016, 45, 3781–3810. [Google Scholar] [CrossRef]
- Strmcnik, D.; Lopes, P.P.; Genorio, B.; Stamenkovic, V.R.; Markovic, N.M. Design principles for hydrogen evolution reaction catalyst materials. Nano Energy 2016, 29, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Nakibli, Y.; Mazal, Y.; Dubi, Y.; Wächtler, M.; Amirav, L. Size Matters: Cocatalyst Size Effect on Charge Transfer and Photocatalytic Activity. Nano Lett. 2017, 18, 357–364. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, H.; Li, J.; Du, P. Extraordinarily efficient photocatalytic hydrogen evolution in water using semiconductor nanorods integrated with crystalline Ni 2 P cocatalysts. Energy Environ. Sci. 2015, 8, 2668–2676. [Google Scholar] [CrossRef]
- Kalisman, P.; Nakibli, Y.; Amirav, L. Perfect Photon-to-Hydrogen Conversion Efficiency. Nano Lett. 2016, 16, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Talapin, D.V.; Nelson, J.H.; Shevchenko, E.V.; Aloni, S.; Sadtler, B.; Alivisatos, A.P.; Alivisatos, P. Seeded Growth of Highly Luminescent CdSe/CdS Nanoheterostructures with Rod and Tetrapod Morphologies. Nano Lett. 2007, 7, 2951–2959. [Google Scholar] [CrossRef] [Green Version]
- Carbone, L.; Nobile, C.; De Giorgi, M.; Della Sala, F.; Morello, G.; Pompa, P.; Hÿtch, M.; Snoeck, E.; Fiore, A.; Franchini, I.R.; et al. Synthesis and Micrometer-Scale Assembly of Colloidal CdSe/CdS Nanorods Prepared by a Seeded Growth Approach. Nano Lett. 2007, 7, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Kodaimati, M.S.; McClelland, K.P.; He, C.; Lian, S.; Jiang, Y.; Zhang, Z.; Weiss, E.A. Viewpoint: Challenges in Colloidal Photocatalysis and Some Strategies for Addressing Them. Inorg. Chem. 2018, 57, 3659–3670. [Google Scholar] [CrossRef] [Green Version]
- Susumu, K.; Uyeda, H.T.; Medintz, I.L.; Pons, T.; Delehanty, J.B.; Mattoussi, H. Enhancing the Stability and Biological Functionalities of Quantum Dots via Compact Multifunctional Ligands. J. Am. Chem. Soc. 2007, 129, 13987–13996. [Google Scholar] [CrossRef]
- Zhang, Y.; Clapp, A. Overview of Stabilizing Ligands for Biocompatible Quantum Dot Nanocrystals. Sensors 2011, 11, 11036–11055. [Google Scholar] [CrossRef]
- Ansar, S.M.; Chakraborty, S.; Kitchens, C.L. pH-Responsive Mercaptoundecanoic Acid Functionalized Gold Nanoparticles and Applications in Catalysis. Nanomaterials 2018, 8, 339. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shahar, Y.; Scotognella, F.; Waiskopf, N.; Kriegel, I.; Conte, S.D.; Cerullo, G.; Banin, U. Effect of Surface Coating on the Photocatalytic Function of Hybrid CdS-Au Nanorods. Small 2014, 11, 462–471. [Google Scholar] [CrossRef]
- Wolff, C.M.; Frischmann, P.; Schulze, M.; Bohn, B.J.; Wein, R.; Livadas, P.; Carlson, M.T.; Jäckel, F.; Feldmann, J.; Würthner, F.; et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 2018, 3, 862–869. [Google Scholar] [CrossRef]
- Simon, T.; Bouchonville, N.; Berr, M.J.; Vaneski, A.; Adrović, A.; Volbers, D.; Wyrwich, R.; Döblinger, M.; Susha, A.S.; Rogach, A.L.; et al. Redox shuttle mechanism enhances photocatalytic H2 generation on Ni-decorated CdS nanorods. Nat. Mater. 2014, 13, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Vaneski, A.; Pesch, G.R.; Susha, A.S.; Teoh, W.Y.; Rogach, A.L. Enhanced hydrogen evolution rates at high pH with a colloidal cadmium sulphide–platinum hybrid system. APL Mater. 2014, 2, 126102. [Google Scholar] [CrossRef] [Green Version]
