
Silencing of Taxol-Sensitizer Genes in Cancer Cells: Lack of Sensitization Effects

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Drug-Resistant Cell Lines
2.3. Cell Viability Assay
2.4. Quantitative PCR Analysis
2.5. Knockdown Assay
2.6. Statistical Analysis
3. Results
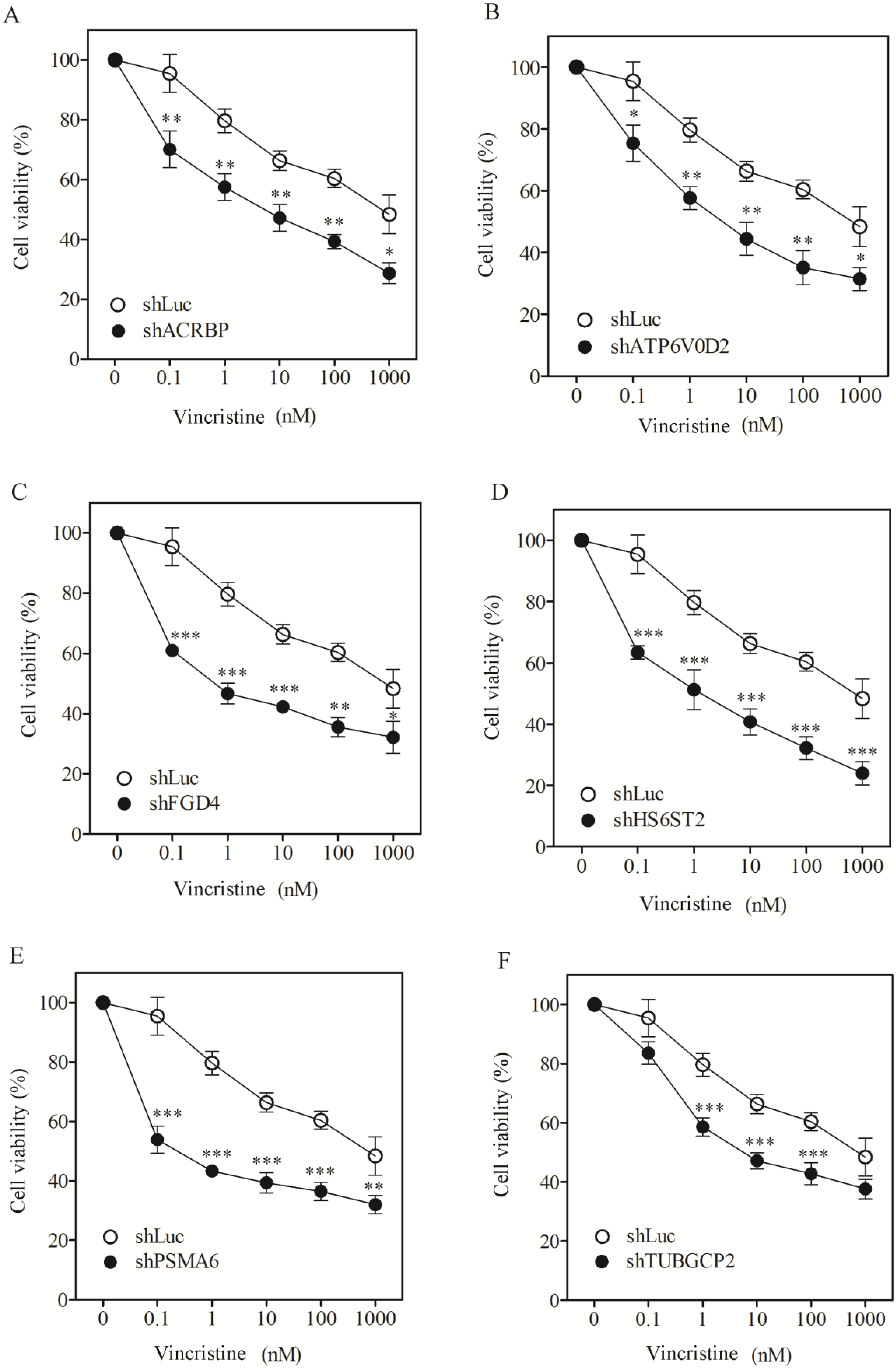
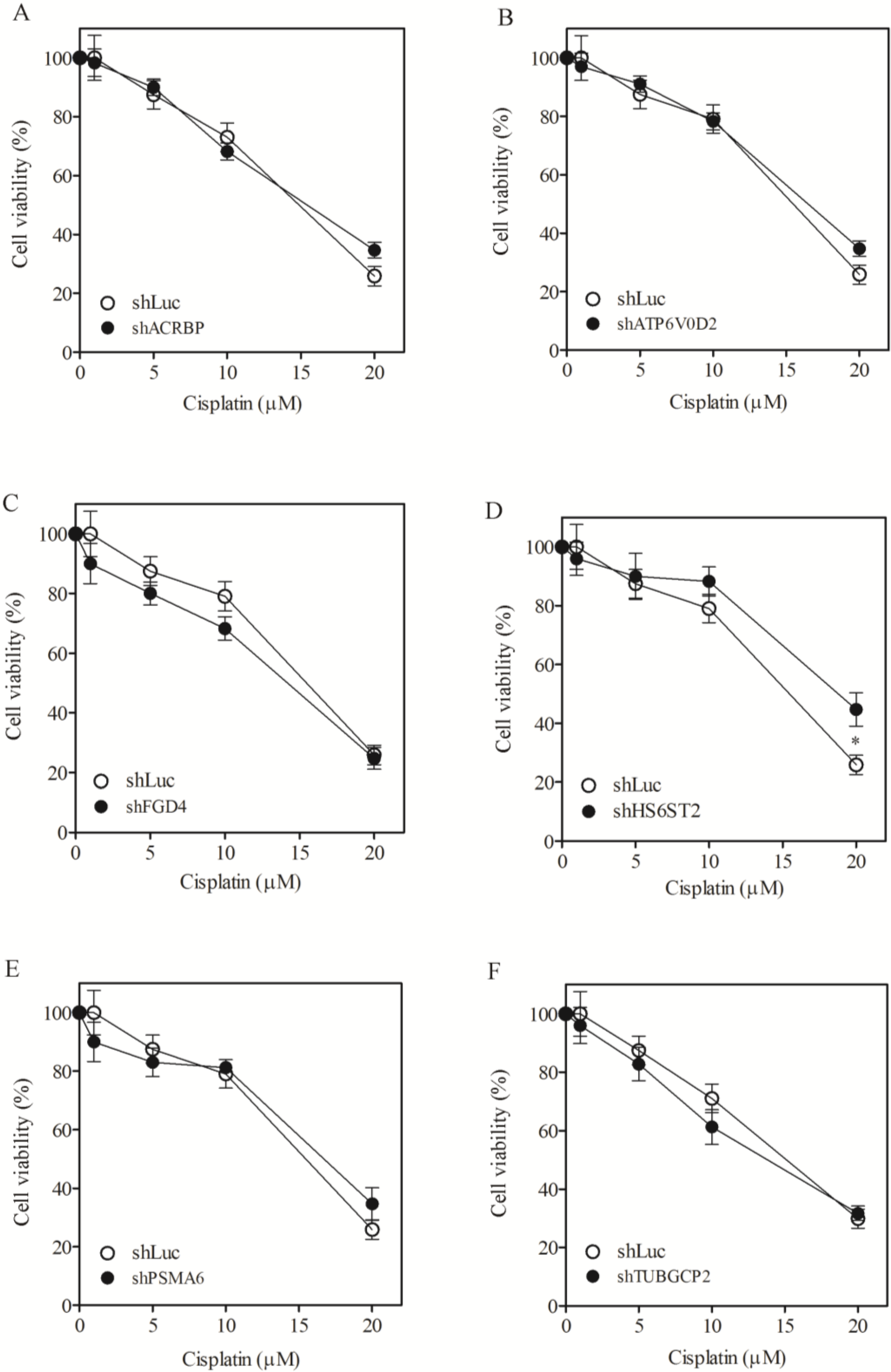
3.1. Sensitization of H1155 Cells to Taxol Following Silencing of Chemosensitizer Genes
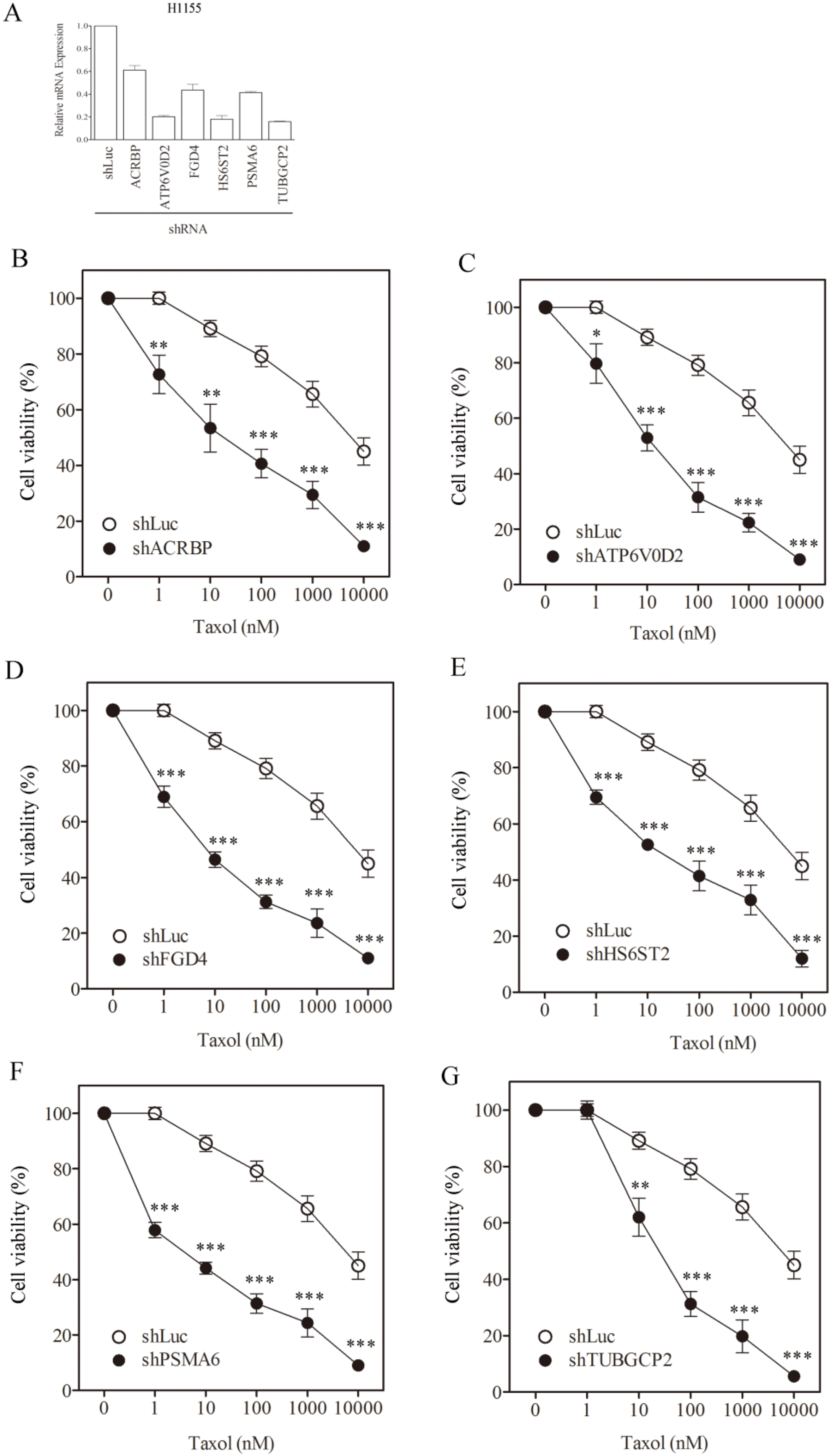

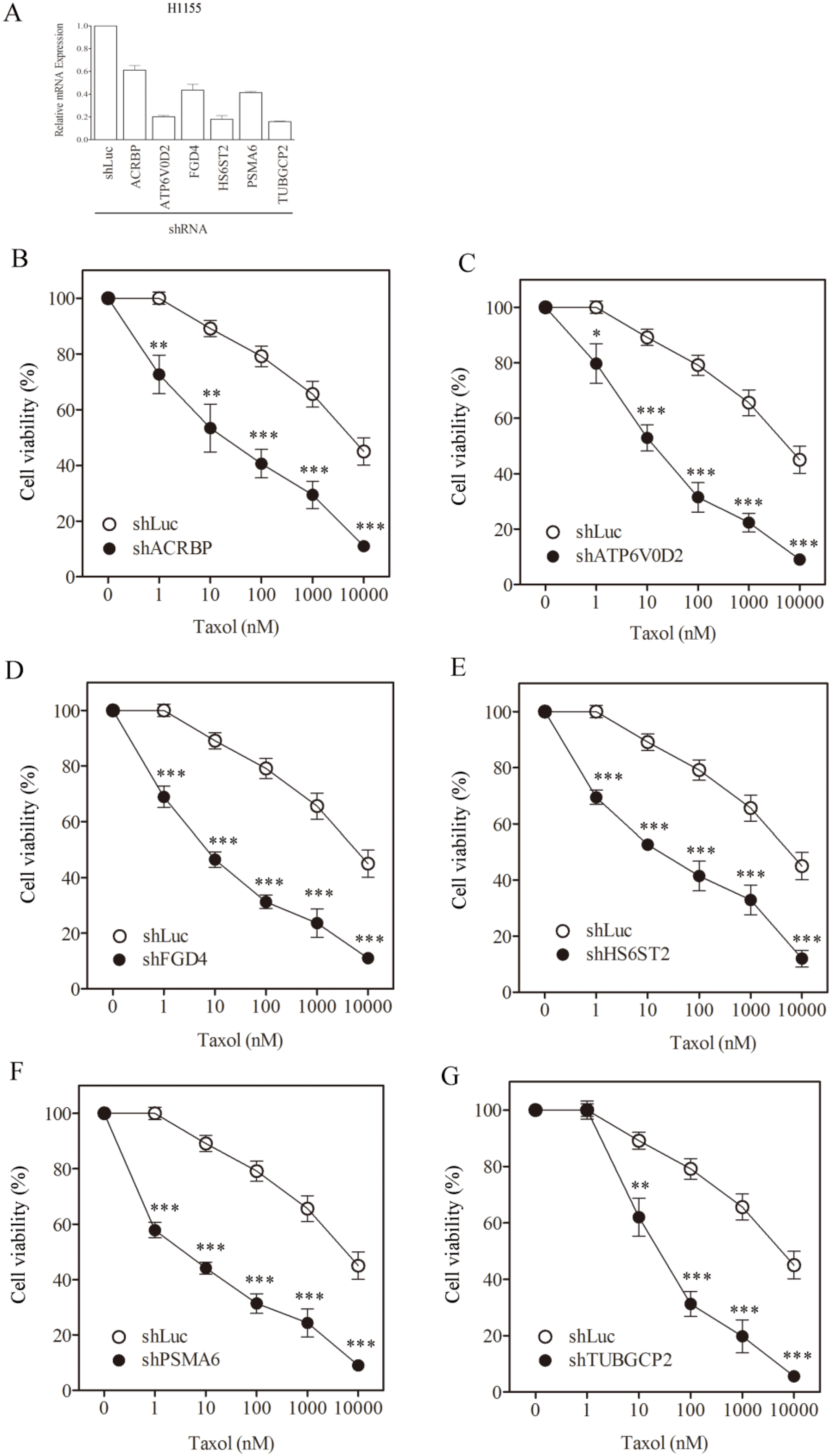{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H1155, IC50 (SF) | |||
|---|---|---|---|
| Taxol (nM) | Vincristine (nM) | Cisplatin (µM) | |
| shLuc | 7817.1 ± 601 | 876.3 ± 61.4 | 14.9 ± 2.1 |
| shACRBP | 34.0 ± 1.6 (230.1) | 7.5 ± 0.8 (116.1) | 15.4 ± 1.1 (ND) |
| shATP6V0D2 | 22.1 ± 1.1 (354.1) | 6.2 ± 0.4 (141.3) | 16.5 ± 2.3 (ND) |
| shFGD4 | 8.6 ± 1.1 (911.4) | 0.8 ± 0.0 (1105.2) | 14.2 ± 1.0 (ND) |
| shHS6ST2 | 31.2 ± 2.9 (250.7) | 2.1 ± 0.4 (419.0) | 18.8 ± 3.4 (ND) |
