
The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
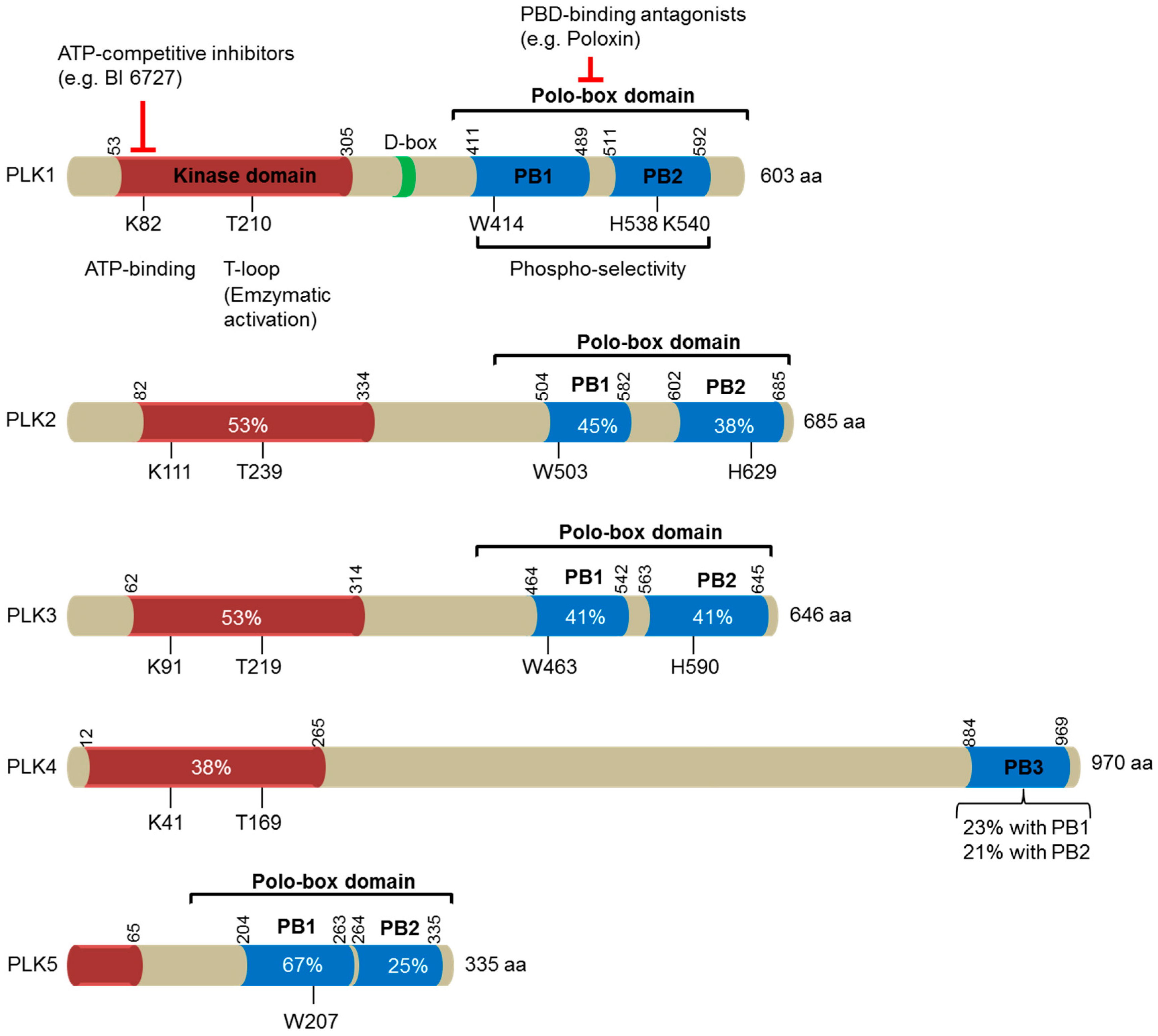
2. PLK1 in Tumor Development
2.1. PLK1 Expression in Human Cancers
2.2. PLK1 and Oncogenic Pathways
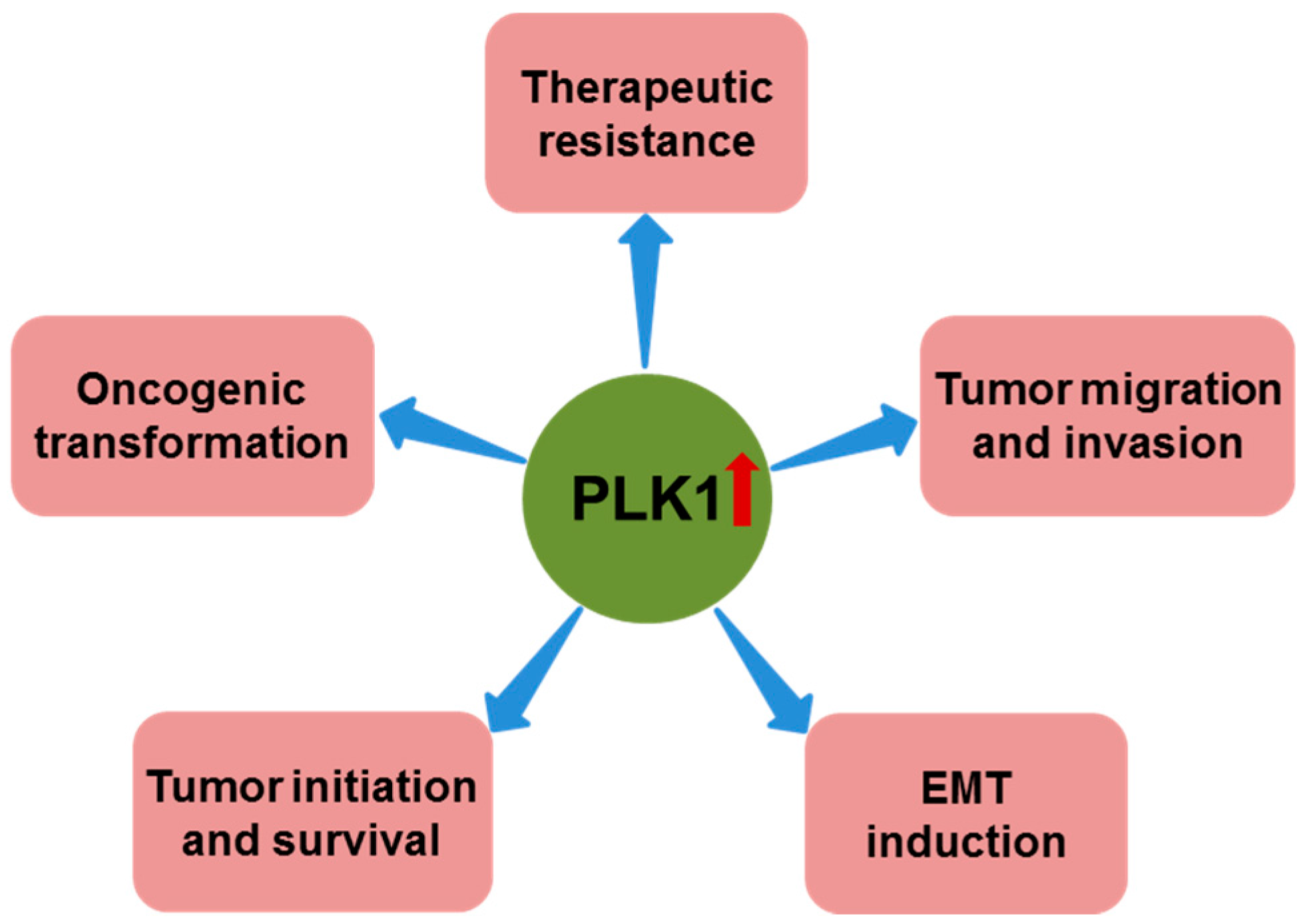
2.3. PLK1 and Oncogenic Transformation
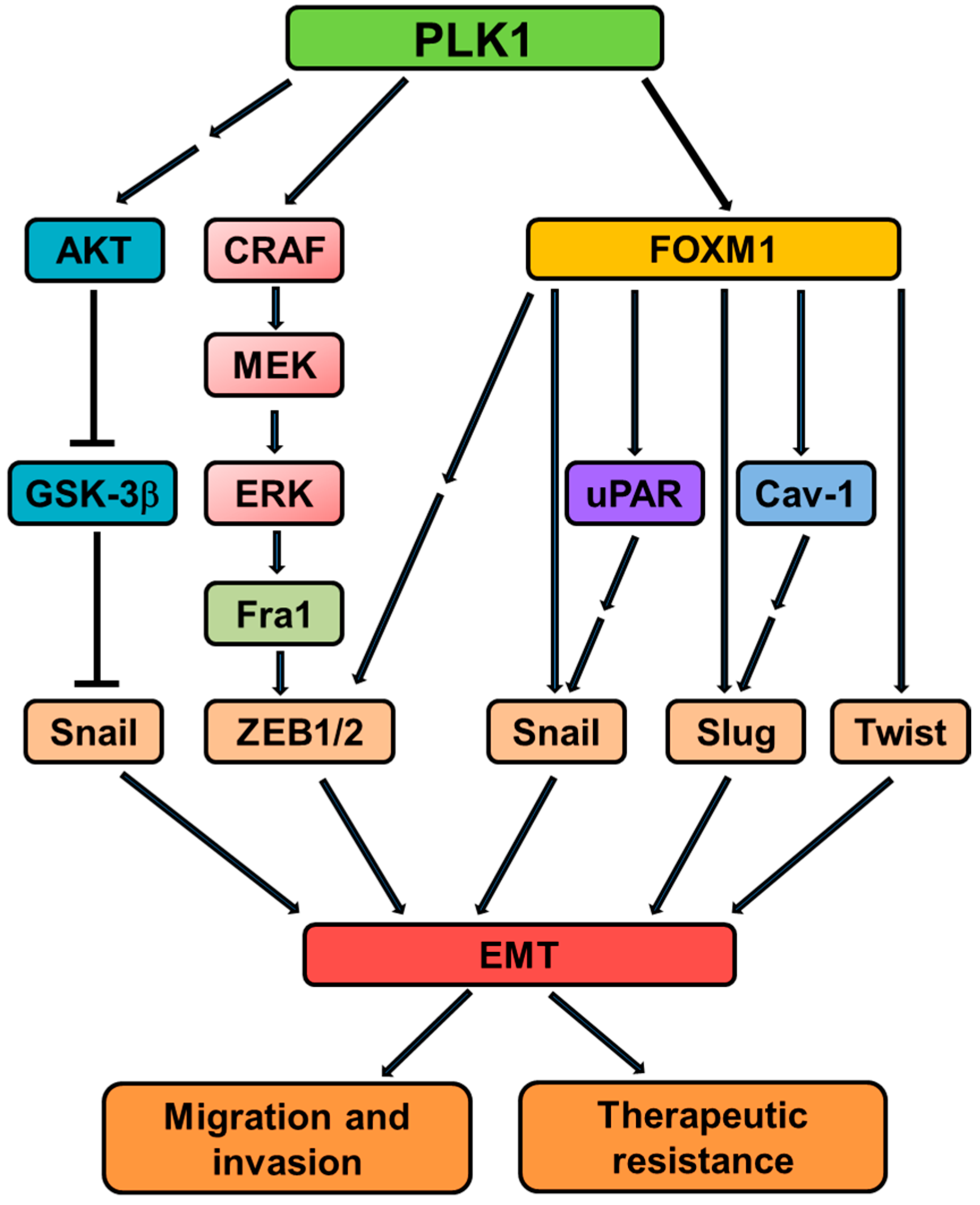
2.4. PLK1 and EMT
3. PLK1 in Tumor Invasion and Metastasis
4. PLK1 as a Key Target for Cancer Therapy
5. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Archambault, V.; Glover, D.M. Polo-like kinases: Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009, 10, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Sillje, H.H.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- van de Weerdt, B.C.; Medema, R.H. Polo-like kinases: A team in control of the division. Cell Cycle 2006, 5, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Sunkel, C.E.; Glover, D.M. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. 1988, 89, 25–38. [Google Scholar] [PubMed]
- de Carcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Winkles, J.A.; Alberts, G.F. Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 2005, 24, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.; Maloid, S.; Smith, M.R.; O’Connell, C.D.; Longo, D.L.; Ferris, D.K. Cloning and characterization of human and murine homologues of the Drosophila polo serine-threonine kinase. Cell Growth Differ. 1994, 5, 249–257. [Google Scholar] [PubMed]
- Elia, A.E.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Donaldson, M.M.; Tavares, A.A.; Hagan, I.M.; Nigg, E.A.; Glover, D.M. The mitotic roles of Polo-like kinase. J. Cell Sci. 2001, 114, 2357–2358. [Google Scholar] [PubMed]
- Lee, K.S.; Grenfell, T.Z.; Yarm, F.R.; Erikson, R.L. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc. Natl. Acad. Sci. USA 1998, 95, 9301–9306. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Soung, N.K.; Johmura, Y.; Kang, Y.H.; Liao, C.; Lee, K.H.; Park, C.H.; Nicklaus, M.C.; Lee, K.S. Polo-box domain: A versatile mediator of polo-like kinase function. Cell Mol. Life Sci. 2010, 67, 1957–1970. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Lin, C.Y.; Ma, S.; Erikson, R.L. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Song, B.; Liu, X. The substrates of Plk1, beyond the functions in mitosis. Protein Cell 2010, 1, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Liu, X.S.; Davis, K.; Liu, X. Plk1 phosphorylation of Orc2 promotes, D.N.A replication under conditions of stress. Mol. Cell. Biol. 2011, 31, 4844–4856. [Google Scholar] [CrossRef] [PubMed]
