Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer

Abstract
:1. Introduction
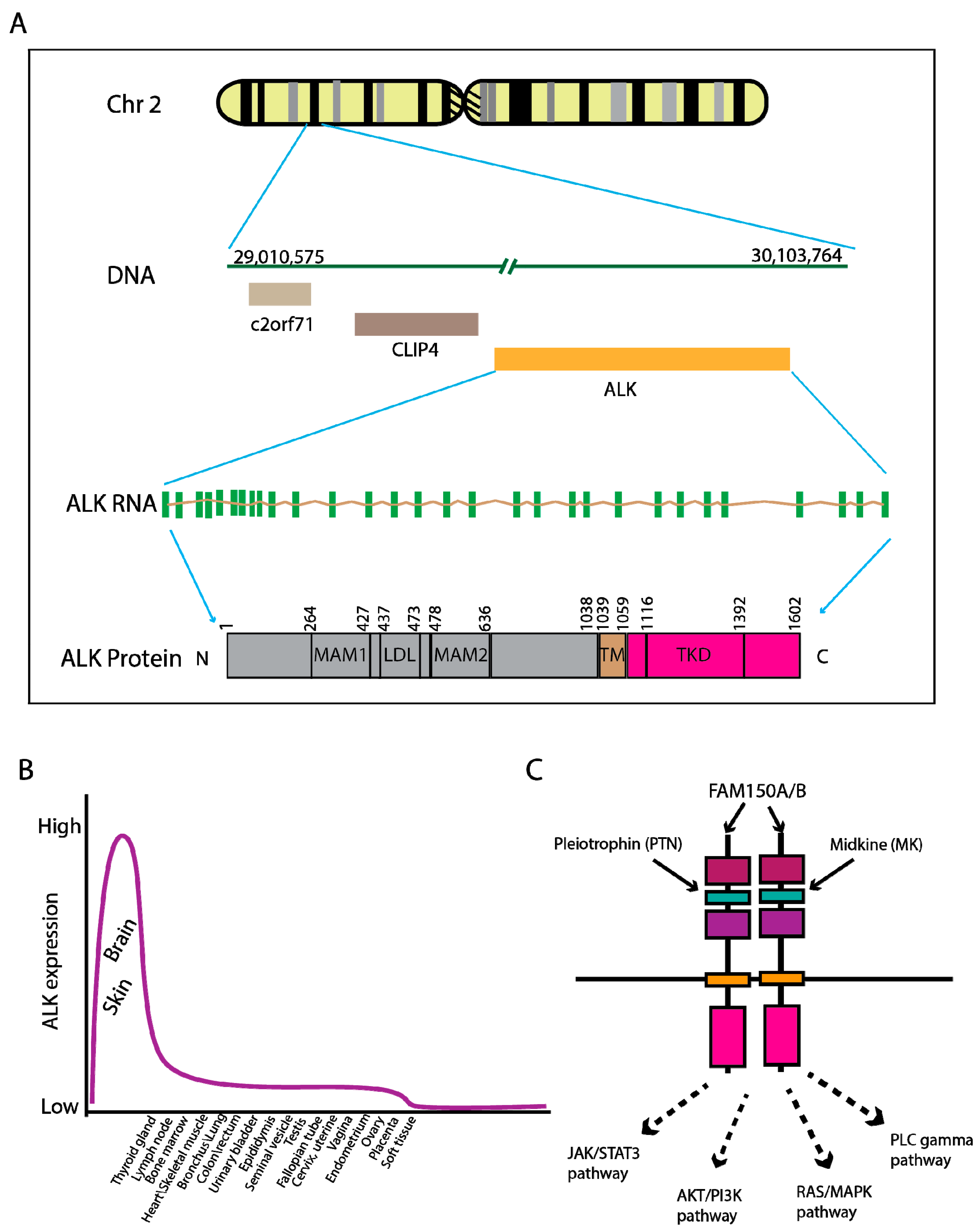
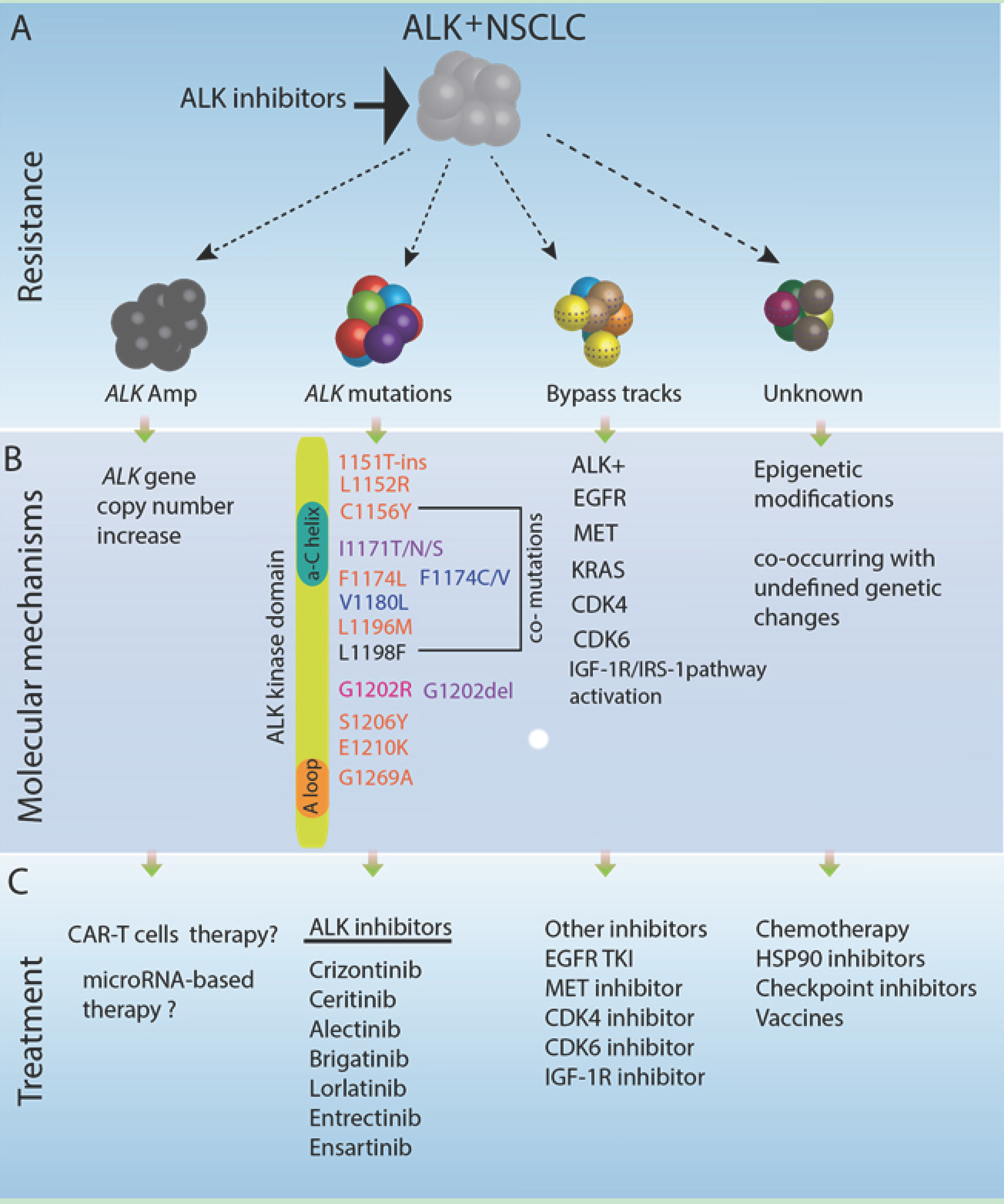
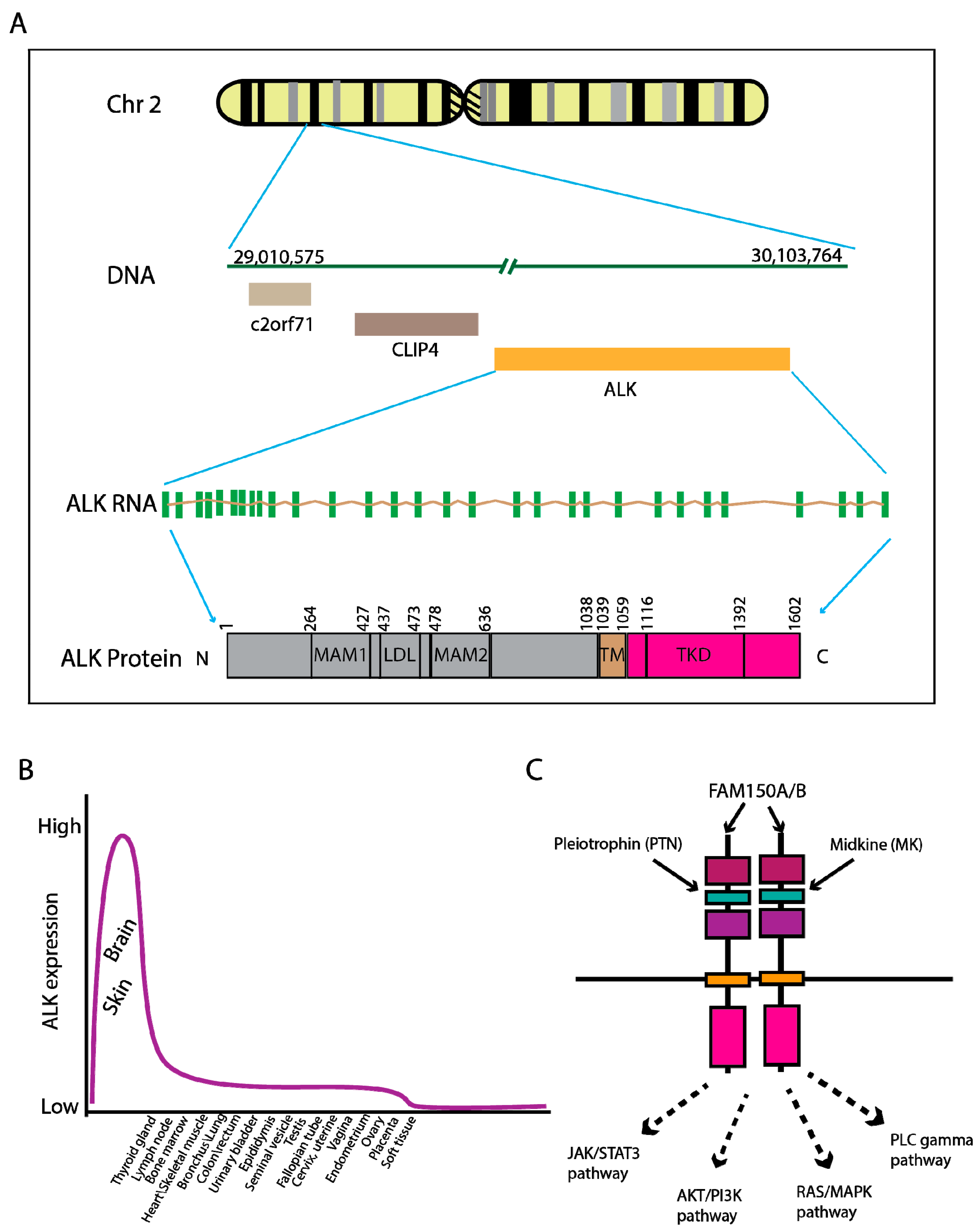
2. Genomic Characteristics of ALK Fusion-Driven NSCLC