| shPSMA6 | 6.1 ± 0.4 (1272.5) | 0.4 ± 0.0 (2050.9) | 16.7 ± 2.2 (ND) |
| shTUBGCP2 | 45.1 ± 5.6 (173.3) | 7.8 ± 0.9 (112.9) | 13.8 ± 0.9 (ND) |
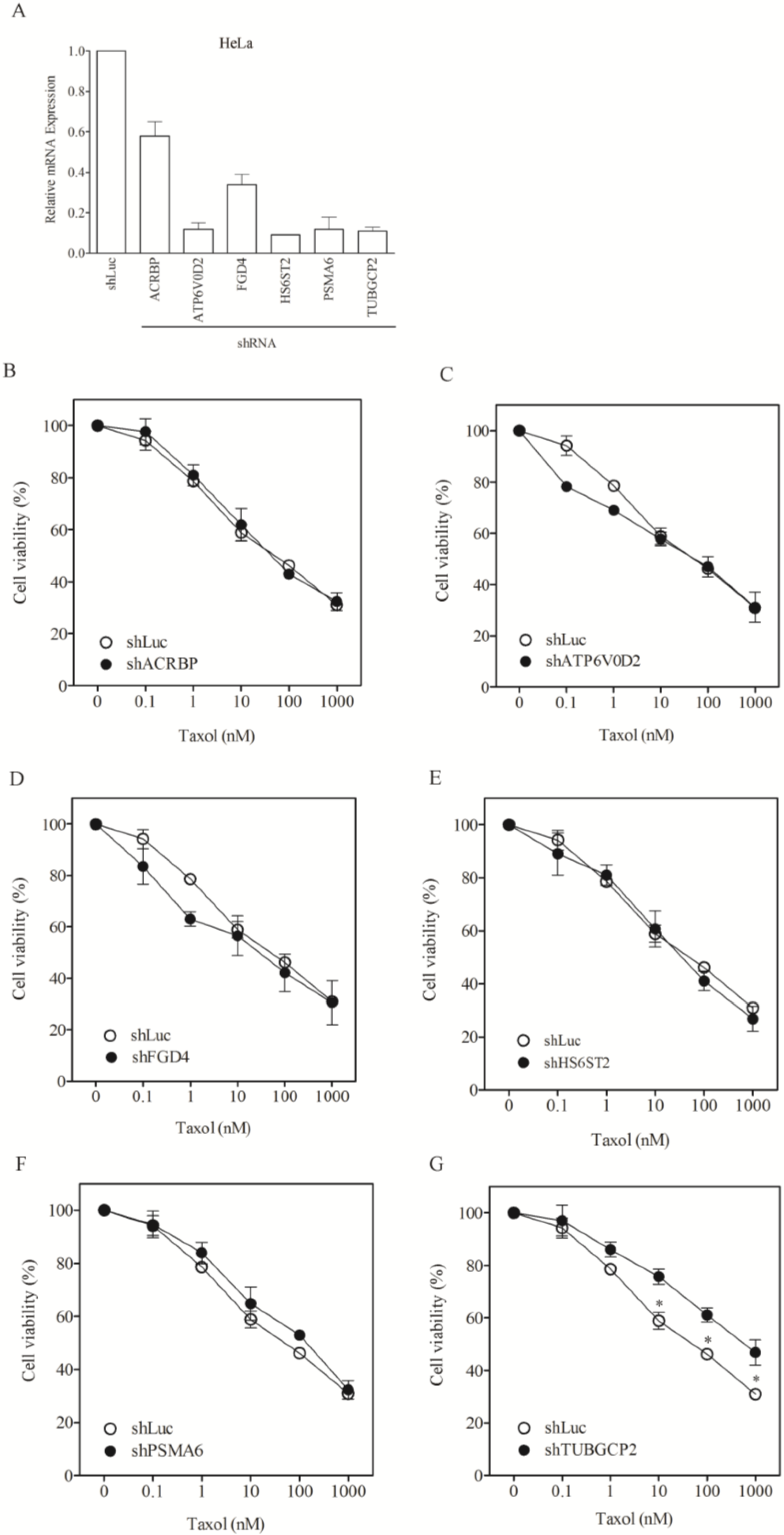
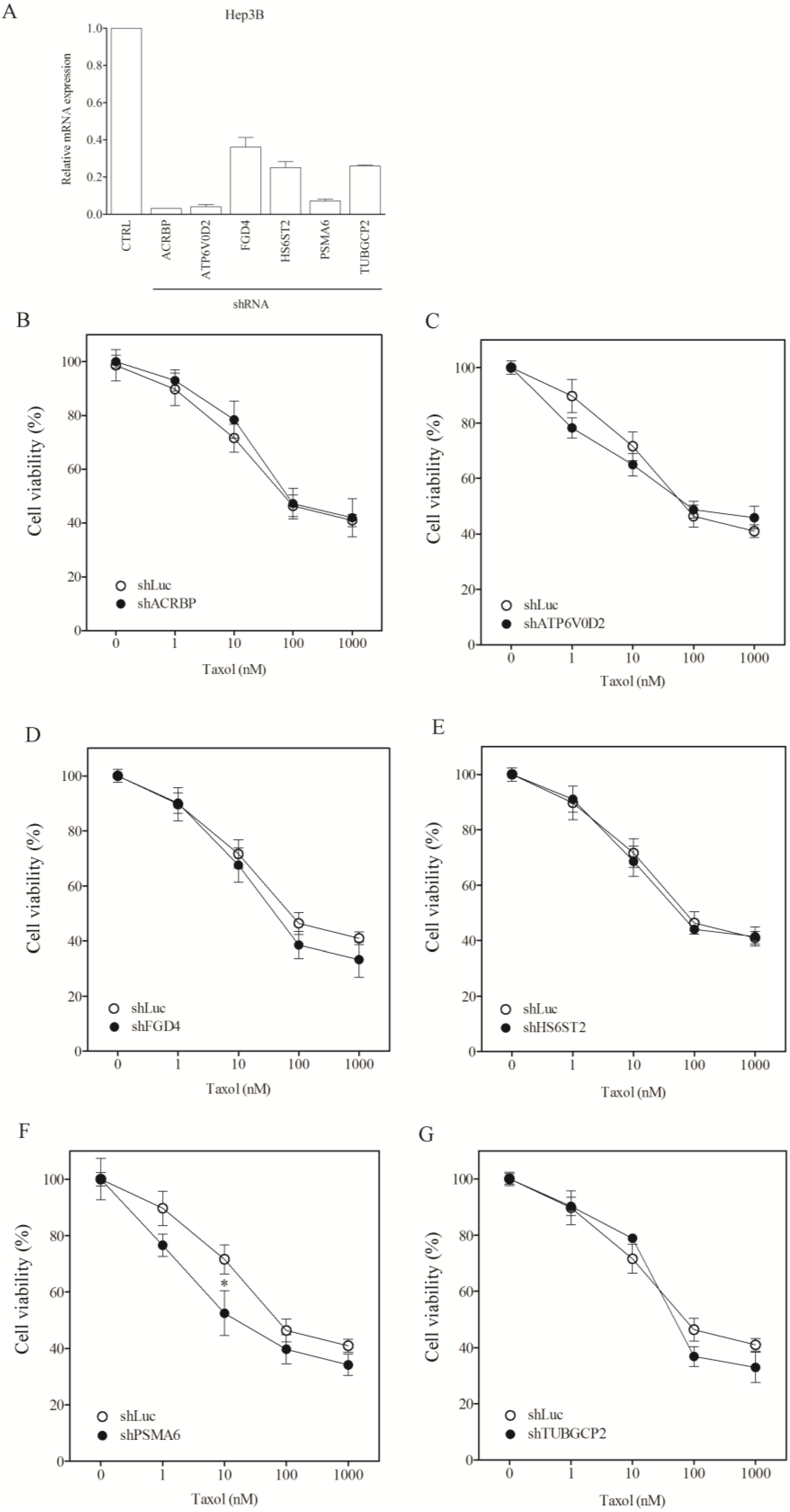
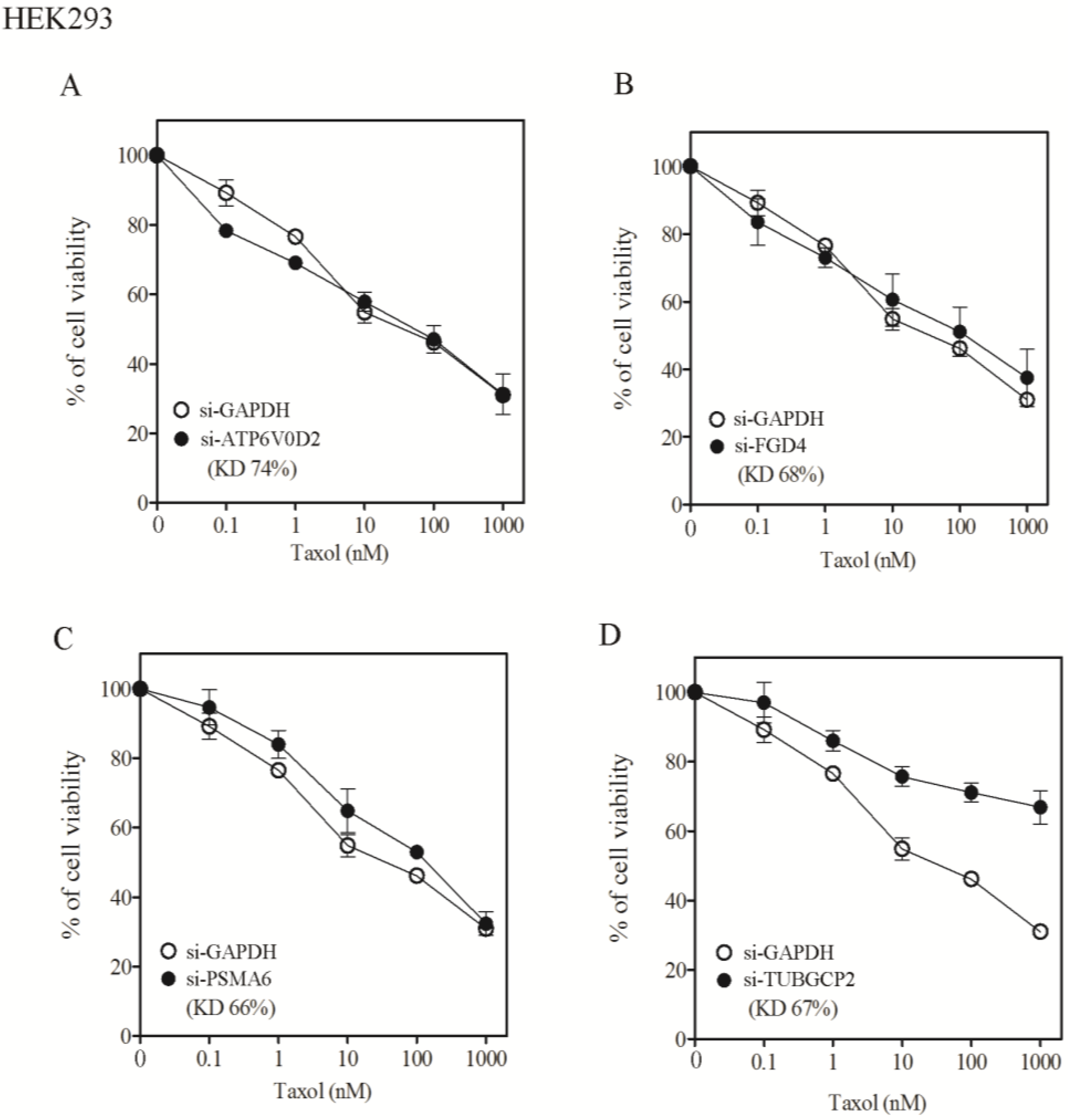
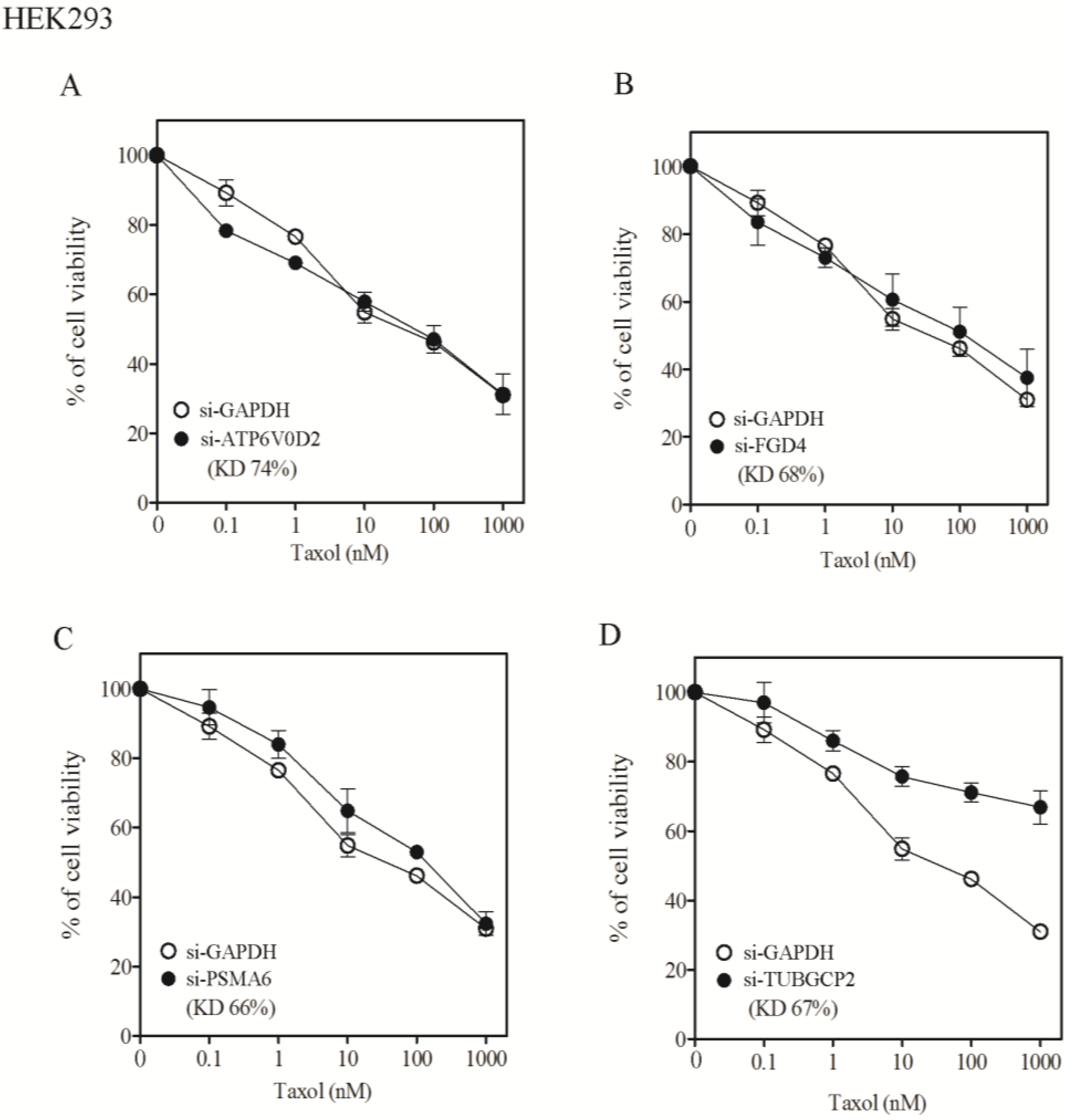
3.2. Lack of Sensitization of Non-H1155 Cells to Taxol Following Silencing of Chemosensitizer Genes
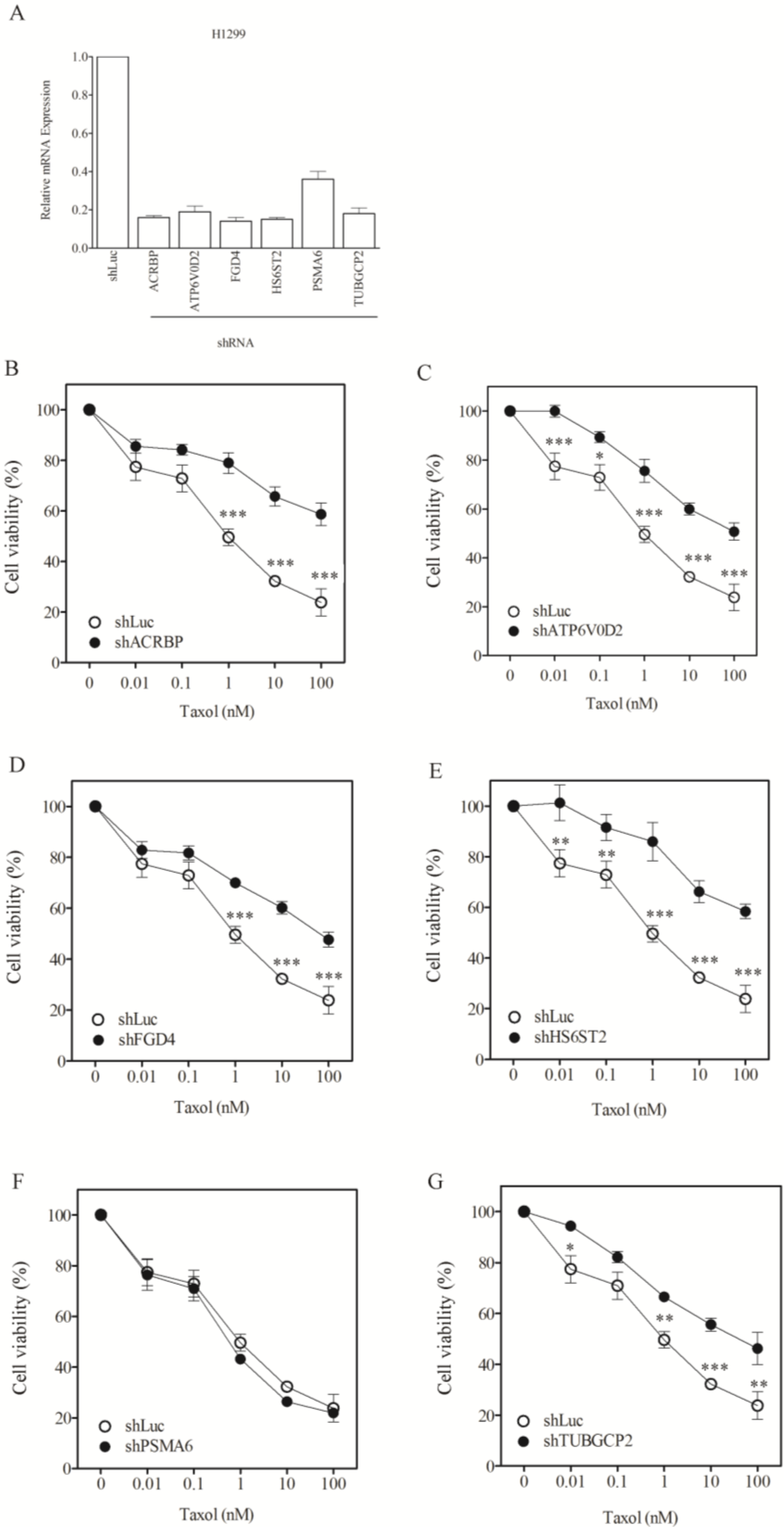
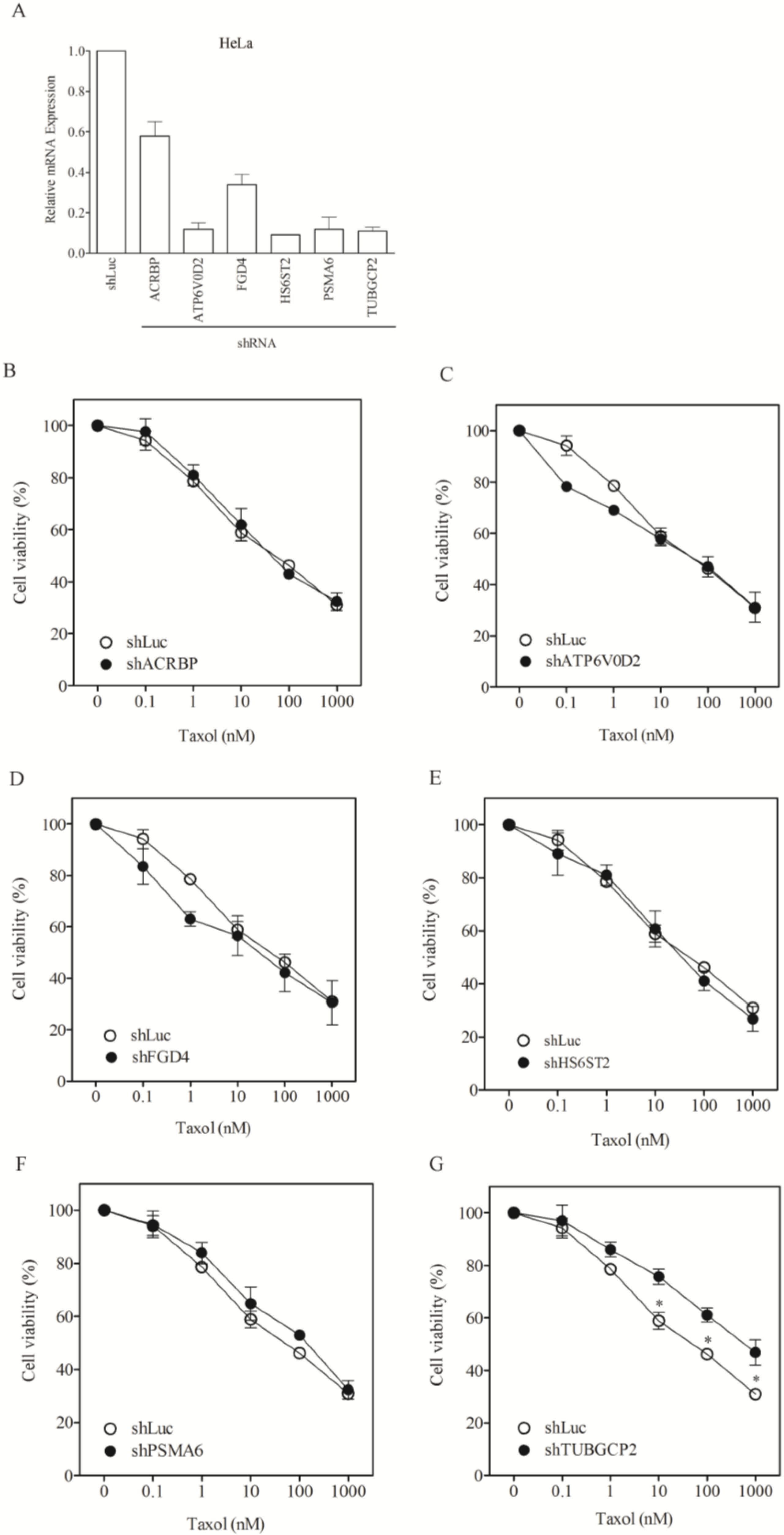

| Taxol nM, IC50 (SF) | |||
|---|---|---|---|
| H1299 | HeLa | Hep3B | |
| shLuc | 1.0 ± 0.1 | 72.7 ± 3.4 | 87.0 ± 8.1 |
| shACRBP | 656.4 ± 51.3 (0.00) * | 66.4 ± 3.9 (1.10) | 92.1 ± 5.6 (0.94) * |
| shATP6V0D2 | 150.3 ± 21.6 (0.01) * | 75.0 ± 6.1.0 (0.96) * | 92.7 ± 11.0 (0.94) * |
| shFGD4 | 83.0 ± 9.6 (0.01) * | 50.8 ± 4.4 (1.43) | 76.1 ± 6.1(1.14) |
| shHS6ST2 | 637.8 ± 71.2 (0.00) * | 67.3 ± 9.1 (1.08) | 78.1 ± 9.0 (1.11) |
| shPSMA6 | 0.8 ± 0.06 (1.25) | 228.5 ± 13.9 (0.32) * | 27.4 ± 1.2 (3.18) |