- Cholewa, B.D.; Liu, X.; Ahmad, N. The role of polo-like kinase 1 in carcinogenesis: Cause or consequence? Cancer Res. 2013, 73, 6848–6855. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Hamanaka, R.; Yoshimatsu, J.; Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 2005, 24, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Deeraksa, A.; Pan, J.; Sha, Y.; Liu, X.D.; Eissa, N.T.; Lin, S.H.; Yu-Lee, L.Y. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 2012, 32, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, X. Polo-like kinase 1, on the rise from cell cycle regulation to prostate cancer development. Protein Cell 2012, 3, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Golsteyn, R.M.; Mundt, K.E.; Fry, A.M.; Nigg, E.A. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J. Cell Biol. 1995, 129, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Yuan, Y.L.; Kuriyama, R.; Erikson, R.L. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol. Cell. Biol. 1995, 15, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Schmidt, M.; Gekeler, V.; Denkert, C.; Stephan, C.; Jung, K.; Loening, S.; Dietel, M.; Kristiansen, G. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004, 60, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rubsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Knecht, R.; Elez, R.; Oechler, M.; Solbach, C.; von Ilberg, C.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 1999, 59, 2794–2797. [Google Scholar] [PubMed]
- Knecht, R.; Oberhauser, C.; Strebhardt, K. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int. J. Cancer 2000, 89, 535–536. [Google Scholar] [CrossRef]
- Tokumitsu, Y.; Mori, M.; Tanaka, S.; Akazawa, K.; Nakano, S.; Niho, Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int. J. Oncol. 1999, 15, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Kneisel, L.; Linhart, C.; Bernd, A.; Kaufmann, R. Prognostic value of pololike kinase expression in melanomas. JAMA 2000, 283, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Hildenbrand, R.; Schwar, C.; Grobholz, R.; Kaufmann, M.; Stutte, H.J.; Strebhardt, K.; Bleyl, U. Polo-like kinase: A novel marker of proliferation: Correlation with estrogen-receptor expression in human breast cancer. Pathol. Res. Pract. 2000, 196, 753–759. [Google Scholar] [PubMed]
- Weichert, W.; Denkert, C.; Schmidt, M.; Gekeler, V.; Wolf, G.; Kobel, M.; Dietel, M.; Hauptmann, S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br. J. Cancer 2004, 90, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Miyazaki, T.; Fujisawa, K.; Nasu, K.; Hamanaka, R.; Miyakawa, I. Polo-like kinase (PLK) expression in endometrial carcinoma. Cancer Lett. 2001, 169, 41–49. [Google Scholar] [CrossRef]
