
Does Hypoxia Cause Carcinogenic Iron Accumulation in Alcoholic Liver Disease (ALD)?


Abstract
1. Introduction
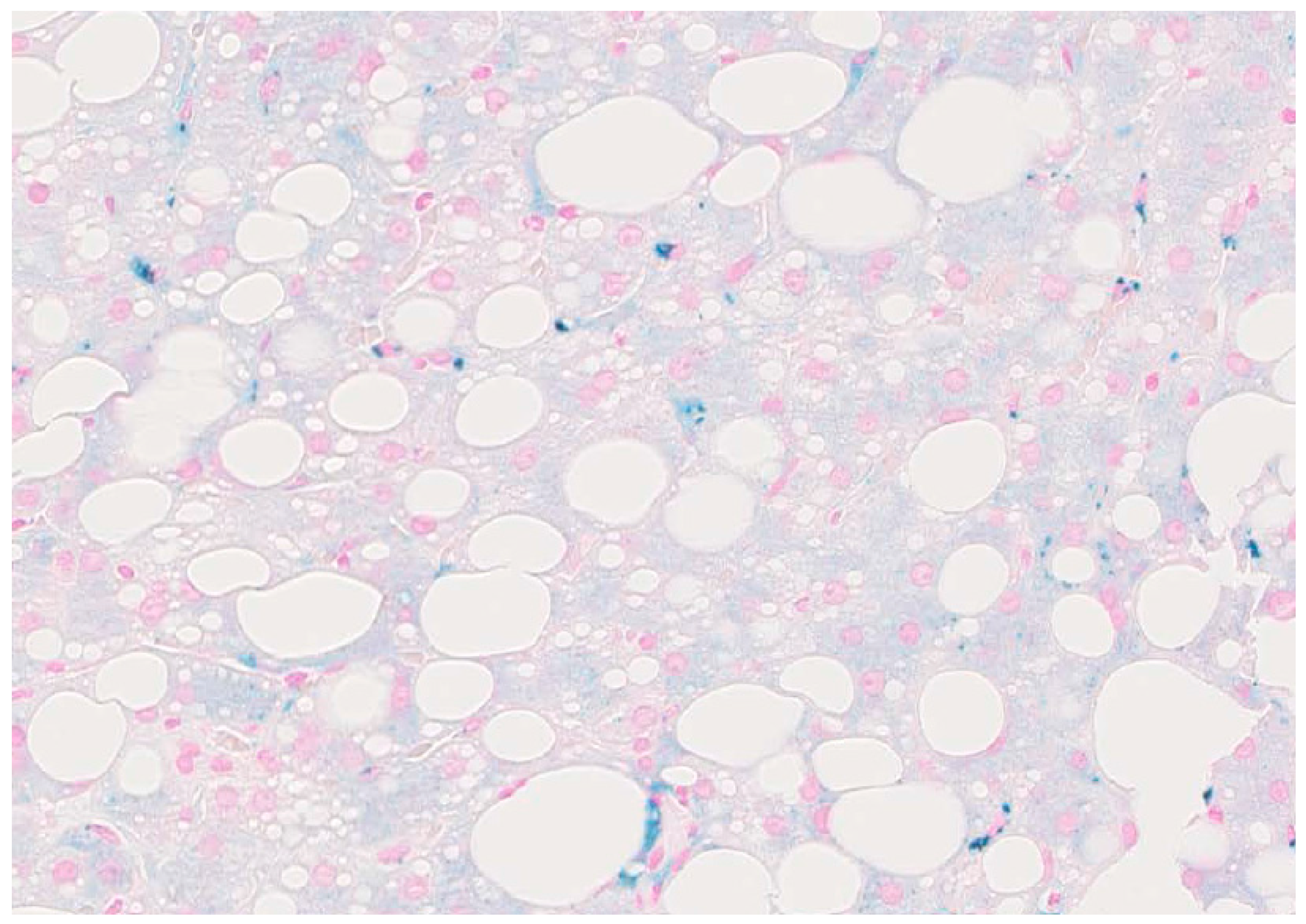
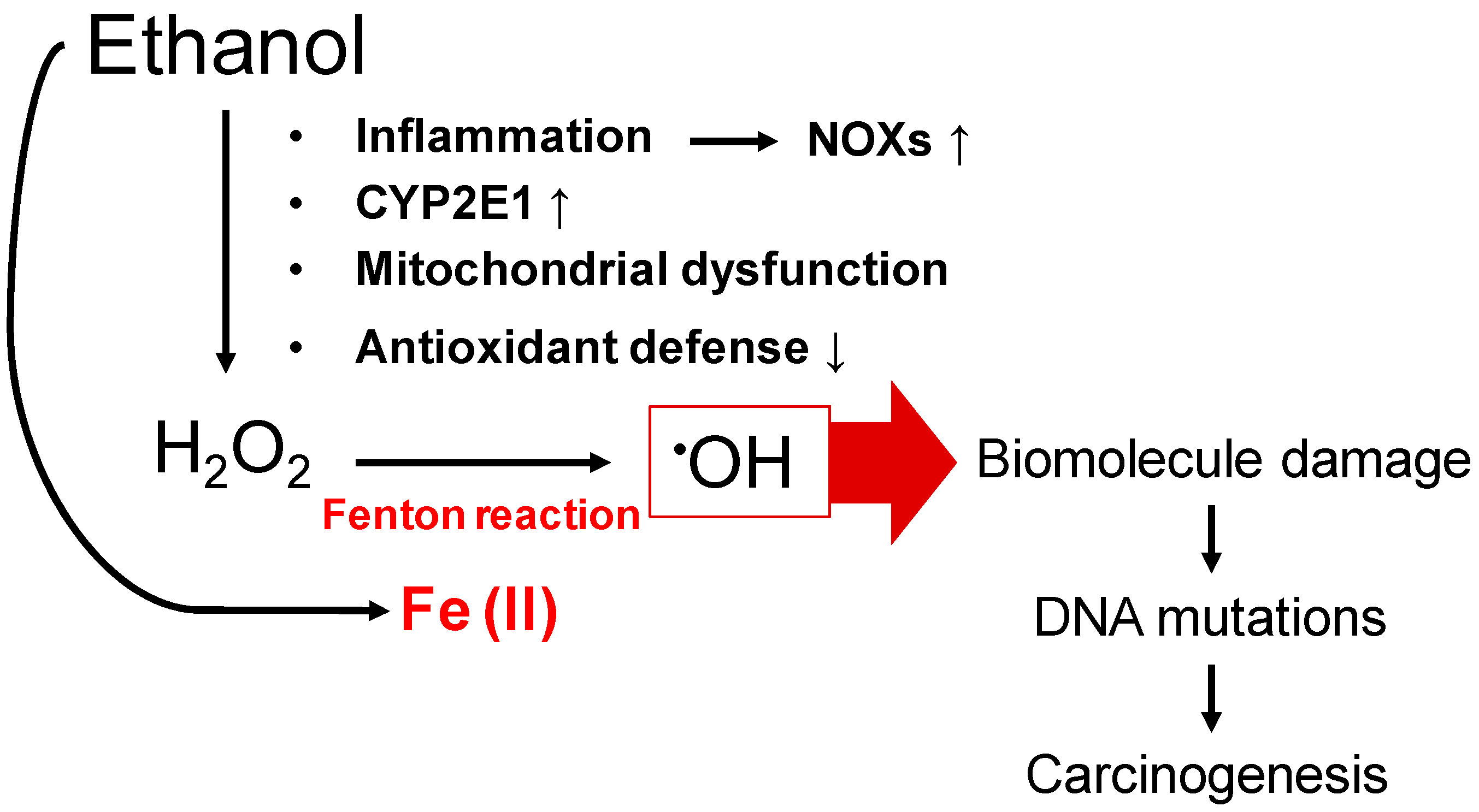
2. Hepatic Iron Overload in ALD and Carcinogenesis
3. Control of Iron Homeostasis
3.1. Cellular Regulation of Iron
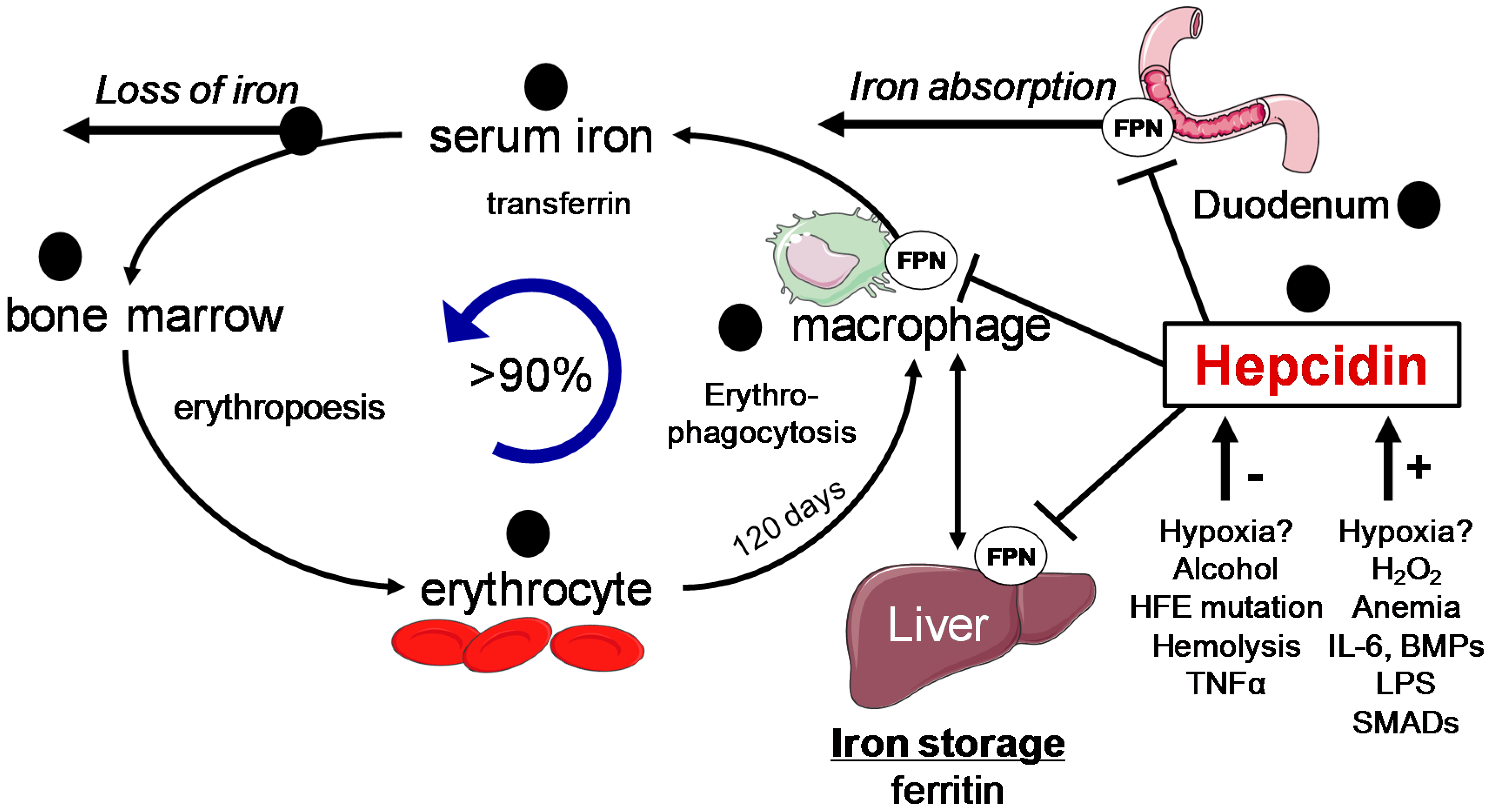
3.2. Systemic Iron Control by Hepcidin
4. Transcriptional Regulation of Hepcidin
5. Regulation of Hepcidin by Hypoxia
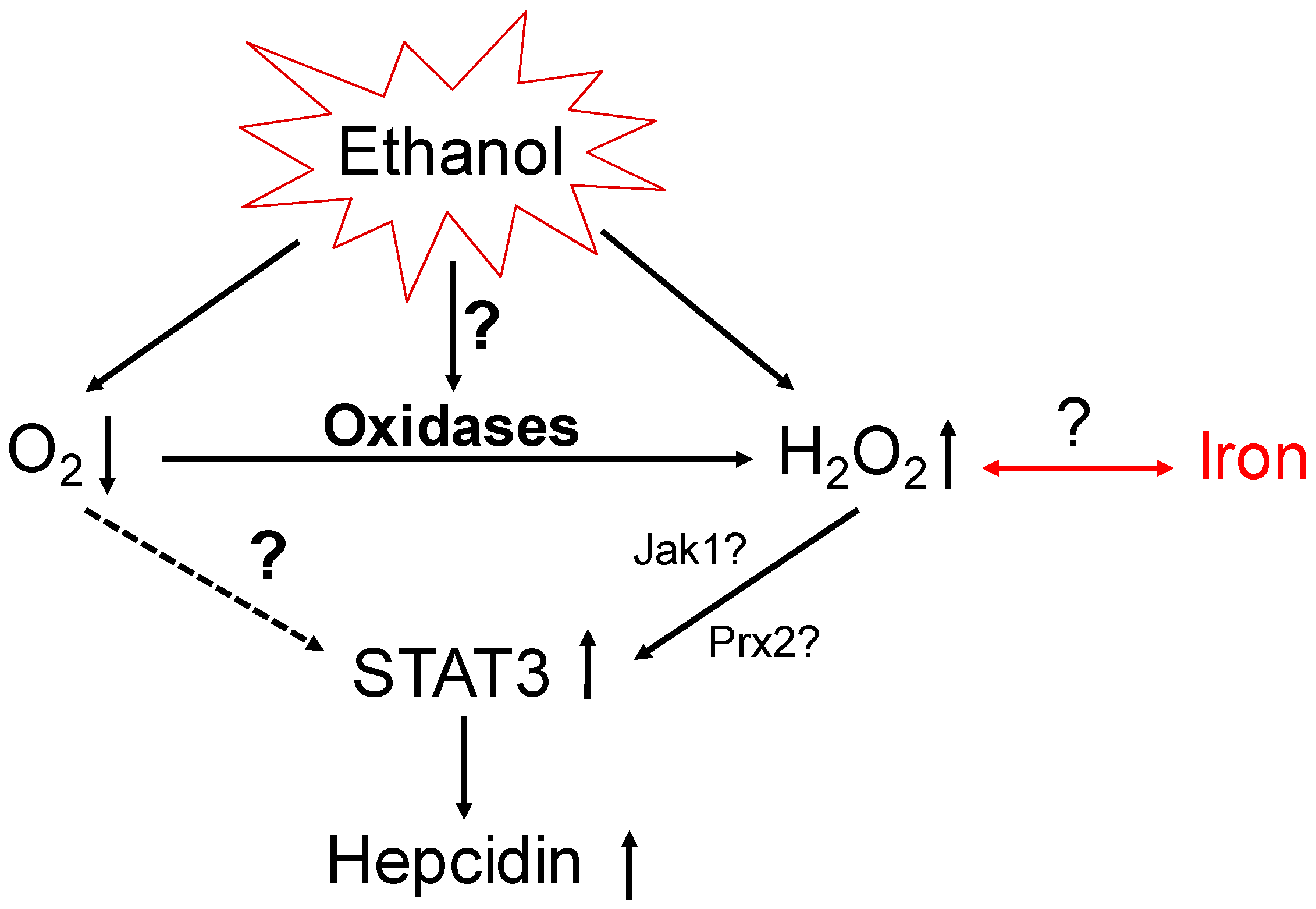
6. Hepatic Oxygen Levels and Methodological Challenges to Studying Hypoxia
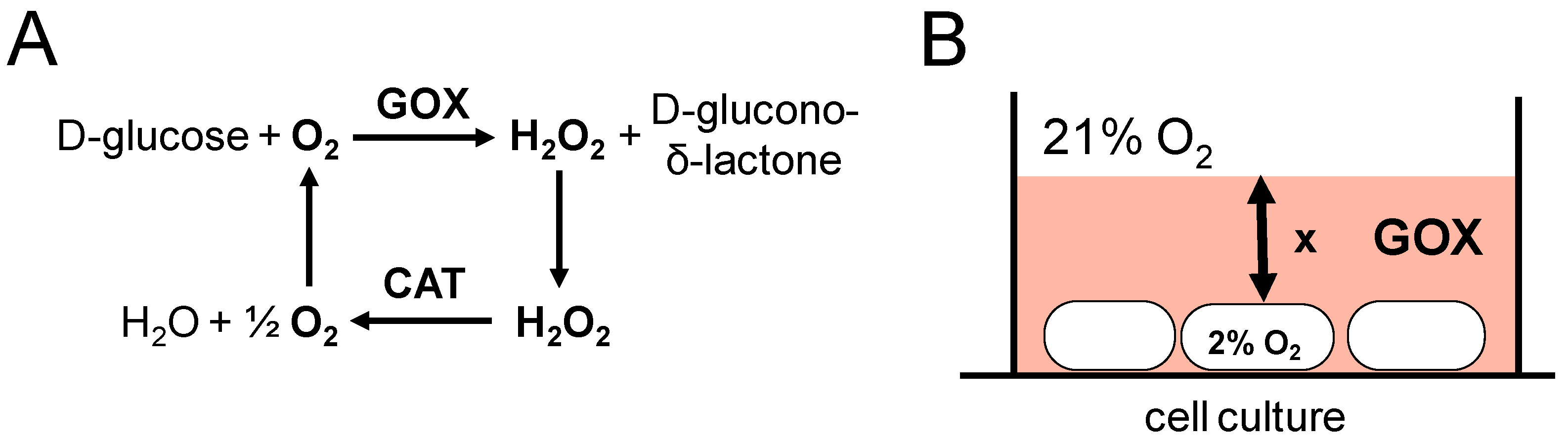
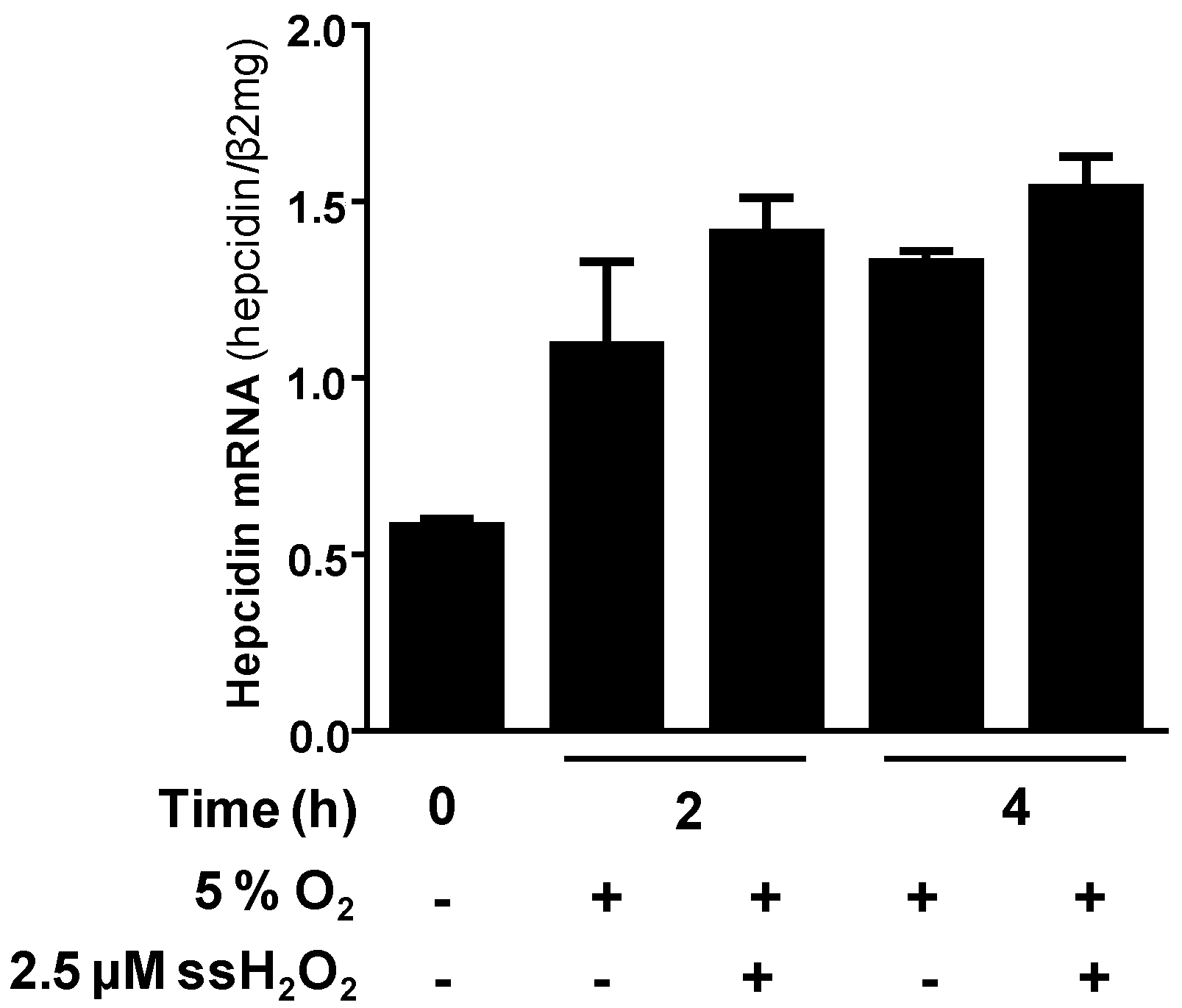
7. The GOX/CAT System Allows for Independent Study of Hypoxia and Hydrogen Peroxide
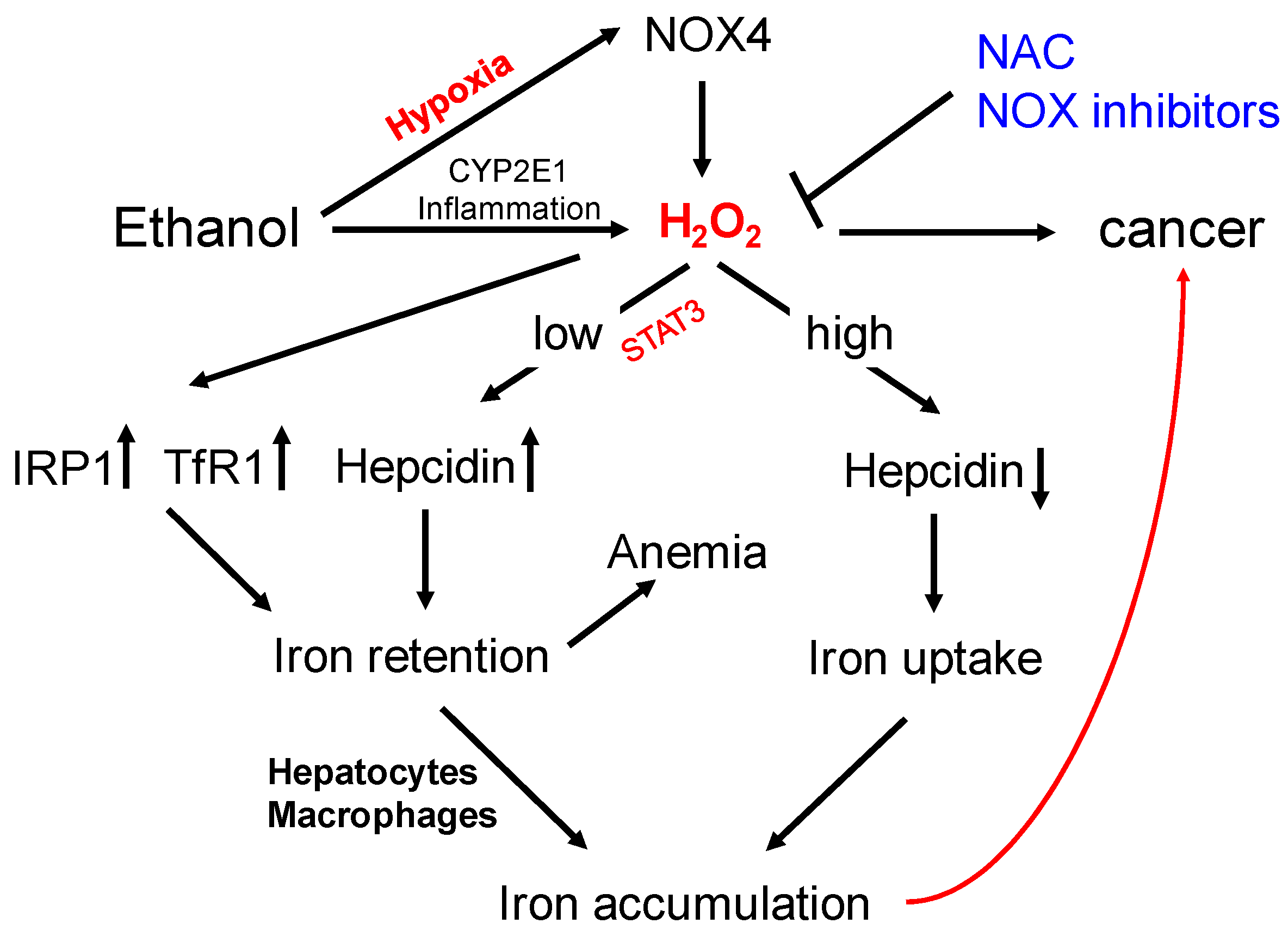
8. Could NOX4 Be Responsible for Iron Overload in ALD?
9. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the global burden of disease study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Rausch, V. The role of iron in alcohol-mediated hepatocarcinogenesis. Adv. Exp. Med. Biol. 2015, 815, 89–112. [Google Scholar] [PubMed]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Harrison-Findik, D.D.; Schafer, D.; Klein, E.; Timchenko, N.A.; Kulaksiz, H.; Clemens, D.; Fein, E.; Andriopoulos, B.; Pantopoulos, K.; Gollan, J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J. Biol. Chem. 2006, 281, 22974–22982. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.W. The role of alcoholism in hepatic iron storage disease. Ann. N. Y. Acad. Sci. 1975, 252, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Ganne-Carrie, N.; Christidis, C.; Chastang, C.; Ziol, M.; Chapel, F.; Imbert-Bismut, F.; Trinchet, J.C.; Guettier, C.; Beaugrand, M. Liver iron is predictive of death in alcoholic cirrhosis: A multivariate study of 229 consecutive patients with alcoholic and/or hepatitis c virus cirrhosis: A prospective follow up study. Gut 2000, 46, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.C.; Agnew, S. Alcoholism in hereditary hemochromatosis revisited: Prevalence and clinical consequences among homozygous siblings. Hepatology 1996, 23, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol. Chem. 2006, 387, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Tirnitz-Parker, J.E.; Glanfield, A.; Olynyk, J.K.; Ramm, G.A. Iron and hepatic carcinogenesis. Crit. Rev. Oncog. 2013, 18, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Szweda, L.I.; Chae, H.Z.; Stadtman, E.R. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 8742–8746. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prev. 2004, 28, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Kohgo, Y.; Ohtake, T.; Ikuta, K.; Suzuki, Y.; Hosoki, Y.; Saito, H.; Kato, J. Iron accumulation in alcoholic liver diseases. Alcohol Clin. Exp. Res. 2005, 29, 189S–193S. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Otsuki, Y. Triggering of parkin mitochondrial translocation in mitophagy: Implications for liver diseases. Front. Pharmacol. 2016, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, O.; Fischer, H.; Hasegawa, T.; Maisel, M.; Leitner, P.; Miesel, F.; Sharma, M.; Bornemann, A.; Berg, D.; Gasser, T.; et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009, 18, 3832–3850. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Beaumont, C.; Richardson, D.R. Function and regulation of transferrin and ferritin. Semin. Hematol. 1998, 35, 35–54. [Google Scholar] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of iron homeostasis: New players in metabolism, cell death, and disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies ncoa4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective vps34 inhibitor blocks autophagy and uncovers a role for ncoa4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. Ncoa4 deficiency impairs systemic iron homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Wallander, M.L.; Leibold, E.A.; Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 2006, 1763, 668–689. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.R.; Kuhn, L.C. Differential modulation of the RNA-binding proteins IRP-1 and IRP-2 in response to iron. IRP-2 inactivation requires translation of another protein. J. Biol. Chem. 1995, 270, 20509–20515. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.N.; Ponka, P. Identification of an erythroid active element in the transferrin receptor gene. J. Biol. Chem. 2000, 275, 24185–24190. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B.; Hegedusch, S.; Mangin, J.; Riedel, H.D.; Hebling, U.; Wang, J.; Pantopoulos, K.; Mueller, S. Sustained hydrogen peroxide induces iron uptake by transferrin receptor-1 independent of the iron regulatory protein/iron-responsive element network. J. Biol. Chem. 2007, 282, 20301–20308. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Pantopoulos, K.; Hubner, C.A.; Stremmel, W.; Hentze, M.W. Irp1 activation by extracellular oxidative stress in the perfused rat liver. J. Biol. Chem. 2001, 276, 23192–23196. [Google Scholar] [CrossRef] [PubMed]
- Mutze, S.; Hebling, U.; Stremmel, W.; Wang, J.; Arnhold, J.; Pantopoulos, K.; Mueller, S. Myeloperoxidase-derived hypochlorous acid antagonizes the oxidative stress-mediated activation of iron regulatory protein 1. J. Biol. Chem. 2003, 278, 40542–40549. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S. Iron regulatory protein 1 as a sensor of reactive oxygen species. Biofactors 2005, 24, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Lesbordes-Brion, J.C.; Viatte, L.; Bennoun, M.; Lou, D.Q.; Ramey, G.; Houbron, C.; Hamard, G.; Kahn, A.; Vaulont, S. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 2006, 108, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA 2002, 99, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.; Nemeth, E. Manipulation of the hepcidin pathway for therapeutic purposes. Haematologica 2013, 98, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Harrison-Findik, D.D.; Klein, E.; Crist, C.; Evans, J.; Timchenko, N.; Gollan, J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007, 46, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Gerjevic, L.N.; Liu, N.; Lu, S.; Harrison-Findik, D.D. Alcohol activates tgf-beta but inhibits bmp receptor-mediated smad signaling and smad4 binding to hepcidin promoter in the liver. Int. J. Hepatol. 2012, 2012, 459278. [Google Scholar] [CrossRef] [PubMed]
- Costa-Matos, L.; Batista, P.; Monteiro, N.; Simoes, M.; Egas, C.; Pereira, J.; Pinho, H.; Santos, N.; Ribeiro, J.; Cipriano, M.A.; et al. Liver hepcidin mrna expression is inappropriately low in alcoholic patients compared with healthy controls. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Nahon, P.; Nuraldeen, R.; Rufat, P.; Sutton, A.; Trautwein, C.; Strnad, P. In alcoholic cirrhosis, low-serum hepcidin levels associate with poor long-term survival. Liver Int. 2016, 36, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Millonig, G.; Waite, G.N.; Longerich, T.; Schirmacher, P.; Muckenthaler, M.U.; Seitz, H.K.; Mueller, S. Hepatic iron overload in patients with alcoholic liver disease is due to inadequate hepcidin induction: Role of h2o2 in hepcidin regulation. J. Hepatol. 2010, 52, S444. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the iron homeostatic hormone hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.G.; Wang, Y.; Wu, Q.; Cheng, W.H.; Liu, W.; Zhao, Y.; Mayeur, C.; Schmidt, P.J.; Yu, P.B.; Wang, F.; et al. Hfe interacts with the bmp type I receptor ALK3 to regulate hepcidin expression. Blood 2014, 124, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins hfe, tfr2, and hjv form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, T.; Vujic Spasic, M.; Altamura, S.; Breitkopf-Heinlein, K.; Altenoder, J.; Weiss, T.S.; Dooley, S.; Muckenthaler, M.U. Transforming growth factor beta1 (TGF-beta1) activates hepcidin mRNA expression in hepatocytes. J. Biol. Chem. 2016, 291, 13160–13174. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N.K.; Chen, K.; Cherayil, B.J. Commensal bacteria-induced interleukin 1beta (il-1beta) secreted by macrophages up-regulates hepcidin expression in hepatocytes by activating the bone morphogenetic protein signaling pathway. J. Biol. Chem. 2015, 290, 30637–30647. [Google Scholar] [CrossRef] [PubMed]
