
Integrins as Therapeutic Targets: Successes and Cancers


{kind=link}
Abstract
1. Introduction
1.1. Issues in Translating Integrin Preclinical Data to the Clinic
1.2. TGFβ Activation and Integrins: An Emerging Strategy?
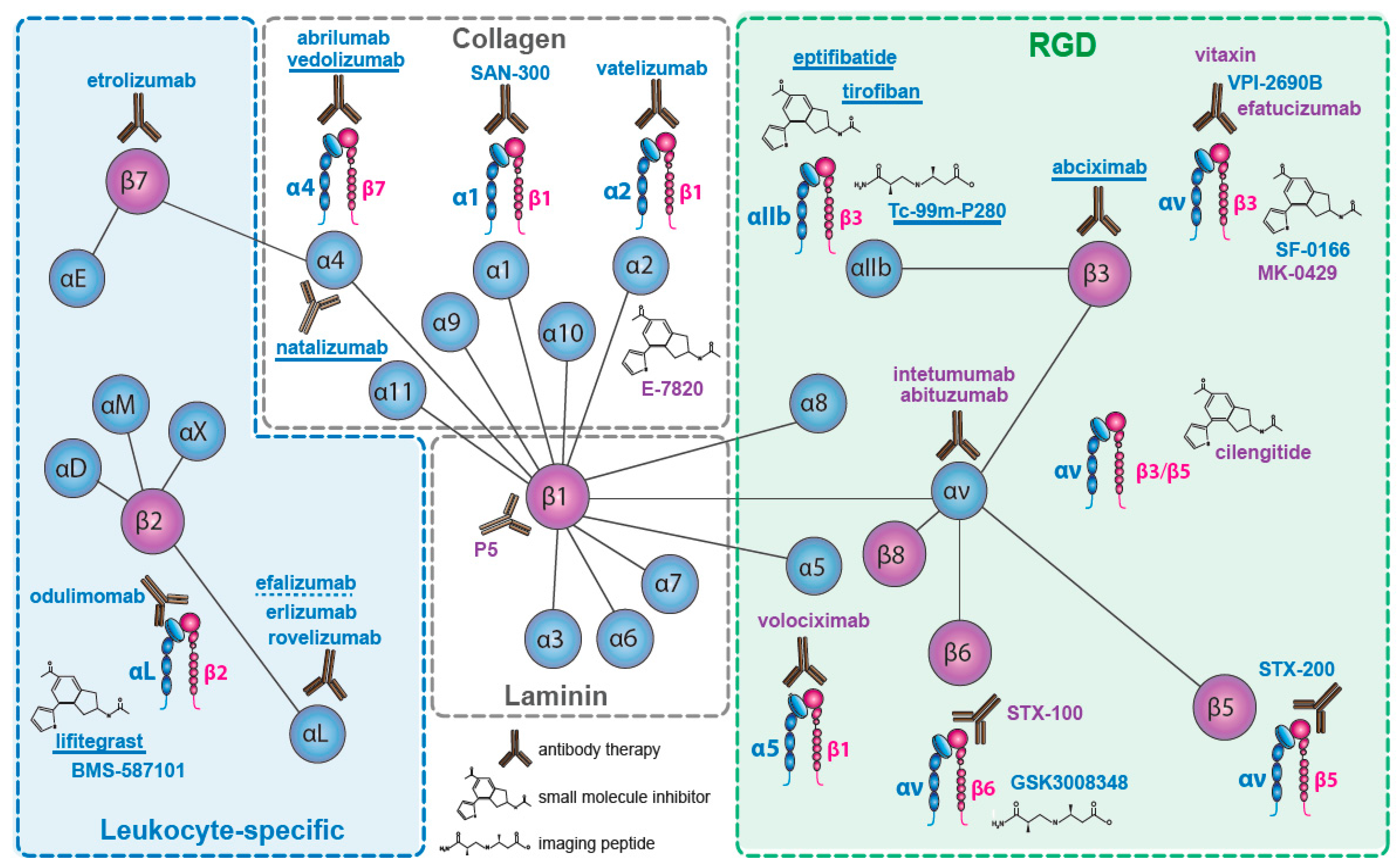
2. Integrins Targeted in Clinical Trials
2.1. The Collagen Receptors
2.2. The Fibronectin Receptors
2.3. The αv—Integrins
2.4. The Laminin Receptors
2.5. The Leukocyte Integrins
2.5.1. β2 Integrins
2.5.2. β7 Integrins
2.6. β3—Inhibition on Platelets
3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Goodman, S.L.; Xiong, J.P. Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 2007, 19, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Pietila, M.; Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 2016, 115, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J. The molecular basis and specificity of integrin-ligand interactions. J. Cell Sci. 1990, 97, 585–592. [Google Scholar] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Blandin, A.F.; Renner, G.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. Beta1 integrins as therapeutic targets to disrupt hallmarks of cancer. Front. Pharmacol. 2015, 6, 279. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, D.; Unseld, M.; Prager, G.W. Integrins in the spotlight of cancer. Int. J. Mol. Sci. 2016, 17, 2037. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ye, F.; Ginsberg, M.H. Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 2011, 27, 321–345. [Google Scholar] [CrossRef] [PubMed]
- Coller, B.S.; Shattil, S.J. The GPIIb/IIIa (integrin {alpha}iib{beta}3) odyssey: A technology-driven saga of a receptor with twists, turns, and even a bend. Blood 2008, 112, 3011–3025. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Ivaska, J. Endosomes: Emerging platforms for integrin-mediated fak signalling. Trends Cell. Biol. 2016, 26, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Van der Meel, R.; Vehmeijer, L.J.; Kok, R.J.; Storm, G.; van Gaal, E.V. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv. Drug Deliv. Rev. 2013, 65, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Currier, N.V.; Ackerman, S.E.; Kintzing, J.R.; Chen, R.; Filsinger Interrante, M.; Steiner, A.; Sato, A.K.; Cochran, J.R. Targeted drug delivery with an integrin-binding knottin-fc-mmaf conjugate produced by cell-free protein synthesis. Mol. Cancer Ther. 2016, 15, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Albelda, S.M.; Mette, S.A.; Elder, D.E.; Stewart, R.; Damjanovich, L.; Herlyn, M.; Buck, C.A. Integrin distribution in malignant melanoma: Association of the beta 3 subunit with tumor progression. Cancer Res. 1990, 50, 6757–6764. [Google Scholar] [PubMed]
- Gladson, C.L.; Cheresh, D.A. Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J. Clin. investig. 1991, 88, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, A.M.; Reisfeld, R.A.; Cheresh, D.A. Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc. Natl. Acad. Sci. USA 1994, 91, 8856–8860. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef]
- Brooks, P.C.; Stromblad, S.; Klemke, R.; Visscher, D.; Sarkar, F.H.; Cheresh, D.A. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J. Clin. investig. 1995, 96, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Brooks, P.C.; Shaffer, R.W.; Kincaid, C.M.; Varner, J.A.; Cheresh, D.A. Definition of two angiogenic pathways by distinct alpha v integrins. Science 1995, 270, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Gutheil, J.C.; Campbell, T.N.; Pierce, P.R.; Watkins, J.D.; Huse, W.D.; Bodkin, D.J.; Cheresh, D.A. Targeted antiangiogenic therapy for cancer using vitaxin: A humanized monoclonal antibody to the integrin alphavbeta3. Clin. Cancer Res. 2000, 6, 3056–3061. [Google Scholar] [PubMed]
- Delbaldo, C.; Raymond, E.; Vera, K.; Hammershaimb, L.; Kaucic, K.; Lozahic, S.; Marty, M.; Faivre, S. Phase i and pharmacokinetic study of etaracizumab (abegrin), a humanized monoclonal antibody against alphavbeta3 integrin receptor, in patients with advanced solid tumors. investig. New Drugs 2008, 26, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.E.; Wyder, L.; Lively, J.C.; Taverna, D.; Robinson, S.D.; Huang, X.; Sheppard, D.; Hynes, R.O.; Hodivala-Dilke, K.M. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 2002, 8, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Reynolds, L.E.; Nagel, T.E.; Lively, J.C.; Robinson, S.D.; Hicklin, D.J.; Bodary, S.C.; Hodivala-Dilke, K.M. Elevated flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 2004, 64, 8643–8650. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of rgd-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sasich, L.D.; Sukkari, S.R. The us fdas withdrawal of the breast cancer indication for avastin (bevacizumab). Saudi Pharm. J. 2012, 20, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Moreno, T.A.; Kim, S.J. Ranibizumab (lucentis) versus bevacizumab (avastin) for the treatment of age-related macular degeneration: An economic disparity of eye health. Semin. Ophthalmol. 2016, 31, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Duro-Castano, A.; Gallon, E.; Decker, C.; Vicent, M.J. Modulating angiogenesis with integrin-targeted nanomedicines. Adv. Drug Deliv. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J. Switching tgfbeta from a tumor suppressor to a tumor promoter. Curr. Opin. Genet. Dev. 2011, 21, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moses, H.L. Transforming growth factor beta: Tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. 2008, 68, 9107–9111. