
Aberrant Promoter Methylation of the Tumour Suppressor RASSF10 and Its Growth Inhibitory Function in Breast Cancer



Abstract
:1. Introduction
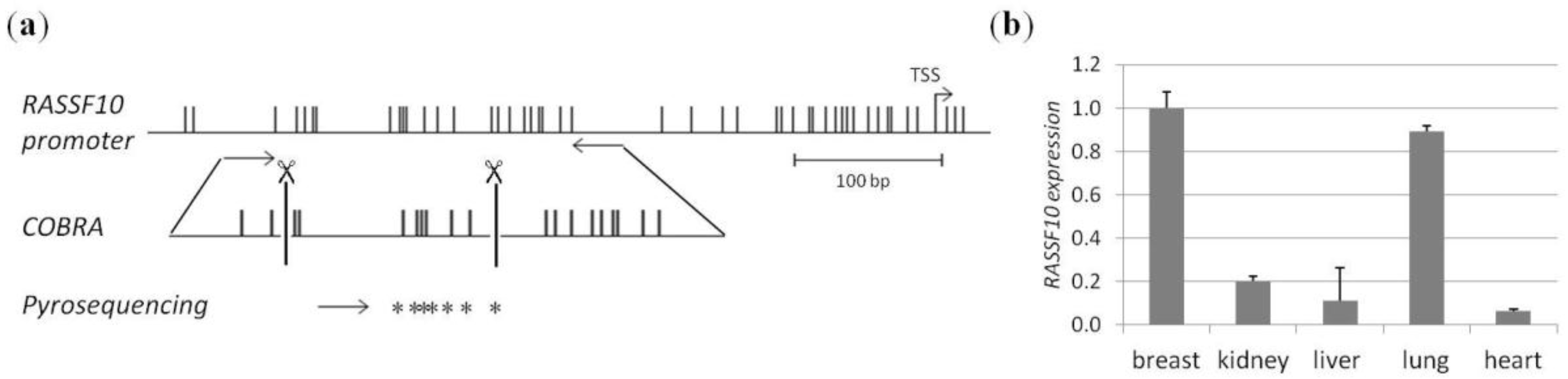
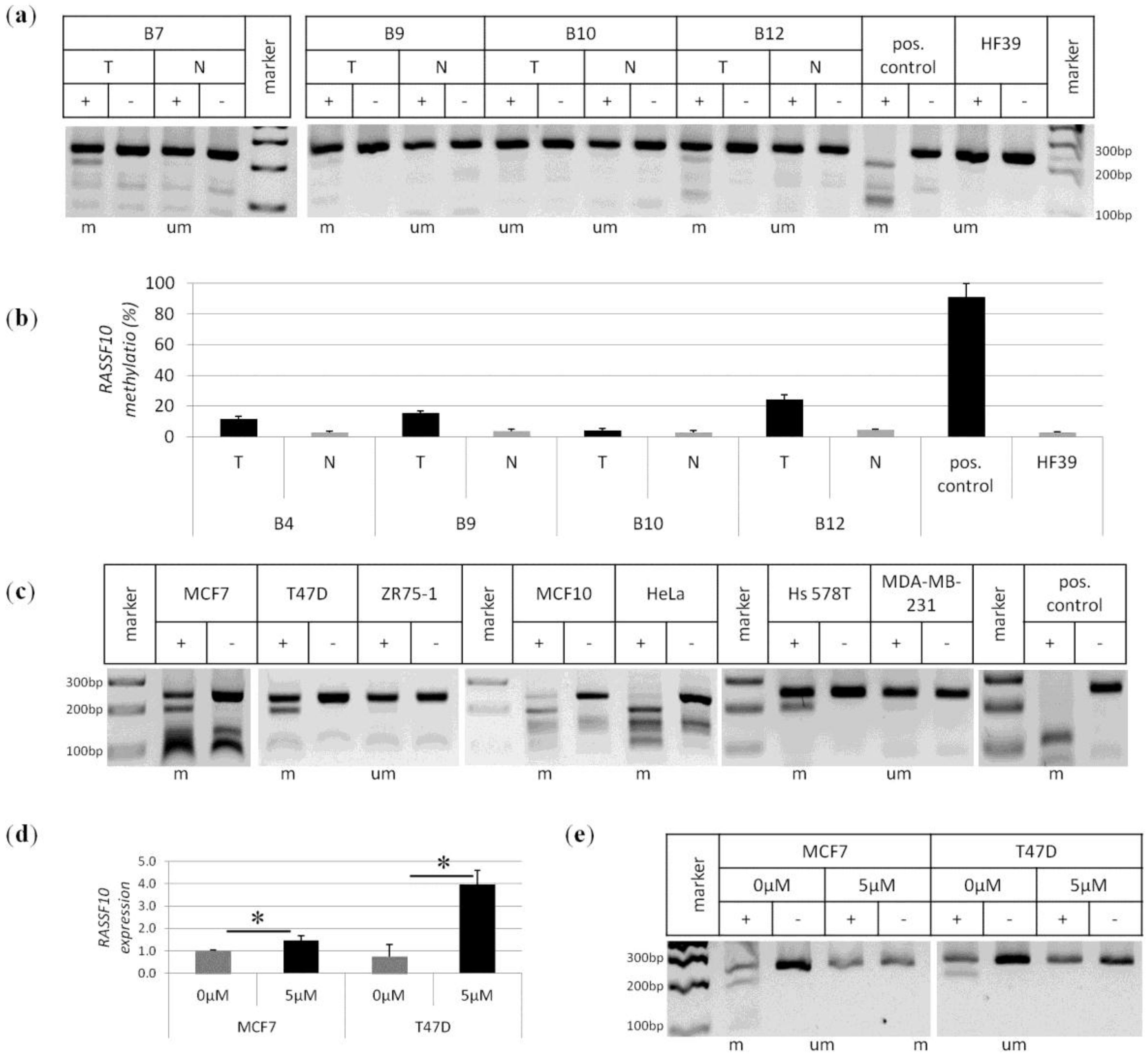
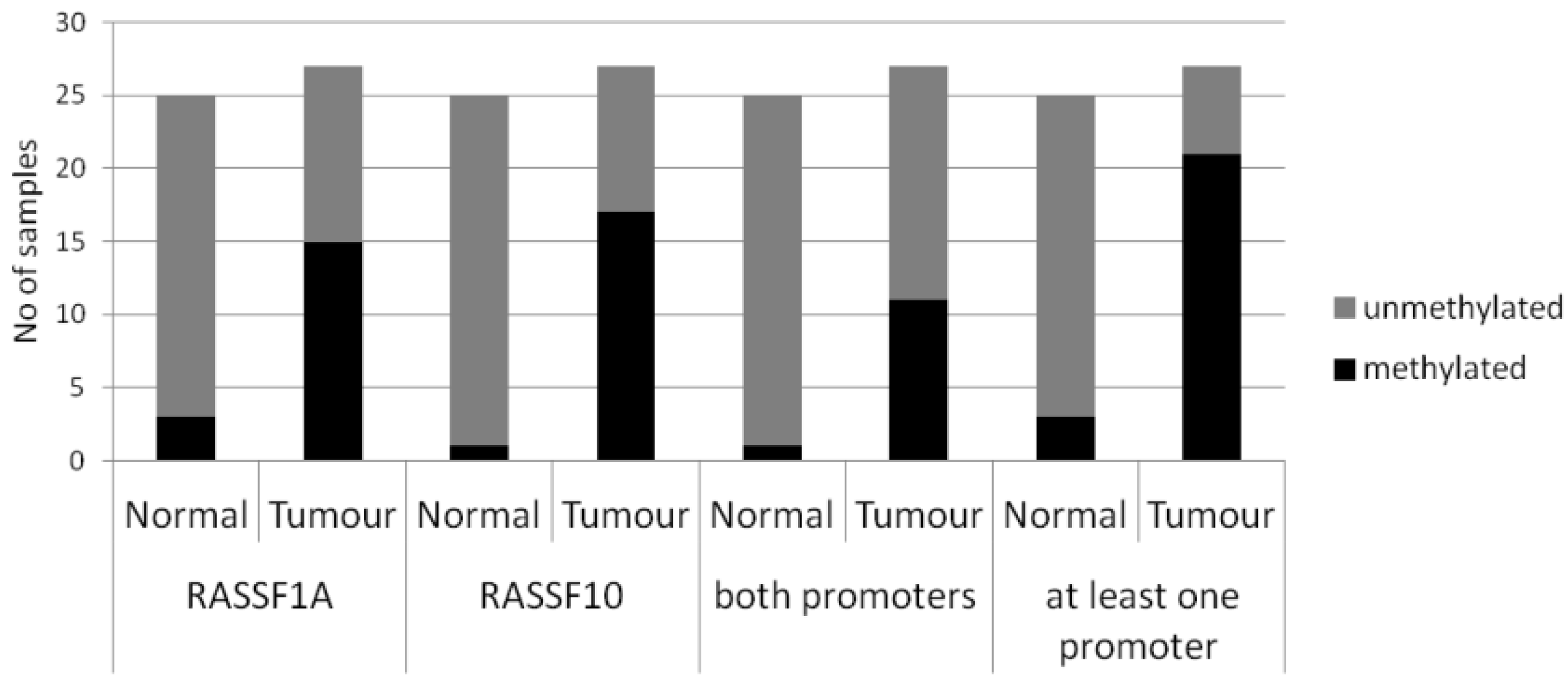
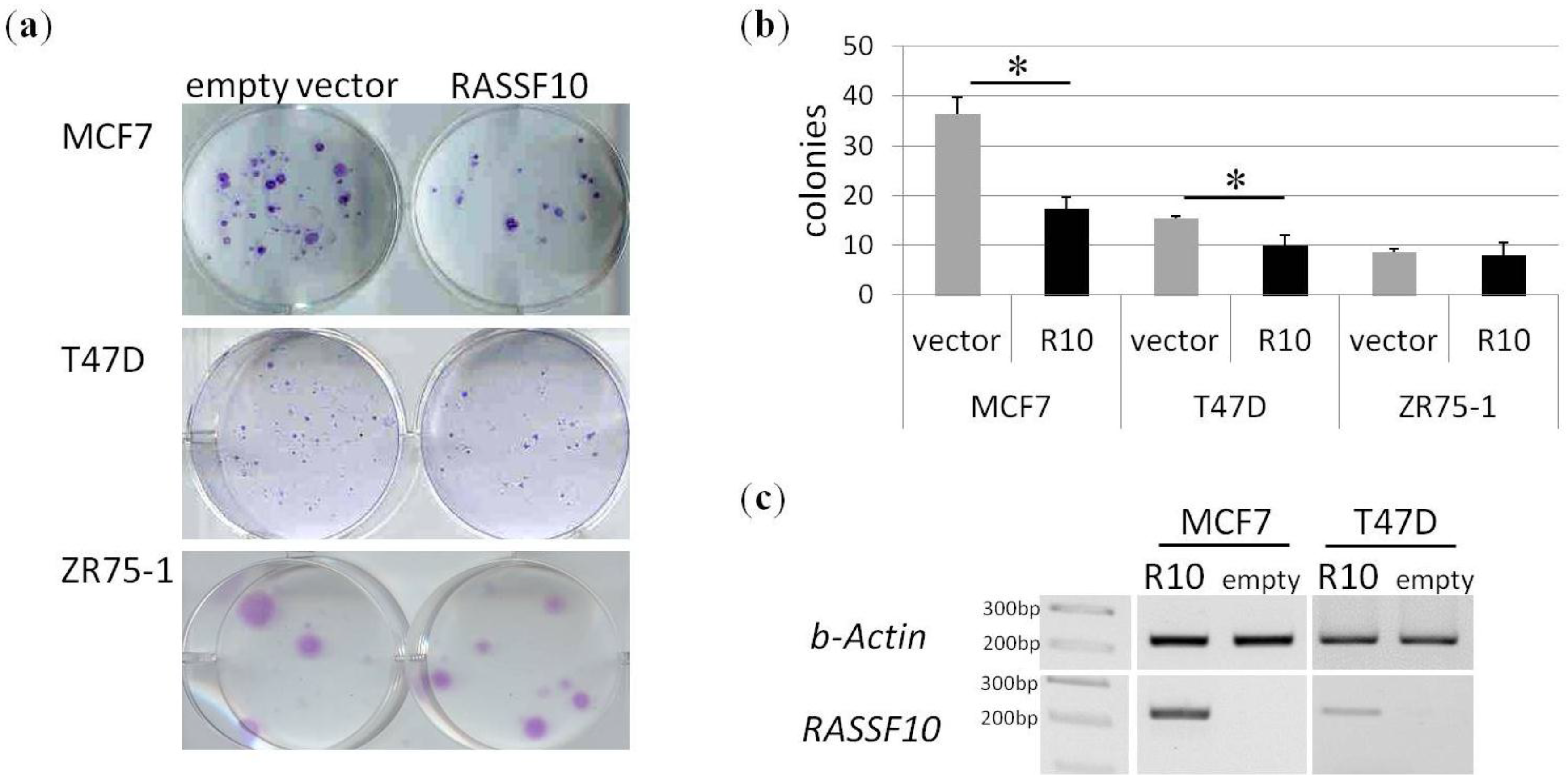
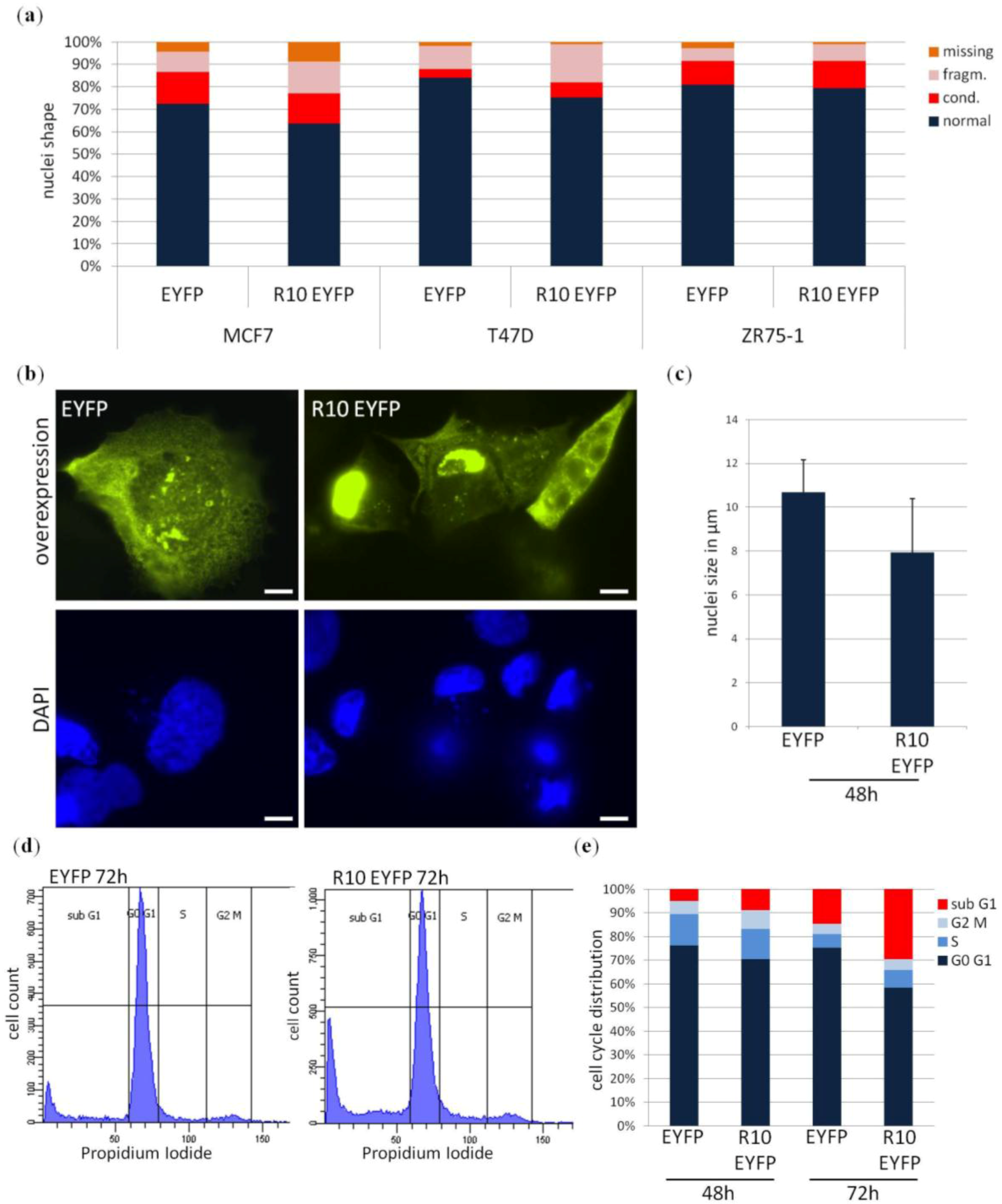
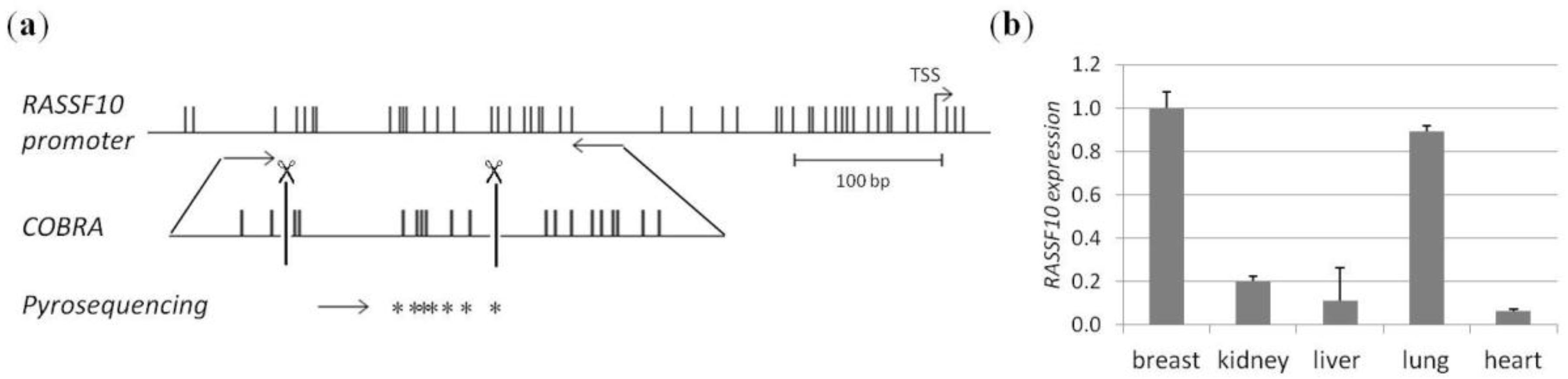
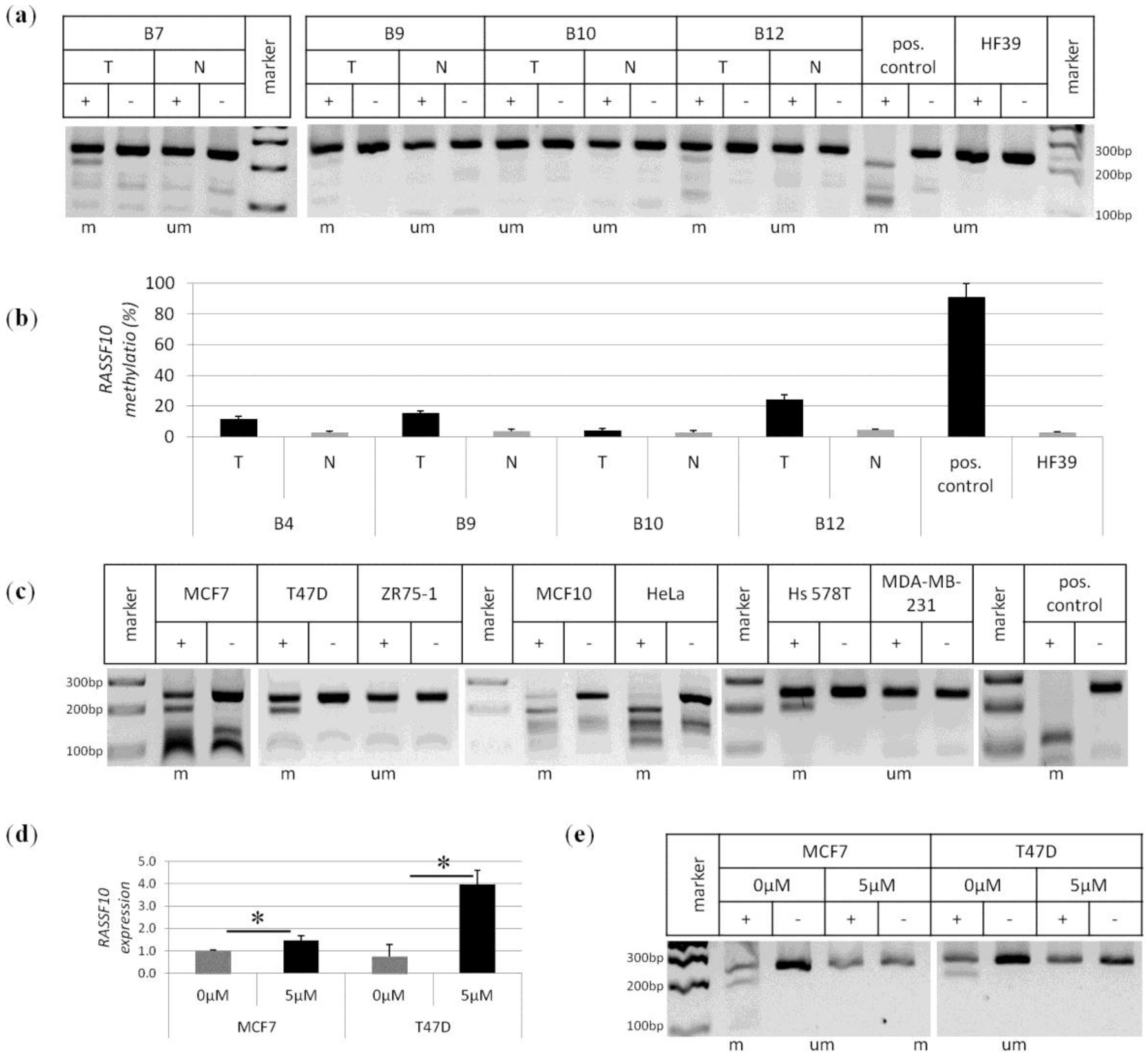
2. Results
3. Discussion
4. Materials and Methods
4.1. CpG Island Prediction, PCR Product Size and Digestion Products
4.2. Breast Cancer Tissues and Controls
4.3. DNA Isolation
4.4. Methylation Analysis by COBRA and Pyrosequencing
4.5. RNA Expression Analysis
4.6. Cell Culture, Aza Treatment and Colony Formation Analysis
4.7. Apoptosis Assays
4.8. Plasmids
4.9. Primers
Author Contributions
Conflicts of Interest
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Type | Details Histology | Grade | Promoter Methylation | |
|---|---|---|---|---|---|
| RASSF1A | RASSF10 | ||||
| B1 | Tumor | infl. poor diff ductal BrCA | 3 | m | m |
| B2 | Tumor | infl. poor diff ductal BrCA | n.a. | u | u |
