The Positive Effect of Iron Doping in the Electrocatalytic Activity of Cobalt Hexacyanoferrate



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Electrode Preparation
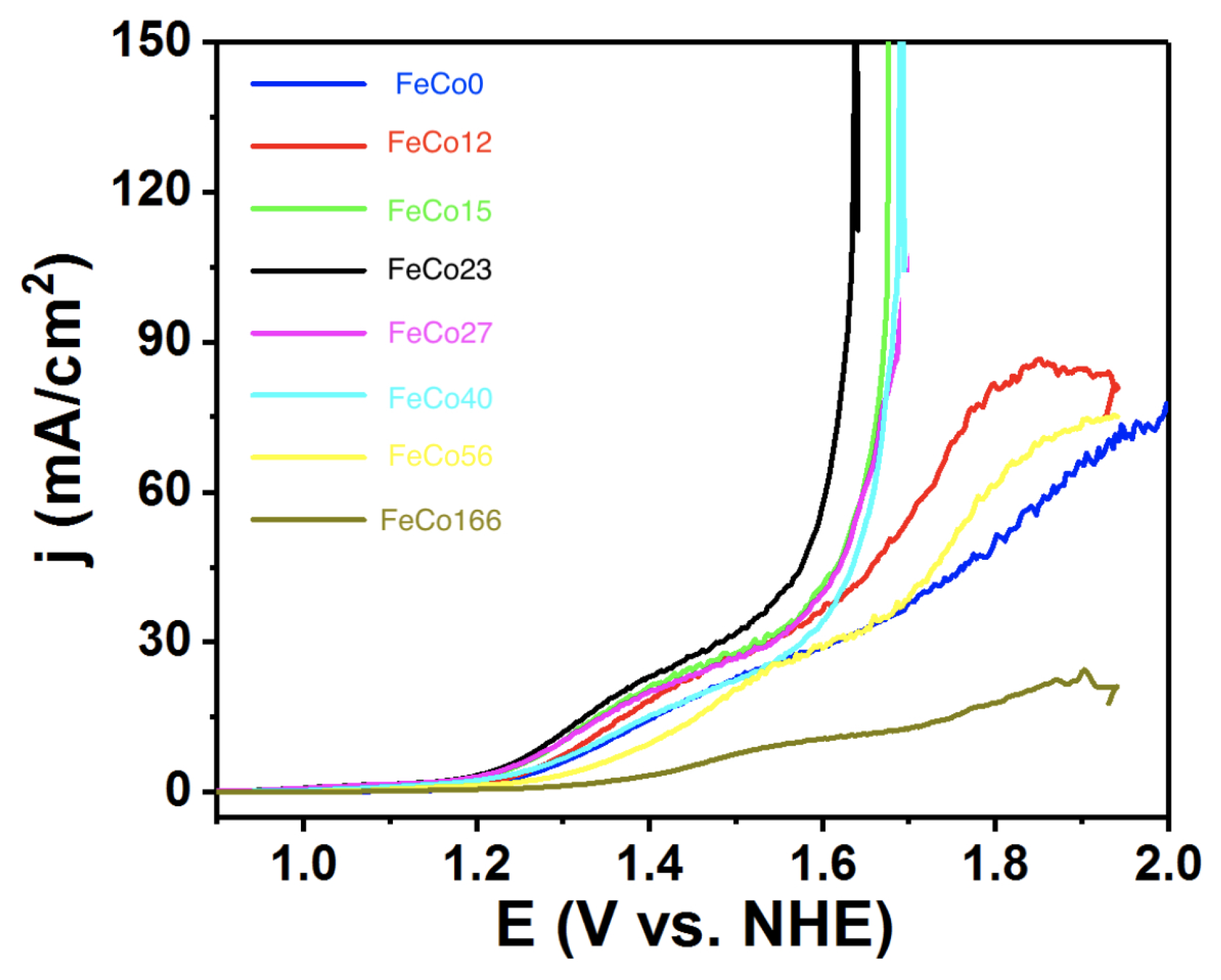
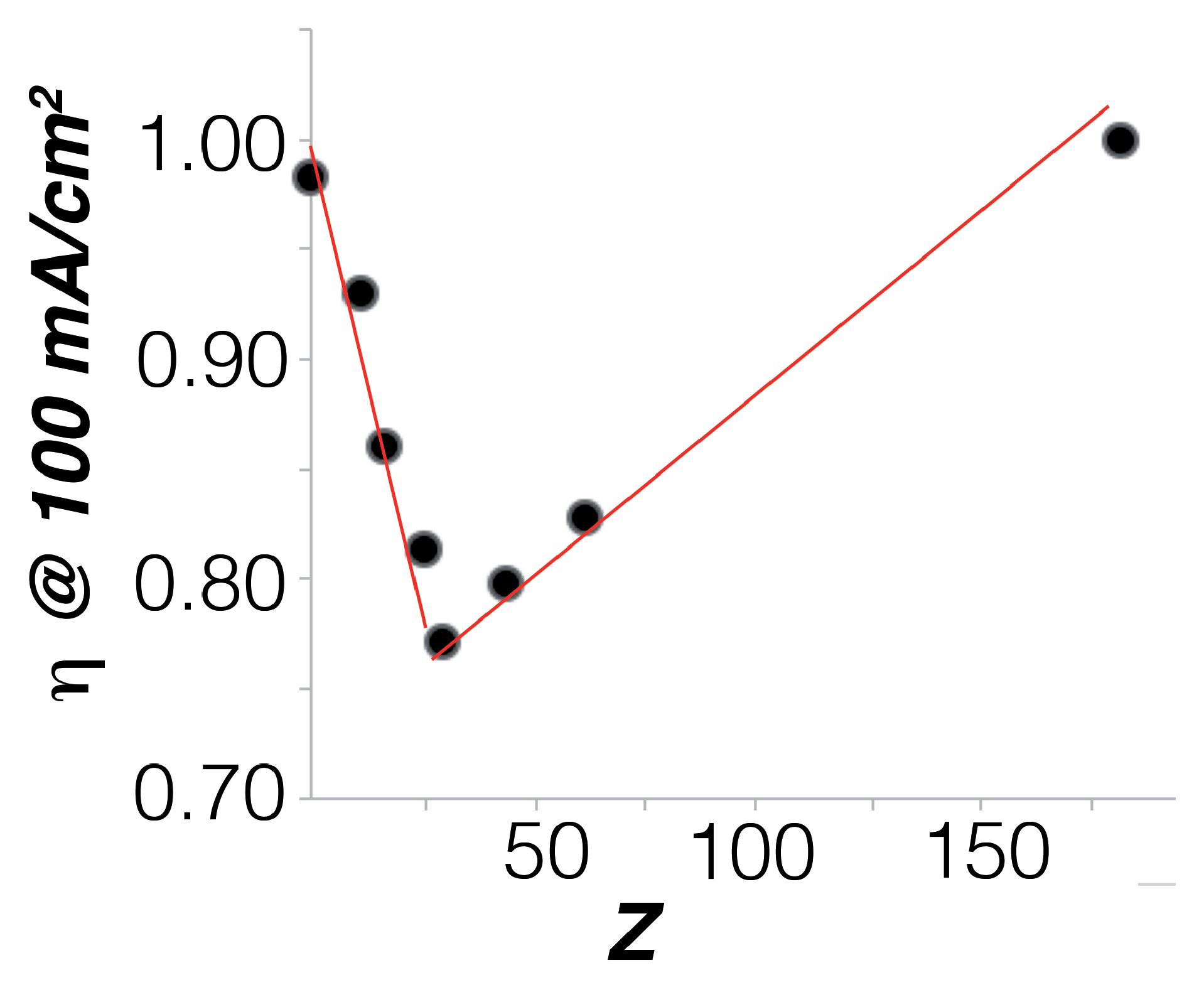
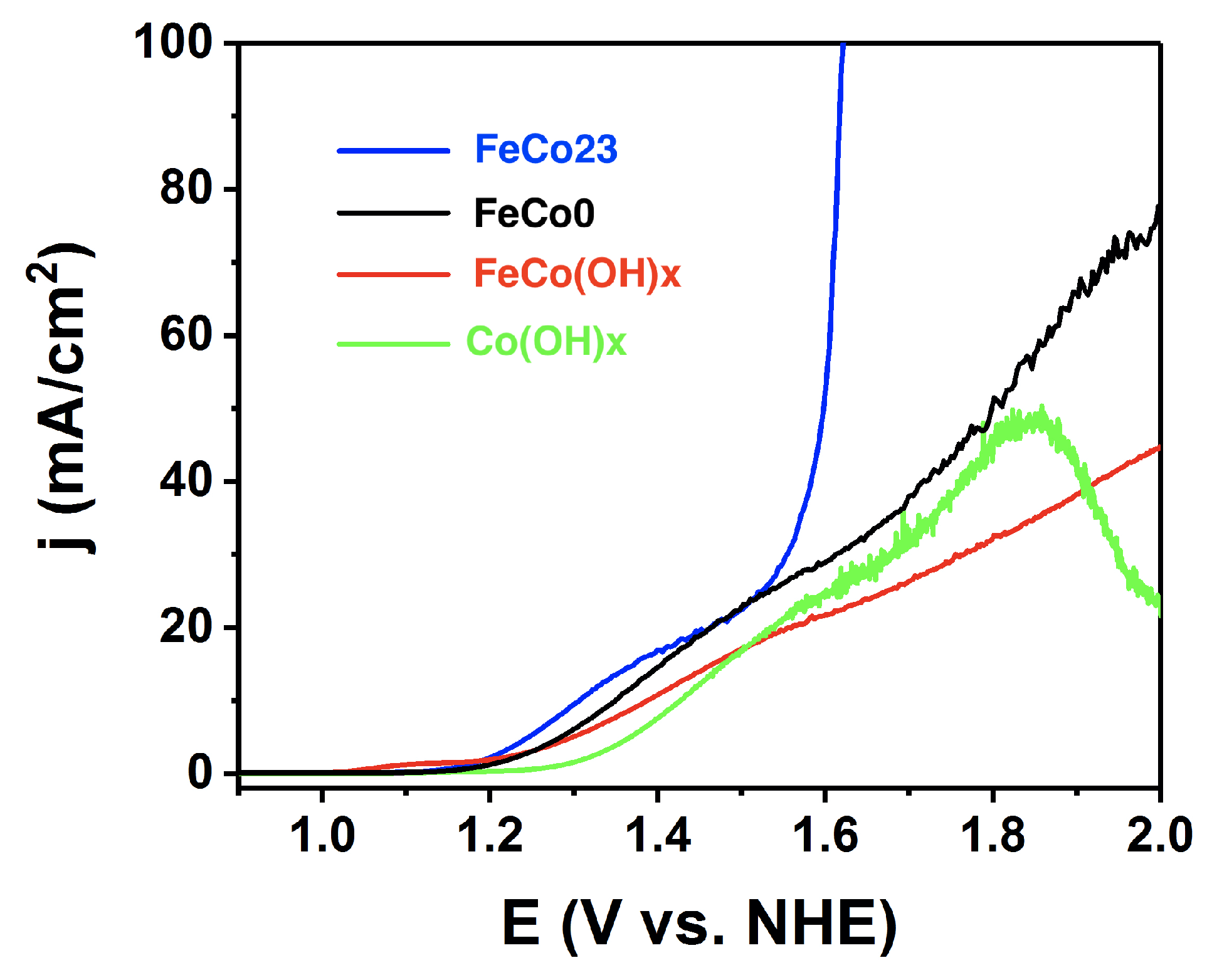
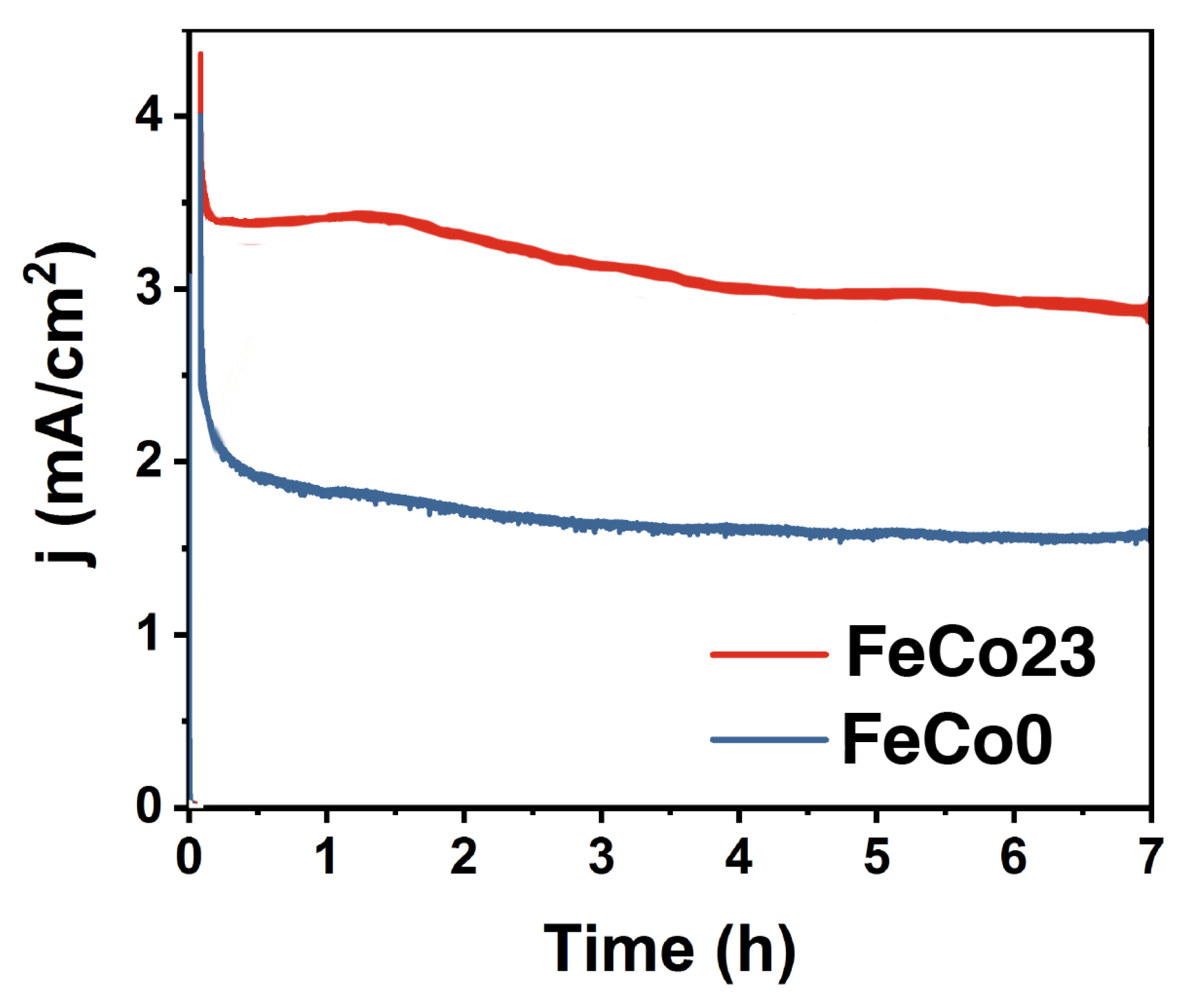
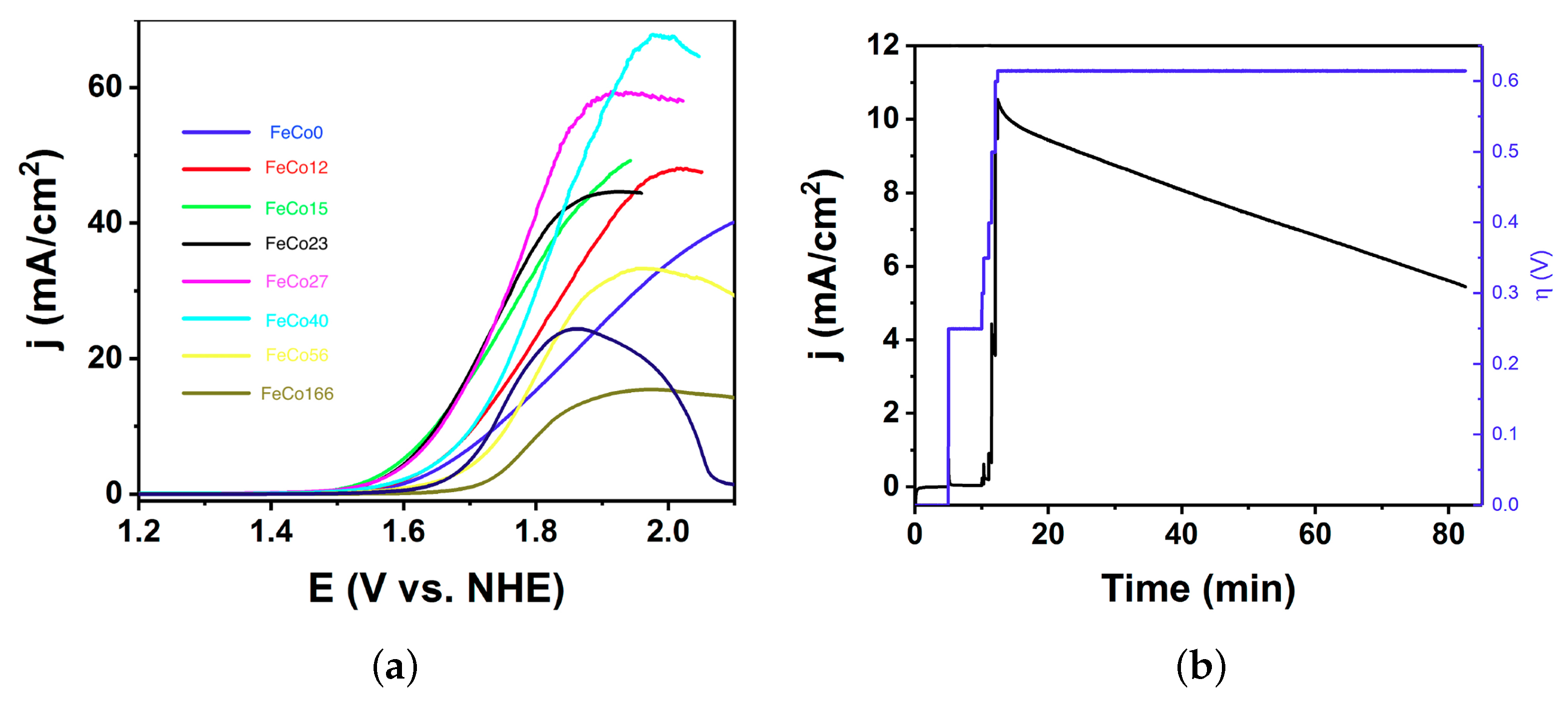
2.2. Electrocatalysis in Neutral pH
2.3. Electrocatalysis in Acidic pH
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PBA | Prussian blue analogues |
| OER | Oxygen evolution reaction |
| FTO | Fluoride-doped tin oxide |
References
- Karyakin, A.A. Prussian Blue and Its Analogues: Electrochemistry and Analytical Applications. Electroanalysis 2001, 13, 813–819. [Google Scholar] [CrossRef]
- de Tacconi, N.R.; Rajeshwar, K.; Lezna, R.O. Metal Hexacyanoferrates:? Electrosynthesis, in Situ Characterization, and Applications. Chem. Mater. 2003, 15, 3046–3062. [Google Scholar] [CrossRef]