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of tgf-beta targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-beta-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent tgf beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through mt1-mmp-dependent activation of tgf-beta1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Puthawala, K.; Hadjiangelis, N.; Jacoby, S.C.; Bayongan, E.; Zhao, Z.; Yang, Z.; Devitt, M.L.; Horan, G.S.; Weinreb, P.H.; Lukashev, M.E.; et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Hahm, K.; Lukashev, M.E.; Luo, Y.; Yang, W.J.; Dolinski, B.M.; Weinreb, P.H.; Simon, K.J.; Chun Wang, L.; Leone, D.R.; Lobb, R.R.; et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in alport mouse. Am. J. Pathol. 2007, 170, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Lou, J.; Seed, R.I.; Cormier, A.; Wu, S.; Cheng, Y.; Murray, L.; Tsui, P.; Connor, J.; Herbst, R.; et al. Selective targeting of TGF-beta activation to treat fibroinflammatory airway disease. Sci. Transl. Med. 2014, 6, 241ra279. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra279. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lau, W.L.; Jo, H.; Tsujino, K.; Gewin, L.; Reed, N.I.; Atakilit, A.; Nunes, A.C.; DeGrado, W.F.; Sheppard, D. Pharmacologic blockade of alphavbeta1 integrin ameliorates renal failure and fibrosis in vivo. J. Am. Soc. Nephrol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Bellovin, D.I.; Brown, C.; Maynard, E.; Wu, B.; Kawakatsu, H.; Sheppard, D.; Oettgen, P.; Mercurio, A.M. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. investig. 2005, 115, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Elayadi, A.N.; Samli, K.N.; Prudkin, L.; Liu, Y.H.; Bian, A.; Xie, X.J.; Wistuba, I.I.; Roth, J.A.; McGuire, M.J.; Brown, K.C. A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007, 67, 5889–5895. [Google Scholar] [CrossRef] [PubMed]
- Hazelbag, S.; Kenter, G.G.; Gorter, A.; Dreef, E.J.; Koopman, L.A.; Violette, S.M.; Weinreb, P.H.; Fleuren, G.J. Overexpression of the alpha v beta 6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J. Pathol. 2007, 212, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Xu, K.S.; Wang, J.S.; Yang, G.Y.; Wang, W.; Wang, J.Y.; Niu, W.B.; Liu, E.Y.; Mi, Y.T.; Niu, J. Integrin alphanvbeta6 acts as a prognostic indicator in gastric carcinoma. Clin. Oncol. 2008, 20, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Lian, P.L.; Liu, Z.; Yang, G.Y.; Zhao, R.; Zhang, Z.Y.; Chen, Y.G.; Zhuang, Z.N.; Xu, K.S. Integrin alphavbeta6 and matrix metalloproteinase 9 correlate with survival in gastric cancer. World J. Gastroenterol. 2016, 22, 3852–3859. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.M.; Thomas, G.J.; Duffy, S.W.; Warwick, J.; Gabe, R.; Chou, P.; Ellis, I.O.; Green, A.R.; Haider, S.; Brouilette, K.; et al. Therapeutic targeting of integrin alphavbeta6 in breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Van Aarsen, L.A.; Leone, D.R.; Ho, S.; Dolinski, B.M.; McCoon, P.E.; LePage, D.J.; Kelly, R.; Heaney, G.; Rayhorn, P.; Reid, C.; et al. Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res. 2008, 68, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.; Kendrew, J.; McDaid, K.; Alfred, A.; Kang, J.S.; Jacobs, V.N.; Ross, S.J.; Rooney, C.; Smith, N.R.; Rinkenberger, J.; et al. A human monoclonal antibody 264rad targeting alphavbeta6 integrin reduces tumour growth and metastasis, and modulates key biomarkers in vivo. Oncogene 2013, 32, 4406–4416. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Ellison, D.; Thomas, G.J.; Vallath, S.; Mather, S.J.; Hart, I.R.; Marshall, J.F. High-resolution in vivo imaging of breast cancer by targeting the pro-invasive integrin alphavbeta6. J. Pathol. 2010, 222, 52–63. [Google Scholar] [PubMed]
- Elez, E.; Kocakova, I.; Hohler, T.; Martens, U.M.; Bokemeyer, C.; Van Cutsem, E.; Melichar, B.; Smakal, M.; Csoszi, T.; Topuzov, E.; et al. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with kras wild-type metastatic colorectal cancer: The randomised phase I/II poseidon trial. Ann. Oncol. 2015, 26, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Sipos, B.; Hahn, D.; Carceller, A.; Piulats, J.; Hedderich, J.; Kalthoff, H.; Goodman, S.L.; Kosmahl, M.; Kloppel, G. Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 2004, 45, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Deshpande, V.; Zimmerman, S.M.; Contino, G.; Alagesan, B.; O'Dell, M.R.; Rivera, L.B.; Harper, J.; Lonning, S.; Brekken, R.A.; et al. TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 2012, 72, 4840–4845. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Meckel, K.F.; Laird, L.S.; Chia, C.Y.; Park, J.J.; Olino, K.L.; Tsunedomi, R.; Harada, T.; Iizuka, N.; Hazama, S.; et al. Integrin alpha2 mediates selective metastasis to the liver. Cancer Res. 2009, 69, 7320–7328. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; et al. Loss of e-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008, 68, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Landowski, T.H.; Gard, J.; Pond, E.; Pond, G.D.; Nagle, R.B.; Geffre, C.P.; Cress, A.E. Targeting integrin alpha6 stimulates curative-type bone metastasis lesions in a xenograft model. Mol. Cancer Ther. 2014, 13, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Sheldrake, H.M.; Patterson, L.H. Strategies to inhibit tumor associated integrin receptors: Rationale for dual and multi-antagonists. J. Med. Chem. 2014, 57, 6301–6315. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Layani-Bazar, A.; Skornick, I.; Berrebi, A.; Pauker, M.H.; Noy, E.; Silberman, A.; Albeck, M.; Longo, D.L.; Kalechman, Y.; Sredni, B. Redox modulation of adjacent thiols in vla-4 by as101 converts myeloid leukemia cells from a drug-resistant to drug-sensitive state. Cancer Res. 2014, 74, 3092–3103. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Qu, X.J.; Gao, Z.H. Arginine-glycine-aspartate-binding integrins as therapeutic and diagnostic targets. Am. J. Ther. 2016, 23, e198–e207. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, F.; Liu, S. Radiolabeled cyclic rgd peptides as radiotracers for tumor imaging. Biophys. Rep. 2016, 2, 1–20. [Google Scholar] [CrossRef] [PubMed]
