
Melanoma and the Unfolded Protein Response

Abstract
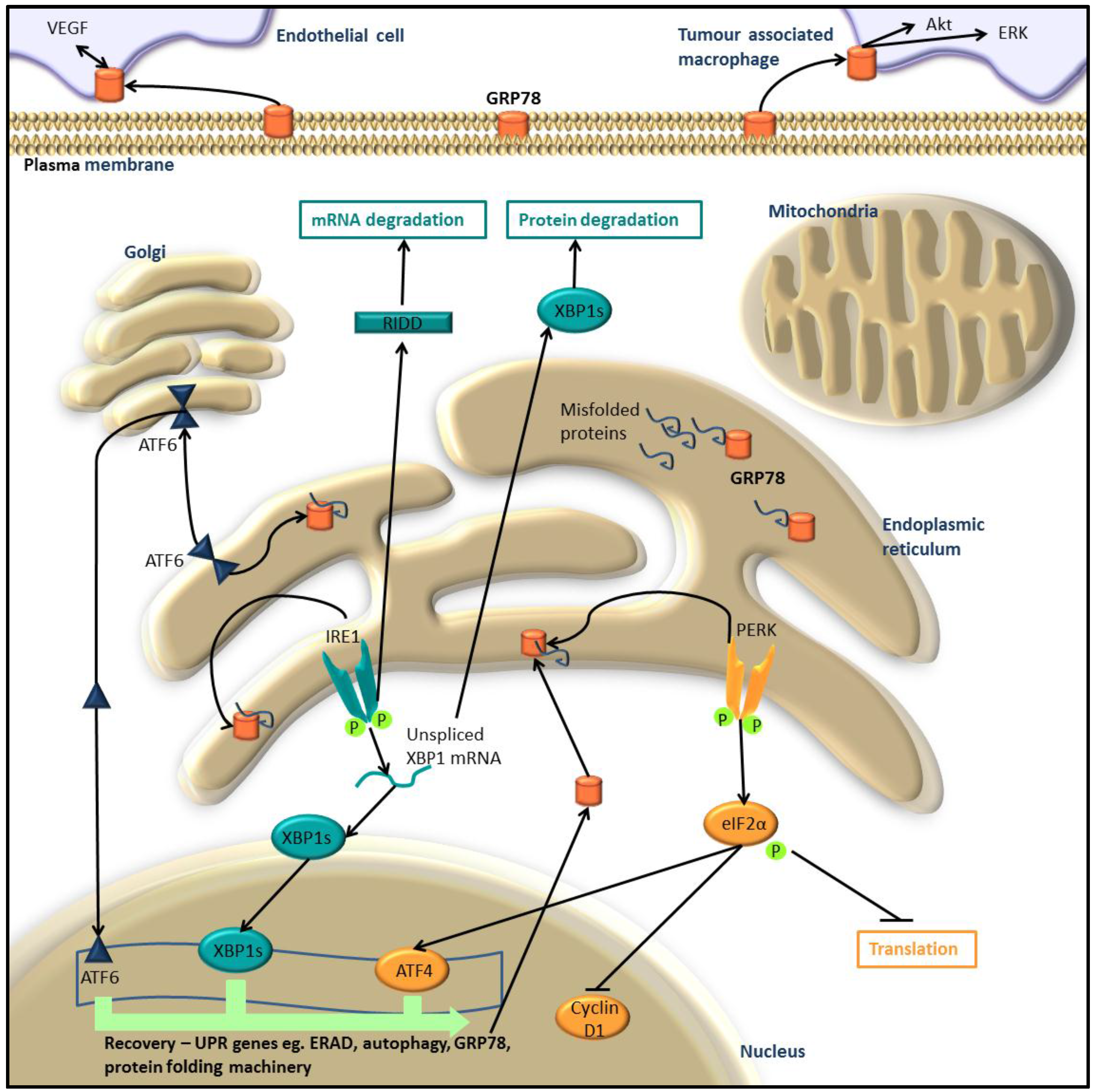
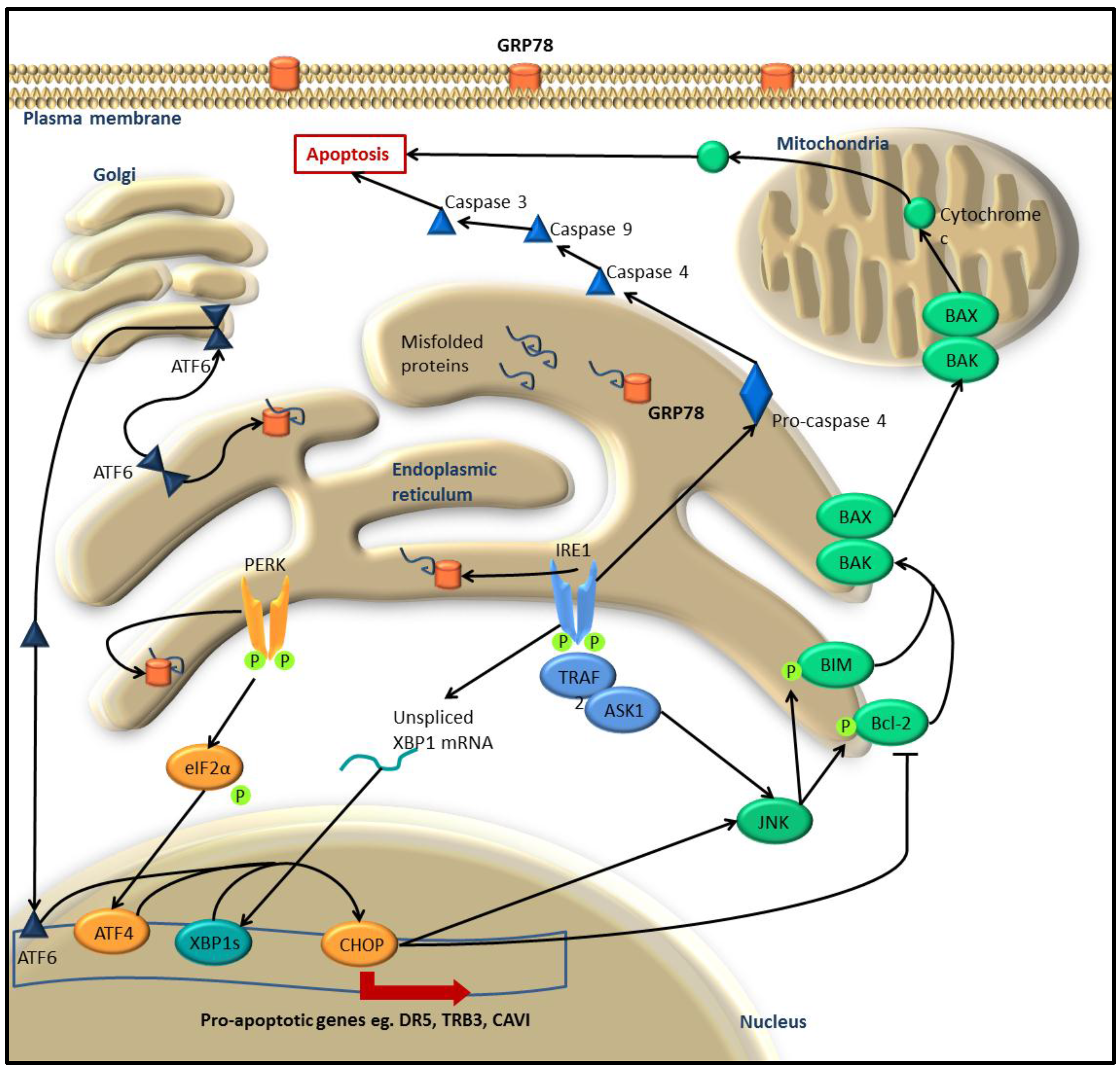
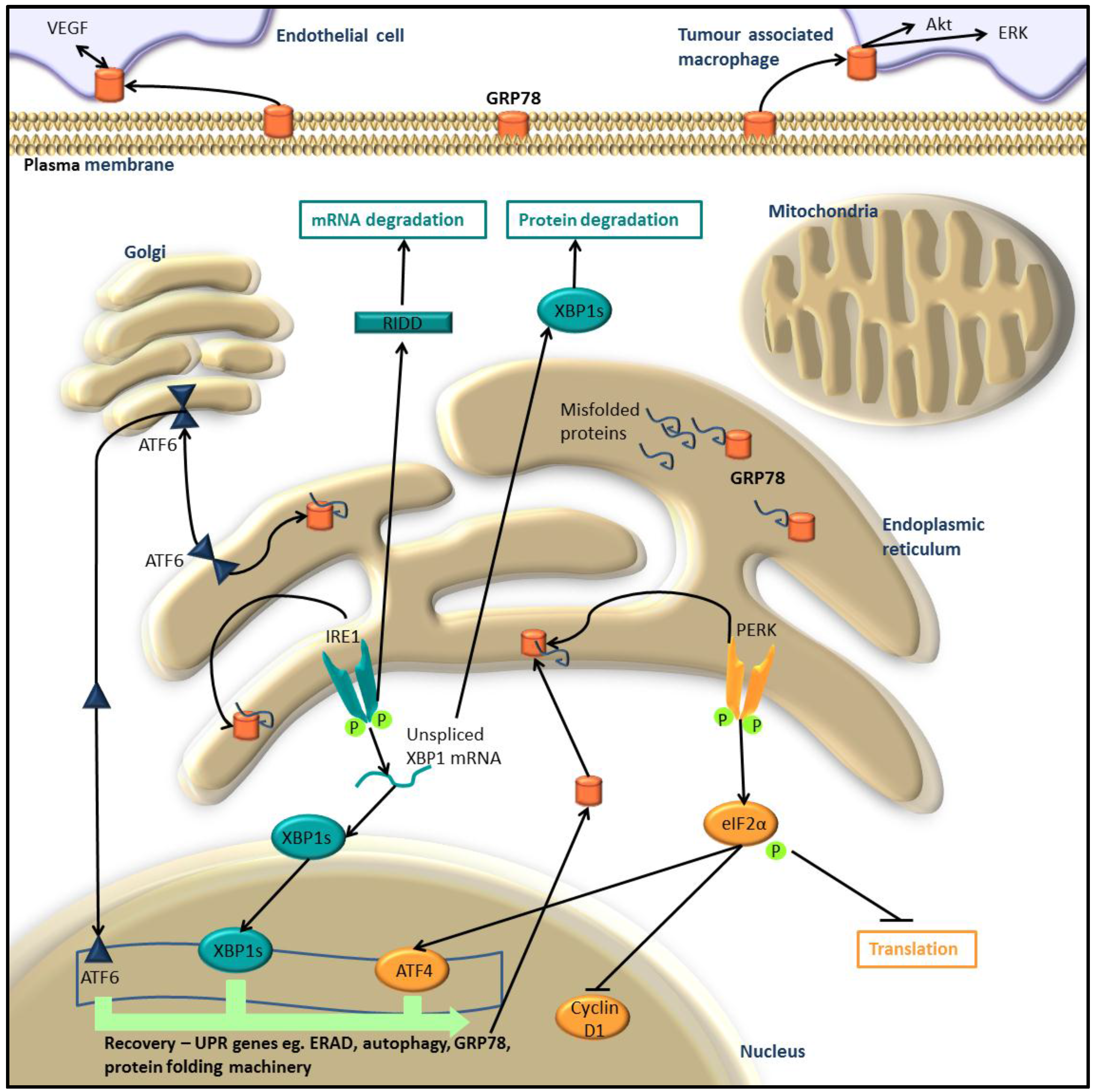
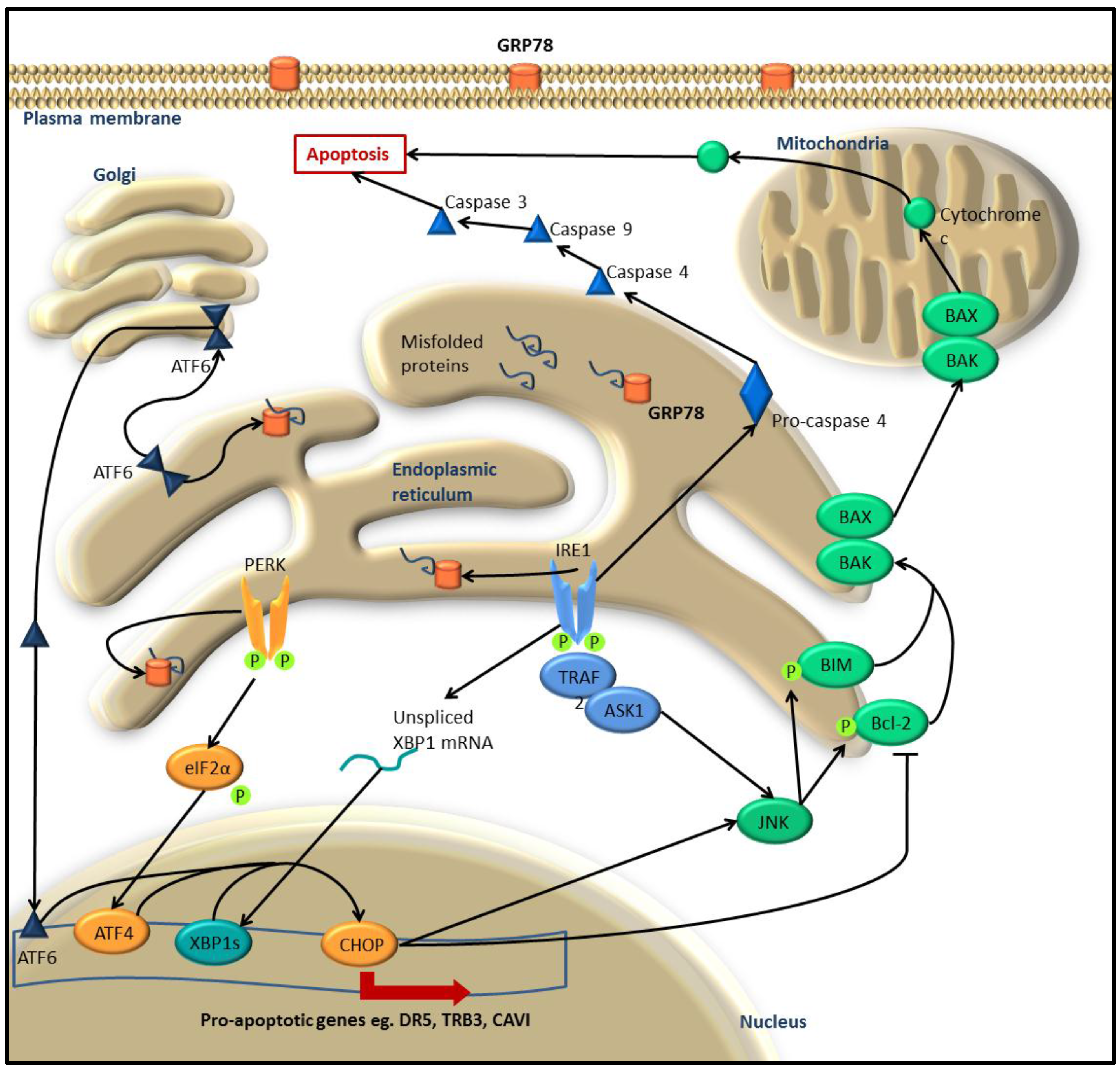
:1. The Unfolded Protein Response
1.1. Activation of the UPR
1.2. Return to Homeostasis
1.3. UPR-Induced Apoptosis
2. UPR in Melanoma and Other Cancers
UPR and MEK/ERK
3. UPR and Chemotherapy
3.1. Drug Resistance
3.2. The UPR: As a Drug Target
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Almen, M.S.; Nordstrom, K.J.; Fredriksson, R.; Schioth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator grp78/bip in development, cancer, and neurological disorders. Antioxid. Redox Signal 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Prischi, F.; Nowak, P.R.; Carrara, M.; Ali, M.M.U. Phosphoregulation of ire1 rnase splicing activity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Kondratyev, M.; Avezov, E.; Shenkman, M.; Groisman, B.; Lederkremer, G.Z. Perk-dependent compartmentalization of erad and unfolded protein response machineries during er stress. Exp. Cell Res. 2007, 313, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for xbp1/hac1 splicing and ridd. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, T.; Milani, M.; Singleton, D.C.; Harris, A.L. Role of atf4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009, 8, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by gadd34-mediated dephosphorylation of eif2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eif2α dephosphorylation protects cells from er stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. Er stress regulation of atf6 localization by dissociation of bip/grp78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eif2 kinase perk and the integrated stress response facilitate activation of atf6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. Xbp1 mrna is induced by atf6 and spliced by ire1 in response to er stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. Ire1: Er stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Eif2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Anderson, P. Reprogramming mrna translation during stress. Curr. Opin. Cell Biol. 2008, 20, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. Xbp1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Prywes, R. Er stress signaling by regulated proteolysis of atf6. Methods 2005, 35, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Shan, J.; Su, N. Atf4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metabol. 2009, 20, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, J.G.; Carlisle, R.E.; Jerome, D.E.; Mohammed-Ali, Z.; Jiang, H.; Yang, G.; Mani, S.; Garg, S.K.; Banerjee, R.; Kaufman, R.J.; et al. Integrated stress response modulates cellular redox state via induction of cystathionine γ-lyase: Cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem. 2012, 287, 7603–7614. [Google Scholar] [CrossRef] [PubMed]
- Merksamer, P.I.; Trusina, A.; Papa, F.R. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 2008, 135, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. The unfolding tale of the unfolded protein response. Cell 2001, 107, 827–830. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and er-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Ishiguro, M.; Niinuma, Y.; Uesugi, M.; Nomura, Y. Human hrd1 protects against er stress-induced apoptosis through er-associated degradation. FEBS Lett. 2002, 532, 147–152. [Google Scholar] [CrossRef]