| B3 | Tumor | infl. ductal BrCA | 3 | m | u |
| B4T | Tumor | infl. ductal BrCA | 2-3 | u | m |
| B4N | Normal | u | u | ||
| B5 | Tumor | infl. ductal BrCA | 3 | u | u |
| B6 | Tumor | infl. poor diff ductal BrCA | 3 | m | m |
| B7T | Tumor | breast cancer | n.a. | m | m |
| B7N | Normal | u | u | ||
| B8T | Tumor | BrCA | n.a. | m | m |
| B9T | Tumor | ductal AdenoCA | n.a. | u | m |
| B9N | Normal | u | u | ||
| B10T | Tumor | well diff. infl. ductal AdenoCA | 1 | m | u |
| B10N | Normal | u | u | ||
| B12T | Tumor | infl. ductal BrCA | 3 | m | m |
| B12N | Normal | u | u | ||
| B28T | Tumor | infl. moderatly diff ductal CA | 2 | u | m |
| B28N | Normal | u | u | ||
| B33T | Tumor | colloid BrCA | n.a. | m | m |
| B33N | Normal | u | u | ||
| B34T | Tumor | invasive BrCA | 3 | m | m |
| B34N | Normal | u | u | ||
| B36T | Tumor | infl. moderatly diff ductal CA | 2 | m | m |
| B36N | Normal | m | m | ||
| B38T | Tumor | infl. moderatly diff ductal CA | 2 | m | m |
| B38N | Normal | u | u | ||
| B47T | Tumor | infl. ductal BrCA | 3 | m | u |
| B47N | Normal | u | u | ||
| B26T | Tumor | invasive mammary ductal | n.a. | m | m |