- White, D.J.; Puranen, S.; Johnson, M.S.; Heino, J. The collagen receptor subfamily of the integrins. Int. J. Biochem. Cell Biol. 2004, 36, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz, C.; Weinreb, P.H.; Calvete, J.J.; Kisiel, D.G.; Mousa, S.A.; Tuszynski, G.P.; Lobb, R.R. Obtustatin: A potent selective inhibitor of alpha1beta1 integrin in vitro and angiogenesis in vivo. Cancer Res. 2003, 63, 2020–2023. [Google Scholar] [PubMed]
- The New Valeant: Delivering on Our Commitments, Proceedings of the 35th Annual JP Morgan Healthcare Conference, San Francisco, CA, USA, 9–12 January 2017; Valeant Pharmaceuticals: West Laval, QC, Canada, 2017.
- Mattila, E.; Pellinen, T.; Nevo, J.; Vuoriluoto, K.; Arjonen, A.; Ivaska, J. Negative regulation of EGFR signalling through integrin-alpha1beta1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 2005, 7, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Alberio, L.; Dale, G.L. Review article: Platelet-collagen interactions: Membrane receptors and intracellular signalling pathways. Eur. J. Clin. investig. 1999, 29, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Vankooyk, Y.; Kemenade, P.V.; Weder, P.; Kuijpers, T.W.; Figdor, C.G. Enhancement of lfa-1-mediated cell adhesion by triggering through cd2 or cd3 on lymphocytes-t. Nature 1989, 342, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Vanhoorelbeke, K.; Ulrichts, H.; Schoolmeester, A.; Deckmyn, H. Inhibition of platelet adhesion to collagen as a new target for antithrombotic drugs. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2003, 3, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Gendron, S.; Couture, J.; Aoudjit, F. Integrin alpha2beta1 inhibits fas-mediated apoptosis in t lymphocytes by protein phosphatase 2a-dependent activation of the mapk/erk pathway. J. Biol. Chem. 2003, 278, 48633–48643. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, L.; Ojala, M.; Langen, B.; Dost, R.; Pihlavisto, M.; Kapyla, J.; Marjamaki, A.; Heino, J. Sulfonamide inhibitors of alpha2beta1 integrin reveal the essential role of collagen receptors in in vivo models of inflammation. Pharmacol. Res. Perspect. 2015, 3, e00146. [Google Scholar] [CrossRef] [PubMed]
- Naci, D.; Vuori, K.; Aoudjit, F. Alpha2beta1 integrin in cancer development and chemoresistance. Semin. Cancer Biol. 2015, 35, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Sugi, N.H.; Semba, T.; Yamamoto, Y.; Hamaoka, S.; Tsukahara-Tamai, N.; Ozawa, Y.; Tsuruoka, A.; Nara, K.; Takahashi, K.; et al. Sulfonamide derivative, e7820, is a unique angiogenesis inhibitor suppressing an expression of integrin α2 subunit on endothelium. Cancer Res. 2002, 62, 6116–6123. [Google Scholar] [PubMed]
- Milojkovic, K.B.; Slater, S.; Flynn, M.; Greystoke, A.; Witteveen, P.O.; Megui-Roelvink, M.; de Vos, F.; Dean, E.; Reyderman, L.; Ottesen, L.; et al. A phase i, dose escalation, pharmacodynamic, pharmacokinetic, and food-effect study of alpha2 integrin inhibitor e7820 in patients with advanced solid tumors. investig. New Drugs 2016, 34, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell. Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Mould, A.P.; Askari, J.A.; Craig, S.E.; Garratt, A.N.; Clements, J.; Humphries, M.J. Integrin alpha 4 beta 1-mediated melanoma cell adhesion and migration on vascular cell adhesion molecule-1 (vcam-1) and the alternatively spliced iiics region of fibronectin. J. Biol. Chem. 1994, 269, 27224–27230. [Google Scholar] [PubMed]
- Clements, J.M.; Newham, P.; Shepherd, M.; Gilbert, R.; Dudgeon, T.J.; Needham, L.A.; Edwards, R.M.; Berry, L.; Brass, A.; Humphries, M.J. Identification of a key integrin-binding sequence in vcam-1 homologous to the ldv active site in fibronectin. J. Cell Sci. 1994, 107, 2127–2135. [Google Scholar] [PubMed]
- Hamann, A.; Andrew, D.P.; Jablonski-Westrich, D.; Holzmann, B.; Butcher, E.C. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J. Immunol. 1994, 152, 3282–3293. [Google Scholar] [PubMed]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin madcam-1. Cell 1993, 74, 185. [Google Scholar] [CrossRef]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of functional suppression by cd4+cd25+ regulatory t cells in patients with multiple sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Elices, M.J.; Osborn, L.; Takada, Y.; Crouse, C.; Luhowskyj, S.; Hemler, M.E.; Lobb, R.R. Vcam-1 on activated endothelium interacts with the leukocyte integrin vla-4 at a site distinct from the vla-4/fibronectin binding site. Cell 1990, 60, 577–584. [Google Scholar] [CrossRef]
- Makker, J.; Hommes, D.W. Etrolizumab for ulcerative colitis: The new kid on the block? Expert. Opin. Biol. Ther. 2016, 16, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Natalizumab and pml. Nat. Neurosci. 2005, 8, 1275. [Google Scholar] [CrossRef] [PubMed]
- Singer, E. Tysabri withdrawal calls entire class into question. Nat. Med. 2005, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Ateeq, H.S.; Hsiung, S.H.; Chong, L.T.; Zimmerman, C.N.; Castro, A.; Lee, W.C.; Hammond, C.E.; Kalkunte, S.; Chen, L.L.; et al. Selective, tight-binding inhibitors of integrin alpha4beta1 that inhibit allergic airway responses. J. Med. Chem. 1999, 42, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Leger, O.J.; Yednock, T.A.; Tanner, L.; Horner, H.C.; Hines, D.K.; Keen, S.; Saldanha, J.; Jones, S.T.; Fritz, L.C.; Bendig, M.M. Humanization of a mouse antibody against human alpha-4 integrin: A potential therapeutic for the treatment of multiple sclerosis. Hum. Antibodies 1997, 8, 3–16. [Google Scholar] [PubMed]