- Takahashi, T.; Sano, B.; Nagata, T.; Kato, H.; Sugiyama, Y.; Kunieda, K.; Kimura, M.; Okano, Y.; Saji, S. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003, 94, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Dietzmann, K.; Kirches, E.; von, B.; Jachau, K.; Mawrin, C. Increased human polo-like kinase-1 expression in gliomas. J. Neurooncol. 2001, 53, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Miyoshi, E.; Sasaki, N.; Kakudo, K.; Yoshida, H.; Tomoda, C.; Uruno, T.; Takamura, Y.; Miya, A.; Kobayashi, K.; et al. Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br. J. Cancer 2004, 90, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Mok, W.C.; Wasser, S.; Tan, T.; Lim, S.G. Polo-like kinase 1, a new therapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 3527–3536. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.B.; Lin, D.C.; Shi, Z.Z.; Wang, X.C.; Shen, X.M.; Zhang, Y.; Du, X.L.; Luo, M.L.; Xu, X.; Han, Y.L.; et al. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of survivin level in esophageal squamous cell carcinoma. Int J. Cancer 2009, 124, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, S.; Saito, H.; Tsujitani, S.; Matsumoto, S.; Tatebe, S.; Kondo, A.; Ozaki, M.; Ito, H.; Ikeguchi, M. Expression of polo-like kinase 1 (PLK1) protein predicts the survival of patients with gastric carcinoma. Oncology 2006, 70, 126–133. [Google Scholar] [CrossRef] [PubMed]
- King, S.I.; Purdie, C.A.; Bray, S.E.; Quinlan, P.R.; Jordan, L.B.; Thompson, A.M.; Meek, D.W. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcom. Breast Cancer Res. 2012, 14, R40. [Google Scholar] [CrossRef] [PubMed]
- Tut, T.G.; Lim, S.H.; Dissanayake, I.U.; Descallar, J.; Chua, W.; Ng, W.; de Souza, P.; Shin, J.S.; Lee, C.S. Upregulated Polo-Like Kinase 1 Expression Correlates with Inferior Survival Outcomes in Rectal Cancer. PLoS ONE 2015, 10, e0129313. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Kristiansen, G.; Schmidt, M.; Gekeler, V.; Noske, A.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase 1 expression is a prognostic factor in human colon cancer. World J. Gastroenterol. 2005, 11, 5644–5650. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Kristiansen, G.; Winzer, K.J.; Schmidt, M.; Gekeler, V.; Noske, A.; Muller, B.M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Ohira, M.; Horie, H.; Ando, K.; Takayasu, H.; Suzuki, Y.; Sugano, S.; Hirata, T.; Goto, T.; Matsunaga, T.; et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: Identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene 2004, 23, 5901–5911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, G.; Kong, C. High expression of polo-like kinase 1 is associated with the metastasis and recurrence in urothelial carcinoma of bladder. Urol. Oncol. 