- Kong, B.; Wu, W.; Valkovska, N.; Jäger, C.; Hong, X.; Nitsche, U.; Friess, H.; Esposito, I.; Erkan, M.; Kleeff, J.; et al. A common genetic variation of melanoma inhibitory activity-2 labels a subtype of pancreatic adenocarcinoma with high endoplasmic reticulum stress levels. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-H.; Piao, S.; Wang, D.; Mcafee, Q.W.; Nathanson, K.L.; Lum, J.J.; Li, L.Z.; Amaravadi, R.K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Minami, M.; Takeda, K.; Sakao, Y.; Akira, S. Ectopic expression of chop (gadd153) induces apoptosis in m1 myeloblastic leukemia cells. FEBS Lett. 1996, 395, 143–147. [Google Scholar] [CrossRef]
- Maytin, E.V.; Ubeda, M.; Lin, J.C.; Habener, J.F. Stress-inducible transcription factor chop/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001, 267, 193–204. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic bax and bak modulate the unfolded protein response by a direct interaction with ire1alpha. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the er to activation of jnk protein kinases by transmembrane protein kinase ire1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (er) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the er stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [PubMed]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein grp78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Lai, M.; Wang, Y.; Xu, E.; Huang, Q. Overexpression of glucose-regulated protein 78 in colon cancer. Clin. Chim. Acta 2006, 364, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, Y.; Jia, Z.; Li, Q.; Gong, W.; Wang, L.; Wei, D.; Yao, J.; Fang, S.; Xie, K. Association of elevated grp78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metast. 2006, 23, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Bini, L.; Magi, B.; Marzocchi, B.; Arcuri, F.; Tripodi, S.; Cintorino, M.; Sanchez, J.C.; Frutiger, S.; Hughes, G.; Pallini, V. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis 1997, 18, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene grp78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Koomägi, R.; Mattern, J.; Volm, M. Glucose-related protein (grp78) and its relationship to the drug-resistance proteins p170, gst-pi, lrp56 and angiogenesis in non-small cell lung carcinomas. Anticancer Res. 1998, 19, 4333–4336. [Google Scholar]
- Zhuang, L.; Scolyer, R.A.; Lee, C.S.; McCarthy, S.W.; Cooper, W.A.; Zhang, X.D.; Thompson, J.F.; Hersey, P. Expression of glucose-regulated stress protein grp78 is related to progression of melanoma. Histopathol 2009, 54, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Chen, X.; Ma, Y.; Tang, L.; Guan, L.; Ren, X.; Yu, B.; Zhang, W.; Su, B. Mda-9 and grp78 as potential diagnostic biomarkers for early detection of melanoma metastasis. Tumor Biol. 2015, 36, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Tay, K.H.; Luan, Q.; Croft, A.; Jiang, C.C.; Jin, L.; Zhang, X.D.; Tseng, H.Y. Sustained ire1 and atf6 signaling is important for survival of melanoma cells undergoing er stress. Cell. Signal. 2014, 26, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wey, S.; Wang, M.; Ye, R.; Liao, C.-P.; Roy-Burman, P.; Lee, A.S. Pten null prostate tumorigenesis and akt activation are blocked by targeted knockout of er chaperone grp78/bip in prostate epithelium. Proc. Natl. Acad. Sci. USA 2008, 105, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L. Critical role of the stress chaperone grp78/bip in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Croft, A.; Tay, K.H.; Boyd, S.C.; Guo, S.T.; Jiang, C.C.; Lai, F.; Tseng, H.Y.; Jin, L.; Rizos, H.; Hersey, P.; et al. Oncogenic activation of mek/erk primes melanoma cells for adaptation to endoplasmic reticulum stress. J. Invest. Dermatol. 2014, 134, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. Xbp1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ramirez, L.; Cao, H.; Regalado, M.P.; Kambham, N.; Siemann, D.; Kim, J.J.; Le, Q.T.; Koong, A.C. X box-binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl. Oncol. 