| B26N | Normal | u | u | ||
| B27T | Tumor | invasive mammary ductal | n.a. | m | u |
| B27N | Normal | u | u | ||
| B20N | Normal | m | u | ||
| B23N | Normal | u | u | ||
| B29N | Normal | u | u | ||
| B31N | Normal | u | u | ||
| B40N | Normal | u | u | ||
| B41N | Normal | u | u | ||
| B43N | Normal | u | u | ||
| B44N | Normal | u | u | ||
| B45N | Normal | u | u | ||
| B46N | Normal | u | u | ||
| B50N | Normal | m | u | ||
| B51N | Normal | u | u | ||
| B15T | Tumor | infl. ductal BrCA | 3 | u | u |
| B18T | Tumor | infl. lobular BrCA | 2 | u | u |
| B21T | Tumor | ductal AdenoCA | 3 | u | u |
| B24T | Tumor | invasive mod. diff. ductal AdenoCA | 2 | u | u |
| B30T | Tumor | infl. mod.diff. ductal AdenoCA | 2 | u | m |
| B32T | Tumor | infl. ductal carcinoma | 3 | m | m |
| B37T | Tumor | infil. ductal AdenoCA | 3 | u | m |
| B39T | Tumor | infil. ductal AdenoCA | 2 | u | m |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Peterse, J.L.; van ’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Nounou, M.I.; ElAmrawy, F.; Ahmed, N.; Abdelraouf, K.; Goda, S.; Syed-Sha-Qhattal, H. Breast