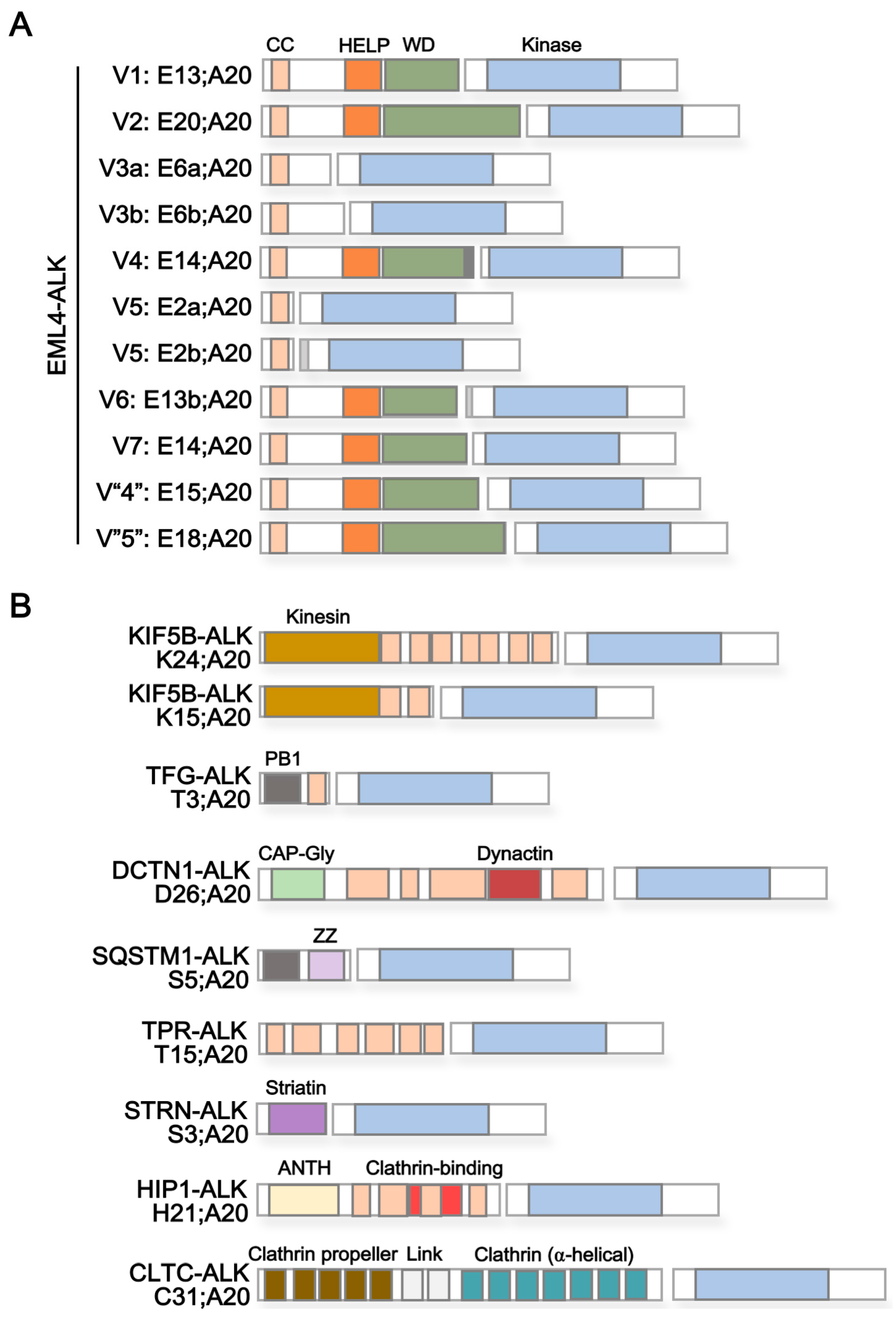
3. ALK Fusion Partner Proteins as Potential Therapeutic Targets
4. Immunotherapy for ALK+ Lung Cancer
5. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALCL | Anaplastic Large Cell Lymphoma |
| ALK | Anaplastic Lymphoma Kinase |
| CAR-T | Chimeric Antigen Receptor-T cell |
| NSCLC | Non-Small Cell Lung Cancer |
| TKI | Tyrosine Kinase Inhibitor |
References
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000171094-ALK/tissue (accessed on 5 November 2017).