2013, 31, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Han, D.P.; Zhu, Q.L.; Cui, J.T.; Wang, P.X.; Qu, S.; Cao, Q.F.; Zong, Y.P.; Feng, B.; Zheng, M.H.; Lu, A.G. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med. Sci. Monit. 2012, 18, BR237–BR246. [Google Scholar] [CrossRef] [PubMed]
- Rizki, A.; Mott, J.D.; Bissell, M.J. Polo-like kinase 1 is involved in invasion through extracellular matrix. Cancer Res. 2007, 67, 11106–11110. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ivanov, A.I.; Fisher, P.B.; Fu, Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016, 5, e10734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Z.; Liu, Z. Polo-like kinase 1 is overexpressed in renal cancer and participates in the proliferation and invasion of renal cancer cells. Tumour Biol. 2013, 34, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.G.; Lu, X.F.; Jiao, X.M.; Chen, B.; Wu, J.X. PLK1 gene suppresses cell invasion of undifferentiated thyroid carcinoma through the inhibition of CD44v6, MMP-2 and MMP-9. Exp. Ther. Med. 2012, 4, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.S.; Hogan, C.; Ochocka, A.M.; Meek, D.W. Polo-like kinase-1 phosphorylates MDM2 at Ser260 and stimulates MDM2-mediated p53 turnover. FEBS Lett. 2009, 583, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Zhou, Z.; Wang, W.H.; Deng, A.; Andrisani, O.; Liu, X. Plk1-mediated phosphorylation of Topors regulates p53 stability. J. Biol. Chem. 2009, 284, 18588–18592. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Li, H.; Song, B.; Liu, X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. 2010, 11, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Pagano, M.; Dai, W. Plk1 protein phosphorylates phosphatase and tensin homolog (PTEN) and regulates its mitotic activity during the cell cycle. J. Biol. Chem. 2014, 289, 14066–14074. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Bi, P.; Lu, Y.; Burcham, G.; Elzey, B.D.; Ratliff, T.; Konieczny, S.F.; Ahmad, N.; Kuang, S.; et al. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol. Cell. Biol. 2014, 34, 3642–3661. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Song, B.; Elzey, B.D.; Ratliff, T.L.; Konieczny, S.F.; Cheng, L.; Ahmad, N.; Liu, X. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J. Biol. Chem. 2011, 286, 35795–35800. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, L.; Xu, X.; Li, J.; Stern, D.F. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J. Biol. Chem. 2003, 278, 8468–8475. [Google Scholar] [CrossRef] [PubMed]