- Jian, J.-X.; Liu, Q.; Li, Z.-J.; Wang, F.; Li, X.-B.; Li, C.-B.; Liu, B.; Meng, Q.-Y.; Chen, B.; Feng, K.; et al. Chitosan confinement enhances hydrogen photogeneration from a mimic of the diiron subsite of [FeFe]-hydrogenase. Nat. Commun. 2013, 4, 2695. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Li, X.-B.; Jian, J.-X.; Wang, X.-Z.; Wu, H.-L.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Secondary coordination sphere accelerates hole transfer for enhanced hydrogen photogeneration from [FeFe]-hydrogenase mimic and CdSe QDs in water. Sci. Rep. 2016, 6, 29851. [Google Scholar] [CrossRef] [Green Version]
- Jian, J.-X.; Ye, C.; Wang, X.-Z.; Wen, M.; Li, Z.-J.; Chen, B.; Tung, C.; Wu, L.-Z. Comparison of H 2 photogeneration by [FeFe]-hydrogenase mimics with CdSe QDs and Ru(bpy) 3 Cl 2 in aqueous solution. Energy Environ. Sci. 2016, 9, 2083–2089. [Google Scholar] [CrossRef]
- Kong, D.; Cha, J.J.; Wang, H.; Lee, H.R.; Cui, Y. First-row transition metal dichalcogenide catalysts for hydrogen evolution reaction. Energy Environ. Sci. 2013, 6, 3553–3558. [Google Scholar] [CrossRef]
- Archana, B.; Kottam, N.; Nayak, S.; Chandrasekhar, K.B.; Sreedhara, M.B. Superior Photocatalytic Hydrogen Evolution Performances of WS2 over MoS2 Integrated with CdS Nanorods. J. Phys. Chem. C 2020, 124, 14485–14495. [Google Scholar] [CrossRef]
- Ali, M.; Zayed, D.; Ramadan, W.; Kamel, O.A.; Shehab, M.; Ebrahim, S. Synthesis, characterization and cytotoxicity of polyethylene glycol-encapsulated CdTe quantum dots. Int. Nano Lett. 2019, 9, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Palui, G.; Bin Na, H.; Mattoussi, H. Poly(ethylene glycol)-Based Multidentate Oligomers for Biocompatible Semiconductor and Gold Nanocrystals. Langmuir 2012, 28, 2761–2772. [Google Scholar] [CrossRef]
- Susumu, K.; Mei, B.C.; Mattoussi, H. Multifunctional ligands based on dihydrolipoic acid and polyethylene glycol to promote biocompatibility of quantum dots. Nat. Protoc. 2009, 4, 424–436. [Google Scholar] [CrossRef]
- Liang, W.-J.; Wang, F.; Wen, M.; Jian, J.-X.; Wang, X.-Z.; Chen, B.; Tung, C.; Wu, L.-Z. Branched Polyethylenimine Improves Hydrogen Photoproduction from a CdSe Quantum Dot/[FeFe]-Hydrogenase Mimic System in Neutral Aqueous Solutions. Chem.—A Eur. J. 2015, 21, 3187–3192. [Google Scholar] [CrossRef] [PubMed]
- Mohs, A.M.; Duan, H.; Kairdolf, B.A.; Smith, A.M.; Nie, S. Proton-resistant quantum dots: Stability in gastrointestinal fluids and implications for oral delivery of nanoparticle agents. Nano Res. 2009, 2, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, J.-M.; Yan, X.-P. Self-Assembly of Folate onto Polyethyleneimine-Coated CdS/ZnS Quantum Dots for Targeted Turn-On Fluorescence Imaging of Folate Receptor Overexpressed Cancer Cells. Anal. Chem. 2012, 85, 228–234. [Google Scholar] [CrossRef]