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Aigner, A.; Sunitha, I.; Souttou, B.; Malerczyk, C.; Caughey, D.J.; Wen, D.; Karavanov, A.; Riegel, A.T.; et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J. Biol. Chem. 2001, 276, 16772–16779. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Powers, C.; Bowden, E.T.; Sale, E.B.; Riegel, A.T.; Wellstein, A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J. Biol. Chem. 2002, 277, 35990–35998. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. eLife 2015, 4, e09811. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.; Menez, C.; Girre, C.; Bodenez, P.; Hispard, E.; Menez, J.F. Decrease in cytochrome P4502E1 as assessed by the rate of chlorzoxazone hydroxylation in alcoholics during the withdrawal phase. Alcohol. Clin. Exp. Res. 1995, 19, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.W.; Lee, M.S.; Lee, S.E.; Song, J.Y.; Shin, H.T.; Kim, Y.J.; Oh, D.Y.; Jung, K.; Sung, M.; Kim, M.; et al. Molecular breakdown: A comprehensive view of anaplastic lymphoma kinase (ALK)-rearranged non-small cell lung cancer. J. Pathol. 2017, 243, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Takeuchi, K.; Togashi, Y.; Hatano, S.; Ninomiya, H.; Motoi, N.; Mun, M.Y.; Sakao, Y.; Okumura, S.; Nakagawa, K.; et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod. Pathol. 2009, 22, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Lung cancer: Understanding its molecular pathology and the 2015 WHO classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yang, J.M.; Jin, Y.; Jheon, S.; Kim, K.; Lee, C.T.; Chung, J.H.; Paik, J.H. MicroRNA expression profiles and clinicopathological implications in lung adenocarcinoma according to EGFR, KRAS, and ALK status. Oncotarget 2017, 8, 8484–8498. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Lovly, C.M.; Shaw, A.T. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: A paradigm for precision cancer medicine. Clin. Cancer Res. 2015, 21, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R. Therapeutic strategies and mechanisms of drug resistance in anaplastic lymphoma kinase (ALK)-rearranged lung cancer. Pharmacol. Ther. 2017, 177, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Sweis, R.F.; Thomas, S.; Bank, B.; Fishkin, P.; Mooney, C.; Salgia, R. Concurrent EGFR mutation and ALK translocation in non-small cell lung cancer. Cureus 2016, 8, e513. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Song, Y.J.; Liu, X.L. Adenocarcinoma of the lung with concomitant ALK fusion gene and EGFR gene mutation: A case report and literature review. Mol. Clin. Oncol. 2016, 4, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.-W.; Kim, D.W.; Thomas, M.; Pantano, S.; Wang, Y.; Lukasz Szpakowski, S.; Javier Yovine, A.; Mehra, R.; Laura, Q.; Sunil Sharma, C.; et al. Genetic landscape of ALK+ non-small cell lung cancer (NSCLC) patients (pts) and response to ceritinib in ASCEND-1. J. Clin. Oncol. 2016, 34, 9064. [Google Scholar]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretic, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Gautschi, O.; Rothschild, S.; Mark, M.; Froesch, P.; Klingbiel, D.; Reichegger, H.; Jochum, W.; Diebold, J.; Fruh, M. Clinical outcome of ALK-positive non-small cell lung cancer (NSCLC) patients with de novo EGFR or KRAS co-mutations receiving tyrosine kinase inhibitors (TKIs). J. Thorac. Oncol. 2017, 12, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Deng, X.; Yoshioka, Y.; Vougiouklakis, T.; Park, J.H.; Suzuki, T.; Dohmae, N.; Ueda, K.; Hamamoto, R.; Nakamura, Y. Effects of SMYD2-mediated EML4-ALK methylation on the signaling pathway and growth in non-small-cell lung cancer cells. Cancer Sci. 2017, 108, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Cascione, L.; Landi, L.; Carasi, S.; Lovat, F.; Tibaldi, C.; Ali, G.; D’Incecco, A.; Minuti, G.; Chella, A.; et al. microRNA classifiers are powerful diagnostic/prognostic tools in ALK-, EGFR-, and KRAS-driven lung cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 14924–14929. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Diagnostic and therapeutic potential of microRNAs in lung cancer. Cancers 2017, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Takeuchi, K.; Soda, M.; Inamura, K.; Togashi, Y.; Hatano, S.; Enomoto, M.; Hamada, T.; Haruta, H.; Watanabe, H.; et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008, 68, 4971–4976. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.; Leung, E.L.; So, K.K.; Tam, I.Y.; Sihoe, A.D.; Cheng, L.C.; Ho, K.K.; Au, J.S.; Chung, L.P.; Pik Wong, M.; et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009, 115, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Choi, Y.L.; Soda, M.; Inamura, K.; Togashi, Y.; Hatano, S.; Enomoto, M.; Takada, S.; Yamashita, Y.; Satoh, Y.; et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin. Cancer Res. 2008, 14, 6618–6624. [Google Scholar] [CrossRef] [PubMed]
- Iyevleva, A.G.; Raskin, G.A.; Tiurin, V.I.; Sokolenko, A.P.; Mitiushkina, N.V.; Aleksakhina, S.N.; Garifullina, A.R.; Strelkova, T.N.; Merkulov, V.O.; Ivantsov, A.O.; et al. Novel ALK fusion partners in lung cancer. Cancer Lett. 2015, 362, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Lira, M.E.; Hong, M.; Kim, R.N.; Choi, S.J.; Song, J.Y.; Pandy, K.; Mann, D.L.; Stahl, J.A.; Peckham, H.E.; et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Majewski, I.J.; Mittempergher, L.; Davidson, N.M.; Bosma, A.; Willems, S.M.; Horlings, H.M.; de Rink, I.; Greger, L.; Hooijer, G.K.; Peters, D.; et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J. Pathol. 2013, 230, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Kim, R.N.; Song, J.Y.; Choi, S.J.; Oh, E.; Lira, M.E.; Mao, M.; Takeuchi, K.; Han, J.; Kim, J.; et al. HIP1-ALK, a novel fusion protein identified in lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Soda, M.; Sakata, S.; Sugawara, E.; Hatano, S.; Asaka, R.; Nakajima, T.; Mano, H.; Takeuchi, K. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 2012, 7, e31323. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, W.-X.; Fang, M.; Chen, Y.-P.; Chen, Y.; Zhong, L.-H.; Chen, F.-F.; Zhuang, W.; Lin, G.; Chen, X.-H.; et al. Parallel VENTANA IHC and RT-PCR of ALK status in non-small cell lung cancer and response to crizotinib. J. Clin. Oncol. 2017, 35, 11623. [Google Scholar]
- Wong, D.W.; Leung, E.L.; Wong, S.K.; Tin, V.P.; Sihoe, A.D.; Cheng, L.C.; Au, J.S.; Chung, L.P.; Wong, M.P. A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer 2011, 117, 2709–2718. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, P.; Jung, Y.; Keum, J.; Kim, S.N.; Choi, Y.S.; Do, I.G.; Lee, J.; Choi, S.J.; Kim, S.; et al. Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosom. Cancer 2012, 51, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.; Duplantier, M.M.; Trempat, P.; Hieblot, C.; Lamant, L.; Espinos, E.; Racaud-Sultan, C.; Allouche, M.; Campo, E.; Delsol, G.; et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene 2004, 23, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.S.A.B.; Gowen, K.; Stephens, P.J.; Ross, J.S.; Johnson, M.L. Association of ALK resistance mutations by EML4-ALK variant (v3 vs. non-v3) in ALK+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35, 9010. [Google Scholar] [CrossRef]
- Heuckmann, J.M.; Balke-Want, H.; Malchers, F.; Peifer, M.; Sos, M.L.; Koker, M.; Meder, L.; Lovly, C.M.; Heukamp, L.C.; Pao, W.; et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 2012, 18, 4682–4690. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Oya, Y.; Tanaka, K.; Shimizu, J.; Horio, Y.; Kuroda, H.; Sakao, Y.; Hida, T.; Yatabe, Y. Differential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 3383–3389. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Shaw, A.T. Differential sensitivity to crizotinib: Does EML4-ALK fusion variant matter? J. Clin. Oncol. 2016, 34, 3363–3365. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.G.; Seo, S.; Kim, S.W.; Jang, S.J.; Park, K.S.; Song, J.Y.; Lee, B.; Richards, M.W.; Bayliss, R.; Lee, D.H.; et al. Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Ann. Oncol. 2017, 28, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; Law, E.W.; Rennalls, L.P.; Busacca, S.; O’Regan, L.; Fry, A.M.; Fennell, D.A.; Bayliss, R. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc. Natl. Acad. Sci. USA 2014, 111, 5195–5200. [Google Scholar] [CrossRef] [PubMed]
- Pfam. Available online: http://pfam.xfam.org (accessed on 9 April 2017).