2009, 2, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Drogat, B.; Auguste, P.; Nguyen, D.T.; Bouchecareilh, M.; Pineau, R.; Nalbantoglu, J.; Kaufman, R.J.; Chevet, E.; Bikfalvi, A.; Moenner, M. Ire1 signaling is essential for ischemia-induced vascular endothelial growth factor-a expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007, 67, 6700–6707. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. Er stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. Embo. J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Mintz, P.J.; Kim, J.; Do, K.A.; Wang, X.; Zinner, R.G.; Cristofanilli, M.; Arap, M.A.; Hong, W.K.; Troncoso, P.; Logothetis, C.J.; et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotech. 2003, 21, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chinni, S.R.; Falchetto, R.; Gercel-Taylor, C.; Shabanowitz, J.; Hunt, D.F.; Taylor, D.D. Humoral immune responses to cathepsin d and glucose-regulated protein 78 in ovarian cancer patients. Clin. Cancer Res. 1997, 3, 1557–1564. [Google Scholar] [PubMed]
- Selim, M.A.; Burchette, J.L.; Bowers, E.V.; de Ridder, G.G.; Mo, L.; Pizzo, S.V.; Gonzalez-Gronow, M. Changes in oligosaccharide chains of autoantibodies to grp78 expressed during progression of malignant melanoma stimulate melanoma cell growth and survival. Melanoma Res. 2011, 21, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; Dennert, G.; Lee, A.S. Inhibition of tumor progression by suppression of stress protein grp78/bip induction in fibrosarcoma b/c10me. Proc. Natl. Acad. Sci. USA 1996, 93, 7690–7694. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Lee, C.S.; Scolyer, R.A.; McCarthy, S.W.; Zhang, X.D.; Thompson, J.F.; Screaton, G.; Hersey, P. Progression in melanoma is associated with decreased expression of death receptors for tumor necrosis factor–related apoptosis-inducing ligand. Hum. Pathol. 2006, 37, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hua, J.; Wang, Q.; Xu, W.; Zhang, J.; Zhang, J.; Kang, J.; Li, M. Expressions of grp78 and bax associate with differentiation, metastasis, and apoptosis in non-small cell lung cancer. Mol. Biol. Rep. 2012, 39, 6753–6761. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Chen, W.Y.; Huang, C.Y.; Liu, H.H.; Wei, P.L. Glucose-regulated protein 78 (grp78) regulates colon cancer metastasis through emt biomarkers and the nrf-2/ho-1 pathway. Tumour Biol. 2015, 36, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Kang, J.; Jiao, K.; Xu, G.; Yang, L.; Tang, S.; Zhang, H.; Wang, Y.; Nie, Y.; Wu, K.; et al. High expression of grp78 promotes invasion and metastases in patients with esophageal squamous cell carcinoma. Dig. Dis. Sci. 2015, 60, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Ren, X.; Li, H.; Shull, A.; Kim, J.; Cowell, J.K. Mitochondrial atad3a combines with grp78 to regulate the wasf3 metastasis-promoting protein. Oncogene 2016, 35, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-X.; Li, H.-D.; Zhao, S.; Zhao, L.; Song, H.-J.; Wang, G.; Guo, Q.-J.; Luan, Z.-D.; Su, R.-J. The cell surface grp78 facilitates the invasion of hepatocellular carcinoma cells. Bio. Med. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, L.; Zhao, Y.; Li, H.; Xiao, H.; Fu, R.; Zhao, C.; Wu, H.; Li, Z. Cell-surface grp78 facilitates colorectal cancer cell migration and invasion. Int. J. Biochem. Cell Biol. 2013, 45, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Li, Z.; Li, H.; Song, H.; Bao, C.; Wei, J.; Cheng, L. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Papalas, J.A.; Vollmer, R.T.; Gonzalez-Gronow, M.; Pizzo, S.V.; Burchette, J.; Youens, K.E.; Johnson, K.B.; Selim, M.A. Patterns of grp78 and mtj1 expression in primary cutaneous malignant melanoma. Mod. Pathol. 