
Neural Network Methodology for the Identification and Classification of Lipopeptides Based on SMILES Annotation

Abstract
1. Introduction
2. Materials and Method
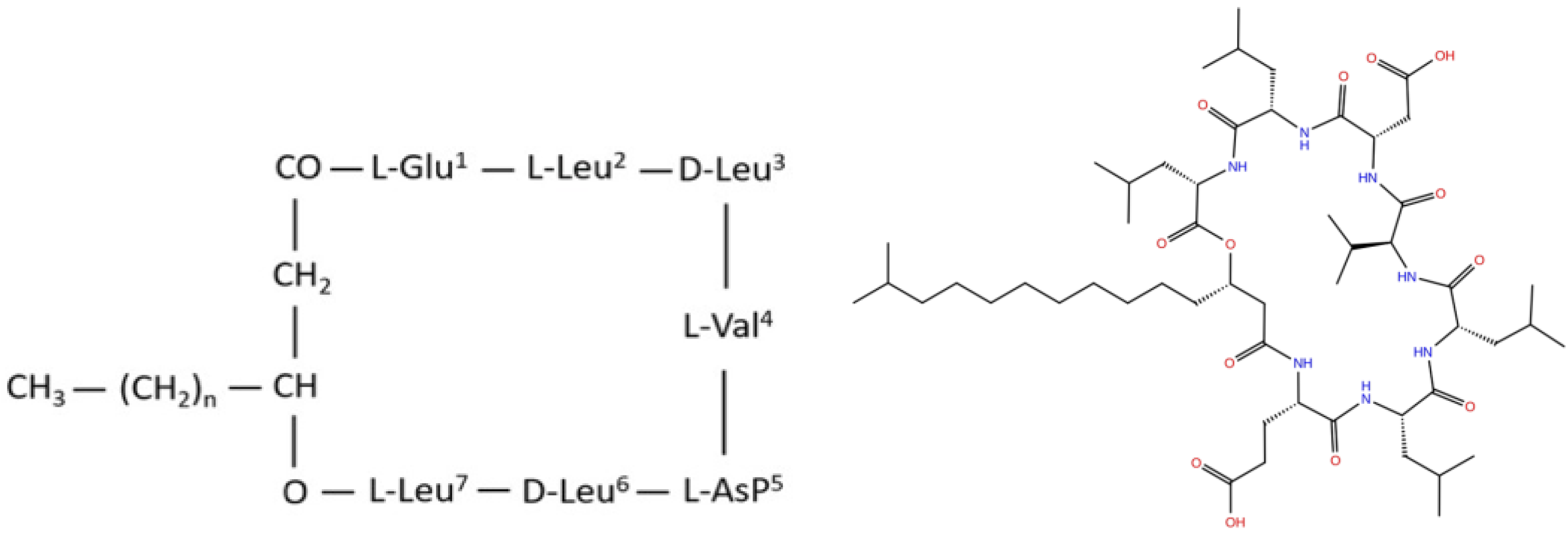
2.1. Data Collection
2.2. Data Preparation
2.3. Experimental Protocol
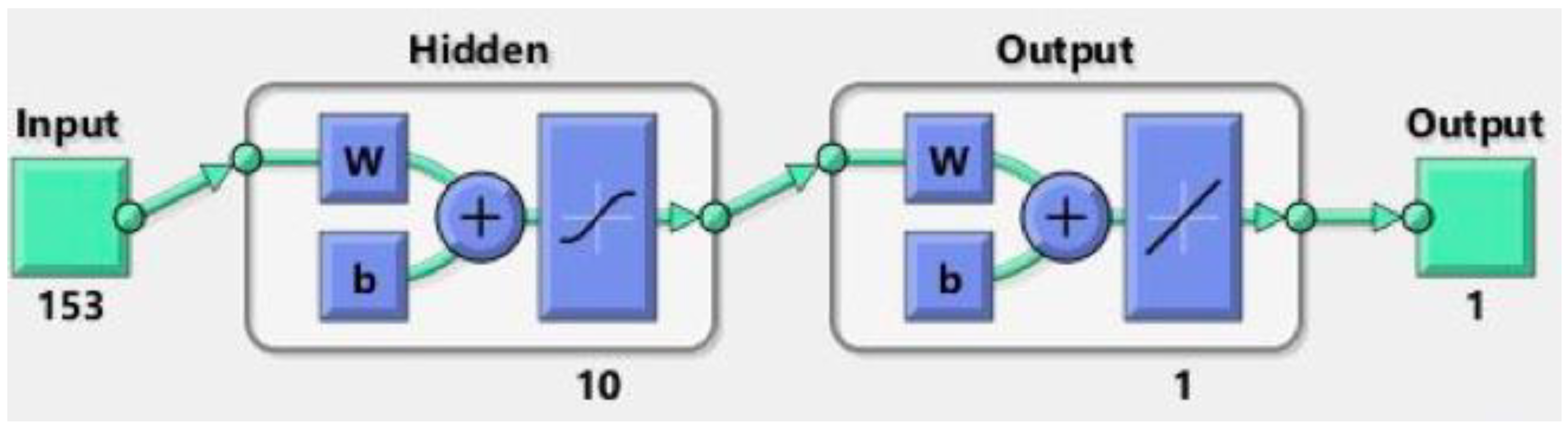
2.4. Neural Network Methodology
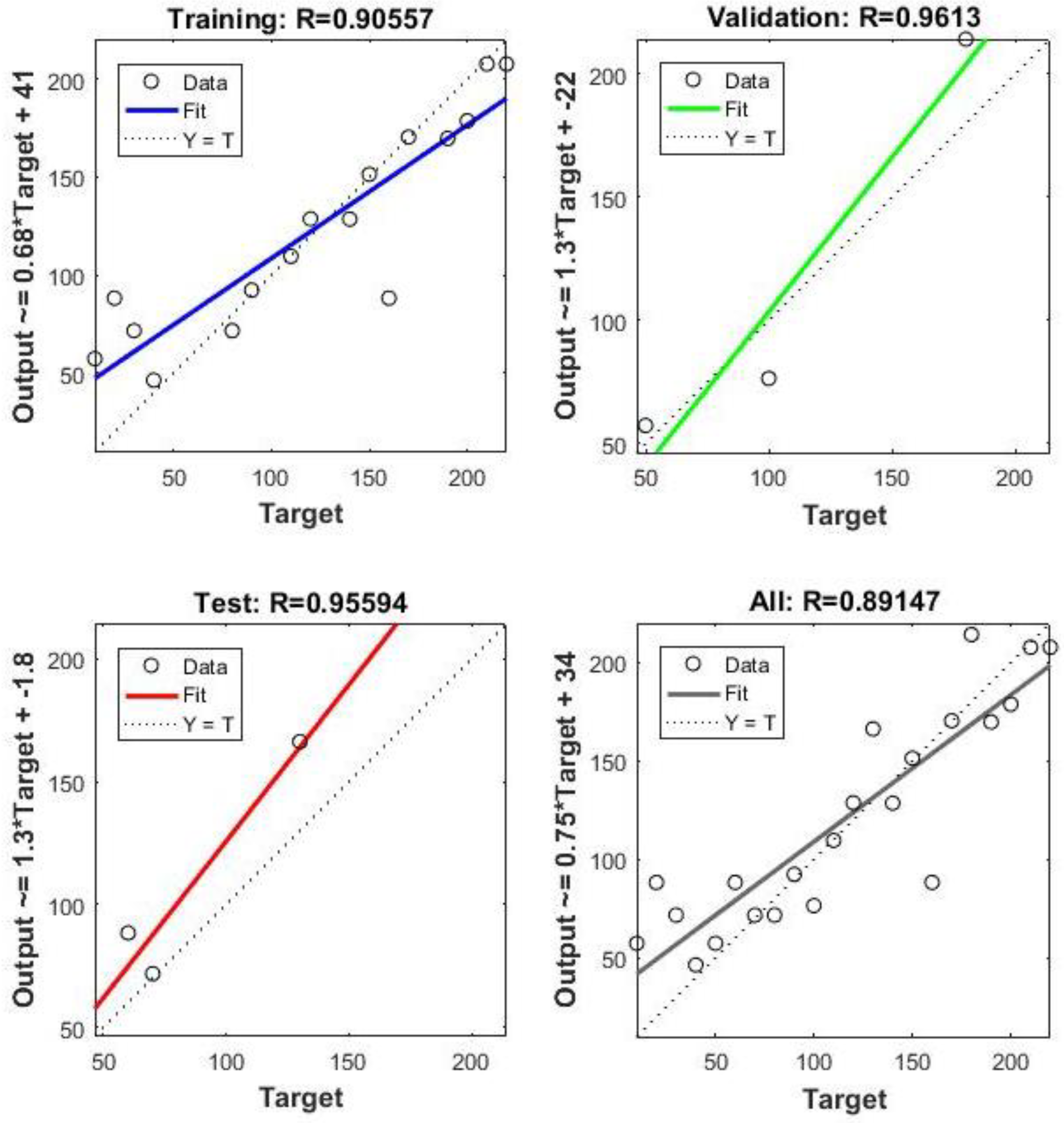
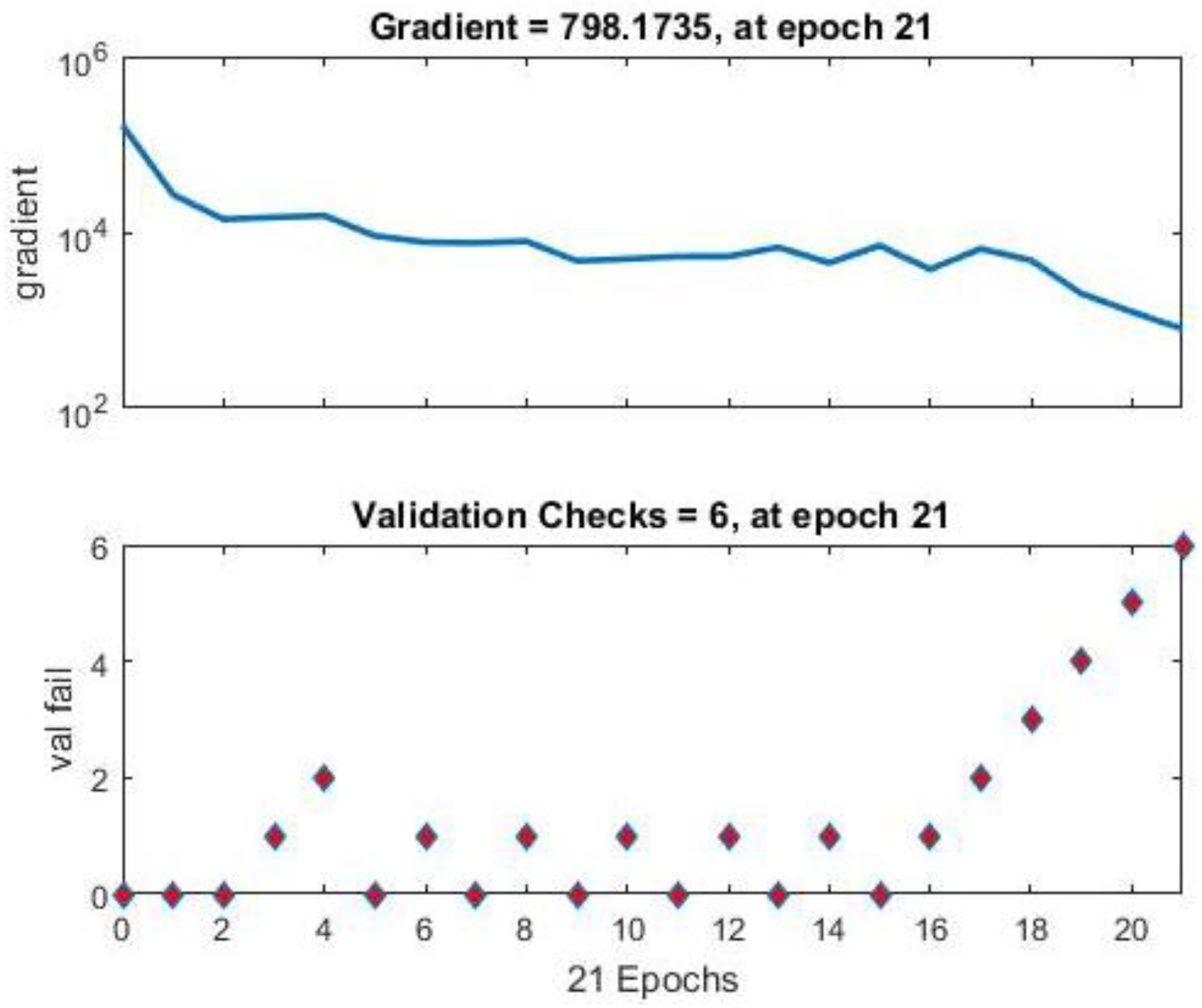
3. Results and Discussion
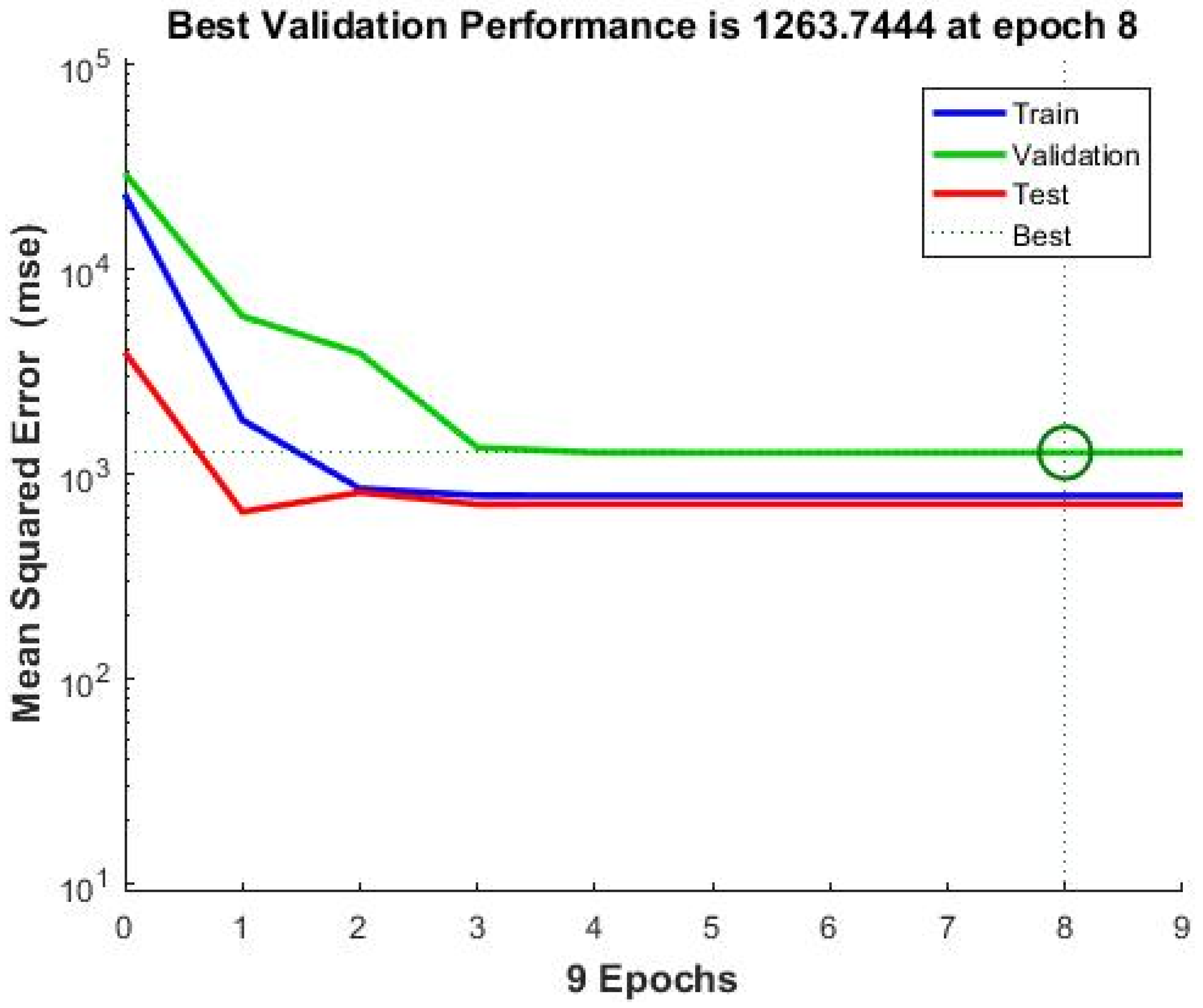
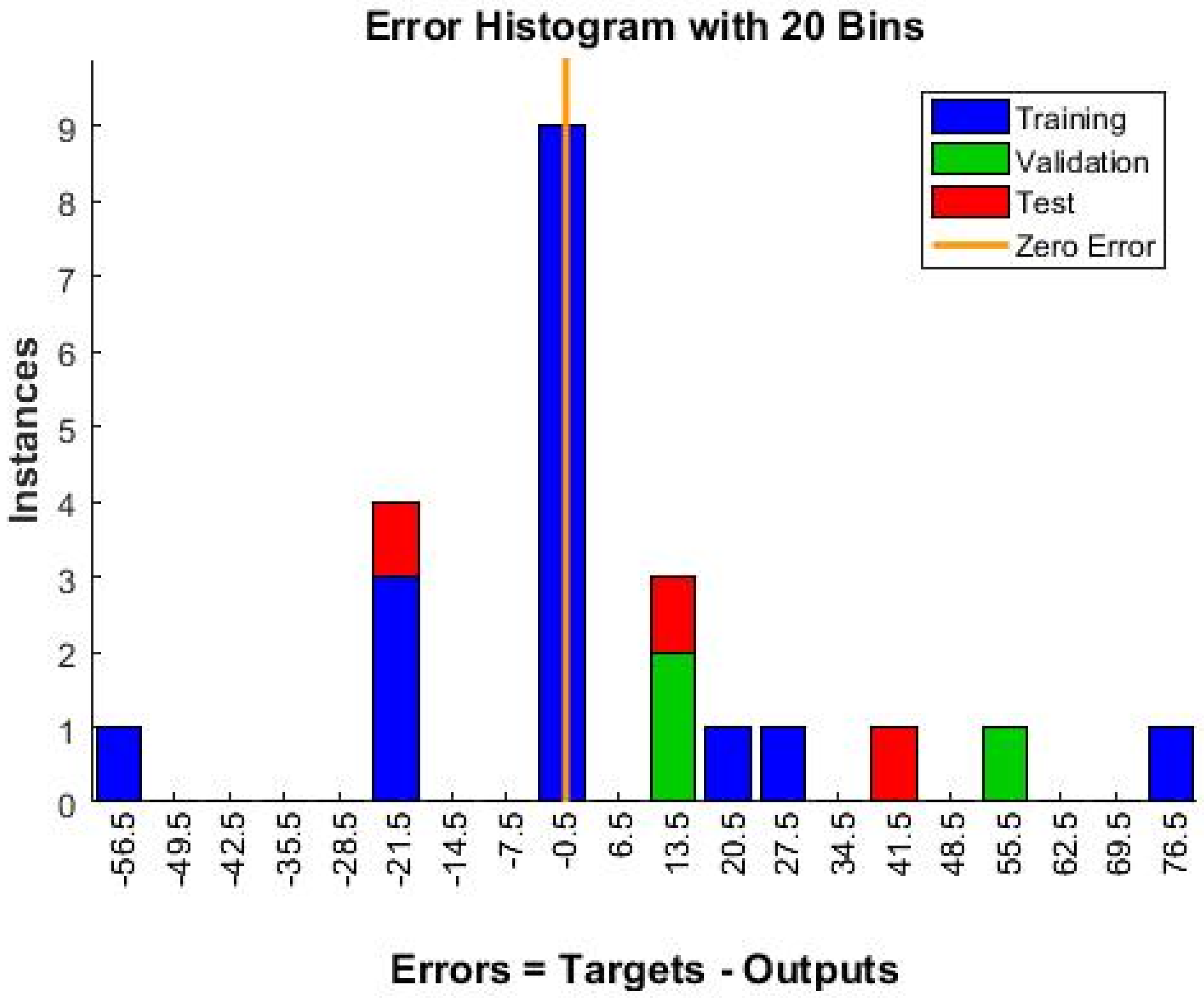
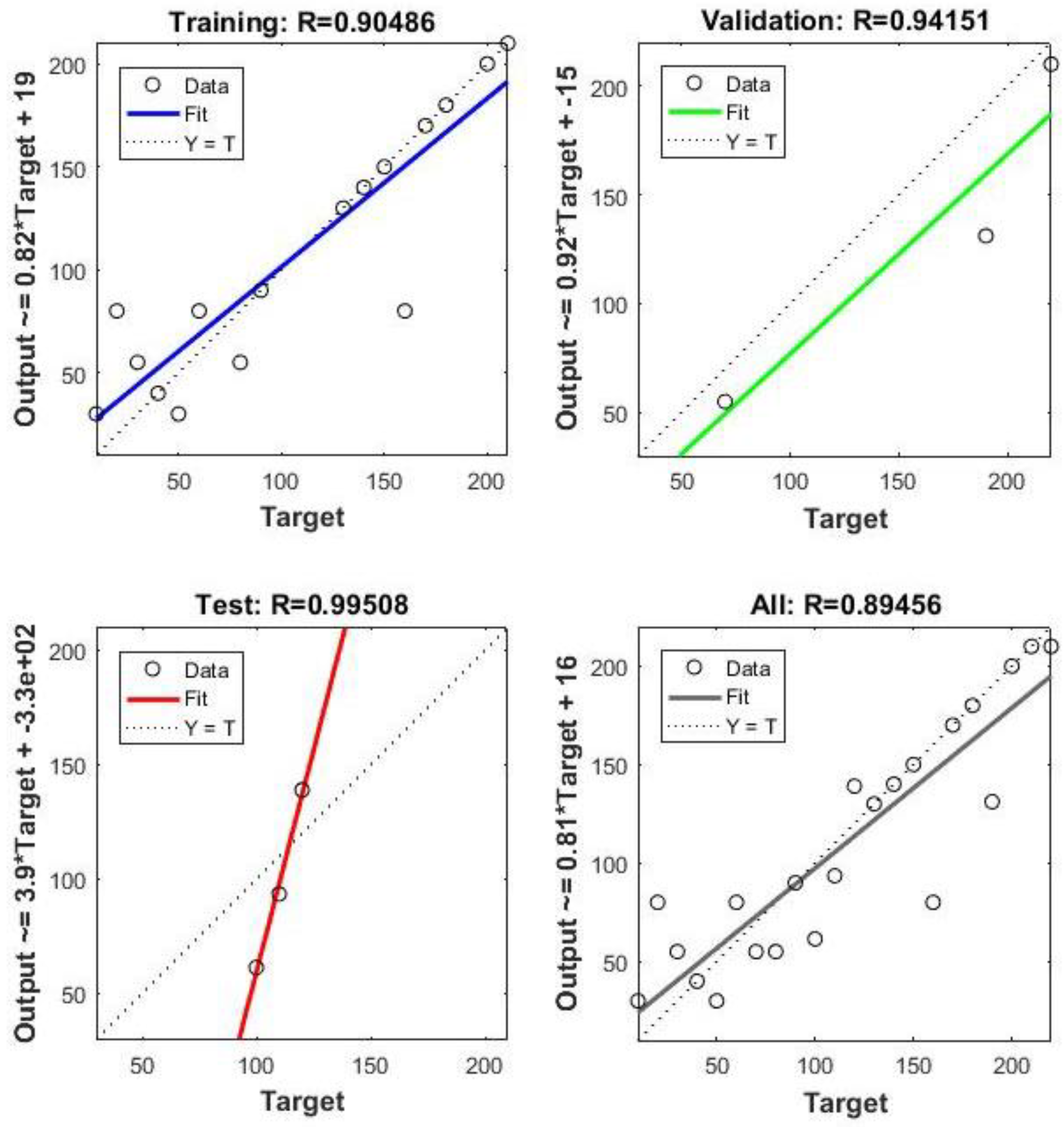
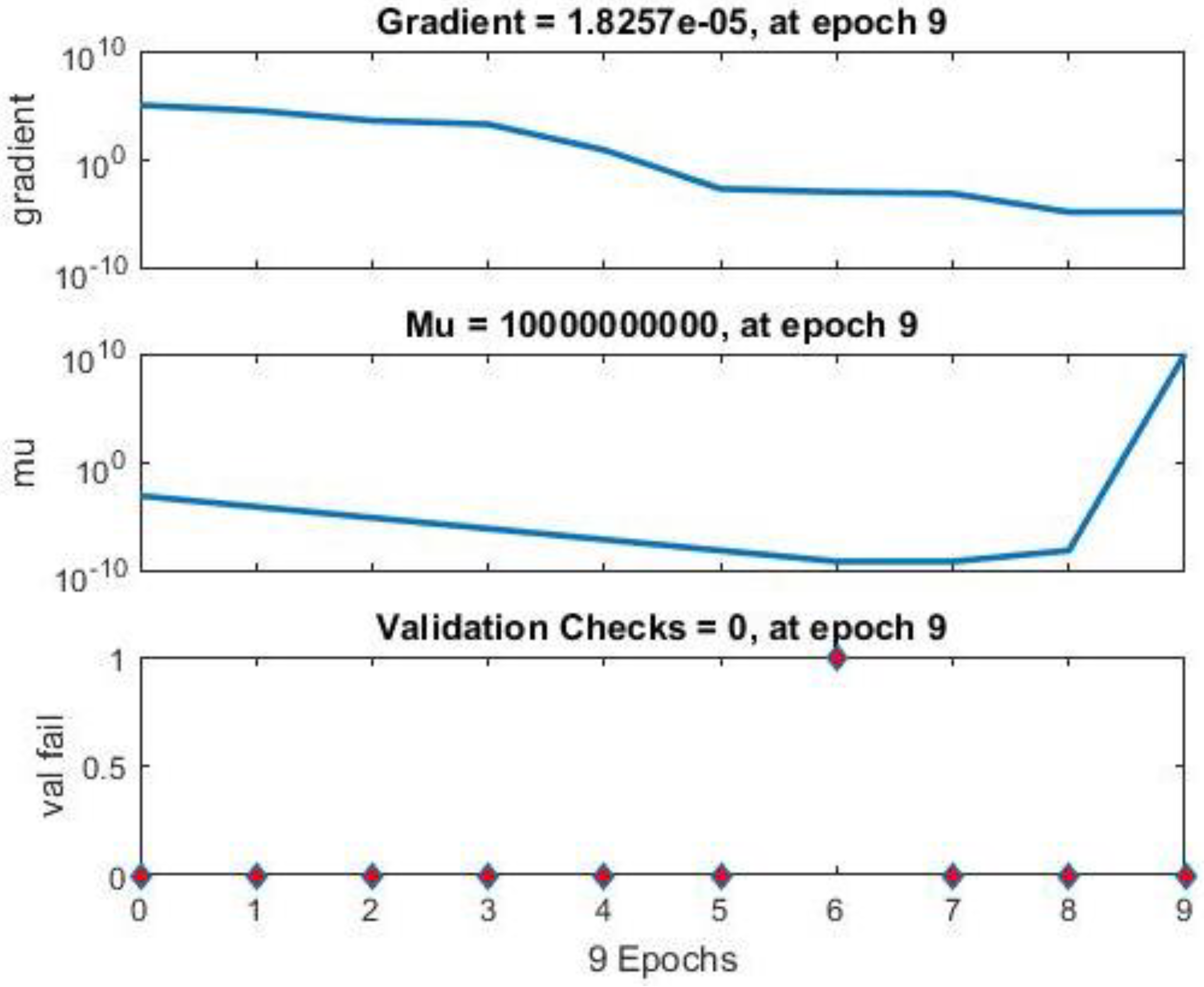
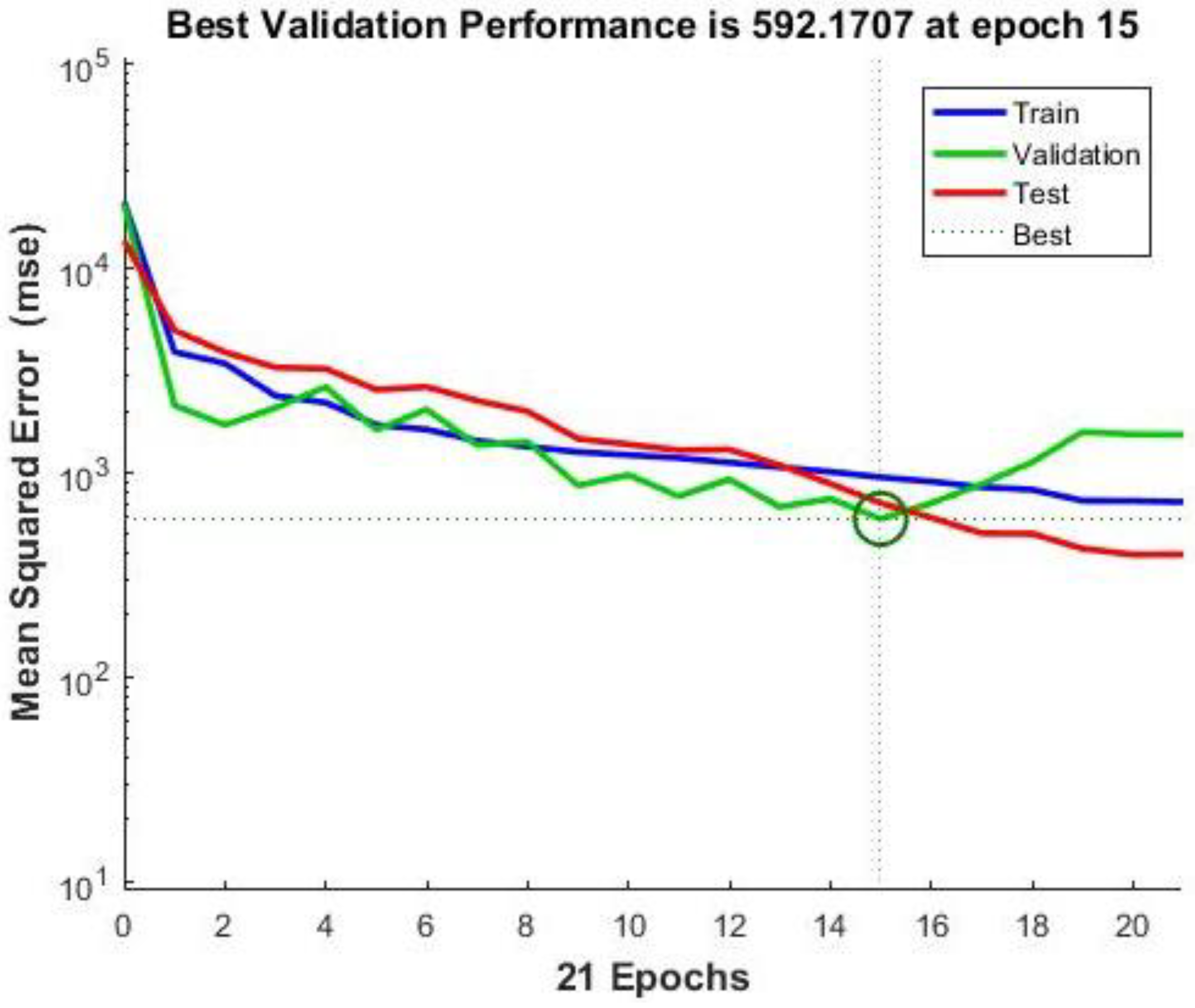
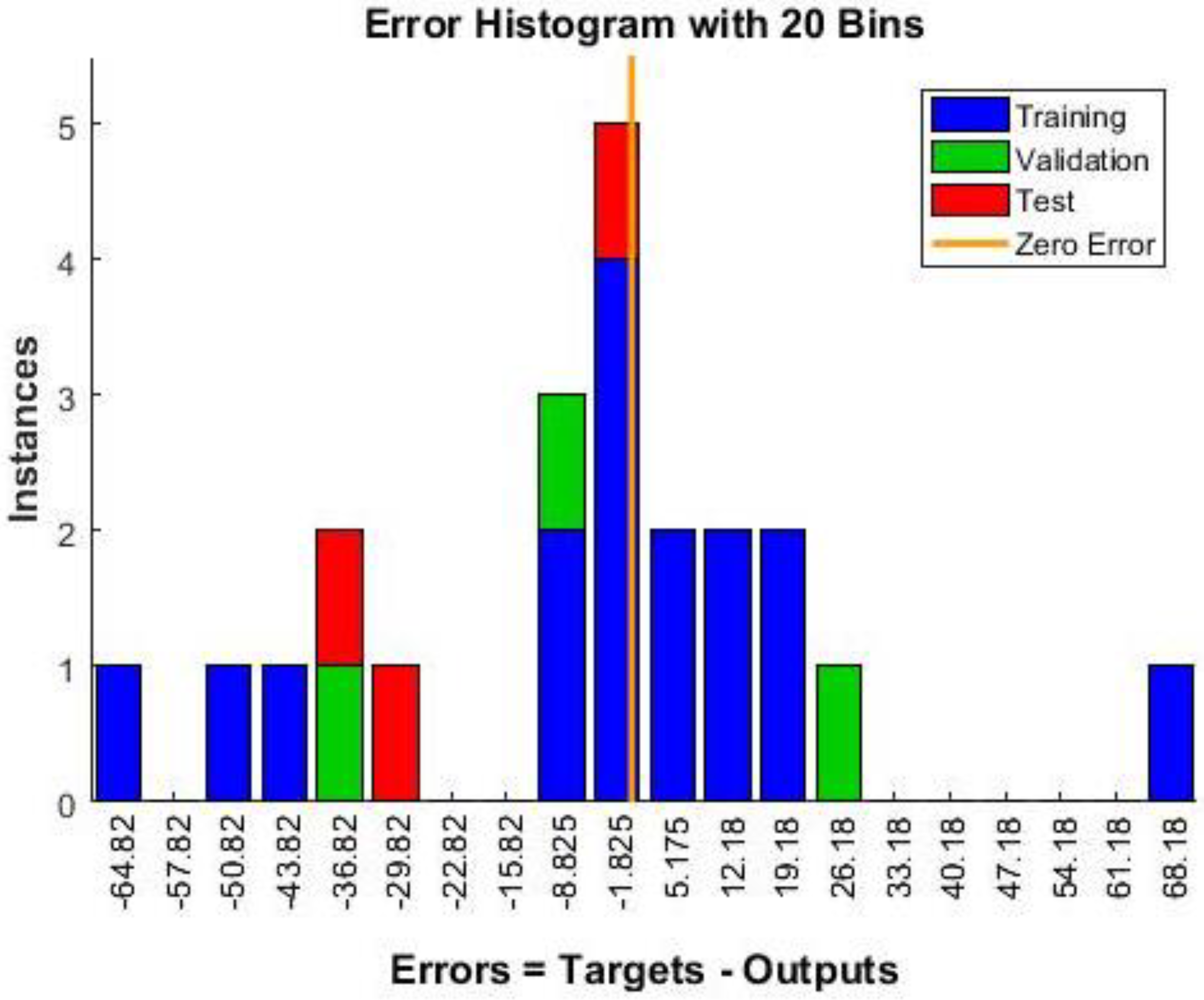