- Zhang, G.; Scarborough, H.; Kim, J.; Rozhok, A.I.; Chen, Y.A.; Zhang, X.; Song, L.; Bai, Y.; Fang, B.; Liu, R.Z.; et al. Coupling an EML4-ALK-centric interactome with RNA interference identifies sensitizers to ALK inhibitors. Sci. Signal. 2016, 9, rs12. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, P.; Stetefeld, J.; Strelkov, S.V. Coiled coils: A highly versatile protein folding motif. Trends Cell Biol. 2001, 11, 82–88. [Google Scholar] [CrossRef]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M. Transport of c-MYC by Kinesin-1 for proteasomal degradation in the cytoplasm. Biochim. Biophys. Acta 2014, 1843, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.C.; Vale, R.D.; Kuntz, I.D. Inhibitors of kinesin activity from structure-based computer screening. Biochemistry 2000, 39, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Gaetz, J.; Kapoor, T.M. Dynein/dynactin regulate metaphase spindle length by targeting depolymerizing activities to spindle poles. J. Cell Biol. 2004, 166, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Cavolo, S.L.; Zhou, C.; Ketcham, S.A.; Suzuki, M.M.; Ukalovic, K.; Silverman, M.A.; Schroer, T.A.; Levitan, E.S. Mycalolide B dissociates dynactin and abolishes retrograde axonal transport of dense-core vesicles. Mol. Biol. Cell 2015, 26, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Wuo, M.G.; Mahon, A.B.; Arora, P.S. An effective strategy for stabilizing minimal coiled coil mimetics. J. Am. Chem. Soc. 2015, 137, 11618–11621. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.M.; Wuo, M.G.; Arora, P.S. Protein-protein interactions mediated by helical tertiary structure motifs. J. Am. Chem. Soc. 2015, 137, 11622–11630. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.S.; Pendley, S.S.; Bruno, B.J.; Woessner, D.W.; Shimpi, A.A.; Cheatham, T.E., III; Lim, C.S. Disruption of Bcr-Abl coiled coil oligomerization by design. J. Biol. Chem. 2011, 286, 27751–27760. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Karasaki, T.; Nagayama, K.; Kawashima, M.; Hiyama, N.; Murayama, T.; Kuwano, H.; Nitadori, J.; Anraku, M.; Sato, M.; Miyai, M.; et al. Identification of individual cancer-specific somatic mutations for neoantigen-based immunotherapy of lung cancer. J. Thorac. Oncol. 2016, 11, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Kudo-Saito, C.; Muramatsu, R.; Fujita, T.; Saito, M.; Nagumo, H.; Sakurai, T.; Noji, S.; Takahata, E.; Yaguchi, T.; et al. Determination of poor prognostic immune features of tumour microenvironment in non-smoking patients with lung adenocarcinoma. Eur. J. Cancer 2017, 86, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Colli, L.M.; Machiela, M.J.; Myers, T.A.; Jessop, L.; Yu, K.; Chanock, S.J. Burden of nonsynonymous mutations among TCGA cancers and candidate immune checkpoint inhibitor responses. Cancer Res. 2016, 76, 3767–3772. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Menotti, M.; Mastini, C.; Di Giacomo, F.; Longo, D.L.; Castella, B.; Merlo, M.E.; Ambrogio, C.; Wang, Q.; Minero, V.G.; et al. Efficacy of a cancer vaccine against ALK-rearranged lung tumors. Cancer Immunol. Res. 2015, 3, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Keskin, D.B.; DeLuca, D.S.; Alonso, A.; Zhang, W.; Zhang, G.L.; Hammond, N.N.; Nardi, V.; Stone, R.M.; Neuberg, D.; et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin. Cancer Res. 2012, 18, 5761–5772. [Google Scholar] [CrossRef] [PubMed]
- Pulford, K.; Falini, B.; Banham, A.H.; Codrington, D.; Roberton, H.; Hatton, C.; Mason, D.Y. Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood 2000, 96, 1605–1607. [Google Scholar] [PubMed]
- Passoni, L.; Scardino, A.; Bertazzoli, C.; Gallo, B.; Coluccia, A.M.; Lemonnier, F.A.; Kosmatopoulos, K.; Gambacorti-Passerini, C. ALK as a novel lymphoma-associated tumor antigen: Identification of 2 HLA-A2.1-restricted CD8+ T-cell epitopes. Blood 2002, 99, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Martinengo, C.; Mastini, C.; Ambrogio, C.; D’Escamard, V.; Forni, G.; Inghirami, G. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat. Med. 2008, 14, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Chen, N.; Fang, W.; Zhan, J.; Liu, Q.; Kang, S.; He, X.; Liu, L.; Zhou, T.; Huang, J.; et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology 2016, 5, e1094598. [Google Scholar] [CrossRef] [PubMed]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, M.A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001115. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2016, 3, 16006. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, D.C.; Auf der Maur, A.; Kodack, D.P.; Henke, R.T.; Hohn, S.; Toretsky, J.A.; Riegel, A.T.; Wellstein, A. Effect of single-chain antibody targeting of the ligand-binding domain in the anaplastic lymphoma kinase receptor. Oncogene 2009, 28, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- Creative Biolabs. Available online: http://www.creative-biolabs.com/car-t/alk-alk48-h-cd28–41bb-cd3%CE%B6-car-1124.htm (accessed on 29 September 2017).