- Chabalier-Taste, C.; Brichese, L.; Racca, C.; Canitrot, Y.; Calsou, P.; Larminat, F. Polo-like kinase 1 mediates BRCA1 phosphorylation and recruitment at DNA double-strand breaks. Oncotarget 2016, 7, 2269–2283. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Daniels, M.J.; Venkitaraman, A.R. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 2004, 23, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hwang, H.I.; Jang, Y.J. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle 2010, 9, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Deming, P.B.; Flores, K.G.; Downes, C.S.; Paules, R.S.; Kaufmann, W.K. ATR enforces the topoisomerase II-dependent G2 checkpoint through inhibition of Plk1 kinase. J. Biol. Chem. 2002, 277, 36832–36838. [Google Scholar] [CrossRef] [PubMed]
- Elowe, S.; Hummer, S.; Uldschmid, A.; Li, X.; Nigg, E.A. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007, 21, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Matsumoto, Y.; Ikeuchi, T.; Saya, H.; Kajii, T.; Matsuura, S. BubR1 localizes to centrosomes and suppresses centrosome amplification via regulating Plk1 activity in interphase cells. Oncogene 2009, 28, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Sowa, M.E.; Nalepa, G.; Gygi, S.P.; Harper, J.W.; Elledge, S.J. The tumor suppressor CYLD regulates entry into mitosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8869–8874. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.L.; Mondal, G.; Hartman, J.K.; Tyagi, S.; Kurley, S.J.; Bland, C.S.; Hsu, T.Y.; Renwick, A.; Fang, J.E.; Migliaccio, I.; et al. The oncogenic STP axis promotes triple-negative breast cancer via degradation of the REST tumor suppressor. Cell Rep. 2014, 9, 1318–1332. [Google Scholar] [CrossRef] [PubMed]
- Astrinidis, A.; Senapedis, W.; Henske, E.P. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum. Mol. Genet. 2006, 15, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Hanneder, M.; Siegel, N.; Valli, A.; Hengstschlager, M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 2008, 658, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. FOX(M1) news—It is cancer. Mol. Cancer Ther. 2013, 12, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell. Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Li, X.; Bieberich, C.J. Protein kinase A regulates MYC protein through transcriptional and post-translational mechanisms in a catalytic subunit isoform-specific manner. J. Biol. Chem. 2013, 288, 14158–14169. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.; Wee, Z.N.; et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Yue, M.; Su, H.; Ren, P.; Jiang, J.; Li, F.; Hu, Y.; Du, H.; Liu, H.; Qing, G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol. Cell 2016, 64, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Wilson, M.L.; Hamanaka, R.; Chase, D.; Kung, H.; Longo, D.L.; Ferris, D.K. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem. Biophys. Res. Commun. 1997, 234, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial—Mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J. Cell Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Zhau, H.Y.; Chang, S.M.; Chen, B.Q.; Wang, Y.; Zhang, H.; Kao, C.; Sang, Q.A.; Pathak, S.J.; Chung, L.W. Androgen-repressed phenotype in human prostate cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 15152–15157. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, R.; Xie, Z.H.; Odero-Marah, V.; Pathak, S.; Multani, A.; Chung, L.W.; Zhau, H.E. Prostate cancer metastasis: Role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate 2006, 66, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.; Kramerov, D.; Grigorian, M.; Berezin, V.; Bock, E.; Lukanidin, E.; Tulchinsky, E. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol. Cell. Biol. 1998, 18, 7095–7105. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K. The Fos family of transcription factors and their role in tumourigenesis. Eur. J. Cancer 2005, 41, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Young, M.R.; Colburn, N.H. Fra-1 a target for cancer prevention or intervention. Gene 2006, 379, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.P.; Chen, L.D.; Song, H.B.; Zhang, C.X.; Yuan, Z.W.; Xiang, Z.X. PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am. J. Transl. Res. 2016, 8, 4172–4183. [Google Scholar] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J. Cell Biochem. 2011, 112, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, H.; Tan, G.; Gao, W.; Cheng, L.; Jiang, X.; Yu, L.; Tan, Y. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett. 2013, 340, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, B.; Zhang, M.; Fu, S.; Gao, H.; Peng, R.; Zhang, L.; Tang, J. Increased FoxM1 expression is a target for metformin in the suppression of EMT in prostate cancer. Int. J. Mol. Med. 2014, 33, 1514–1522. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Xiong, X.; Lin, Y.; Cheng, Y.; Lu, J.; Zhang, J.; Cheng, N. Down-regulation of FoxM1 leads to the inhibition of the epithelial-mesenchymal transition in gastric cancer cells. Cancer Genet. 