Cancer: Conventional Diagnosis and Treatment Modalities and Recent Patents and Technologies. Breast Cancer 2015, 9, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [PubMed]
- Dawood, S.; Broglio, K.; Buzdar, A.U.; Hortobagyi, G.N.; Giordano, S.H. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: An institutional-based review. J. Clin. Oncol. 2010, 28, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Fischer, C. Breast cancer risks and risk prediction models. Breast. Care. 2015, 10, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Wong, E.M.; Southey, M.C.; Fox, S.B.; Brown, M.A.; Dowty, J.G.; Jenkins, M.A.; Giles, G.G.; Hopper, J.L.; Dobrovic, A. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer. Prev. Res. 2011, 4, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Yang, G.; Pfeifer, G.P. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 2001, 61, 3105–3109. [Google Scholar] [PubMed]
- Paska, A.V.; Hudler, P. Aberrant methylation patterns in cancer: A clinical view. Biochem. Med. 2015, 25, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The RASSF proteins in cancer: From epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Recino, A.; Jeffries, A.; Ward, A.; Chalmers, A.D. The N-terminal RASSF family: A new group of Ras-association-domain-containing proteins, with emerging links to cancer formation. Biochem. J. 2010, 425, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Benjamin, D.R. A novel family of Ras-binding domains. Trends. Biochem. Sci. 1996, 21, 422–425. [Google Scholar] [CrossRef]
- Scheel, H.; Hofmann, K. A novel interaction motif, SARAH, connects three classes of tumor suppressor. Curr. Biol. 2003, 13, R899–R900. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Walesch, S.K.; Wurl, P.; Taubert, H.; Dammann, R.H. The tumor suppressor RASSF10 is upregulated upon contact inhibition and frequently epigenetically silenced in cancer. Oncogenesis 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [PubMed]
- Wei, Z.; Chen, X.; Chen, J.; Wang, W.; Xu, X.; Cai, Q. RASSF10 is epigenetically silenced and functions as a tumor suppressor in gastric cancer. Biochem. Biophys. Res. Commun. 