2009, 23, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, X.; Gao, Y.; Wu, J.; Zeng, F.; Song, F. Xbp1 induces snail expression to promote epithelial-to-mesenchymal transition and invasion of breast cancer cells. Cell. Signal. 2015, 27, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic emt-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Alonso, S.R.; Tracey, L.; Ortiz, P.; Pérez-Gómez, B.; Palacios, J.; Pollán, M.; Linares, J.; Serrano, S.; Sáez-Castillo, A.I.; Sánchez, L.; et al. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007, 67, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Arap, M.A.; Lahdenranta, J.; Mintz, P.J.; Hajitou, A.; Sarkis, Á.S.; Arap, W.; Pasqualini, R. Cell surface expression of the stress response chaperone grp78 enables tumor targeting by circulating ligands. Cancer cell 2004, 6, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhang, Y.; Lee, A. Beyond the endoplasmic reticulum: Atypical grp78 in cell viability, Signal. and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, P.; Neshat, A.; Mokhtari, M.; Rajabi, M.A.; Eftekhari, M.; Tavakoli, P. The role of vegf in melanoma progression. J. Res. Med. Sci. 2012, 17, 534–539. [Google Scholar] [PubMed]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. Vegf signals through atf6 and perk to promote endothelial cell survival and angiogenesis in the absence of er stress. Mol. Cell. 2014, 54, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Untergasser, G.; Zenzmaier, C.; Sarg, B.; Gastl, G.; Gunsilius, E.; Steurer, M. Grp-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 2009, 114, 3960–3967. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of vegf-a by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef] [PubMed]
- Katanasaka, Y.; Ishii, T.; Asai, T.; Naitou, H.; Maeda, N.; Koizumi, F.; Miyagawa, S.; Ohashi, N.; Oku, N. Cancer antineovascular therapy with liposome drug delivery systems targeted to bip/grp78. Int. J. Cancer 2010, 127, 2685–2698. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Dubeau, L.; Bading, J.; Nguyen, K.; Luna, M.; Yu, H.; Gazit-Bornstein, G.; Gordon, E.M.; Gomer, C.; Hall, F.L.; et al. Spontaneous and controllable activation of suicide gene expression driven by the stress-inducible grp78 promoter resulting in eradication of sizable human tumors. Hum. Gene Therapy 2004, 15, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Weissman, A.M. The unfolded protein response, degradation from the endoplasmic reticulum, and cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, L.; Aguirre-Ghiso, J.A. Dormancy of metastatic melanoma. Pigment. Cell Melanoma Res. 2010, 23, 41. [Google Scholar] [CrossRef] [PubMed]
- Eskelin, S.; Pyrhonen, S.; Summanen, P.; Hahka-Kemppinen, M.; Kivela, T. Tumor doubling times in metastatic malignant melanoma of the uvea: Tumor progression before and after treatment. Ophthalmology 2000, 107, 1443–1449. [Google Scholar] [CrossRef]
- Logan, P.T.; Fernandes, B.F.; Di Cesare, S.; Marshall, J.C.; Maloney, S.C.; Burnier, M.N., Jr. Single-cell tumor dormancy model of uveal melanoma. Clin. Exp. Metast. 2008, 25, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.; Gauss, R.; Horn, S.C.; Neuber, O.; Sommer, T. The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Platz, A.; Egyhazi, S.; Ringborg, U.; Hansson, J. Human cutaneous melanoma; a review of nras and braf mutation frequencies in relation to histogenetic subclass and body site. Mol. Oncol. 2008, 1, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the braf gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Stokoe, D. Mechanisms of translational deregulation in human tumors and therapeutic intervention strategies. Oncogene 2007, 26, 5973–5990. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Chen, L.H.; Gillespie, S.; Wang, Y.F.; Kiejda, K.A.; Zhang, X.D.; Hersey, P. Inhibition of mek sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 2007, 67, 9750–9761. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.; Niessner, H.; Smalley, K.S.; Flaherty, K.; Paraiso, K.H.; Busch, C.; Sinnberg, T.; Vasseur, S.; Iovanna, J.L.; Driessen, S.; et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in brafv600e melanoma cells. Sci. Signal 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Al-Rawashdeh, F.Y.; Scriven, P.; Cameron, I.C.; Vergani, P.V.; Wyld, L. Unfolded protein response activation contributes to chemoresistance in hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ko, B.; Baumeister, P.; Swenson, S.; Costa, F.; Markland, F.; Stiles, C.; Patterson, J.B.; Bates, S.E.; Lee, A.S. Vascular targeting and antiangiogenesis agents induce drug resistance effector grp78 within the tumor microenvironment. Cancer Res. 2005, 65, 5785–5791. [Google Scholar] [CrossRef] [PubMed]
- Vincent, L.-A.; Attaoua, C.; Bellis, M.; Rozkydalova, L.; Hadj-Kaddour, K.; Vian, L.; Cuq, P. Lysosomes and unfolded protein response, determinants of differential resistance of melanoma cells to vinca alkaloids. Fundam. Clin. Pharmacol. 2015, 29, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. British J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Mathieu, V.; Kiss, R. Galectin-1-mediated biochemical controls of melanoma and glioma aggressive behavior. World J. Biol. Chem. 2011, 2, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choy, M.L.; Marks, P.A. Mechanisms of resistance to histone deacetylase inhibitors. Adv. Cancer Res. 2012, 116, 39–86. [Google Scholar]
- Fiskus, W.; Rao, R.; Fernandez, P.; Herger, B.; Yang, Y.; Chen, J.; Kolhe, R.; Mandawat, A.; Wang, Y.; Joshi, R.; et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor–resistant acute myeloid leukemia cells. Blood 2008, 112, 2896–2905. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Dedes, I.; Imesch, P.; von Bueren, A.O.; Fink, D.; Fedier, A. Acquired vorinostat resistance shows partial cross-resistance to 'second-generation' hdac inhibitors and correlates with loss of histone acetylation and apoptosis but not with altered hdac and hat activities. Anti-Cancer Drugs 2009, 20, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Guo, S.T.; Jin, L.; Jiang, C.C.; Wang, C.Y.; Croft, A.; Chi, M.N.; Tseng, H.Y.; Farrelly, M.; Atmadibrata, B.; et al. Cotargeting histone deacetylases and oncogenic braf synergistically kills human melanoma cells by necrosis independently of ripk1 and ripk3. Cell Death Dis. 2013, 4, e655. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Jin, L.; Gallagher, S.; Mijatov, B.; Zhang, X.D.; Hersey, P. Histone deacetylases (hdacs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective braf inhibitors. Advances Pharmacol. 2012, 65, 27–43. [Google Scholar]
- Baumeister, P.; Dong, D.; Fu, Y.; Lee, A.S. Transcriptional induction of grp78/bip by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis. Mol. Cancer Ther. 2009, 8, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Hensel, F.; Eckstein, M.; Rosenwald, A.; Brandlein, S. Early development of pat-sm6 for the treatment of melanoma. Melanoma Res. 2013, 23, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Morgner, C.; Chatterjee, M.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; Brändlein, S. The natural human igm antibody pat-sm6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein grp78. PLoS ONE 2013, 8, e63414. [Google Scholar] [CrossRef] [PubMed]