- Duan, H.; Nie, S. Cell-Penetrating Quantum Dots Based on Multivalent and Endosome-Disrupting Surface Coatings. J. Am. Chem. Soc. 2007, 129, 3333–3338. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Duan, H.; Rhyner, M.N.; Ruan, G.; Nie, S. A systematic examination of surface coatings on the optical and chemical properties of semiconductor quantum dots. Phys. Chem. Chem. Phys. 2006, 8, 3895–3903. [Google Scholar] [CrossRef]
- Tang, H.; Wu, R.; Mao, M.; Li, L.S.; Zhou, C.; Shen, H. The enhanced fluorescence properties & colloid stability of aqueous CdSe/ZnS QDs modified with N-alkylated poly(ethyleneimine). New J. Chem. 2015, 39, 4334–4342. [Google Scholar] [CrossRef]
- Zhuang, Z.; Lu, X.; Peng, Q.; Li, Y.D. Direct Synthesis of Water-Soluble Ultrathin CdS Nanorods and Reversible Tuning of the Solubility by Alkalinity. J. Am. Chem. Soc. 2010, 132, 1819–1821. [Google Scholar] [CrossRef]
- Peterson, M.D.; Cass, L.C.; Harris, R.D.; Edme, K.; Sung, K.; Weiss, E.A. The Role of Ligands in Determining the Exciton Relaxation Dynamics in Semiconductor Quantum Dots. Annu. Rev. Phys. Chem. 2014, 65, 317–339. [Google Scholar] [CrossRef]
- Wilker, M.B.; Utterback, J.K.; Greene, S.; Brown, K.A.; Mulder, D.W.; King, P.W.; Dukovic, G. Role of Surface-Capping Ligands in Photoexcited Electron Transfer between CdS Nanorods and [FeFe] Hydrogenase and the Subsequent H2 Generation. J. Phys. Chem. C 2017, 122, 741–750. [Google Scholar] [CrossRef]
- Yang, W.; VanSuch, G.E.; Liu, Y.; Jin, T.; Liu, Q.; Ge, A.; Sanchez, M.L.K.; Haja, D.K.; Adams, M.W.W.; Dyer, R.; et al. Surface-Ligand “Liquid” to “Crystalline” Phase Transition Modulates the Solar H2 Production Quantum Efficiency of CdS Nanorod/Mediator/Hydrogenase Assemblies. ACS Appl. Mater. Interfaces 2020, 12, 35614–35625. [Google Scholar] [CrossRef]
- Uyeda, H.T.; Medintz, I.L.; Jaiswal, J.; Simon, S.M.; Mattoussi, H. Synthesis of Compact Multidentate Ligands to Prepare Stable Hydrophilic Quantum Dot Fluorophores. J. Am. Chem. Soc. 2005, 127, 3870–3878. [Google Scholar] [CrossRef]
- Nann, T. Phase-transfer of CdSe@ZnS quantum dots using amphiphilic hyperbranched polyethylenimine. Chem. Commun. 2005, 1735. [Google Scholar] [CrossRef]
- Young, A.G.; Green, D.P.; McQuillan, A.J. Infrared Spectroscopic Studies of Monothiol Ligand Adsorption on CdS Nanocrystal Films in Aqueous Solutions. Langmuir 2006, 22, 11106–11112. [Google Scholar] [CrossRef] [PubMed]
- Young, A.G.; Green, D.P.; McQuillan, A.J. IR Spectroscopic Studies of Adsorption of Dithiol-Containing Ligands on CdS Nanocrystal Films in Aqueous Solutions. Langmuir 2007, 23, 12923–12931. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Rodríguez-Córdoba, W.; Liu, Z.; Zhu, H.; Lian, T. Beyond Band Alignment: Hole Localization Driven Formation of Three Spatially Separated Long-Lived Exciton States in CdSe/CdS Nanorods. ACS Nano 2013, 7, 7173–7185. [Google Scholar] [CrossRef] [PubMed]
- Grennell, A.; Utterback, J.K.; Pearce, O.M.; Wilker, M.B.; Dukovic, G. Relationships between Exciton Dissociation and Slow Recombination within ZnSe/CdS and CdSe/CdS Dot-in-Rod Heterostructures. Nano Lett. 2017, 17, 3764–3774. [Google Scholar] [CrossRef] [PubMed]
