
Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors

Abstract
:
1. Introduction
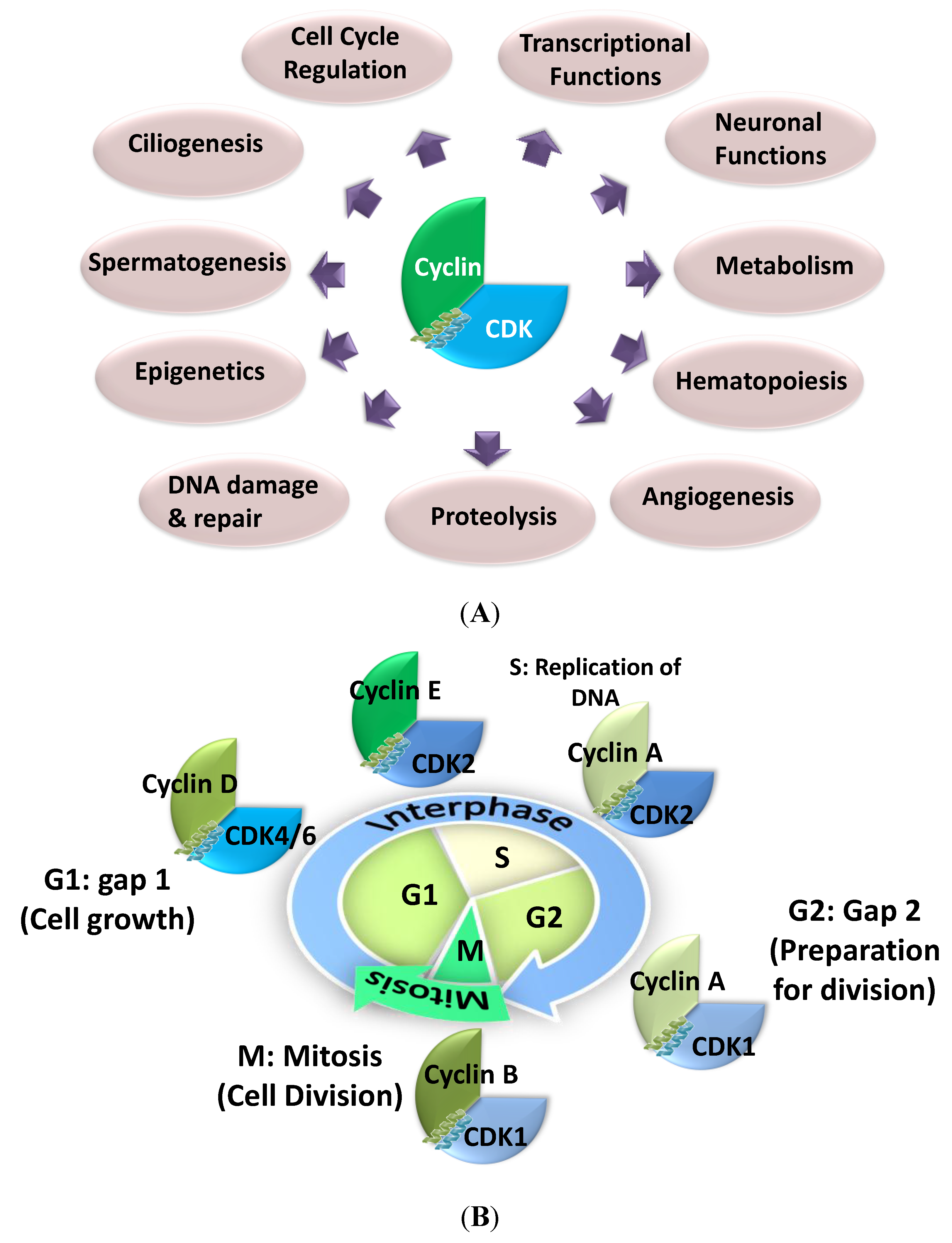
1.1. Cyclin-Dependent Kinases—From Cell Cycle Control to Physiological Regulation
1.1.1. Bona Fide Cell Cycle CDK/Cyclins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDK | In complex with | Cell cycle function | Transcriptional function | Other functions | References |
|---|---|---|---|---|---|
| CDK1 | Cyclin B | mitosis | + | stem cell self-renewal | [23,24,25,26,27,28,29,30,31,32,33,34,35] |
| DNA damage repair | |||||
| epigenetic regulation | |||||
| CDK2 | Cyclin E | G1/S transition | + | stem cell self-renewal | [26,28,31,35,36,37,38] |
| Cyclin A | S phase | epigenetic regulation | |||
| CDK3 | Cyclin C | G1 phase | + | DNA damage repair | [39,40,41] |