Neural Network Training Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirohara, M.; Saito, Y.; Koda, Y.; Sato, K.; Sakakibara, Y. Convolutional neural network based on SMILES representation of compounds for detecting chemical motif. BMC Bioinform. 2018, 19, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Richards, W.G. Ultrafast shape recognition to search compound databases for similar molecular shapes. J. Comput. Chem. 2007, 28, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Helma, C. Predictive Toxicology; Taylor and Francis: Washington, DC, USA, 2005. [Google Scholar]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure—Activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016, 26, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Lanchantin, J.; Singh, R.; Wang, B.; Qi, Y. Deep motif dashboard: Visualizing and understanding genomic sequences using deep neural networks. Pac. Symp. Biocomput. 2017, 22, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Edwards, M.D.; Liu, G.; Gifford, D.K. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics 2016, 32, i121–i127. [Google Scholar] [CrossRef]
- Jujjavarapu, S.E.; Deshmukh, S. Artificial neural network as a classifier for the identification of hepatocellular carcinoma through prognosticgene signatures. Curr. Genom. 2018, 19, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Meena, K.R.; Kanwar, S.S. Lipopeptides as the antifungal and antibacterial agents: Applications in food safety and therapeutics. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jujjavarapu, S.E.; Dhagat, S.; Yadav, M. Computer-Aided Design of Antimicrobial Lipopeptides as Prospective Drug Candidates; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Kracht, M.; Rokos, H.; Özel, M.; Kowall, M.; Pauli, G.; Vater, J. Antiviral and hemolytic activities of surfactin isoforms and their methyl ester derivatives. J. Antibiot. 1999, 52, 613–619. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Unraveling the bioactivity of anticancer peptides as deduced from machine learning. EXCLI J. 2018, 17, 734–752. [Google Scholar]