- Adel, P.; Bloh, J.; Hinrichs, D.; Kodanek, T.; Dorfs, D. Determination of all Dimensions of CdSe Seeded CdS Nanorods Solely via their UV/Vis Spectra. Z. Phys. Chem. 2017, 231, 93–106. [Google Scholar] [CrossRef]
- Mooney, J.; Kambhampati, P. Get the Basics Right: Jacobian Conversion of Wavelength and Energy Scales for Quantitative Analysis of Emission Spectra. J. Phys. Chem. Lett. 2013, 4, 3316–3318. [Google Scholar] [CrossRef]
- Jakubek, Z.J.; Devries, J.; Lin, S.; Ripmeester, J.; Yu, K. Exciton Recombination and Upconverted Photoluminescence in Colloidal CdSe Quantum Dots. J. Phys. Chem. C 2008, 112, 8153–8158. [Google Scholar] [CrossRef]
- Woodall, D.L.; Tobias, A.K.; Jones, M. Resolving carrier recombination in CdS quantum dots: A time-resolved fluorescence study. Chem. Phys. 2016, 471, 2–10. [Google Scholar] [CrossRef]
- Fitzmorris, B.C.; Cooper, J.K.; Edberg, J.; Gul, S.; Guo, J.; Zhang, J.Z. Synthesis and Structural, Optical, and Dynamic Properties of Core/Shell/Shell CdSe/ZnSe/ZnS Quantum Dots. J. Phys. Chem. C 2012, 116, 25065–25073. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Pędziwiatr, M.; Grzyb, J. Quantum dots use both LUMO and surface trap electrons in photoreduction process. J. Lumin. 2017, 183, 401–409. [Google Scholar] [CrossRef]
- Rabouw, F.T.; Lunnemann, P.; Van Dijk-Moes, R.J.A.; Frimmer, M.; Pietra, F.; Koenderink, A.F.; Vanmaekelbergh, D.; Koenderink, A.F. Reduced Auger Recombination in Single CdSe/CdS Nanorods by One-Dimensional Electron Delocalization. Nano Lett. 2013, 13, 4884–4892. [Google Scholar] [CrossRef] [PubMed]
- Geißler, D.; Würth, C.; Wolter, C.; Weller, H.; Resch-Genger, U. Excitation wavelength dependence of the photoluminescence quantum yield and decay behavior of CdSe/CdS quantum dot/quantum rods with different aspect ratios. Phys. Chem. Chem. Phys. 2017, 19, 12509–12516. [Google Scholar] [CrossRef]
- Coropceanu, I.; Rossinelli, A.A.; Caram, J.R.; Freyria, F.S.; Bawendi, M.G. Slow-Injection Growth of Seeded CdSe/CdS Nanorods with Unity Fluorescence Quantum Yield and Complete Shell to Core Energy Transfer. ACS Nano 2016, 10, 3295–3301. [Google Scholar] [CrossRef] [Green Version]
- Green, M. A new approach to the formal classification of covalent compounds of the elements. J. Organomet. Chem. 1995, 500, 127–148. [Google Scholar] [CrossRef]
- Owen, J.S. The coordination chemistry of nanocrystal surfaces. Sci. 2015, 347, 615–616. [Google Scholar] [CrossRef]
- Boles, M.A.; Ling, D.; Hyeon, T.; Talapin, D.V. The surface science of nanocrystals. Nat. Mater. 2016, 15, 141–153. [Google Scholar] [CrossRef]
- Houtepen, A.J.; Hens, Z.; Owen, J.S.; Infante, I. On the Origin of Surface Traps in Colloidal II–VI Semiconductor Nanocrystals. Chem. Mater. 2017, 29, 752–761. [Google Scholar] [CrossRef]
- Wenger, W.N.; Bates, F.S.; Aydil, E.S. Functionalization of Cadmium Selenide Quantum Dots with Poly(ethylene glycol): Ligand Exchange, Surface Coverage, and Dispersion Stability. Langmuir 2017, 33, 8239–8245. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Xie, B.; Yin, Z.-G.; Fang, S.-M.; Zhao, J.-B. Synthesis of Highly Stable CdTe/CdS Quantum Dots with Biocompatibility. Eur. J. Inorg. Chem. 2010, 2010, 1501–1506. [Google Scholar] [CrossRef]