| CDK4 | Cyclin D | G1 phase | epigenetic regulation | [19,36,42] | |
| CDK5 | p35 | + | neuronal functions | [35,43,44,45,46,47,48,49] | |
| epigenetic regulation | |||||
| glycogen synthesis | |||||
| insulin secretion | |||||
| CDK6 | Cyclin D | G1 phase | [15,36] | ||
| CDK7 | Cyclin H | CDK-activating | + | [50] | |
| CDK8 | Cyclin C | regulator of multiple steps | + | Wnt/β-catenin signaling | [51,52,53,54] |
| inhibition of lipogenesis | |||||
| CDK9 | Cyclin T, K | + (cyclin T) | DNA damage repair (cyclin K) | [55,56] | |
| CDK10 | Cyclin M | G2/M transition | + | [57,58,59] | |
| CDK11 | Cyclin L | splicing regulation | [60] | ||
| CDK12 | Cyclin K, L | + (cyclin K) | splicing regulation (cyclin L) | [61,62,63,64] | |
| DNA damage repair (cyclin K) | |||||
| CDK13 | Cyclin K, L | splicing regulation (cyclin L) | [65] | ||
| CDK14 | Cyclin Y | Wnt/β-catenin signaling | [66] | ||
| CDK15 | |||||
| CDK16 | Cyclin Y | trafficking of synaptic proteins and synapse remodeling | [67,68,69] | ||
| spermatogenesis |
1.1.2. Transcriptional CDKs
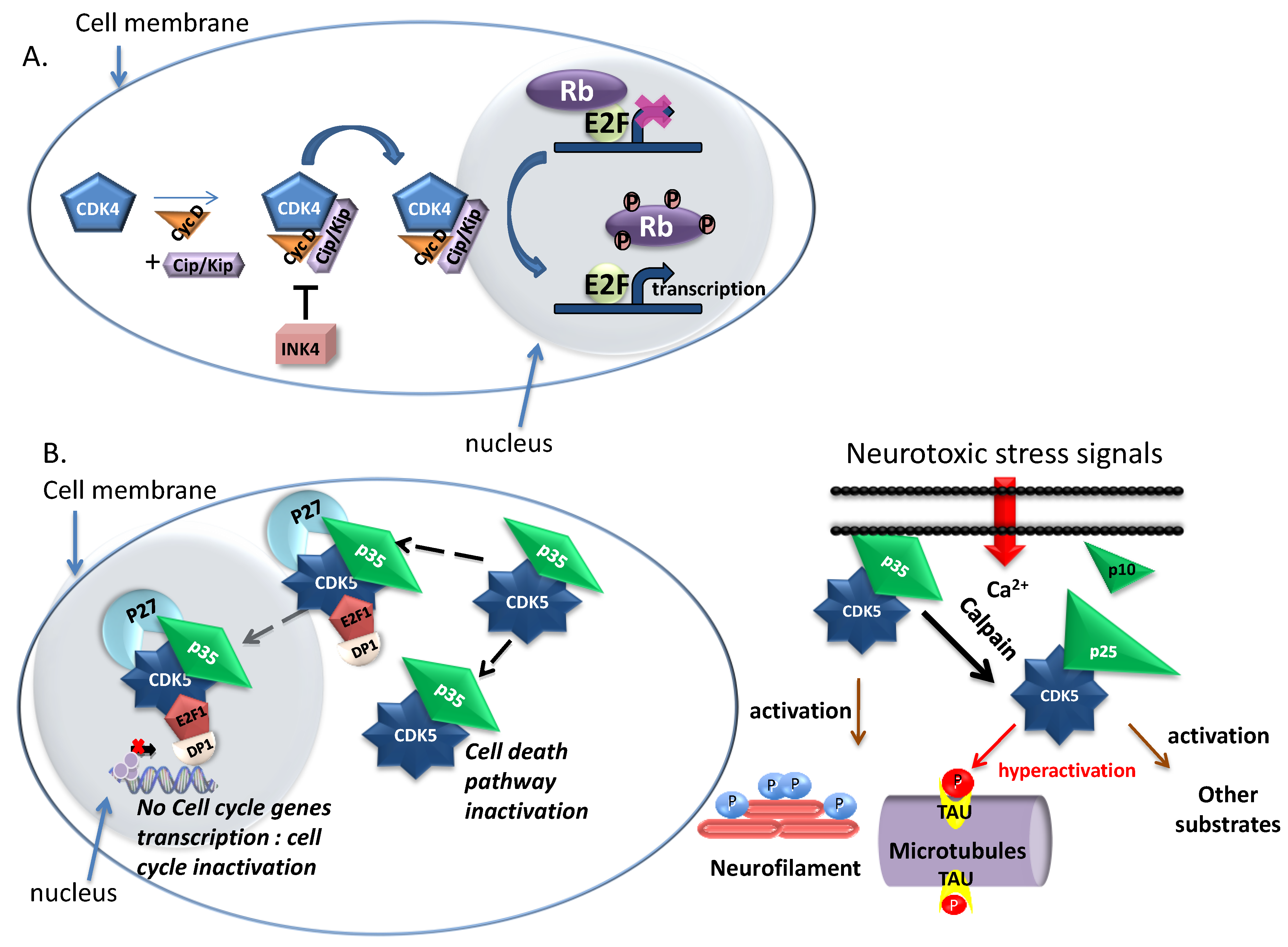
1.1.3. CDK5—Neuronal and Non Neuronal Functions
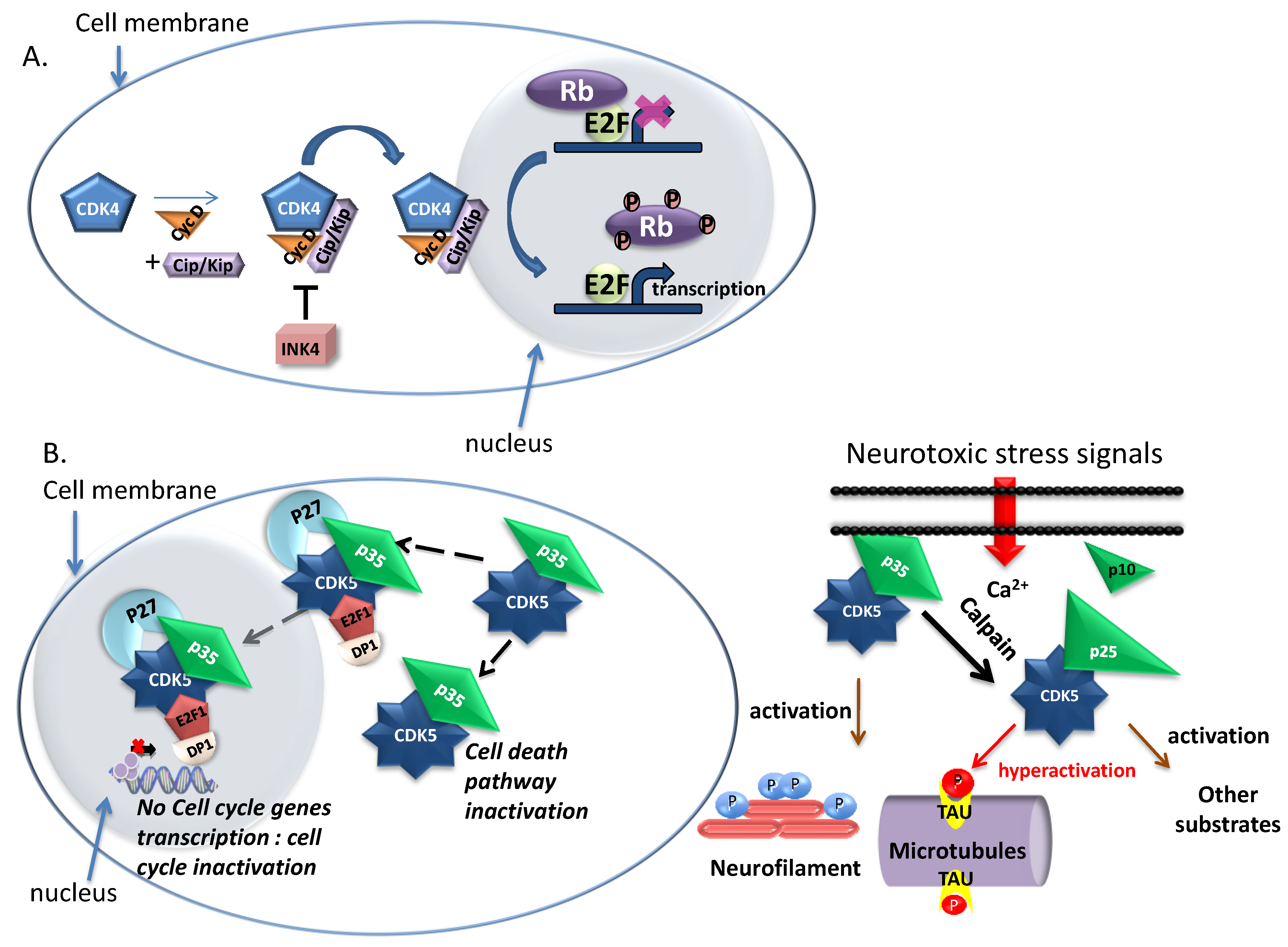
1.1.4. Other Non Cell Cycle CDKs
1.1.5. Functional Redundancy of CDK/Cyclins
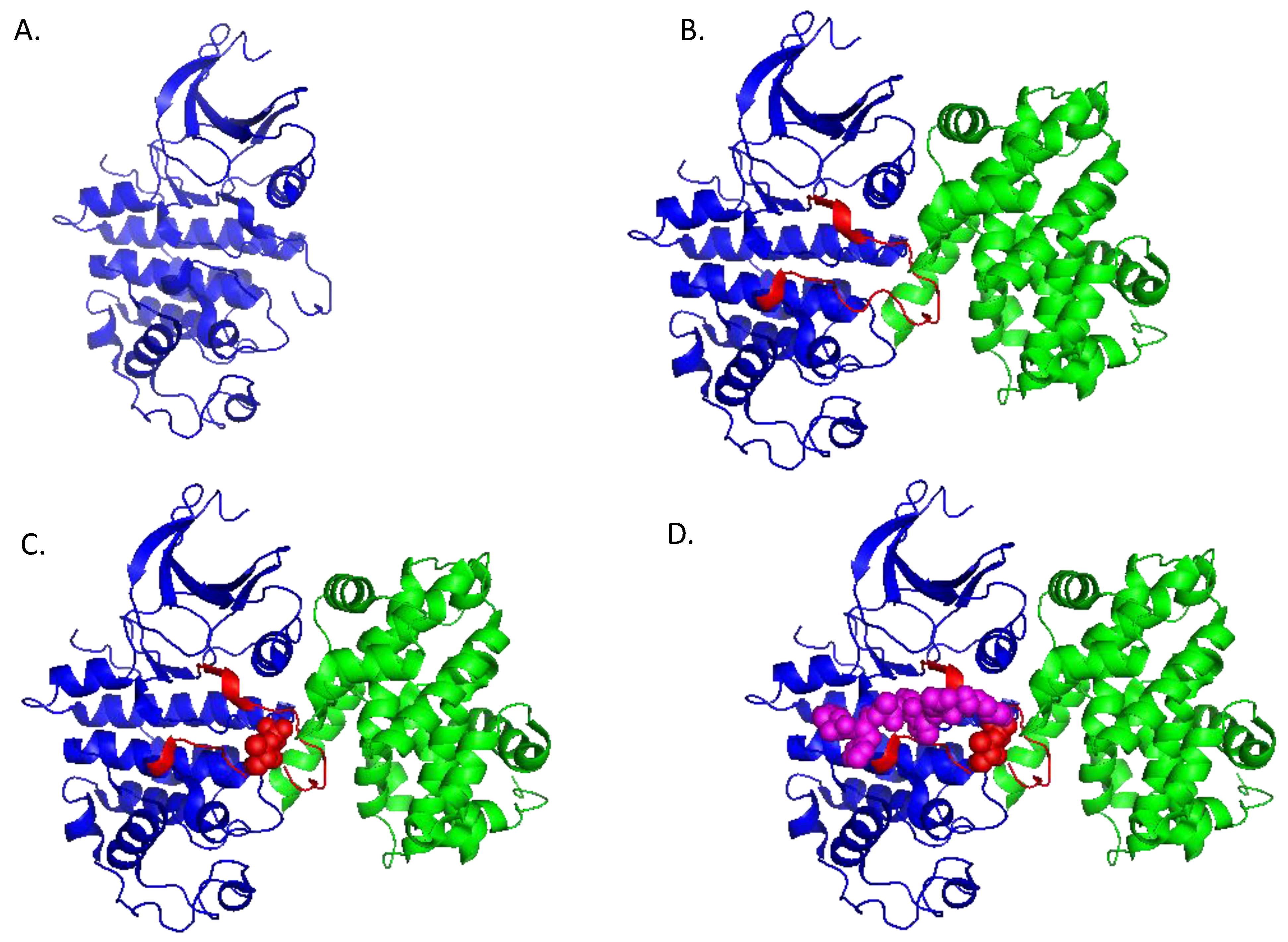
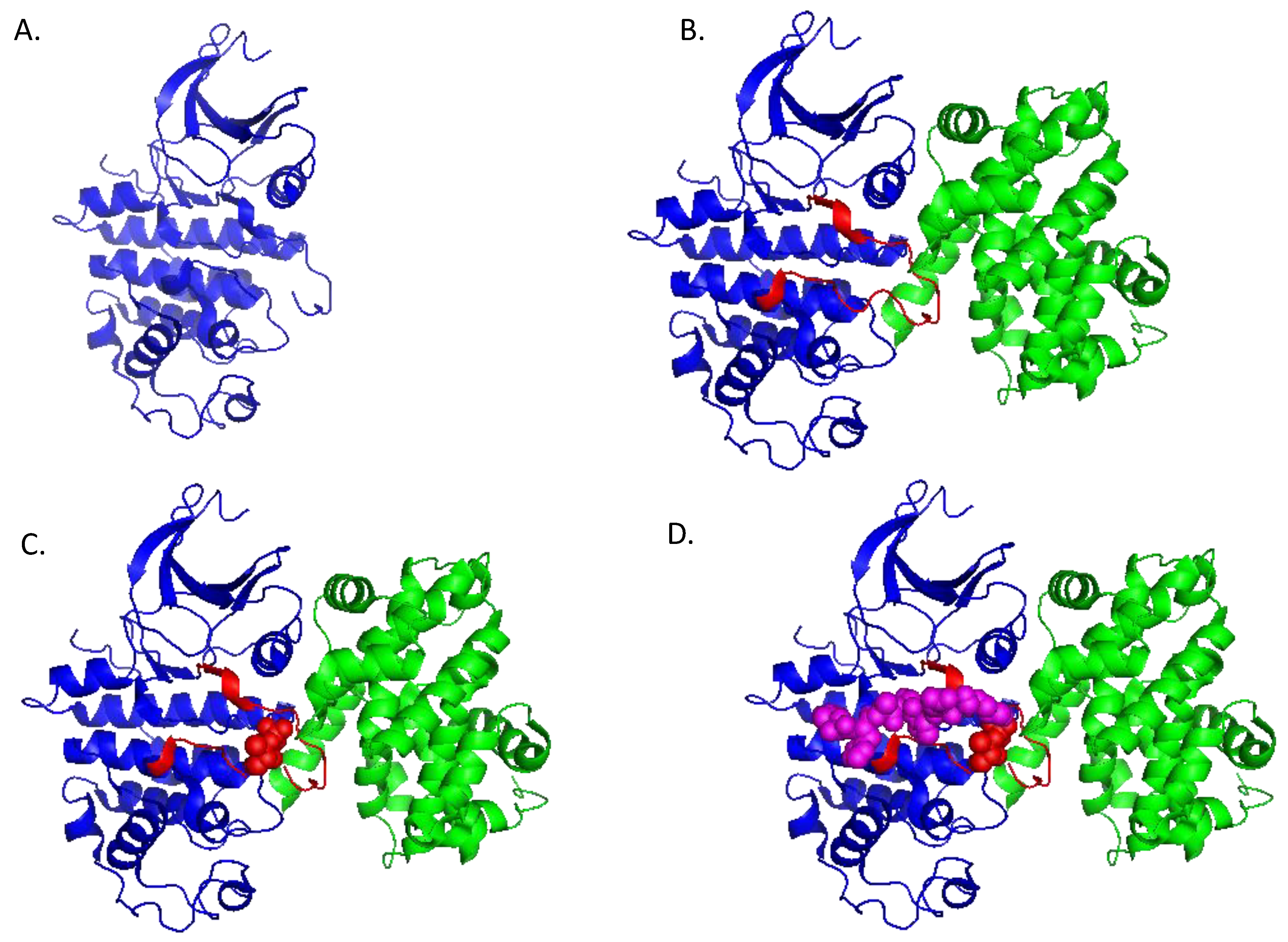
1.2. Structure and Regulation of CDK/Cyclins
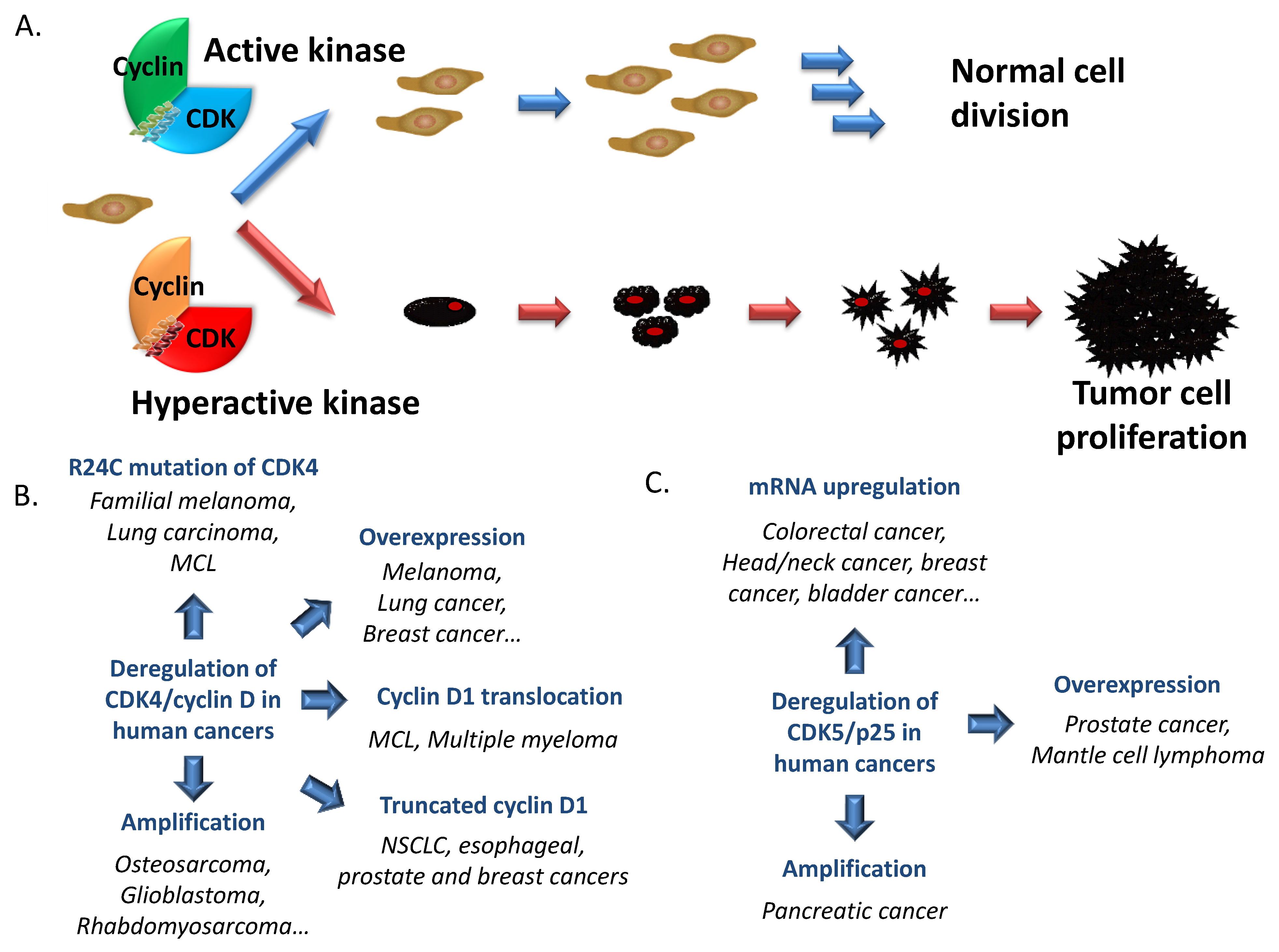
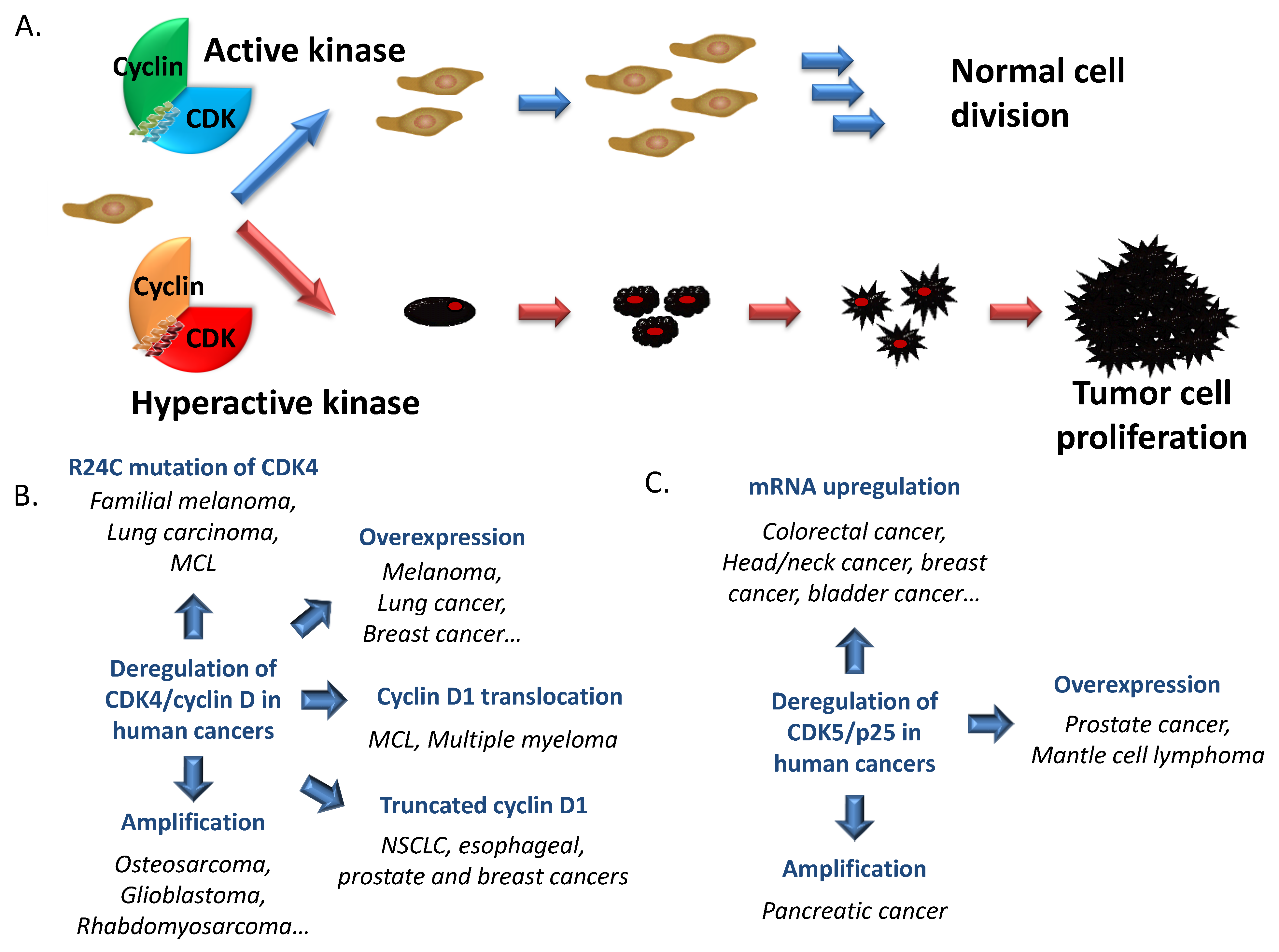
2. Cyclin-Dependent Kinases in Cancer
2.1. Cell Cycle CDKs
| Target | Deregulation | Cancer | Reference |