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- UniProt. Available online: http://www.uniprot.org (accessed on 9 April 2017).

{kind=link}
{kind=link}
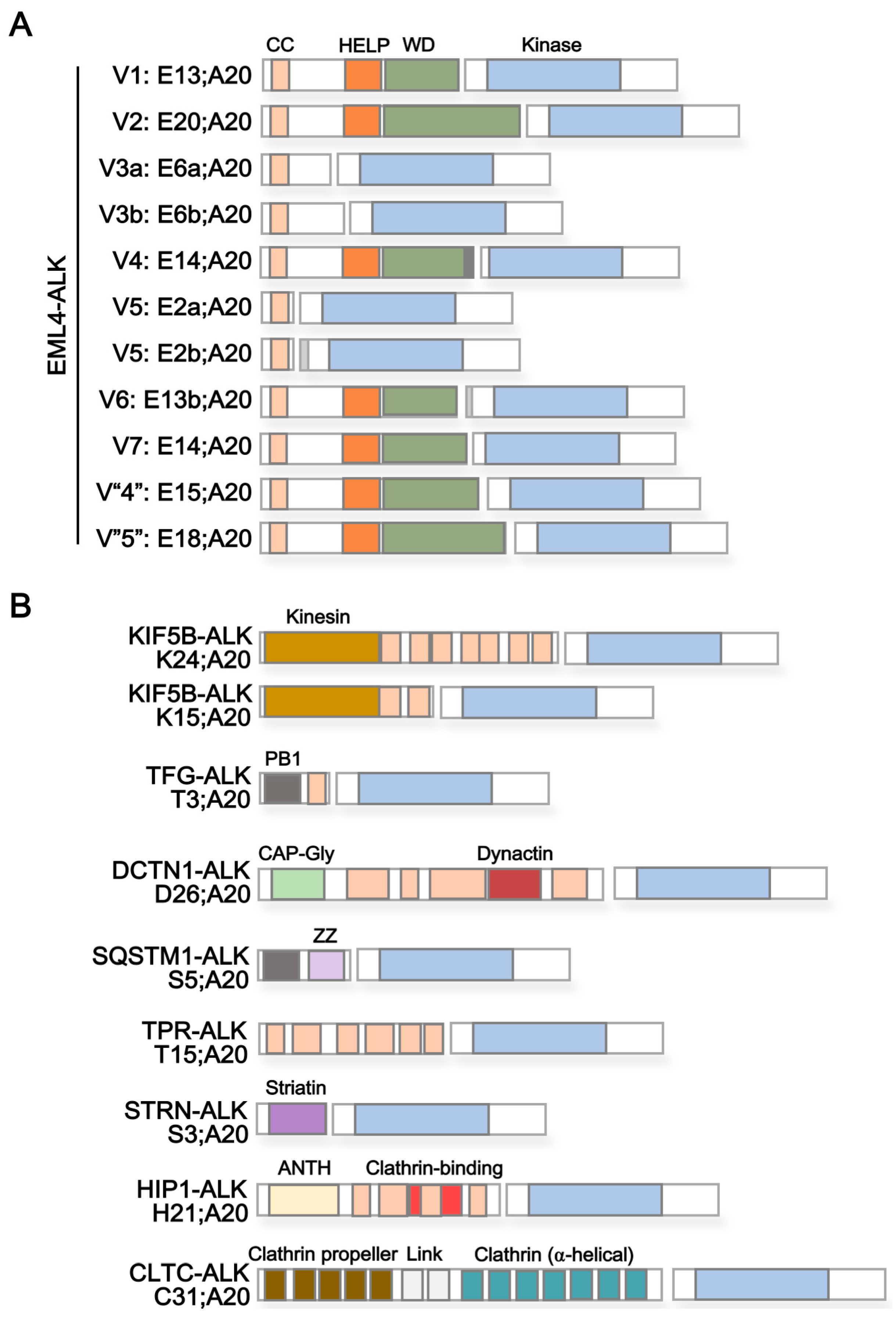{kind=link}
| Protein Name 1 | Reported ALK Fusion | Uniprot ID | Molecular Function 2 | Subcellular Localization 3 | Protein Domains 4 | |
|---|---|---|---|---|---|---|
| Echinoderm microtubule-associated protein-like 4 (EML4) | E2a/b;A20 | [36] | Q9HC35 | Microtubule binding and assembly | Cytoplasm > cytoskeleton | Coiled coil [aa17–51] HELP [aa225–297] WD40 repeats [aa229–347, 499–537, 583–620, 711–749, 824–863] |
| E6a/b;A20 | [32] | |||||
| E13;A20 | [20] | |||||
| E13b;A20 | [33] | |||||
| E14;A20 | [33,36] | |||||
| E15;A20 | [34] | |||||
| E18;A20 | [35] | |||||
| E20;A20 | [20] | |||||
| Kinesin-1 heavy chain (KIF5B) | K24;A20 K15;A20 | [33] [43] | P33176 | Microtubule-associated motor protein | Cytoplasm > cytoskeleton | Kinesin [aa14–325] Coiled coil [aa330–364, 418–543, 594–684, 691–711, 716–757, 768–802, 825–912] |
| TRK-fused gene (TFG) | T3;A20 | [21] | Q92734 | Dynamic interaction of endoplasmic reticulum and microtubules | Endoplasmic reticulum (ER) | PB1 [aa10–93] Coiled coil [aa102–122] |
| Dynactin subunit 1 (DCTN1) | D26;A20 | [37] | Q14203 | Dynein-mediated retrograde transport of vesicles and organelles along microtubules | Cytoplasm > cytoskeleton > microtubule | CAP-Gly [aa29–94] Coiled coil [aa217–352, 360–383,388–540, 952–1043, 1185–1205] Dynactin [aa527–805] |
| Sequestosome-1 (SQSTM1) | S5;A20 | [37] | Q13501 | Autophagy receptor, endosome organization | Cytoplasm > late endosome, autolysosome/-phagosome | PB1 [aa21–102] Zinc finger [aa122–165] UBA [aa379–440] |
| Nucleoprotein TPR | T15;A20 | [38] | P12270 | Scaffolding element in nuclear pore complex, nucleocytoplasmic transport | Nucleus > Nuclear pore complex | Coiled coil [aa29–49, 61–172, 219–281, 293–366, 427–513, 543–570, 576–596, 665–752, 760–801, 827–868, 887–914, 934–982, 997–1071, 1096–1116, 1149–1169, 1221–1238, 1269–1303, 1311–1345, 1354–1416, 1469–1552, 1564–1598, 1603–1630] TPR/MLP1/MLP2 [aa1038–1165] |
| Cysteine-rich motor neuron 1 protein (CRIM1) | n/a | [26] | Q9NZV1 | Tissue development, interaction with transforming growth factor beta family proteins | Cell membrane | Single pass type I transmembrane [aa1–34] IGFBP [aa37–90] VWC [aa336–390, 403–456, 608–662, 679–734, 753–808, 819–873] Antistatin [aa469–498, 505–532, 539–564, 567–592] |