- Canali, S.; Core, A.B.; Zumbrennen-Bullough, K.B.; Merkulova, M.; Wang, C.Y.; Schneyer, A.L.; Pietrangelo, A.; Babitt, J.L. Activin b induces noncanonical SMAD1/5/8 signaling via bmp type I receptors in hepatocytes: Evidence for a role in hepcidin induction by inflammation in male mice. Endocrinology 2016, 157, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S.; Anderson, S.A.; Wang, J.; Yang, F.; DeMaster, K.; Ahmed, R.; Nizzi, C.P.; Eisenstein, R.S.; Tsukamoto, H.; Enns, C.A. Suppression of hepatic hepcidin expression in response to acute iron deprivation is associated with an increase of matriptase-2 protein. Blood 2011, 117, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, S.R.; McHugh, K.; Drakesmith, H. Regulation of hepcidin by erythropoiesis: The story so far. Annu. Rev. Nutr. 2016, 36, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and host defense against infectious diseases. PLoS Pathog. 2015, 11, e1004998. [Google Scholar] [CrossRef] [PubMed]
- Millonig, G.; Ganzleben, I.; Peccerella, T.; Casanovas, G.; Brodziak-Jarosz, L.; Breitkopf-Heinlein, K.; Dick, T.P.; Seitz, H.K.; Muckenthaler, M.U.; Mueller, S. Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3). J. Biol. Chem. 2012, 287, 37472–37482. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; De Falco, L.; Federti, E.; Cozzi, A.; Iolascon, A.; Levi, S.; Mohandas, N.; Zamo, A.; Bruno, M.; Leboeuf, C.; et al. Peroxiredoxin-2: A novel regulator of iron homeostasis in ineffective erythropoiesis. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Cairo, G. Mutual cross talk between iron homeostasis and erythropoiesis. Vitam. Horm. 2017, 105, 143–160. [Google Scholar] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Srai, S.K.; Mascarenhas, M.; Churchill, L.J.; Debnam, E.S. Increased duodenal iron uptake and transfer in a rat model of chronic hypoxia is accompanied by reduced hepcidin expression. Gut 2005, 54, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Braliou, G.G.; Verga Falzacappa, M.V.; Chachami, G.; Casanovas, G.; Muckenthaler, M.U.; Simos, G. 2-oxoglutarate-dependent oxygenases control hepcidin gene expression. J. Hepatol. 2008, 48, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Volke, M.; Gale, D.P.; Maegdefrau, U.; Schley, G.; Klanke, B.; Bosserhoff, A.K.; Maxwell, P.H.; Eckardt, K.U.; Warnecke, C. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS ONE 2009, 4, e7875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Mathieu, J.R.; Delga, S.; Mayeux, P.; Vaulont, S.; Peyssonnaux, C. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica 2012, 97, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Goetze, O.; Schmitt, J.; Spliethoff, K.; Theurl, I.; Weiss, G.; Swinkels, D.W.; Tjalsma, H.; Maggiorini, M.; Krayenbuhl, P.; Rau, M.; et al. Adaptation of iron transport and metabolism to acute high-altitude hypoxia in mountaineers. Hepatology 2013, 58, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.P.; Lakhal, S.; Smith, T.G.; Privat, C.; Nickol, A.H.; Rivera-Ch, M.; Leon-Velarde, F.; Dorrington, K.L.; Mole, D.R.; Robbins, P.A. Regulation of hepcidin expression at high altitude. Blood 2012, 119, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Sonnweber, T.; Nachbaur, D.; Schroll, A.; Nairz, M.; Seifert, M.; Demetz, E.; Haschka, D.; Mitterstiller, A.M.; Kleinsasser, A.; Burtscher, M.; et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor bb. Gut 2014, 63, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Camaschella, C. Furin-mediated release of soluble hemojuvelin: A new link between hypoxia and iron homeostasis. Blood 2008, 111, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Lakhal, S.; Schodel, J.; Townsend, A.R.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. Regulation of type ii transmembrane serine proteinase tmprss6 by hypoxia-inducible factors: New link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 2011, 286, 4090–4097. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.R.; Taylor, M.; Xue, X.; Martin, A.; Moons, D.S.; Omary, M.B.; Shah, Y.M. The hypoxia-inducible factor-c/ebpalpha axis controls ethanol-mediated hepcidin repression. Mol. Cell Biol. 2012, 32, 4068–4077. [Google Scholar] [CrossRef] [PubMed]
- Chaston, T.B.; Matak, P.; Pourvali, K.; Srai, S.K.; McKie, A.T.; Sharp, P.A. Hypoxia inhibits hepcidin expression in huh7 hepatoma cells via decreased smad4 signaling. Am. J. Physiol. Cell Physiol. 2011, 300, C888–C895. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.O.; Cho, Y.S.; Kim, H.L.; Park, J.W. Ros mediate the hypoxic repression of the hepcidin gene by inhibiting c/ebpalpha and STAT-3. Biochem. Biophys. Res. Commun. 2007, 356, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Jungermann, K.; Kietzmann, T. Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology 2000, 31, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Iimuro, Y.; Yin, M.; Raleigh, J.A.; Thurman, R.G. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology 1997, 25, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Raleigh, J.A.; Bradford, B.U.; Thurman, R.G. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: Role of kupffer cells. Am. J. Physiol. 1996, 271, G494–G500. [Google Scholar] [PubMed]
- Nath, B.; Szabo, G. Alcohol-induced modulation of signaling pathways in liver parenchymal and nonparenchymal cells: Implications for immunity. Semin. Liver Dis. 2009, 29, 166–177. [Google Scholar] [CrossRef] [PubMed]