- Kent, S.J.; Karlik, S.J.; Cannon, C.; Hines, D.K.; Yednock, T.A.; Fritz, L.C.; Horner, H.C. A monoclonal antibody to [alpha]4 integrin suppresses and reverses active experimental allergic encephalomyelitis. J. Neuroimmunol. 1995, 58, 1–10. [Google Scholar] [CrossRef]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integri. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Schurpf, T.; Springer, T.A. How natalizumab binds and antagonizes alpha4 integrins. J. Biol. Chem. 2013, 288, 32314–32325. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.A.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Eng. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O'Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Eng. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005, 353, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Tyler, K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 2005, 353, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, I.; Rodriguez de, A.L.; Orviz, A.; Moreno-Garcia, S.; Valle-Arcos, M.D.; Matias-Guiu, J.A.; Valencia, C.; Jorquera, M.M.; Oreja-Guevara, C. Catastrophic outcome of patients with a rebound after natalizumab treatment discontinuation. Brain Behav. 2017, 7, e00671. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.; Veltkamp, R.; Montaner, J.; Johnston, S.C.; Singhal, A.B.; Becker, K.; Lansberg, M.G.; Tang, W.; Chang, I.; Muralidharan, K.; et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (action): A randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2017, 16, 217–226. [Google Scholar] [CrossRef]
- Macdonald, J.K.; McDonald, J.W. Natalizumab for induction of remission in crohn's disease. Cochrane Database Syst. Rev. 2006, CD006097. [Google Scholar]
- Huggett, B. How tysabri survived. Nat. Biotechnol. 2009, 27, 986. [Google Scholar] [CrossRef] [PubMed]
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A phase ii, single-arm study of the anti-alpha5beta1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Almokadem, S.; Belani, C.P. Volociximab in cancer. Expert. Opin. Biol. Ther. 2012, 12, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.; Bachelot, T.; Hudis, C.A.; Curigliano, G.; Reynolds, A.R.; Petrioli, R.; Generali, D. The role of bevacizumab in solid tumours: A literature based meta-analysis of randomised trials. Eur. J. Cancer 2017, 75, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Cho, W.D.; Hong, K.P.; Choi da, B.; Hong, J.W.; Kim, S.; Moon, Y.R.; Son, S.M.; Lee, O.J.; Lee, H.C.; et al. Novel monoclonal antibody against beta 1 integrin enhances cisplatin efficacy in human lung adenocarcinoma cells. J. Biomed. Res. 2016, 30, 217–224. [Google Scholar] [PubMed]
- Cirkel, G.A.; Kerklaan, B.M.; Vanhoutte, F.; Van der Aa, A.; Lorenzon, G.; Namour, F.; Pujuguet, P.; Darquenne, S.; de Vos, F.Y.; Snijders, T.J.; et al. A dose escalating phase i study of glpg0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. investig. New Drugs 2016, 34, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Amend, S.R.; Weilbaecher, K.N. Integrins and bone metastasis: Integrating tumor cell and stromal cell interactions. Bone 2011, 48, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Vogetseder, A.; Thies, S.; Ingold, B.; Roth, P.; Weller, M.; Schraml, P.; Goodman, S.L.; Moch, H. Alphav-integrin isoform expression in primary human tumors and brain metastases. Int. J. Cancer 2013, 133, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Le, M.S.; Gimmi, C.; Bruns, R.; Straub, J.; Miller, K. Differential effect on bone lesions of targeting integrins: Randomized phase ii trial of abituzumab in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Mitjans, F.; Sander, D.; Adan, J.; Sutter, A.; Martinez, J.M.; Jaggle, C.S.; Moyano, J.M.; Kreysch, H.G.; Piulats, J.; Goodman, S.L. An anti-alpha v-integrin antibody that blocks integrin function inhibits the development of a human melanoma in nude mice. J. Cell Sci. 1995, 108, 2825–2838. [Google Scholar] [PubMed]
- O'Day, S.; Pavlick, A.; Loquai, C.; Lawson, D.; Gutzmer, R.; Richards, J.; Schadendorf, D.; Thompson, J.A.; Gonzalez, R.; Trefzer, U.; et al. A randomised, phase ii study of intetumumab, an anti-alphav-integrin mab, alone and with dacarbazine in stage IV melanoma. Br. J. Cancer 2011, 105, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, B.; Van Agthoven, J.F.; Xiong, J.P.; Alonso, J.L.; Adair, B.D.; Rui, X.; Anand, S.; Mehrbod, M.; Mofrad, M.R.; Burger, C.; et al. Atomic basis for the species-specific inhibition of alphav integrins by mab 17e6 is revealed by the crystal structure of alphavbeta3 ectodomain-17e6 fab complex. J. Biol. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mitjans, F.; Meyer, T.; Fittschen, C.; Goodman, S.; Jonczyk, A.; Marshall, J.F.; Reyes, G.; Piulats, J. In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int. J. Cancer 2000, 87, 716–723. [Google Scholar] [CrossRef]
- Chen, Q.; Manning, C.D.; Millar, H.; McCabe, F.L.; Ferrante, C.; Sharp, C.; Shahied-Arruda, L.; Doshi, P.; Nakada, M.T.; Anderson, G.M. Cnto 95, a fully human anti alphav integrin antibody, inhibits cell signaling, migration, invasion, and spontaneous metastasis of human breast cancer cells. Clin. Exp. Metastasis 2008, 25, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Trikha, M.; Zhou, Z.; Nemeth, J.A.; Chen, Q.; Sharp, C.; Emmell, E.; Giles-Komar, J.; Nakada, M.T. Cnto 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int. J. Cancer 2004, 110, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Heidenreich, A.; Gschwend, J.E.; Gil, T.; Zastrow, S.; Laniado, M.; Gerloff, J.; Zuhlsdorf, M.; Mordenti, G.; Uhl, W.; et al. A multicenter phase 1 study of emd 525797 (di17e6), a novel humanized monoclonal antibody targeting alphav integrins, in progressive castration-resistant prostate cancer with bone metastases after chemotherapy. Eur. Urol. 2014, 65, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Grote, H.J.; Wilm, C. Matched rabbit monoclonal antibodies against alphav-series integrins reveal a novel alphavbeta3-libs epitope, and permit routine staining of archival paraffin samples of human tumors. Biol. Open 2012, 1, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Agrez, M.V.; Bates, R.C.; Mitchell, D.; Wilson, N.; Ferguson, N.; Anseline, P.; Sheppard, D. Multiplicity of fibronectin-binding alpha v integrin receptors in colorectal cancer. Br. J. Cancer 1996, 73, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-tgf-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.Y.; Morris, S.M.; Campbell, J.; Fausto, N.; Yeh, M.M.; Grady, W.M. Tgf-beta inactivation and tgf-alpha overexpression cooperate in an in vivo mouse model to induce hepatocellular carcinoma that recapitulates molecular features of human liver cancer. Int. J. Cancer 2010, 127, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Scar wars: Is tgfbeta the phantom menace in scleroderma? Arthritis Res. Ther. 2006, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, T.R.; Violette, S.M.; Sheppard, D. Blocking tgfbeta via inhibition of the alphavbeta6 integrin: A possible therapy for systemic sclerosis interstitial lung disease. Int. J. Rheumatol. 2011, 2011, 208219. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Rawal, S.K.; Szkarlat, K.; Bogdanova, N.; Dirix, L.; Stenzl, A.; Welslau, M.; Wang, G.; Dawkins, F.; de Boer, C.J.; et al. A randomized, double-blind, multicenter, phase 2 study of a human monoclonal antibody to human alphanu integrins (intetumumab) in combination with docetaxel and prednisone for the first-line treatment of patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2013, 24, 329–336. [Google Scholar] [PubMed]
- Hersey, P.; Sosman, J.; O'Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or − dacarbazine in patients with stage iv metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.A.; Davidson, P.; Rolland, F.; Campone, M.; Xue, L.; Han, T.H.; Mehta, A.; Berd, Y.; He, W.; Lombardi, A. Evaluation of the safety, pharmacokinetics and treatment effects of an alpha(v)beta(3) integrin inhibitor on bone turnover and disease activity in men with hormone-refractory prostate cancer and bone metastases. Asia Pac. J. Clin. Oncol. 2010, 6, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Nandrot, E.F.; Chang, Y.; Finnemann, S.C. Alphavbeta5 integrin receptors at the apical surface of the rpe: One receptor, two functions. Adv. Exp. Med. Biol. 2008, 613, 369–375. [Google Scholar] [PubMed]
- Chiu, C.Y.; Mathias, P.; Nemerow, G.R.; Stewart, P.L. Structure of adenovirus complexed with its internalization receptor, alphavbeta5 integrin. J. Virol. 1999, 73, 6759–6768. [Google Scholar] [PubMed]
- Dechantsreiter, M.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-methylated cyclic rgd peptides as highly active and selective αvβ3 integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Bretschi, M.; Cheng, C.; Witt, H.; Dimitrakopoulou-Strauss, A.; Strauss, L.G.; Semmler, W.; Bauerle, T. Cilengitide affects tumor compartment, vascularization and microenvironment in experimental bone metastases as shown by longitudinal (18)f-fdg pet and gene expression analysis. J. Cancer Res. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Bauerle, T.; Komljenovic, D.; Merz, M.; Berger, M.R.; Goodman, S.L.; Semmler, W. Cilengitide inhibits progression of experimental breast cancer bone metastases as imaged noninvasively using vct, mri and dce-mri in a longitudinal in vivo study. Int. J. Cancer 2011, 128, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.; Brodie, C.; Finniss, S.; Berens, M.E.; Rennert, J.L.; Nelson, K.; Lemke, N.; Brown, S.L.; Hahn, D.; Neuteboom, B.; et al. Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency. Int. J. Cancer 2009, 124, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Prieto, V.; Joseph, R.W.; Diwan, A.H.; Gallick, G.E.; Papadopoulos, N.E.; Bedikian, A.Y.; Camacho, L.H.; Hwu, P.; Ng, C.S.; et al. A randomized phase ii study of cilengitide (emd 121974) in patients with metastatic melanoma. Melanoma Res. 2012, 22, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Langrehr, J.M.; Oettle, H.; Raedle, J.; Niedergethmann, M.; Dittrich, C.; Hossfeld, D.K.; Stoger, H.; Neyns, B.; Herzog, P.; et al. A randomized multi-center phase ii trial of the angiogenesis inhibitor cilengitide (emd 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer 2006, 6, 285. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.Y.; et al. Phase i/iia study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Nabors, L.B.; Gorlia, T.; Leske, H.; Rushing, E.; Bady, P.; Hicking, C.; Perry, J.; Hong, Y.K.; Roth, P.; et al. Cilengitide in newly diagnosed glioblastoma: Biomarker expression and outcome. Oncotarget 2016, 7, 15018–15032. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated mgmt promoter (centric eortc 26071–22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated mgmt gene promoter: Results of the open-label, controlled, randomized phase ii core study. Neuro Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.P. End of the road: Confounding results of the core trial terminate the arduous journey of cilengitide for glioblastoma. Neuro Oncol. 2015, 17, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: Results of an open-label, randomized, controlled phase ii study (certo). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Lee, A.; McCowatt, S.; Kerestes, Z.; Buyse, M.E.; Back, M.; Kichenadasse, G.; Ackland, S.; Wheeler, H. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated mgmt gene promoter in excentric, an open-label phase ii trial. J. Neurooncol. 2016, 128, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Ye, X.; Duda, D.G.; Levine, M.A.; Mikkelsen, T.; Kaley, T.J.; Olson, J.J.; Nabors, B.L.; Ahluwalia, M.S.; Wen, P.Y.; et al. A phase i study of cediranib in combination with cilengitide in patients with recurrent glioblastoma. Neuro Oncol. 2015, 17, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Haddad, T.; Qin, R.; Lupu, R.; Satele, D.; Eadens, M.; Goetz, M.P.; Erlichman, C.; Molina, J. A phase i study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Marosi, C.; Preusser, M. Milestones of the last 10 years: CNS cancer. Memo 2017, 10, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Eskens, F.A.; Dumez, H.; Hoekstra, R.; Perschl, A.; Brindley, C.; Bottcher, S.; Wynendaele, W.; Drevs, J.; Verweij, J.; van Oosterom, A.T. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of cilengitide (emd 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur. J. Cancer 2003, 39, 917–926. [Google Scholar] [CrossRef]