- Piernas-Muñoz, M.J.; Castillo Martínez, E. Prussian Blue and Its Analogues.Structure, Characterization and Applications. In Prussian Blue Based Batteries; Springer: Cham, Switzerland, 2007; pp. 9–22. [Google Scholar]
- Karyakin, A.A. Advances of Prussian blue and its analogues in (bio)sensors. Curr. Opin. Electrochem. 2017, 5, 92–98. [Google Scholar] [CrossRef]
- Komkova, M.A.; Karyakina, E.E.; Karyakin, A.A. Catalytically Synthesized Prussian Blue Nanoparticles Defeating Natural Enzyme Peroxidase. J. Am. Chem. Soc. 2018, 140, 11302–11307. [Google Scholar] [CrossRef] [PubMed]
- De Lara González, G.L.; Kahlert, H.; Scholz, F. Catalytic reduction of hydrogen peroxide at metal hexacyanoferrate composite electrodes and applications in enzymatic analysis. Electrochim. Acta 2007, 52, 1968–1974. [Google Scholar] [CrossRef]
- Pintado, S.; Goberna-Ferron, S.; Escudero-Adan, E.C.; Galan-Mascaros, J.R. Fast and persistent electrocatalytic water oxidation by Co-Fe Prussian blue coordination polymers. J. Am. Chem. Soc. 2013, 135, 13270–13273. [Google Scholar] [CrossRef]
- Alsac, E.P.; Ulker, E.; Nune, S.V.K.; Dede, Y.; Karadas, F. Tuning the Electronic Properties of Prussian Blue Analogues for Efficient Water Oxidation Electrocatalysis: Experimental and Computational Studies. Chem. Eur. J. 2018, 24, 4856–4863. [Google Scholar] [CrossRef]
- Aksoy, M.; Nune, S.V.K.; Karadas, F. A Novel Synthetic Route for the Preparation of an Amorphous Co/Fe Prussian Blue Coordination Compound with High Electrocatalytic Water Oxidation Activity. Inorg. Chem. 2016, 55, 4301–4307. [Google Scholar] [CrossRef]
- Han, L.; Tang, P.; Reyes-Carmona, A.; Rodríguez-García, B.; Torréns, M.; Morante, J.R.; Arbiol, J.; Galan-Mascaros, J.R. Oxygen Evolution Electrocatalysts Processed by Chemical Etching. J. Am. Chem. Soc. 2016, 138, 16037–16045. [Google Scholar] [CrossRef]