- French, S.W. The role of hypoxia in the pathogenesis of alcoholic liver disease. Hepatol. Res. 2004, 29, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Yuki, T.; Thurman, R.G. The swift increase in alcohol metabolism. Time course for the increase in hepatic oxygen uptake and the involvement of glycolysis. Biochem. J. 1980, 186, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Q.; Je, Y.T.; Kim, M.; Kim, J.H.; Kong, G.; Kang, K.S.; Kim, H.L.; Yoon, B.I.; Lee, M.O.; Lee, B.H. Analysis of hepatic gene expression during fatty liver change due to chronic ethanol administration in mice. Toxicol. Appl. Pharmacol. 2009, 235, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Millonig, G.; Hegedusch, S.; Becker, L.; Seitz, H.K.; Schuppan, D.; Mueller, S. Hypoxia-inducible factor 1 alpha under rapid enzymatic hypoxia: Cells sense decrements of oxygen but not hypoxia per se. Free Radic. Biol. Med. 2009, 46, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Millonig, G.; Waite, G.N. The gox/cat system: A novel enzymatic method to independently control hydrogen peroxide and hypoxia in cell culture. Adv. Med. Sci. 2009, 54, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Askoxylakis, V.; Millonig, G.; Wirkner, U.; Schwager, C.; Rana, S.; Altmann, A.; Haberkorn, U.; Debus, J.; Mueller, S.; Huber, P.E. Investigation of tumor hypoxia using a two-enzyme system for in vitro generation of oxygen deficiency. Radiat. Oncol. 2011, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E. Animal models of alcoholic liver disease. Dig. Dis. 2010, 28, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Britton, R.S.; Bacon, B.R.; Sly, W.S.; Fleming, R.E. Decreased liver hepcidin expression in the hfe knockout mouse. Blood Cells Mol. Dis. 2002, 29, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Niederkofler, V.; Salie, R.; Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Investig. 2005, 115, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Koeffler, H.P.; Kawabata, H.; Britton, R.S.; Bacon, B.R.; Sly, W.S. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA 2002, 99, 10653–10658. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of smad4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. Bmp6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; de Lara, F.M.; Pendas, A.M.; Garabaya, C.; Rodriguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; Lopez-Otin, C.; Velasco, G. Membrane-bound serine protease matriptase-2 (TMPRSS6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Sakamori, R.; Takehara, T.; Tatsumi, T.; Shigekawa, M.; Hikita, H.; Hiramatsu, N.; Kanto, T.; Hayashi, N. Stat3 signaling within hepatocytes is required for anemia of inflammation in vivo. J. Gastroenterol. 2010, 45, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H.; et al. Role of stat-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 2004, 10, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus oncogenic functions of stat3 in liver tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Akira, S. Roles of stat3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.T.; Walsh, M.E.; Van Remmen, H. Mouse models of oxidative