- O'Donnell, P.H.; Undevia, S.D.; Stadler, W.M.; Karrison, T.M.; Nicholas, M.K.; Janisch, L.; Ratain, M.J. A phase i study of continuous infusion cilengitide in patients with solid tumors. investig. New Drugs 2012, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- ten Hagen, T.L.; Seynhaeve, A.L.; de Wiel-Ambagtsheer, G.A.; de Bruijn, E.A.; van Tiel, S.T.; Ruegg, C.; Meyring, M.; Grell, M.; Goodman, S.L.; Eggermont, A.M. The alphavbeta3/alphavbeta5 integrin inhibitor cilengitide augments tumor response to melphalan isolated limb perfusion in a sarcoma model. Int. J. Cancer 2013, 132, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Hodivala-Dilke, K. Alphavbeta3 integrin and tumour blood vessels-learning from the past to shape the future. Curr. Opin. Cell Biol. 2016, 42, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Nisato, R.E.; Tille, J.C.; Jonczyk, A.; Goodman, S.L.; Pepper, M.S. Alphav beta 3 and alphav beta 5 integrin antagonists inhibit angiogenesis in vitro. Angiogenesis 2003, 6, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, J.; Schwab, E.I.; Sperveslage, J.; Tatagiba, M.; Meyermann, R.; Fend, F.; Goodman, S.L.; Sipos, B. Longitudinal expression analysis of alphav integrins in human gliomas reveals upregulation of integrin alphavbeta3 as a negative prognostic factor. J. Neuropathol. Exp. Neurol. 2013, 72, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Ducassou, A.; Uro-Coste, E.; Verrelle, P.; Filleron, T.; Benouaich-Amiel, A.; Lubrano, V.; Sol, J.C.; Delisle, M.B.; Favre, G.; Ken, S.; et al. Alphavbeta3 integrin and fibroblast growth factor receptor 1 (fgfr1): Prognostic factors in a phase I–II clinical trial associating continuous administration of tipifarnib with radiotherapy for patients with newly diagnosed glioblastoma. Eur. J. Cancer 2013, 49, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Lo, D.J.; Farris, A.B.; Song, M.; Leopardi, F.; Anderson, D.J.; Strobert, E.A.; Ramakrishnan, S.; Turgeon, N.A.; Mehta, A.K.; Turnbull, B.; et al. Inhibition of alphavbeta6 promotes acute renal allograft rejection in nonhuman primates. Am. J. Transplant. 2013, 13, 3085–3093. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.M.; Kelly, A.; Shuttleworth, E.E.; Smedley, C.; Atakilit, A.; Powrie, F.; Campbell, S.; Nishimura, S.L.; Sheppard, D.; Levison, S.; et al. Inflammatory cues enhance tgfbeta activation by distinct subsets of human intestinal dendritic cells via integrin alphavbeta8. Mucosal Immunol. 2017, 10, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.L.; Boylen, K.P.; Einheber, S.; Milner, T.A.; Ramos, D.M.; Pytela, R. Synaptic and glial localization of the integrin alphavbeta8 in mouse and rat brain. Brain Res. 1998, 791, 271–282. [Google Scholar] [CrossRef]
- Ramovs, V.; Te Molder, L.; Sonnenberg, A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017, 57–58, 213–243. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sekiguchi, K. Molecular basis of laminin-integrin interactions. Curr. Top. Membr. 2015, 76, 197–229. [Google Scholar] [PubMed]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.J.; Vestweber, D.; Cabanas, C.; Lu, C.; Springer, T.A. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017, 36, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A. Biology and structure of leukocyte beta 2 integrins and their role in inflammation. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Mahalingam, B.; Xiong, J.P. Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell. Dev. Biol. 2005, 21, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S. Structural basis of transmembrane domain interactions in integrin signaling. Cell Adh. Migr. 2010, 4, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Laudanna, C.; Kim, J.Y.; Constantin, G.; Butcher, E. Rapid leukocyte integrin activation by chemokines. Immunol. Rev. 2002, 186, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Xiao, T.; Liu, J.H.; Yang, Y.; Dong, Y.; Jun, C.D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the alpha l i domain and its complex with icam-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef]