- Du, L.; Du, C.; Chen, G.; Kong, F.; Yin, G.; Wang, Y. Metal-Organic Coordination Networks: Prussian Blue and Its Synergy with Pt Nanoparticles to Enhance Oxygen Reduction Kinetics. ACS Appl. Matter. Interfaces 2016, 8, 15250–15257. [Google Scholar] [CrossRef]
- Bui, H.T.; Shrestha, N.K.; Cho, K.; Bathula, C.; Opoku, H.; Noh, Y.Y.; Han, S.H. Oxygen reduction reaction on nickel-based Prussian blue analog frameworks synthesized via electrochemical anodization route. J. Electroanal. Chem. 2018, 828, 80–85. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Lin, D.; Zhao, J.; Liu, C.; Xie, J.; Cui, Y. A Prussian blue route to nitrogen-doped graphene aerogels as efficient electrocatalysts for oxygen reduction with enhanced active site accessibility. Nano Res. 2016, 10, 1213–1222. [Google Scholar] [CrossRef]
- Jia, S.; Zang, J.; Li, W.; Tian, P.; Zhou, S.; Cai, H.; Tian, X.; Wang, Y. A novel synthesis of Prussian blue nanocubes/biomass-derived nitrogen-doped porous carbon composite as a high-efficiency oxygen reduction reaction catalyst. Electrochim. Acta 2018, 289, 56–64. [Google Scholar] [CrossRef]