- Surfactin|C53H93N7O13. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/443592 (accessed on 20 May 2021).
- Liu, J.-F.; Mbadinga, S.M.; Yang, S.-Z.; Gu, J.-D.; Mu, B.-Z. Chemical Structure, property and potential applications of biosurfactants produced by Bacillus subtilis in petroleum recovery and spill mitigation. Int. J. Mol. Sci. 2015, 16, 4814–4837. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, S.A.; Vederas, J.C. Lipopeptides from Bacillus and Paenibacillus spp.: A gold mine of antibiotic candidates. Med. Res. Rev. 2016, 36, 4–31. [Google Scholar] [CrossRef] [PubMed]
- Poroikov, V.V. Computer-aided drug design: From discovery of novel pharmaceutical agents to systems pharmacology. Biochemistry 2020, 66, 30–41. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Canonical SMILES |
|---|---|
| Surfactin A | CC(C)CCCCCCCCCC1CC(=O)NC(C(=O) NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O) NC(C(=O)NC(C(=O)O1)CC(C)C)CC(C)C) CC(=O)O)C(C)C)CC(C)C)CC(C)C)CCC(=O)O |
| Symbol | Replaced with |
|---|---|
| 1 | 1 |
| C | 2 |
| O | 3 |
| N | 4 |
| Na | 5 |
| Ca | 6 |
| S | 7 |
| + | 8 |
| = | 9 |
| ( | 10 |
| ) | 11 |
| . | 12 |
| - | 13 |
| [ | 14 |
| ] | 15 |
| Empty space | 0 |
| Compound Name | Numerical Value |
|---|---|
| Surfactin A | 10 |
| Surfactin B | 20 |
| Surfactin C | 30 |
| Surfactin D | 40 |
| Surfactin-C13 | 50 |
| Surfactin-C14 | 60 |