- Fasano, E.; Serini, S.; Piccioni, E.; Toesca, A.; Monego, G.; Cittadini, A.R.; Ranelletti, F.O.; Calviello, G. Dha induces apoptosis by altering the expression and cellular location of grp78 in colon cancer cell lines. Biochim. Biophys. Acta 2012, 1822, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Sit, W.H.; Fan, S.T.; Man, K.; Jor, I.W.; Wong, L.L.; Wan, M.L.; Tan-Un, K.C.; Wan, J.M. The cell cycle effects of docosahexaenoic acid on human metastatic hepatocellular carcinoma proliferation. Int. J. Oncol. 2010, 36, 991–998. [Google Scholar] [PubMed]
- Bougnoux, P.; Hajjaji, N.; Ferrasson, M.N.; Giraudeau, B.; Couet, C.; Le Floch, O. Improving outcome of chemotherapy of metastatic breast cancer by docosahexaenoic acid: A phase ii trial. Br. J. Cancer 2009, 101, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Albino, A.P.; Juan, G.; Traganos, F.; Reinhart, L.; Connolly, J.; Rose, D.P.; Darzynkiewicz, Z. Cell cycle arrest and apoptosis of melanoma cells by docosahexaenoic acid: Association with decreased prb phosphorylation. Cancer Res. 2000, 60, 4139–4145. [Google Scholar] [PubMed]
- Horrocks, L.A.; Yeo, Y.K. Health benefits of docosahexaenoic acid (dha). Pharmacol. Res. 1999, 40, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kolenic, N.; Pardini, R.S. Docosahexaenoic acid (dha), a primary tumor suppressive omega-3 fatty acid, inhibits growth of colorectal cancer independent of p53 mutational status. Nutr. Cancer 2007, 58, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Iigo, M.; Nakagawa, T.; Ishikawa, C.; Iwahori, Y.; Asamoto, M.; Yazawa, K.; Araki, E.; Tsuda, H. Inhibitory effects of docosahexaenoic acid on colon carcinoma 26 metastasis to the lung. Br. J. Cancer 1997, 75, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Awale, S.; Lu, J.; Kalauni, S.K.; Kurashima, Y.; Tezuka, Y.; Kadota, S.; Esumi, H. Identification of arctigenin as an antitumor agent having the ability to eliminate the tolerance of cancer cells to nutrient starvation. Cancer Res. 2006, 66, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.J.; Kuo, P.L.; Hsu, Y.C.; Huang, Y.F.; Tsai, E.M.; Hsu, Y.L. Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ros/p38 mapk pathway and epigenetic regulation. Free Radic. Biol. Med. 2014, 67, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, X.; Wang, C.; Nawaz, A.; Wei, W.; Li, J.; Wang, L.; Yu, D.H. Arctigenin suppresses unfolded protein response and sensitizes glucose deprivation-mediated cytotoxicity of cancer cells. Planta Med. 2011, 77, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel perk kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1h-indol-5-yl)-7h-p yrrolo[2,3-d]pyrimidin-4-amine (gsk2606414), a potent and selective first-in-class inhibitor of protein kinase r (pkr)-like endoplasmic reticulum kinase (perk). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-R.; Tomida, A.; Sato, S.; Tsukumo, Y.; Yun, J.; Yamori, T.; Hayakawa, Y.; Tsuruo, T.; Shin-ya, K. Effect on tumor cells of blocking survival response to glucose deprivation. J. Nat. Cancer Inst. 2004, 96, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-R.; Furihata, K.; Hayakawa, Y.; Shin-ya, K. Versipelostatin, a novel grp78/bip molecular chaperone down-regulator of microbial origin. Tetrahedron Lett. 2002, 43, 6941–6945. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; DeConti, R.C.; Conry, R.; Agarwala, S.; Papadopoulos, N.; Kim, K.B.; Ernstoff, M. Phase 3 study of docosahexaenoic acid–paclitaxel versus dacarbazine in patients with metastatic malignant melanoma. Ann. Oncol. 2011, 22, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Homsi, J.; Bedikian, A.Y.; Papadopoulos, N.E.; Kim, K.B.; Hwu, W.-J.; Mahoney, S.L.; Hwu, P. Phase 2 open-label study of weekly docosahexaenoic acid–paclitaxel in patients with metastatic uveal melanoma. Melanoma Res. 2010, 20, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zhu, F.; Zhao, Z.; Liu, C.; Luo, L.; Yin, Z. Arctigenin enhances chemosensitivity of cancer cells to cisplatin through inhibition of the stat3 signaling pathway. J. Cell. Biochem. 