- Mousavi Shaegh, S.A.; Nguyen, N.-T.; Mousavi Ehteshami, S.M.; Chan, S.H. A membraneless hydrogen peroxide fuel cell using Prussian Blue as cathode material. Energ. Environ. Sci. 2012, 5, 8225–8228. [Google Scholar] [CrossRef]
- Moss, B.; Hegner, F.S.; Corby, S.; Selim, S.; Francas, L.; López, N.; Gimenez, S.; Galan-Mascaros, J.R.; Durrant, J.R. Unraveling Charge Transfer in CoFe Prussian Blue Modified BiVO4 Photoanodes. ACS Energy Lett. 2019, 4, 337–342. [Google Scholar] [CrossRef]
- Hegner, F.S.; Herraiz-Cardona, I.; Cardenas-Morcoso, D.; López, N.; Galan-Mascaros, J.R.; Gimenez, S. Cobalt Hexacyanoferrate on BiVO4 Photoanodes for Robust Water Splitting. ACS Appl. Mater. Interfaces 2017, 9, 37671–37681. [Google Scholar] [CrossRef]
- Goberna-Ferron, S.; Hernández, W.Y.; Rodríguez-García, B.; Galan-Mascaros, J.R. Light-Driven Water Oxidation with Metal Hexacyanometallate Heterogeneous Catalysts. ACS Catal. 2014, 4, 1637–1641. [Google Scholar] [CrossRef]