| shTUBGCP2 | 63.7 ± 8.1 (0.02) * | 798.1 ± 64.4 (0.09) * | 71.7 ± 4.3 (1.21) |
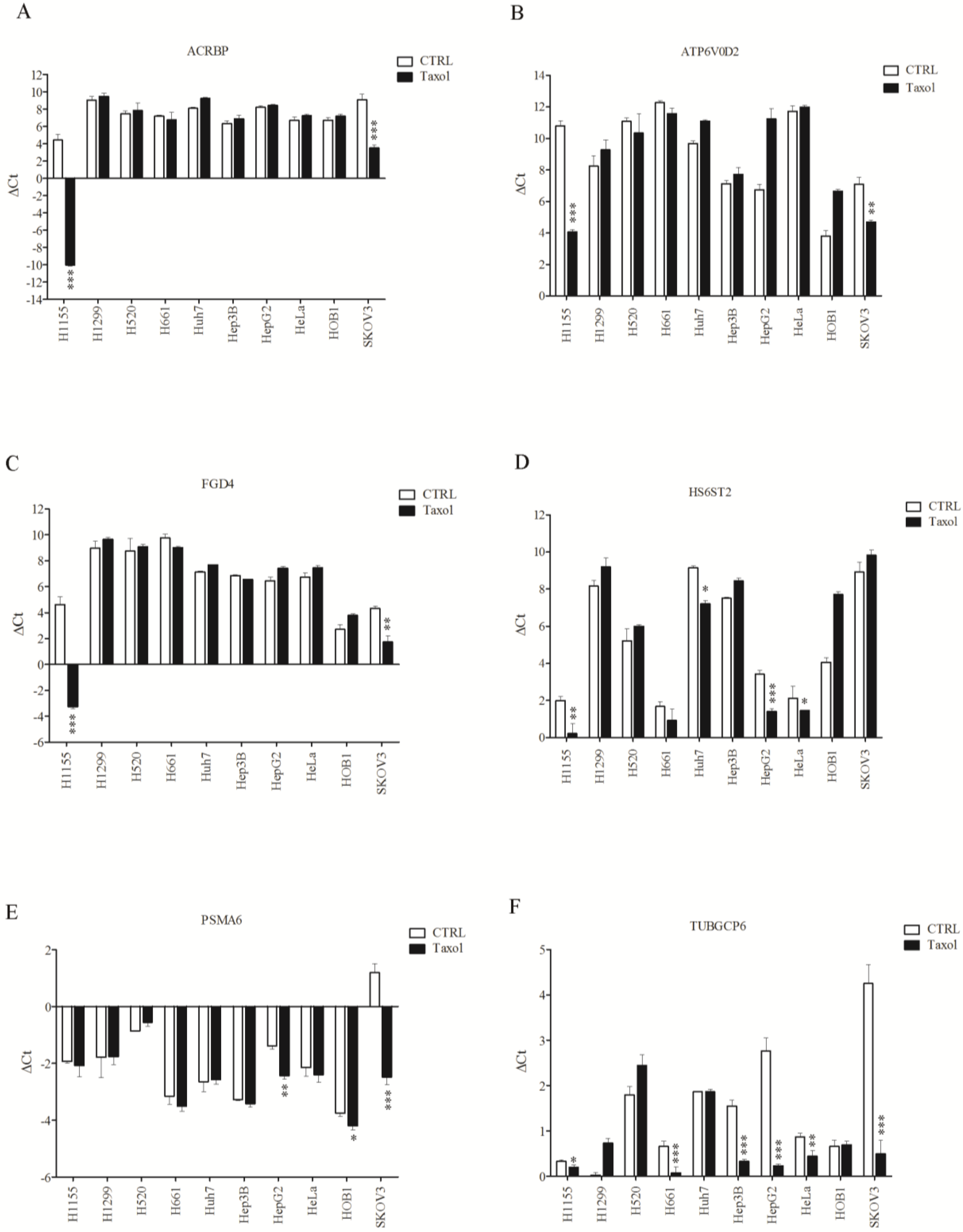
3.3. Sensitive Response of Chemosensitizer Genes to Taxol in H1155 Cells
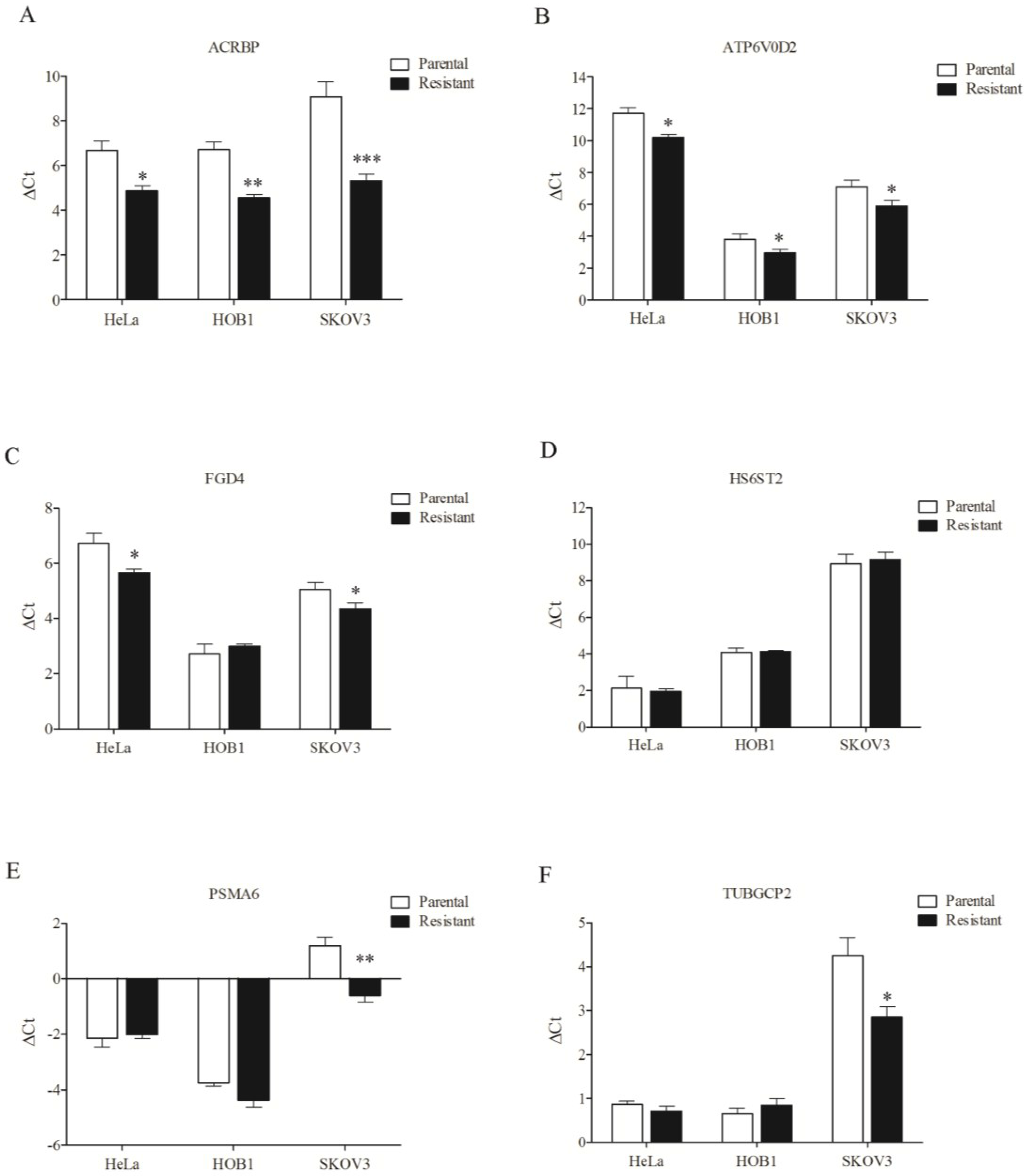
3.4. Overexpression of Chemosensitizer Genes in Drug-Resistant Cells

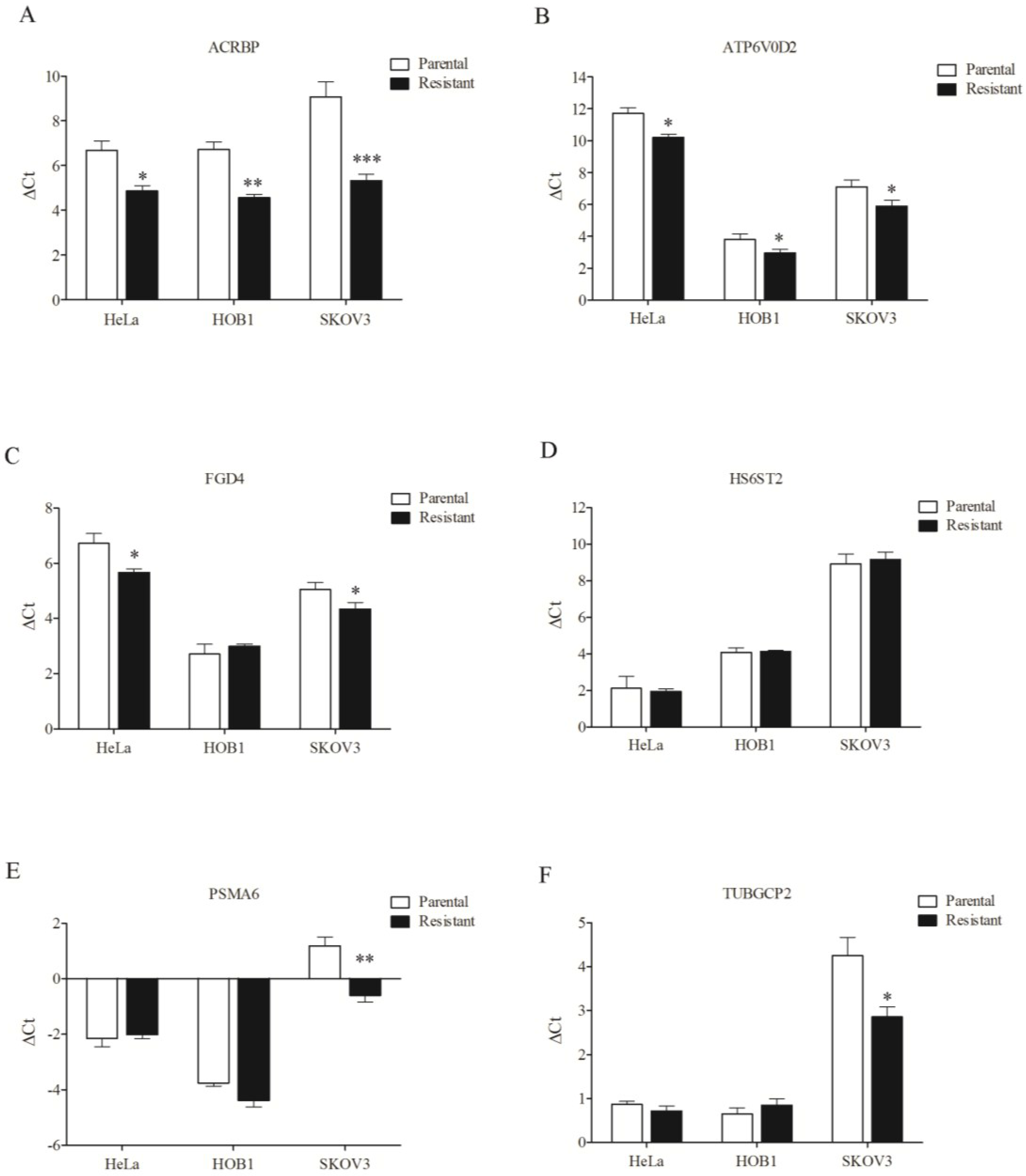
4. Discussion
5. Conclusions
Appendix
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| MDR | multiple drug resistance |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | phosphate-buffered saline |
| PCR | polymerase chain reaction |
| q-PCR | quantitative real-time polymerase chain reaction |
| shRNA | short-hairpin RNA |
References
- Murray, S.; Briasoulis, E.; Linardou, H.; Bafaloukos, D.; Papadimitriou, C. Taxane resistance in breast cancer: Mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 2012, 38, 890–903. [Google Scholar] [CrossRef] [PubMed]