| Striatin (STRN) | S3;A20 | [39] | O43815 | Calmodulin-binding protein involved in scaffolding and signaling | Cytoplasm, Cell membrane | Striatin [aa48–177] Coiled coil [aa67–115] WD40 repeats [aa453–491, 506–544, 559–597, 696–734, 738–779] |
| Huntingtin-interacting protein 1 (HIP1) | H21;A20 | [40] | O00291 | Involved in clathrin-mediated endocytosis and trafficking | Cytoplasm, Endomembrane system, nucleus | ANTH [aa39–308] Coiled coil [aa377–397, 409–488, 496–555, 584–611, 978–1007] Clathrin binding [aa482–580] I/LWEQ [aa862–1010] |
| Tyrosine-protein phosphatase non-receptor type 3 (PTPN3) | Chimeric fusion | [44] | P26045 | Tyrosine phosphatase, interaction with cytoskeleton | Cell membrane, cytoplasm > cytoskeleton | FERM [aa33–97, 113–222, 226–316] PDZ [aa510–595] Y phosphatase [aa670–900] |
| Kinesin light chain 1 (KLC1) | K9;A20 | [41] | Q07866 | Microtubule-associated motor protein | Cytoplasm > cytoskeleton | n/a |
| Clathrin heavy chain 1 (CLTC) | C31;A20 | [22] | Q00610 | Intracellular trafficking and endocytosis | Cell membrane, cytoplasm > cytoskeleton | Clathrin propeller repeat [aa19–56, 148–187, 198–234, 253–288, 296–330] Clathrin (H) link [aa331–354, 356–421] Clathrin [aa537–679, 686–827,835–971, 979–1123, 1128–1268, 1274–1419, 1424–1565] |
| F-box only protein 36 (FBXO36) | n/a | [42] | Q8NEA4 | Substrate recognition in E3 ubiquitin ligase complex | n/a | F-box [aa92–139] Coiled coil [aa166–186] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Haderk, F.; Bivona, T.G. Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer. Cancers 2017, 9, 164. https://doi.org/10.3390/cancers9120164
Wu W, Haderk F, Bivona TG. Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer. Cancers. 2017; 9(12):164. https://doi.org/10.3390/cancers9120164
Chicago/Turabian StyleWu, Wei, Franziska Haderk, and Trever G. Bivona. 2017. "Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer" Cancers 9, no. 12: 164. https://doi.org/10.3390/cancers9120164
APA StyleWu, W., Haderk, F., & Bivona, T. G. (2017). Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer. Cancers, 9(12), 164. https://doi.org/10.3390/cancers9120164