2013, 432, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chang, X.; Dai, D.; Deng, P.; Sun, Q. RASSF10 is an epigenetically silenced tumor suppressor in gastric cancer. Oncol. Rep. 2014, 31, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Zimmermann, T.; Haag, T.; Walesch, S.K.; Dammann, R.H. Promoter methylation status of ras-association domain family members in pheochromocytoma. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Haag, T.; Walesch, S.; Herrmann-Trost, P.; Marsch, W.C.; Kutzner, H.; Helmbold, P.; Dammann, R.H. Aberrant Promoter Hypermethylation of RASSF Family Members in Merkel Cell Carcinoma. Cancers 2013, 5, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Helmbold, P.; Richter, A.M.; Walesch, S.K.; Skorokhod, A.; Marsch, W.C.; Enk, A. RASSF10 promoter hypermethylation is frequent in Malignant Melanoma of the skin but uncommon in nevus cell nevi. J. Investig. Dermatol. 2012, 132, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Schagdarsurengin, U.; Richter, A.M.; Wohler, C.; Dammann, R.H. Frequent epigenetic inactivation of RASSF10 in thyroid cancer. Epigenetics 2009, 4, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Hesson, L.B.; Dunwell, T.L.; Cooper, W.N.; Catchpoole, D.; Brini, A.T.; Chiaramonte, R.; Griffiths, M.; Chalmers, A.D.; Maher, E.R.; Latif, F. The