- Gerken, J.B.; McAlpin, J.G.; Chen, J.Y.C.; Rigsby, M.L.; Casey, W.H.; Britt, R.D.; Stahl, S.S. Electrochemical Water Oxidation with Cobalt-Based Electrocatalysts from pH 0–14: The Thermodynamic Basis for Catalyst Structure, Stability, and Activity. J. Am. Chem. Soc. 2011, 133, 14431–14442. [Google Scholar] [CrossRef]
- You, B.; Sun, Y. Innovative Strategies for Electrocatalytic Water Splitting. Acc. Chem. Res. 2018, 51, 1571–1580. [Google Scholar] [CrossRef]
- Hunter, B.M.; Gray, H.B.; Müller, A.M. Earth-Abundant Heterogeneous Water Oxidation Catalysts. Chem. Rev. 2016, 116, 14120–14136. [Google Scholar] [CrossRef]
- Rodríguez-García, B.; Reyes-Carmona, A.; Jimenez-Morales, I.; Blasco-Ahicart, M.; Cavaliere, S.; Dupont, M.; Jones, D.; Roziere, J.; Galan-Mascaros, J.R.; Jaouen, F. Cobalt hexacyanoferrate supported on Sb-doped SnO2 as a non-noble catalyst for oxygen evolution in acidic medium. Sustain. Energy Fuels 2018, 2, 589–597. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, L.; Galán-Mascarós, J.R. The Positive Effect of Iron Doping in the Electrocatalytic Activity of Cobalt Hexacyanoferrate. Catalysts 2020, 10, 130. https://doi.org/10.3390/catal10010130
Han L, Galán-Mascarós JR. The Positive Effect of Iron Doping in the Electrocatalytic Activity of Cobalt Hexacyanoferrate. Catalysts. 2020; 10(1):130. https://doi.org/10.3390/catal10010130
Chicago/Turabian StyleHan, Lijuan, and José Ramón Galán-Mascarós. 2020. "The Positive Effect of Iron Doping in the Electrocatalytic Activity of Cobalt Hexacyanoferrate" Catalysts 10, no. 1: 130. https://doi.org/10.3390/catal10010130
APA StyleHan, L., & Galán-Mascarós, J. R. (2020). The Positive Effect of Iron Doping in the Electrocatalytic Activity of Cobalt Hexacyanoferrate. Catalysts, 10(1), 130. https://doi.org/10.3390/catal10010130