2011, 112, 2837–2849. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, M.; Gleissman, H.; Ponthan, F.; Castro, J.; Kogner, P.; Johnsen, J.I. Neuroblastoma cell death in response to docosahexaenoic acid: Sensitization to chemotherapy and arsenic-induced oxidative stress. Int. J. Cancer 2006, 118, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Colas, S.; Mahéo, K.; Denis, F.; Goupille, C.; Hoinard, C.; Champeroux, P.; Tranquart, F.; Bougnoux, P. Sensitization by dietary docosahexaenoic acid of rat mammary carcinoma to anthracycline: A role for tumor vascularization. Clin. Cancer Res. 2006, 12, 5879–5886. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, B.A.; Narayanan, N.K.; Desai, D.; Pittman, B.; Reddy, B.S. Effects of a combination of docosahexaenoic acid and 1,4-phenylene bis(methylene) selenocyanate on cyclooxygenase 2, inducible nitric oxide synthase and β-catenin pathways in colon cancer cells. Carcinogenesis 2004, 25, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.C.M.; Tong, K.F.; Ooi, V.E.C. Cytostatic and cytotoxic effects of cyclooxygenase inhibitors and their synergy with docosahexaenoic acid on the growth of human skin melanoma a-375 cells. Biomed. Pharmacother. 2005, 59, S293–S297. [Google Scholar] [CrossRef]
- Newman, M.J. Inhibition of carcinoma and melanoma cell growth by type 1 transforming growth factor beta is dependent on the presence of polyunsaturated fatty acids. Proc. Natl. Acad. Sci. USA 1990, 87, 5543–5547. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Cabello, C.M.; Lamore, S.D.; Lesson, J.L.; Wondrak, G.T. D-penicillamine targets metastatic melanoma cells with induction of the unfolded protein response (upr) and noxa (pmaip1)-dependent mitochondrial apoptosis. Apoptosis 2012, 17, 1079–1094. [Google Scholar] [CrossRef]
- Jiang, C.C.; Mao, Z.G.; Avery-Kiejda, K.A.; Wade, M.; Hersey, P.; Zhang, X.D. Glucose-regulated protein 78 antagonizes cisplatin and adriamycin in human melanoma cells. Carcinogenesis 2009, 30, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Lovat, P.E.; Armstrong, J.L.; Fimia, G.M.; Hill, D.S.; Birch-Machin, M.; Redfern, C.P.; Piacentini, M. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: The role of stress proteins erdj5 and erp57. Br. J. Cancer 2007, 96, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome inhibitors trigger noxa-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [PubMed]
- Amiri, K.I.; Horton, L.W.; LaFleur, B.J.; Sosman, J.A.; Richmond, A. Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: Implication for bortezomib (velcade, ps-341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004, 64, 4912–4918. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Chen, L.H.; Gillespie, S.; Kiejda, K.A.; Mhaidat, N.; Wang, Y.F.; Thorne, R.; Zhang, X.D.; Hersey, P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor–related apoptosis-inducing ligand–induced apoptosis by up-regulation of trail-r2 via the unfolded protein response. Cancer Res. 2007, 67, 5880–5888. [Google Scholar] [CrossRef] [PubMed]
- De Wilt, L.H.A.M.; Kroon, J.; Jansen, G.; de Jong, S.; Peters, G.J.; Kruyt, F.A.E. Bortezomib and trail: A perfect match for apoptotic elimination of tumour cells? Crit. Rev. Oncol./Hematology 2013, 85, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Nikrad, M.; Johnson, T.; Puthalalath, H.; Coultas, L.; Adams, J.; Kraft, A.S. The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand trail via bh3-only proteins bik and bim. Mol. Cancer Ther. 2005, 4, 443–449. [Google Scholar]