- Fan, Z.; Ley, K. Leukocyte arrest: Biomechanics and molecular mechanisms of beta2 integrin activation. Biorheology 2015, 52, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Talamonti, M.; Spallone, G.; Di, S.A.; Costanzo, A.; Chimenti, S. Efalizumab. Expert. Opin. Drug Saf. 2011, 10, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.L.; Pflugfelder, S.C.; Zhang, S.; Shojaei, A.; Haque, R. Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul. Surf. 2016, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Gadek, T.R.; Bui, M.; Shen, W.; Burnier, J.; Barr, K.J.; Hanan, E.J.; Oslob, J.D.; Yu, C.H.; Zhu, J.; et al. Discovery and development of potent lfa-1/icam-1 antagonist sar 1118 as an ophthalmic solution for treating dry eye. ACS Med. Chem. Lett. 2012, 3, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Tauber, J.; Karpecki, P.; Latkany, R.; Luchs, J.; Martel, J.; Sall, K.; Raychaudhuri, A.; Smith, V.; Semba, C.P. Lifitegrast ophthalmic solution 5.0% versus placebo for treatment of dry eye disease results of the randomized phase iii opus-2 study. Ophthalmology 2015, 122, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Seminara, N.M.; Gelfand, J.M. Assessing long-term drug safety: Lessons (re) learned from raptiva. Semin. Cutan. Med. Surg. 2010, 29, 16–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frampton, J.E.; Plosker, G.L. Efalizumab: A review of its use in the management of chronic moderate-to-severe plaque psoriasis. Am. J. Clin. Dermatol. 2009, 10, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Zimmerer, J.M.; Horne, P.H.; Fiessinger, L.A.; Fisher, M.G.; Jayashankar, K.; Garcia, S.F.; Abdel-Rasoul, M.; van, R.N.; Bumgardner, G.L. Inhibition of recall responses through complementary therapies targeting cd8+ t-cell- and alloantibody-dependent allocytotoxicity in sensitized transplant recipients. Cell Transplant. 2013, 22, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Tredget, E.B.; Arefanian, H.; Gill, R.G.; Rajotte, R.V.; Rayat, G.R. Monotherapy with anti-lfa-1 monoclonal antibody promotes long-term survival of rat islet xenografts. Cell Transplant. 2008, 17, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Jones, R. Rovelizumab (icos corp). IDrugs 2000, 3, 442–446. [Google Scholar] [PubMed]
- Qualls, J.E.; Murray, P.J. A double agent in cancer: Stopping macrophages wounds tumors. Nat. Med. 2010, 16, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Suchard, S.J.; Stetsko, D.K.; Davis, P.M.; Skala, S.; Potin, D.; Launay, M.; Dhar, T.G.; Barrish, J.C.; Susulic, V.; Shuster, D.J.; et al. An lfa-1 (alphalbeta2) small-molecule antagonist reduces inflammation and joint destruction in murine models of arthritis. J. Immunol. 2010, 184, 3917–3926. [Google Scholar] [CrossRef] [PubMed]
- Dehnadi, A.; Benedict, C.A.; Neal, S.R.; Li, X.; Alonso, J.L.; Means, T.K.; Arnaout, M.A. Prophylactic orthosteric inhibition of leukocyte integrin cd11b/cd18 prevents long-term fibrotic kidney failure in cynomolgus monkeys. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shimaoka, M.; Salas, A.; Springer, T.A. The binding sites for competitive antagonistic, allosteric antagonistic, and agonistic antibodies to the i domain of integrin lfa-1. J. Immunol. 2004, 173, 3972–3978. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, B.; Ajroud, K.; Alonso, J.L.; Anand, S.; Adair, B.D.; Horenstein, A.L.; Malavasi, F.; Xiong, J.P.; Arnaout, M.A. Stable coordination of the inhibitory ca2+ ion at the metal ion-dependent adhesion site in integrin cd11b/cd18 by an antibody-derived ligand aspartate: Implications for integrin regulation and structure-based drug design. J. Immunol. 2011, 187, 6393–6401. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.; Kornbluth, A. Vedolizumab as a treatment for crohn's disease and ulcerative colitis. Gastroenterol. Hepatol. 2014, 10, 793–800. [Google Scholar]
- Wang, M.C.; Zhang, L.Y.; Han, W.; Shao, Y.; Chen, M.; Ni, R.; Wang, G.N.; Wei, F.X.; Zhang, Y.W.; Xu, X.D.; et al. Prisma--efficacy and safety of vedolizumab for inflammatory bowel diseases: A systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore) 2014, 93, e326. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Lohler, J.; Kunkel, E.J.; Ley, K.; Leung, E.; Krissansen, G.; Rajewsky, K.; Muller, W. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature 1996, 382, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Cyrille, M.; Hansen, M.B.; Feagan, B.G.; Loftus, E.V.; Vermeire, S.; Cruz, M.L.; Mo, M.; Sullivan, B.A.; Reinisch, W. Efficacy and safety of abrilumab (amg 181/medi 7183) therapy for moderate to severe crohn’s disease. Gastroenterology 2017, 152, S598. [Google Scholar]
- Luthra, P.; Peyrin-Biroulet, L.; Ford, A.C. Systematic review and meta-analysis: Opportunistic infections and malignancies during treatment with anti-integrin antibodies in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2015, 41, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Coller, B.S. A new murine monoclonal antibody reports an activation-dependent change in the conformation and/or microenvironment of the platelet glycoprotein iib/iiia complex. J. Clin. investig. 1985, 76, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Stehle, T.; Diefenbach, B.; Zhang, R.; Dunker, R.; Scott, D.L.; Joachimiak, A.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin alpha vbeta3. Science 2001, 294, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin alpha vbeta3 in complex with an arg-gly-asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Goodman, S.L.; Xiong, J.P. Coming to grips with integrin binding to ligands. Curr. Opin. Cell Biol. 2002, 14, 641–651. [Google Scholar] [CrossRef]
- Xiao, T.; Takagi, J.; Coller, B.S.; Wang, J.H.; Springer, T.A. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 2004, 432, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.; Hamad, B.; Syed, B.A. Antithrombotic drugs market. Nat. Rev. Drug Discov. 2014, 13, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Coller, B.S. Blockade of platelet gpiib/iiia receptors as an antithrombotic strategy. Circulation 1995, 92, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.H.; Du, X.; Plow, E.F. Inside-out integrin signalling. Curr. Opin. Cell Biol. 1992, 4, 766–771. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Artoni, A.; Li, J.; Mitchell, B.; Ruan, J.; Takagi, J.; Springer, T.A.; French, D.L.; Coller, B.S. Integrin beta3 regions controlling binding of murine MAB 7e3: Implications for the mechanism of integrin alphaiibbeta3 activation. Proc. Natl. Acad. Sci. USA 2004, 101, 13114–13120. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Springer, T.A. Therapeutic antagonists and conformational regulation of integrin function. Nat. Rev. Drug Discov. 2003, 2, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.A.; Meila, D.; Jacobs, C.; Weber, S.; Kelm, M.; Strauer, B.E.; Zotz, R.B.; Scharf, R.E.; Schrör, K. Low incidence of paradoxical platelet activation by glycoprotein iib/iiia inhibitors. Thromb. Res. 2002, 106, 25–29. [Google Scholar] [CrossRef]