novel RASSF6 and RASSF10 candidate tumour suppressor genes are frequently epigenetically inactivated in childhood leukaemias. Mol. Cancer. 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.K.; Underhill-Day, N.; Krex, D.; Robel, K.; Sangan, C.B.; Summersgill, H.R.; Morris, M.; Gentle, D.; Chalmers, A.D.; Maher, E.R.; et al. Epigenetic inactivation of the RASSF10 candidate tumor suppressor gene is a frequent and an early event in gliomagenesis. Oncogene 2010, 30, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Haag, T.; Herkt, C.E.; Walesch, S.K.; Richter, A.M.; Dammann, R.H. The apoptosis associated tyrosine kinase gene is frequently hypermethylated in human cancer and is regulated by epigenetic mechanisms. Genes Cancer 2014, 5, 365–374. [Google Scholar] [PubMed]
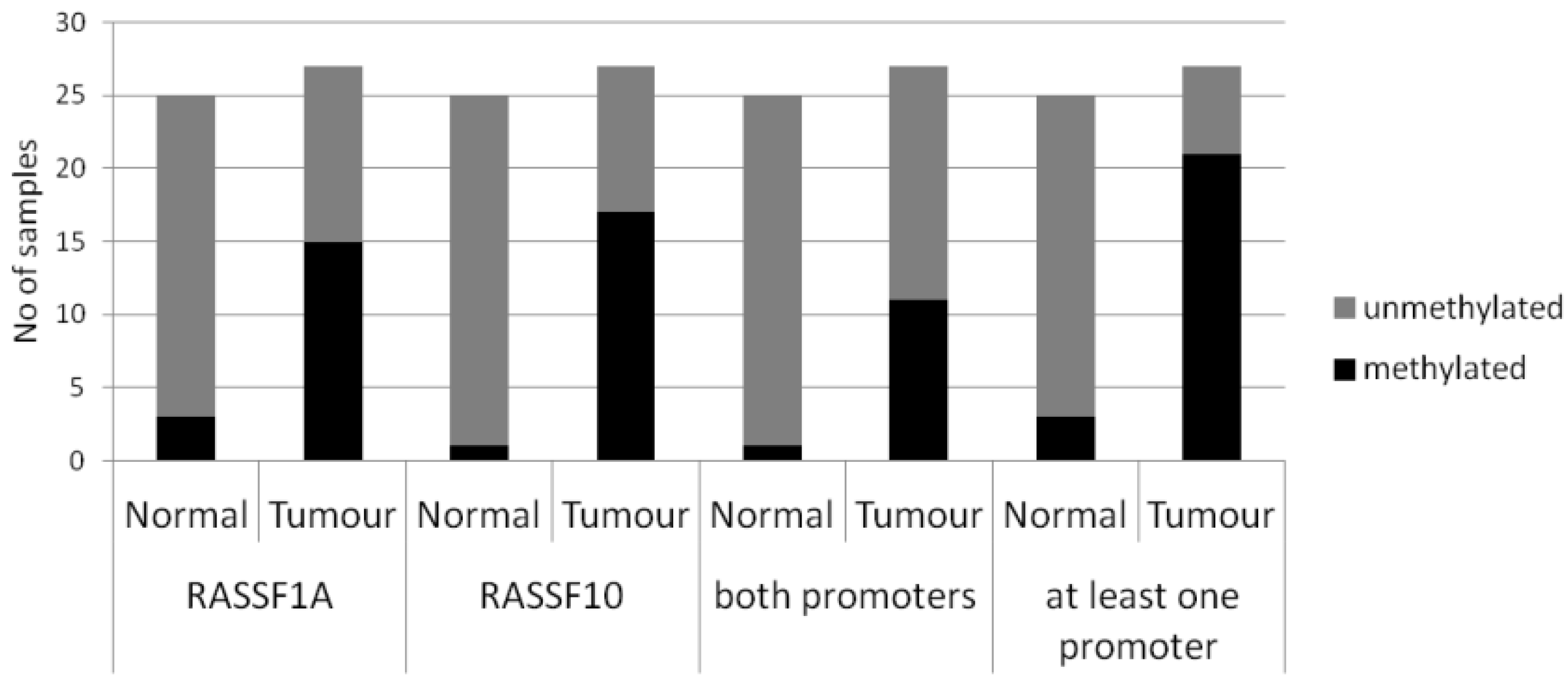
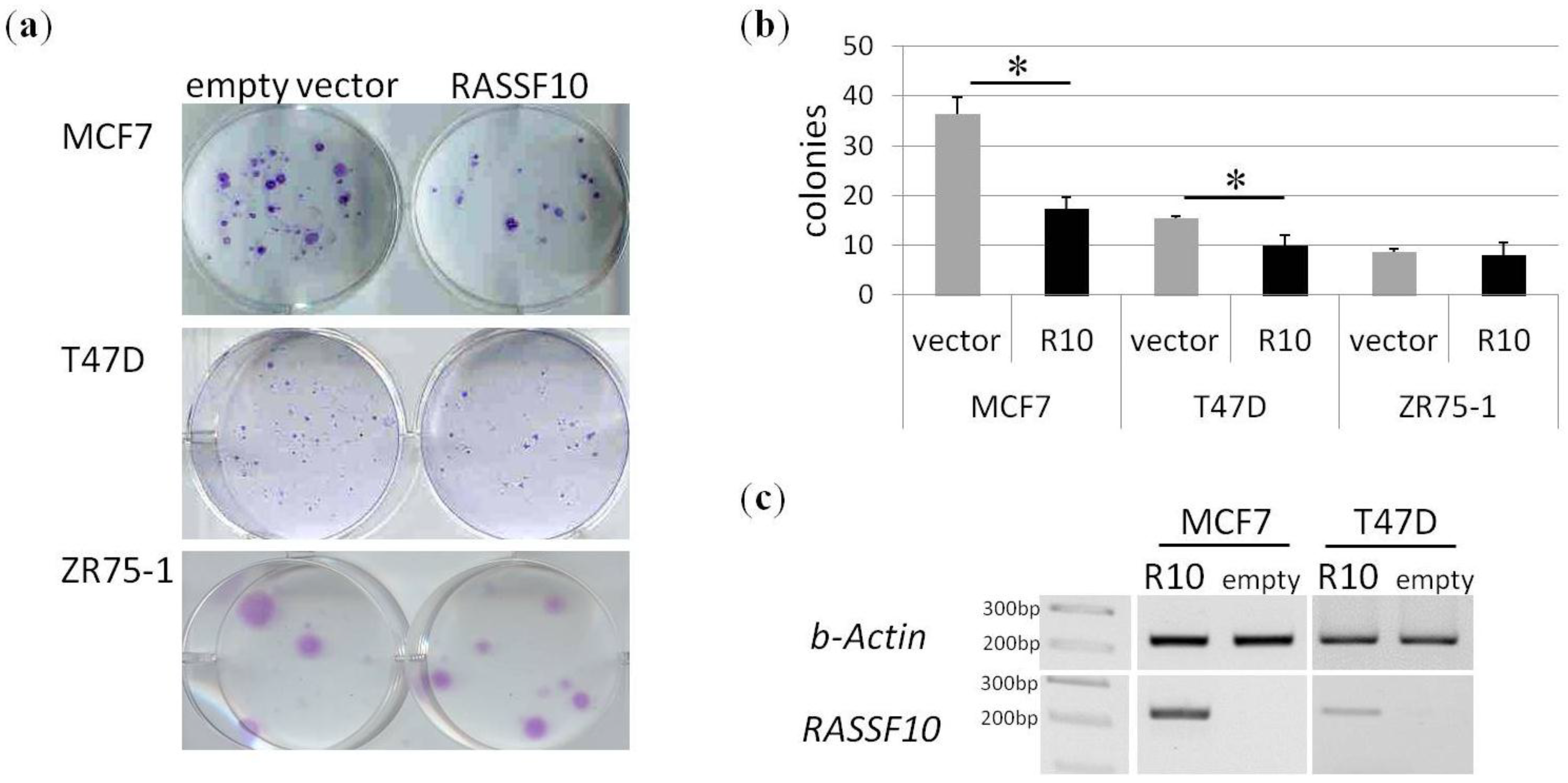
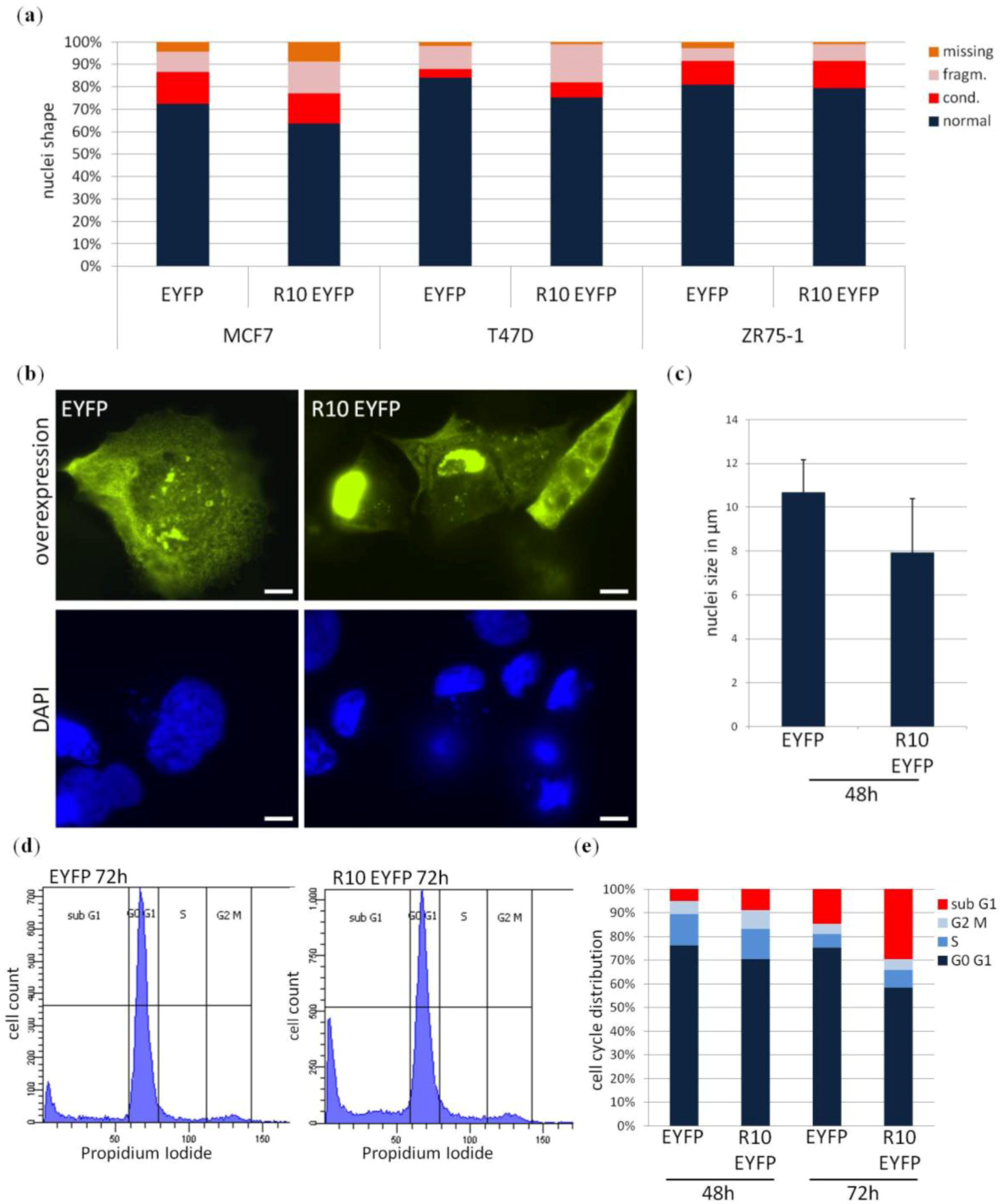
| Primary Breast Tissue | Cases | Promoter Methylation (%) | Statistical Test | |||
|---|---|---|---|---|---|---|
| RASSF1A | RASSF10 | |||||