stress indicate a role for modulating healthy aging. J. Clin. Exp. Pathol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ljungkvist, A.S.; Bussink, J.; Kaanders, J.H.; van der Kogel, A.J. Dynamics of tumor hypoxia measured with bioreductive hypoxic cell markers. Radiat. Res. 2007, 167, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by hif hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Pantopoulos, K.; Hentze, M.W.; Stremmel, W. A chemiluminescence approach to study the regulation of iron metabolism by oxidative stress. In Bioluminescence and Chemiluminescence; Hastings, J.W., Kricka, L.J., Stanley, P.E., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 1997; pp. 338–341. [Google Scholar]
- Pantopoulos, K.; Mueller, S.; Atzberger, A.; Ansorge, W.; Stremmel, W.; Hentze, M.W. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J. Biol. Chem. 1997, 272, 9802–9808. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Hebling, U.; Pons, A.; Mueller, S. Extracellular H2O2 and not superoxide determines the compartment-specific activation of transferrin receptor by iron regulatory protein 1. Free Radic. Res. 2005, 39, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Rausch, V.; Peccerella, T.; Millonig, G.; Mueller, S. Independent and additive induction of hepcidin by hypoxia and H2O2: Evidence for nox4-mediated iron signaling. Zeitschrift für Gastroenterologie 2016, 54, 1343–1404. [Google Scholar]
- Jiang, J.X.; Torok, N.J. Nadph oxidases in chronic liver diseases. Adv. Hepatol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Bland, J.M.; Kleinhenz, D.J.; Mitchell, P.O.; Walp, E.R.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am. J. Respir. Cell Mol. Biol. 2010, 42, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The nadph oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, W.; Zhong, W.; Sun, X.; Zhou, Z. Pharmacological inhibition of nox4 ameliorates alcohol-induced liver injury in mice through improving oxidative stress and mitochondrial function. Biochim. Biophys. Acta 2017, 1861, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tanaka, K.; Masaki, Y.; Miyazaki, M.; Kato, M.; Kotoh, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Intrahepatic microcirculatory disorder, parenchymal hypoxia and nox4 upregulation result in zonal differences in hepatocyte apoptosis following lipopolysaccharide- and d-galactosamine-induced acute liver failure in rats. Int. J. Mol. Med. 2014, 33, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Mueller, S. Established therapies and new therapeutic strategies in alcoholic liver disease. Zeitschrift für Gastroenterologie 2017, 55, 291–303. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Conditions | Hepcidin mRNA | Suggested Mechanism | Reference |
|---|---|---|---|---|
| HepG2 | 1% O2; 24 or 48 h | ↓ | - | [63] |