- Bougie, D.W.; Wilker, P.R.; Wuitschick, E.D.; Curtis, B.R.; Malik, M.; Levine, S.; Lind, R.N.; Pereira, J.; Aster, R.H. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied gpiib/iiia. Blood 2002, 100, 2071. [Google Scholar] [PubMed]
- Schrör, K.; Weber, A.A. Comparative pharmacology of gp iib/iiia antagonists. J. Thromb. Thrombolysis 2003, 15, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Puzon-McLaughlin, W.; Kamata, T.; Takada, Y. Multiple discontinuous ligand-mimetic antibody binding sites define a ligand binding pocket in integrin alpha(iib)beta(3). J. Biol. Chem. 2000, 275, 7795–7802. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Shen, B.; Du, X. Targeting integrin and integrin signaling in treating thrombosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010, 9, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.J.; Byzova, T.V.; Qin, J.; Topol, E.J.; Plow, E.F. Integrin alphaiibbeta3 and its antagonism. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Peerlinck, K.; De Lepeleire, I.; Goldberg, M.; Farrell, D.; Barrett, J.; Hand, E.; Panebianco, D.; Deckmyn, H.; Vermylen, J.; Arnout, J. Mk-383 (l-700,462), a selective nonpeptide platelet glycoprotein IIb/IIIa antagonist, is active in man. Circulation 1993, 88, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Adgey, A.A. An overview of the results of clinical trials with glycoprotein iib/iiia inhibitors. Am. Heart J. 1998, 135, S43–S55. [Google Scholar] [CrossRef]
- Phillips, D.R.; Scarborough, R.M. Clinical pharmacology of eptifibatide. Am. J. Cardiol. 1997, 80, 11B–20B. [Google Scholar] [CrossRef]
- King, A. Eptifibatide is noninferior to abciximab: Implications for clinical practice. Nat. Rev. Cardiol. 2010, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Lele, M.; Sajid, M.; Wajih, N.; Stouffer, G.A. Eptifibatide and 7e3, but not tirofiban, inhibit alpha(v)beta(3) integrin-mediated binding of smooth muscle cells to thrombospondin and prothrombin. Circulation 2001, 104, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Brown, C. Medical isotope supply raises concerns. CMAJ 2016, 188, 181. [Google Scholar] [CrossRef] [PubMed]
- Casserly, I.P.; Topol, E.J. Glycoprotein iib/iiia antagonists—From bench to practice. Cell. Mol. Life Sci. CMLS 2002, 59, 478–500. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Hoxie, J.A.; Cunningham, M.; Brass, L.F. Changes in the platelet membrane glycoprotein iib.Iiia complex during platelet activation. J. Biol. Chem. 1985, 260, 11107–11114. [Google Scholar] [PubMed]
- Sheppard, D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005, 24, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Bader, B.L.; Rayburn, H.; Crowley, D.; Hynes, R.O. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 1998, 95, 507–519. [Google Scholar] [CrossRef]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R.; Ernberg, I. Assessment of tumor characteristic gene expression in cell lines using a tissue similarity index (tsi). Proc. Natl. Acad. Sci. USA 2005, 102, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R.; Ernberg, I. The molecular portrait of in vitro growth by meta-analysis of gene-expression profiles. Genome Biol. 2005, 6, R65. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Auer, J.; Brinkmann, U.; Georges, G.; Tiefenthaler, G. The emerging role of new protein scaffold-based agents for treatment of cancer. Cancer Genomics Proteomics 2013, 10, 155–168. [Google Scholar] [PubMed]
- Tartari, F.; Santoni, M.; Burattini, L.; Mazzanti, P.; Onofri, A.; Berardi, R. Economic sustainability of anti-pd-1 agents nivolumab and pembrolizumab in cancer patients: Recent insights and future challenges. Cancer Treat. Rev. 2016, 48, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Mailankody, S.; Prasad, V. Five years of cancer drug approvals: Innovation, efficacy, and costs. JAMA Oncol. 2015, 1, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve r&d productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Health Economics: The Cancer Drugs Cost Conundrum. Available online: http://www.cancerresearchuk.org/funding-for-researchers/research-features/2016-08-10-health-economics-the-cancer-drugs-cost-conundrum (accessed on 10 August 2016).
- Sarpatwari, A.; Avorn, J.; Kesselheim, A.S. State initiatives to control medication costs—Can transparency legislation help? N. Engl. J. Med. 2016, 374, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Common Cancer Types. Available online: https://www.cancer.gov/types/common-cancers (accessed on 13 February 2017).
- Prasad, V.; De Jesus, K.; Mailankody, S. The high price of anticancer drugs: Origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 2017, 14, 381–390. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as Therapeutic Targets: Successes and Cancers. Cancers 2017, 9, 110. https://doi.org/10.3390/cancers9090110
Raab-Westphal S, Marshall JF, Goodman SL. Integrins as Therapeutic Targets: Successes and Cancers. Cancers. 2017; 9(9):110. https://doi.org/10.3390/cancers9090110
Chicago/Turabian StyleRaab-Westphal, Sabine, John F. Marshall, and Simon L. Goodman. 2017. "Integrins as Therapeutic Targets: Successes and Cancers" Cancers 9, no. 9: 110. https://doi.org/10.3390/cancers9090110
APA StyleRaab-Westphal, S., Marshall, J. F., & Goodman, S. L. (2017). Integrins as Therapeutic Targets: Successes and Cancers. Cancers, 9(9), 110. https://doi.org/10.3390/cancers9090110