- Lecis, D.; Drago, C.; Manzoni, L.; Seneci, P.; Scolastico, C.; Mastrangelo, E.; Bolognesi, M.; Anichini, A.; Kashkar, H.; Walczak, H.; et al. Novel smac-mimetics synergistically stimulate melanoma cell death in combination with trail and bortezomib. Br. J. Cancer 2010, 102, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Lovat, P.E.; Corazzari, M.; Armstrong, J.L.; Martin, S.; Pagliarini, V.; Hill, D.; Brown, A.M.; Piacentini, M.; Birch-Machin, M.A.; Redfern, C.P. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008, 68, 5363–5369. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.S.; Martin, S.; Armstrong, J.L.; Flockhart, R.; Tonison, J.J.; Simpson, D.G.; Birch-Machin, M.A.; Redfern, C.P.F.; Lovat, P.E. Combining the er-stress inducing agents bortezomib and fenretinide as a novel therapeutic strategy for metastatic melanoma. Clin. Cancer Res. 2009, 15, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Hill, D.S.; Paton, J.C.; Paton, A.W.; Birch-Machin, M.A.; Lovat, P.E.; Redfern, C.P.F. Targeting grp78 to enhance melanoma cell death. Pigment. Cell Melanoma Res. 2010, 23, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with braf v600e mutation. New Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in braf v600–mutant advanced melanoma treated with vemurafenib. New Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Hartsough, E.J.; Aplin, A.E. A statement on vemurafenib-resistant melanoma. J. Invest. Dermatology 2013, 133, 1928–1929. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-H.; Piao, S.-F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting er stress–induced autophagy overcomes braf inhibitor resistance in melanoma. J. Clin. Invest. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Suomalainen, M.; Pennauer, M.; Yakimovich, A.; Andriasyan, V.; Hemmi, S.; Greber, U.F. Chemical induction of unfolded protein response enhances cancer cell killing through lytic virus infection. J. Virol. 2014, 88, 13086–13098. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lillo, A.M.; Steiniger, S.C.J.; Liu, Y.; Ballatore, C.; Anichini, A.; Mortarini, R.; Kaufmann, G.F.; Zhou, B.; Felding-Habermann, B.; et al. Targeting heat shock proteins on cancer cells: Selection, characterization, and cell-penetrating properties of a peptidic grp78 ligand. Biochemistry 2006, 45, 9434–9444. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Drug/s | Target | Effects | Study /clinical trial |
|---|---|---|---|
| Versipelostatin | GRP78 and GRP94 |
| Preclinical [119,120] |
| Docosahexaenoic acid | GRP78 |
| Preclinical melanoma [110] Phase II/ III melanoma [121,122] Phase II/III/IV solid tumours |
| PAT-SM6 | GRP78 |
| Phase I melanoma [105] PhaseI/ II multiple myeloma |
| Arctigenin | GRP78 |
| Preclinical [114,115,116] |
| Bortezomib in combination with 1azacytidine, 2decitabine | 26S proteosome |
| FDA-approved multiple myeloma, acute myeloid leukemia, Phase II metastatic melanoma 1,2Phase I multiple myeloma |
| Carfilzomib | 26S proteosome |
| Phase III multiple myeloma Phase II lymphoma |
| GSK2656157 | PERK |
| Preclinical [117,118] |
| ISRIB | ATF4 |
| Preclinical |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sykes, E.K.; Mactier, S.; Christopherson, R.I. Melanoma and the Unfolded Protein Response. Cancers 2016, 8, 30. https://doi.org/10.3390/cancers8030030
Sykes EK, Mactier S, Christopherson RI. Melanoma and the Unfolded Protein Response. Cancers. 2016; 8(3):30. https://doi.org/10.3390/cancers8030030
Chicago/Turabian StyleSykes, Erin K., Swetlana Mactier, and Richard I. Christopherson. 2016. "Melanoma and the Unfolded Protein Response" Cancers 8, no. 3: 30. https://doi.org/10.3390/cancers8030030
APA StyleSykes, E. K., Mactier, S., & Christopherson, R. I. (2016). Melanoma and the Unfolded Protein Response. Cancers, 8(3), 30. https://doi.org/10.3390/cancers8030030