|---|---|---|---|
| CDK1 | Overexpression | B lymphoma, advanced melanoma | [136,137] |
| 1 simple coding mutation/missense mutation (D73H) | ovary carcinoma | [135] | |
| CDK2 | Overexpression | Laryngeal squamous cell cancer, advanced melanoma, breast cancer | [137,144,145,147] |
| 33 simple coding mutation/25 missense mutations/7 synonymous mutations/1 frameshift mutation | wide variety of cancer tissues | [135] | |
| CDK3 | overexpression | glioblastoma | [40] |
| 1 simple coding mutation/missense mutations (S106N) | glioma | [135] | |
| CDK4 | Amplification | refractory rhabdomyosarcoma, osteosarcoma, glioblastoma | [163,164,165] |
| Overexpression | melanoma | [166] | |
| Overexpression | lung cancer | [167] | |
| Amplification/Overexpression | osteosarcoma, sporadic breast carcinoma, uterine cervix cancer | [164,168,169] | |
| R24C mutation | Familial melanoma | [170,171,172,173,174,175,176,177] | |
| R24C mutation | lung carcinoma | [178] | |
| R24C mutation | mantle cell lymphoma | [179] | |
| 38 simple coding mutation/25 missense mutations/12 synonymous mutations | wide variety of cancer tissues | [135] | |
| CDK5 | Amplification/Overexpression | Pancreatic cancer | [180] |
| Overexpression | Breast cancer | [181] | |
| Decreased methylation of promoter leading to overexpression | mantle cell lymphoma | [182] | |
| Single nucleotide polymorphisms (SNPs) in the promoter region | increased risk of lung cancer | [183] | |
| Overexpression | Prostate cancer | [184] | |
| mRNA upregulation | colorectal, head/neck, breast, lung, ovarian, lymphoma, prostatic, sarcoma, myeloma and bladder cancers | [185] | |