| Surfactin-C15 | 70 |
| Surfactin Peptide | 80 |
| Methylated Surfactin | 90 |
| Amidated Surfactin | 100 |
| Linear Surfactin | 110 |
| Sodium Surfactin | 120 |
| Disodium Surfactin | 130 |
| Calcium Surfactin | 140 |
| Aminomethane Sulfonated Surfactin | 150 |
| [Leu-7]Surfactin | 160 |
| [Ile4]Surfactin | 170 |
| [Ile7]Surfactin | 180 |
| [Ile4,7]Surfactin | 190 |
| [Ile2,4,7]Surfactin | 200 |
| 3(R)-Aza-Surfactin | 220 |
| 3(S)-Epi-Aza-Surfactin | 220 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, M.; Jujjavarapu, S.E. Neural Network Methodology for the Identification and Classification of Lipopeptides Based on SMILES Annotation. Computers 2021, 10, 74. https://doi.org/10.3390/computers10060074
Yadav M, Jujjavarapu SE. Neural Network Methodology for the Identification and Classification of Lipopeptides Based on SMILES Annotation. Computers. 2021; 10(6):74. https://doi.org/10.3390/computers10060074
Chicago/Turabian StyleYadav, Manisha, and Satya Eswari Jujjavarapu. 2021. "Neural Network Methodology for the Identification and Classification of Lipopeptides Based on SMILES Annotation" Computers 10, no. 6: 74. https://doi.org/10.3390/computers10060074
APA StyleYadav, M., & Jujjavarapu, S. E. (2021). Neural Network Methodology for the Identification and Classification of Lipopeptides Based on SMILES Annotation. Computers, 10(6), 74. https://doi.org/10.3390/computers10060074