2014, 207, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.F.; Qu, Z.Q.; Yuan, H.H.; Wang, J.Y.; Zhao, M.; Guo, Y.H.; Shi, J.; Gong, X.D.; Zhu, Y.L.; Liu, F.; et al. Overexpression of FOXM1 is associated with EMT and is a predictor of poor prognosis in non-small cell lung cancer. Oncol Rep. 2014, 31, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Zhang, N.; Wang, Y.; Li, D.; Wang, L.; Sun, X.; Shen, C.; Yang, Y.; Zhou, X.; Du, X. FOXM1 promotes lung adenocarcinoma invasion and metastasis by upregulating SNAIL. Int. J. Biol. Sci. 2015, 11, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Wu, S.; Wu, H.; Li, L.; Guo, T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/beta-catenin-FoxM1-Twist signaling. Mol. Carcinog. 2016, 55, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Brassesco, M.S.; Pezuk, J.A.; Morales, A.G.; de Oliveira, J.C.; Roberto, G.M.; da Silva, G.N.; Francisco de Oliveira, H.; Scrideli, C.A.; Tone, L.G. In vitro targeting of Polo-like kinase 1 in bladder carcinoma: Comparative effects of four potent inhibitors. Cancer Biol. Ther. 2013, 14, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer Gene Ther. 2013, 20, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Godde, N.J.; Galea, R.C.; Elsum, I.A.; Humbert, P.O. Cell polarity in motion: Redefining mammary tissue organization through EMT and cell polarity transitions. J. Mammary Gland Biol. Neoplasia 2010, 15, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Kamelgarn, M.; Kyprianou, N. Cytoskeleton targeting value in prostate cancer treatment. Am. J. Clin. Exp. Urol. 2014, 2, 15–26. [Google Scholar] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Efferth, T. Polo-like kinase 1 as target for cancer therapy. Exp. Hematol. Oncol. 2012, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Kappel, S.; Kramer, A.; Sanhaji, M.; Matthess, Y.; Kurunci-Csacsko, E.; Calzada-Wack, J.; Rathkolb, B.; Rozman, J.; Adler, T.; et al. Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat. Commun. 2011, 2, 395. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Hymel, D.; Burke, T.R., Jr.; Lee, K.S. Current progress and future perspectives in the development of anti-polo-like kinase 1 therapeutic agents. F1000Res 2017, 6, 1024. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [PubMed]
- Results of Phase III Study of Volasertib for the Treatment of Acute Myeloid Leukemia Presented at European Hematology Association Annual Meeting. Available online: http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=649494 (accessed on September 2017).