| Normal | n = 25 | 2/25 (8%) | p < 0.005 | 1/25 (4%) | p < 0.005 | Fishers Exact two tailed |
| Tumours | n = 27 | 15/27 (56%) | 17/27 (63%) | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, A.M.; Walesch, S.K.; Dammann, R.H. Aberrant Promoter Methylation of the Tumour Suppressor RASSF10 and Its Growth Inhibitory Function in Breast Cancer. Cancers 2016, 8, 26. https://doi.org/10.3390/cancers8030026
Richter AM, Walesch SK, Dammann RH. Aberrant Promoter Methylation of the Tumour Suppressor RASSF10 and Its Growth Inhibitory Function in Breast Cancer. Cancers. 2016; 8(3):26. https://doi.org/10.3390/cancers8030026
Chicago/Turabian StyleRichter, Antje M., Sara K. Walesch, and Reinhard H. Dammann. 2016. "Aberrant Promoter Methylation of the Tumour Suppressor RASSF10 and Its Growth Inhibitory Function in Breast Cancer" Cancers 8, no. 3: 26. https://doi.org/10.3390/cancers8030026
APA StyleRichter, A. M., Walesch, S. K., & Dammann, R. H. (2016). Aberrant Promoter Methylation of the Tumour Suppressor RASSF10 and Its Growth Inhibitory Function in Breast Cancer. Cancers, 8(3), 26. https://doi.org/10.3390/cancers8030026