- McGrogan, B.T.; Gilmartin, B.; Carney, D.N.; McCann, A. Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta 2008, 1785, 96–132. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Hille, S.; Rein, D.T.; Riffelmann, M.; Neumann, R.; Sartorius, J.; Pfutzner, A.; Kurbacher, C.M.; Schöndorf, T.; Breidenbach, M. Anticancer drugs induce mdr1 gene expression in recurrent ovarian cancer. Anti-Cancer Drugs 2006, 17, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Romero, M.F.; Peng, J.B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins. Pflug. Arch. 2004, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T. Drug transporters as targets for cancer chemotherapy. Cancer Genom. Proteom. 2007, 4, 241–254. [Google Scholar]
- Huang, Y.; Sadee, W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Pharmacogenetics/genomics of membrane transporters in cancer chemotherapy. Cancer Metastasis Rev. 2007, 26, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, A.W.; Bodemann, B.O.; Cardenas, J.; Ferguson, D.; Girard, L.; Peyton, M.; Minna, J.D.; Michnoff, C.; Hao, W.; Roth, M.G.; et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Kurashige, T.; Harada, N.; Noguchi, Y.; Saika, T.; Niikawa, N.; Aoe, M.; Nakamurai, S.; Higashii, T.; Hiraki, A.; et