- Morgan, D.P.; Kelley, D.F. Mechanism of Hole Trap Passivation in CdSe Quantum Dots by Alkylamines. J. Phys. Chem. C 2018, 122, 25661–25667. [Google Scholar] [CrossRef]
- Sowers, K.L.; Hou, Z.; Peterson, J.J.; Swartz, B.; Pal, S.; Prezhdo, O.; Krauss, T.D. Photophysical Properties of CdSe/CdS core/shell quantum dots with tunable surface composition. Chem. Phys. 2016, 471, 24–31. [Google Scholar] [CrossRef]
- Pu, C.; Peng, X. To Battle Surface Traps on CdSe/CdS Core/Shell Nanocrystals: Shell Isolation versus Surface Treatment. J. Am. Chem. Soc. 2016, 138, 8134–8142. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.P.; Kelley, D.F. What Does the Transient Absorption Spectrum of CdSe Quantum Dots Measure? J. Phys. Chem. C 2020, 124, 8448–8455. [Google Scholar] [CrossRef]
- Wu, K.; Zhu, H.; Liu, Z.; Rodríguez-Córdoba, W.; Lian, T. Ultrafast Charge Separation and Long-Lived Charge Separated State in Photocatalytic CdS–Pt Nanorod Heterostructures. J. Am. Chem. Soc. 2012, 134, 10337–10340. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Panov, M.S.; Mereshchenko, A.S.; Tarnovsky, A.N.; Lorek, R.; Perera, D.; Diederich, G.; Lambright, S.; Moroz, P.; Zamkov, M. The Effect of the Charge-Separating Interface on Exciton Dynamics in Photocatalytic Colloidal Heteronanocrystals. ACS Nano 2012, 6, 8156–8165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utterback, J.K.; Ruzicka, J.L.; Hamby, H.; Eaves, J.D.; Dukovic, G. Temperature-Dependent Transient Absorption Spectroscopy Elucidates Trapped-Hole Dynamics in CdS and CdSe Nanorods. J. Phys. Chem. Lett. 2019, 10, 2782–2787. [Google Scholar] [CrossRef] [PubMed]
- Jasrasaria, D.; Philbin, J.P.; Yan, C.; Weinberg, D.; Alivisatos, P.; Rabani, E. Sub-Bandgap Photoinduced Transient Absorption Features in CdSe Nanostructures: The Role of Trapped Holes. J. Phys. Chem. C 2020, 124, 17372–17378. [Google Scholar] [CrossRef]
- Diroll, B.T.; Turk, M.E.; Gogotsi, N.; Murray, C.B.; Kikkawa, J.M. Ultrafast Photoluminescence from the Core and the Shell in CdSe/CdS Dot-in-Rod Heterostructures. ChemPhysChem 2015, 17, 759–765. [Google Scholar] [CrossRef]
- Lupo, M.G.; Della Sala, F.; Carbone, L.; Zavelani-Rossi, M.; Fiore, A.; Luër, L.; Polli, D.; Cingolani, R.; Manna, L.; Lanzani, G. Ultrafast Electron—Hole Dynamics in Core/Shell CdSe/CdS Dot/Rod Nanocrystals. Nano Lett. 2008, 8, 4582–4587. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Rodríguez-Córdoba, W.; Lian, T. Exciton Localization and Dissociation Dynamics in CdS and CdS–Pt Quantum Confined Nanorods: Effect of Nonuniform Rod Diameters. J. Phys. Chem. B 2014, 118, 14062–14069. [Google Scholar] [CrossRef] [PubMed]
- Hewa-Kasakarage, N.N.; El-Khoury, P.Z.; Schmall, N.; Kirsanova, M.; Nemchinov, A.; Tarnovsky, A.N.; Bezryadin, A.; Zamkov, M. The effect of dielectric friction on the rate of charge separation in type II ZnSe/CdS semiconductor nanorods. Appl. Phys. Lett. 2009, 94, 133113. [Google Scholar] [CrossRef]
- Utterback, J.K.; Grennell, A.; Wilker, M.B.; Pearce, O.M.; Eaves, J.D.; Dukovic, G. Observation of trapped-hole diffusion on the surfaces of CdS nanorods. Nat. Chem. 2016, 8, 1061–1066. [Google Scholar] [CrossRef]