| 10, 2 or 0.1% O2; 24 h | ↓ | - | [64] | |
| 1% O2; 12 or 24 h | ↓ | Independent of HIF1 | [77] | |
| 1% O2; 16 h | ↑ | Independent of HIF1 and 2 | [67] | |
| Hep3B | 0.5% O2; 48 h | ↓ | Inhibition of BMP/SMAD pathway by HIF-mediated induction of matriptase-2 | [74] |
| Huh7 | 1% O2; 16 h | ↑ | Independent of HIF | [67] |
| 1% O2; 24 h | ↑ ↓ 1 | Hypoxia inhibits BMP/SMAD signaling pathway | [76] |
| Mice Model | Survival | Implications for Iron Metabolism | Other effects | Reference |
|---|---|---|---|---|
| Hif1α KO 1 | Not evaluated | No effect on hepcidin | - | [65] |
| Hif2α KO 1 | Not evaluated | No effect on hepcidin | - | [69] |
| Vhl KO 1 | 5 to 7 weeks | Decreased hepatic iron, decreased hepcidin, increased IL-6 and IL-1β | Growth deficiency, alopecia, hepatomegaly and splenomegaly. Liver necrosis and steatosis | [65] |
| Vhl/Hif1α KO 1 | 3 to 5 weeks | Low serum iron, decreased hepcidin | Alopecia, weight loss, hepatomegaly and splenomegaly | [69] |
| Hfe KO | Not evaluated | Increases serum and hepatic iron, decreased splenic iron, decreased hepcidin | - | [90] |
| Hjv KO | Not decreased | Sterile males | [91] | |
| Tfr2 KO | Not evaluated | - | [92] | |
| Smad4 KO 1 | Death during development | Weight loss, fibrosis, accumulation of neutrophiles and macrophages in the liver | [93] | |
| Bmp6 KO | Not evaluated | Delay in bone formation | [94] | |
| Tmprss6 KO (matriptase-2) | Not evaluated | Anemia, low iron stores, iron accumulation in enterocytes, increased hepcidin | Growth retardation. alopecia | [95] |
| Stat3 KO 1 | Not evaluated 2 | Increased hepcidin without developing anemia | Insulin resistance, increased gluconeogenesis, higher susceptibility to hepatic damaged | [96,97,98,99] |
| Prx2 KO | Not evaluated | No significant changes in hepcidin expression | Cardiovascular disease, splenomegaly, hemolytic anemia, inflammation, decreased immune function | [60,100] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, I.; Rausch, V.; Seitz, H.-K.; Mueller, S. Does Hypoxia Cause Carcinogenic Iron Accumulation in Alcoholic Liver Disease (ALD)? Cancers 2017, 9, 145. https://doi.org/10.3390/cancers9110145
Silva I, Rausch V, Seitz H-K, Mueller S. Does Hypoxia Cause Carcinogenic Iron Accumulation in Alcoholic Liver Disease (ALD)? Cancers. 2017; 9(11):145. https://doi.org/10.3390/cancers9110145
Chicago/Turabian StyleSilva, Inês, Vanessa Rausch, Helmut-Karl Seitz, and Sebastian Mueller. 2017. "Does Hypoxia Cause Carcinogenic Iron Accumulation in Alcoholic Liver Disease (ALD)?" Cancers 9, no. 11: 145. https://doi.org/10.3390/cancers9110145
APA StyleSilva, I., Rausch, V., Seitz, H.-K., & Mueller, S. (2017). Does Hypoxia Cause Carcinogenic Iron Accumulation in Alcoholic Liver Disease (ALD)? Cancers, 9(11), 145. https://doi.org/10.3390/cancers9110145