al. Identification of proacrosin binding protein sp32 precursor as a human cancerytestis antigen. Proc. Natl. Acad. Sci. USA 2001, 98, 3282–3287. [Google Scholar] [CrossRef] [PubMed]
- Elenich, L.A.; Nandi, D.; Kent, A.E.; McCluskey, T.S.; Cruz, M.; Iyer, M.N.; Woodward, E.C.; Conn, C.W.; Ochoa, A.L.; Ginsburg, D.B.; et al. The complete primary structure of mouse 20S proteasomes. Immunogenetics 1999, 49, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, W.C.; Sillibourne, J.; Rosa, J.; Doxsey, S.J. Mitosis-specific anchoring of α-tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol. Biol. Cell 2004, 15, 3642–3657. [Google Scholar] [CrossRef] [PubMed]
- Backen, A.C.; Cole, C.L.; Lau, S.C.; Clamp, A.R.; McVey, R.; Gallagher, J.T.; Jayson, G.C. Heparan sulphate synthetic and editing enzymes in ovarian cancer. Brit. J. Cancer 2007, 96, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M. Proton translocation driven by ATP hydrolysis in V-ATPases. FEBS Lett. 2003, 545, 76–85. [Google Scholar] [CrossRef]
- Obaishi, H.; Nakanishi, H.; Mandai, K.; Satoh, K.; Satoh, A.; Takahashi, K.; Miyahara, M.; Nishioka, H.; Takaishi, K.; Takai, Y. Frabin, a novel FGD1-related actin filament-binding protein capable of changing cell shape and activating c-Jun N-terminal kinase. J. Biol. Chem. 1998, 273, 18697–18700. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C. Selective drug efflux in multidrug-resistant immunoblastic B lymphoma cells with overexpressed P-glycoprotein. Eur. J. Pharmacol.-Environ. Toxicol. Pharmacol. 1996, 1, 63–72. [Google Scholar] [CrossRef]
- Sun, N.K.; Huang, S.L.; Chang, P.Y.; Lu, H.P.; Chao, C.C. Transcriptomic profiling of taxol-resistant ovarian cancer cells identifies FKBP5 and the androgen receptor as critical markers of chemotherapeutic response. Oncotarget 2014, 5, 11939–11956. [Google Scholar] [PubMed]