- Sadhu, S.; Patra, A. Relaxation Dynamics of Anisotropic Shaped CdS Nanoparticles. J. Phys. Chem. C 2011, 115, 16867–16872. [Google Scholar] [CrossRef]
- Knowles, K.E.; McArthur, E.A.; Weiss, E.A. A Multi-Timescale Map of Radiative and Nonradiative Decay Pathways for Excitons in CdSe Quantum Dots. ACS Nano 2011, 5, 2026–2035. [Google Scholar] [CrossRef]
- Wächtler, M.; Kalisman, P.; Amirav, L. Charge-Transfer Dynamics in Nanorod Photocatalysts with Bimetallic Metal Tips. J. Phys. Chem. C 2016, 120, 24491–24497. [Google Scholar] [CrossRef]
- Balan, A.D.; Olshansky, J.H.; Horowitz, Y.; Han, H.-L.; O’Brien, E.A.; Tang, L.; Somorjai, G.A.; Alivisatos, P. Unsaturated Ligands Seed an Order to Disorder Transition in Mixed Ligand Shells of CdSe/CdS Quantum Dots. ACS Nano 2019, 13, 13784–13796. [Google Scholar] [CrossRef]
- Utterback, J.K.; Ruzicka, J.L.; Keller, H.R.; Pellows, L.M.; Dukovic, G.; Unsleber, J.P.; Reiher, M. Electron Transfer from Semiconductor Nanocrystals to Redox Enzymes. Annu. Rev. Phys. Chem. 2020, 71, 335–359. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Dong, C.; Li, S.; Im, C.; Jin, M.; Yao, S.; Cui, T.; Tian, W.; Liu, Y.; Zhang, H. Effect of Surface Trap States on Photocatalytic Activity of Semiconductor Quantum Dots. J. Phys. Chem. C 2018, 122, 9312–9319. [Google Scholar] [CrossRef]
- Han, Z.; Qiu, F.; Eisenberg, R.; Holland, P.L.; Krauss, T.D. Robust Photogeneration of H2 in Water Using Semiconductor Nanocrystals and a Nickel Catalyst. Science 2012, 338, 1321–1324. [Google Scholar] [CrossRef]
- Liu, C.; Qiu, F.; Peterson, J.J.; Krauss, T.D. Aqueous Photogeneration of H2 with CdSe Nanocrystals and Nickel Catalysts: Electron Transfer Dynamics. J. Phys. Chem. B 2015, 119, 7349–7357. [Google Scholar] [CrossRef]
- Harvie, A.J.; Smith, C.T.; Ahumada-Lazo, R.; Jeuken, L.J.C.; Califano, M.; Bon, R.S.; Hardman, S.J.O.; Binks, D.J.; Critchley, K. Ultrafast Trap State-Mediated Electron Transfer for Quantum Dot Redox Sensing. J. Phys. Chem. C 2018, 122, 10173–10180. [Google Scholar] [CrossRef]
- Olshansky, J.H.; Balan, A.D.; Ding, T.X.; Fu, X.; Lee, Y.; Alivisatos, P. Temperature-Dependent Hole Transfer from Photoexcited Quantum Dots to Molecular Species: Evidence for Trap-Mediated Transfer. ACS Nano 2017, 11, 8346–8355. [Google Scholar] [CrossRef] [PubMed]
- Jasieniak, J.; Smith, L.; Van Embden, J.; Mulvaney, P.; Califano, M. Re-examination of the Size-Dependent Absorption Properties of CdSe Quantum Dots. J. Phys. Chem. C 2009, 113, 19468–19474. [Google Scholar] [CrossRef]
- Rasband, W.S.; Image, J. U. S. National Institutes of Health, Bethesda, Maryland, USA, 1997–2018. Available online: https://imagej.nih.gov/ij/ (accessed on 30 May 2020).
- Porres, L.; Holland, A.; Pålsson, L.-O.; Monkman, A.P.; Kemp, C.; Beeby, A. Absolute Measurements of Photoluminescence Quantum Yields of Solutions Using an Integrating Sphere. J. Fluoresc. 2006, 16, 267–273. [Google Scholar] [CrossRef] [PubMed]
- FluorTools DecayFit—Fluorescence Decay Analysis Software 1.4. 2014. Available online: http://www.fluortools.com/software/decayfit (accessed on 30 May 2020).