- Gjertsen, B.T.; Schoffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Archambault, V.; Normandin, K. Several inhibitors of the Plk1 Polo-Box Domain turn out to be non-specific protein alkylators. Cell Cycle 2017, 16, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Burke, T.R., Jr.; Park, J.E.; Bang, J.K.; Lee, E. Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Park, J.E.; Qian, W.J.; Lim, D.; Graber, M.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat. Chem. Biol. 2011, 7, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Park, J.E.; Qian, W.J.; Lim, D.; Scharow, A.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem. Biol. 2012, 7, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.J.; Park, J.E.; Lim, D.; Lai, C.C.; Kelley, J.A.; Park, S.Y.; Lee, K.W.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Mono-anionic phosphopeptides produced by unexpected histidine alkylation exhibit high Plk1 polo-box domain-binding affinities and enhanced antiproliferative effects in HeLa cells. Biopolymers 2014, 102, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Gu, S.; Reda, M.; Castro, D.J.; Sangvanich, T.; Gray, J.W.; Yantasee, W. Targeted Treatment of Metastatic Breast Cancer by, PLK1 siRNA Delivered by an Antioxidant Nanoparticle Platform. Mol. Cancer Ther. 2017, 16, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Hua, E.; Bisht, K.; Woditschka, S.; Skordos, K.W.; Liewehr, D.J.; Steinberg, S.M.; Brogi, E.; Akram, M.M.; Killian, J.K.; et al. Inhibition of Polo-like kinase 1 prevents the growth of metastatic breast cancer cells in the brain. Clin. Exp. Metastasis 2011, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; Andre, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, R.; Wang, L.; Nilsson, M.; Goonatilake, R.; Tong, P.; Li, L.; Giri, U.; Villalobos, P.; Mino, B.; et al. Polo-like kinase 1 inhibition diminishes acquired resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer with T790M mutations. Oncotarget 2016, 7, 47998–48010. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Rubio-Viqueira, B.; Rajeshkumar, N.V.; Chan, A.; sSolomon, A.; Hidalgo, M. A fine-needle aspirate-based vulnerability assay identifies polo-like kinase 1 as a mediator of gemcitabine resistance in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Spankuch, B.; Heim, S.; Kurunci-Csacsko, E.; Lindenau, C.; Yuan, J.; Kaufmann, M.; Strebhardt, K. Down-regulation of Polo-like kinase 1 elevates drug sensitivity of breast cancer cells in vitro and in vivo. Cancer Res. 2006, 66, 5836–5846. [Google Scholar] [CrossRef] [PubMed]
- Spankuch, B.; Kurunci-Csacsko, E.; Kaufmann, M.; Strebhardt, K. Rational combinations of siRNAs targeting Plk1 with breast cancer drugs. Oncogene 2007, 26, 5793–5807. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, X.; Sun, G.; Guo, X.; Li, H.; You, Y.; Jacobs, J.L.; Gardner, K.; Yuan, D.; Xu, Z.; et al. RNA interference-mediated silencing of the polo-like kinase 1 gene enhances chemosensitivity to gemcitabine in pancreatic adenocarcinoma cells. J. Cell. Mol. Med. 2008, 12, 2334–2349. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Z.; Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers 2017, 9, 131. https://doi.org/10.3390/cancers9100131
Fu Z, Wen D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers. 2017; 9(10):131. https://doi.org/10.3390/cancers9100131
Chicago/Turabian StyleFu, Zheng, and Donghua Wen. 2017. "The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis" Cancers 9, no. 10: 131. https://doi.org/10.3390/cancers9100131
APA StyleFu, Z., & Wen, D. (2017). The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers, 9(10), 131. https://doi.org/10.3390/cancers9100131