- Chao, C.C.; Lee, Y.L.; Cheng, P.W.; Lin-Chao, S. Enhanced host cell reactivation of damaged plasmid DNA in HeLa cells resistant to cis-diamminedichloro- platinum (II). Cancer Res. 1991, 51, 601–605. [Google Scholar] [PubMed]
- Sun, C.L.; Chao, C.C. Cross-resistance to death ligand-induced apoptosis in cisplatin-selected HeLa cells associated with overexpression of DDB2 and subsequent induction of cFLIP. Mol. Pharmacol. 2005, 67, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Z.; Lu, H.P.; Chao, C.C. Identification and functional analysis of genes which confer resistance to cisplatin in tumor cells. Biochem. Pharmacol. 2010, 80, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L.; Maiato, H. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Buys, T.P.; Chari, R.; Lee, E.H.; Zhang, M.; MacAulay, C.; Lam, S.; Lam, W.L.; Ling, V. Genetic changes in the evolution of multidrug resistance for cultured human ovarian cancer cells. Genes Chromosomes Cancer 2007, 46, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Lin-Chao, S.; Chao, C.C.-K. Reduced inhibition of DNA synthesis and G2 arrest in the cell cycle progression of resistant HeLa cells in response to cis-diamminedichloroplatinum(II). J. Biomed. Sci. 1994, 1, 131–138. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.-L.; Chao, C.C.-K. Silencing of Taxol-Sensitizer Genes in Cancer Cells: Lack of Sensitization Effects. Cancers 2015, 7, 1052-1071. https://doi.org/10.3390/cancers7020824
Huang S-L, Chao CC-K. Silencing of Taxol-Sensitizer Genes in Cancer Cells: Lack of Sensitization Effects. Cancers. 2015; 7(2):1052-1071. https://doi.org/10.3390/cancers7020824
Chicago/Turabian StyleHuang, Shang-Lang, and Chuck C.-K. Chao. 2015. "Silencing of Taxol-Sensitizer Genes in Cancer Cells: Lack of Sensitization Effects" Cancers 7, no. 2: 1052-1071. https://doi.org/10.3390/cancers7020824
APA StyleHuang, S.-L., & Chao, C. C.-K. (2015). Silencing of Taxol-Sensitizer Genes in Cancer Cells: Lack of Sensitization Effects. Cancers, 7(2), 1052-1071. https://doi.org/10.3390/cancers7020824