| Sample | Eabs,CdS/eV | Eabs,CdSe/eV | EPL/eV | ΦPL,450 | ΦPL,500 |
|---|---|---|---|---|---|
| TOPO-NR | 2.70 | 2.22 | 2.16 | 0.58 ± 0.09 | 0.77 ± 0.02 |
| MUA-NR | 2.71 | 2.22 | 2.18 | 0.25 ± 0.05 | 0.47 ± 0.02 |
| HS-PEG-NR | 2.71 | 2.23 | 2.17 | 0.21 ± 0.07 | 0.39 ± 0.02 |
| DHLA-NR | 2.70 | 2.23 | 2.17 | 0.04 ± 0.02 | 0.15 ± 0.02 |
| DHLA-PEG-NR | 2.70 | 2.22 | 2.14 | 0.05 ± 0.02 | 0.21 ± 0.02 |
| PEI-NR | 2.71 | 2.24 | 2.19 | 0.91 ± 0.12 | 0.87 ± 0.02 |
| Ligand | Energy Range | τ1/ps | τ2/ps | τ3/ps | τ4/ps |
|---|---|---|---|---|---|
| TOPO | 2.48–2.95 eV | 0.2 ± 0.0 | 1.0 ± 0.2 | 34 ± 8 | 530 ± 70 |
| 1.97–2.34 eV | 0.7 ± 0.0 | 47 ± 12 | 190 ± 50 | 6200 ** | |
| MUA | 2.48–2.95 eV | 0.2 ± 0.0 | 2.5 ± 0.4 | 78 ± 6 | 650 ± 40 |
| 1.97–2.34 eV | 0.5 ± 0.1 | 58 ± 15 | 540 ± 170 | 12200 ** | |
| HS-PEG | 2.48–2.95 eV | 0.2 ± 0.1 | 3.0 ± 0.6 | 68 ± 4 | 780 ± 30 |
| 1.97–2.34 eV | 0.5 ± 0.1 | 42 ± 7 | 450 ± 140 | 4000 ** | |
| DHLA | 2.48–2.95 eV | 0.2 ± 0.0 | 4.8 ± 1.0 | 86 ± 7 | 820 ± 40 |
| 1.97–2.34 eV | 0.5 ± 0.1 | — * | 150 ± 20 | 4000** | |
| DHLA-PEG | 2.48–2.95 eV | 0.2 ± 0.0 | 1.7 ± 0.2 | 60 ± 10 | 610 ± 160 |
| 1.97–2.34 eV | 0.4 ± 0.0 | — * | 190 ± 50 | 2500 ** | |
| PEI | 2.48–2.95 eV | 0.2 ± 0.0 | 1.3 ± 0.3 | 25 ± 1 | 470 ± 70 |
| 1.97–2.34 eV | 1.0 ± 0.2 | 74 ± 11 | 270 ± 30 | 4100 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheel, M.; Liu, B.; Wächtler, M. Influence of Surface Ligands on Charge-Carrier Trapping and Relaxation in Water-Soluble CdSe@CdS Nanorods. Catalysts 2020, 10, 1143. https://doi.org/10.3390/catal10101143
Micheel M, Liu B, Wächtler M. Influence of Surface Ligands on Charge-Carrier Trapping and Relaxation in Water-Soluble CdSe@CdS Nanorods. Catalysts. 2020; 10(10):1143. https://doi.org/10.3390/catal10101143
Chicago/Turabian StyleMicheel, Mathias, Bei Liu, and Maria Wächtler. 2020. "Influence of Surface Ligands on Charge-Carrier Trapping and Relaxation in Water-Soluble CdSe@CdS Nanorods" Catalysts 10, no. 10: 1143. https://doi.org/10.3390/catal10101143
APA StyleMicheel, M., Liu, B., & Wächtler, M. (2020). Influence of Surface Ligands on Charge-Carrier Trapping and Relaxation in Water-Soluble CdSe@CdS Nanorods. Catalysts, 10(10), 1143. https://doi.org/10.3390/catal10101143