| 24 simple coding mutation/15 missense mutations/7 synonymous mutations/1 deletion frameshift | wide variety of cancer tissues | [135] | |
| CDK6 | translocation | splenic marginal zone lymphoma | [186] |
| amplification | squamous cell carcinoma, glioma and lymphoma | [187,188] | |
| D32Y mutation | neuroblastoma | [189] | |
| sumoylation | glioblastoma | [190] | |
| overexpression | medulloblastoma | [191] | |
| 33 simple coding mutation/1 nonsensense substitution/18 missense mutations/11 synonymous mutations/1 complex mutation | wide variety of cancer tissues | [135] | |
| CDK7 | 24 simple coding mutation/1 nonsensense substitution/19 missense mutations/3 synonymous mutations | wide variety of cancer tissues | [135] |
| CDK8 | overexpression | colon cancer | [192] |
| amplification and overexpression | colorectal cancer | [52,193,194] | |
| gastric | gastric cancers | [195] | |
| upregulation upon loss of macroH2A histone variant | melanoma | [196] | |
| siRNA-mediated silencing inhibits proliferation | breast cancer | [197] | |
| tumor-suppressive function | endometrial cancer | [198] | |
| 65 simple coding mutation/9 nonsensense substitution/42 missense mutations/12 synonymous mutations/2 inframe deletions | wide variety of cancer tissues | [135] | |
| CDK9 | highly expressed | chronic lymphocytic leukemia and multiple myeloma | [199] |
| differential expression correlating with lymphoid differentiation/activation and malignant transformation | lymphoma | [200] | |
| expression correlates with differentiation grade | neuroblastoma and primary neuroectodermal tumours | [201] | |
| 1 simple coding mutation/missense mutation (D323N) | lung adenocarcinoma | [135] | |
| CDK10 | downregulation | biliary tract cancer | [202] |
| downregulation | hepatocellular carcinoma | [203] | |
| 1 simple coding mutation/missense mutation (N157S) | ovary carcinoma | [135] | |
| CDK11 | Gene deletion/translocation | neuroblastoma | [204] |
| Loss of one allele of Cdc2L/reduced CDK11 expression | melanoma | [205] | |
| overexpression | osteosarcoma | [206] | |
| essential for growth of liposarcoma cells | liposarcoma | [207] | |
| CDK11A | 43 simple coding mutation/2 nonsensense substitution/30 missense mutations/8 synonymous mutations/2 inframe deletions | wide variety of cancer tissues | [135] |
| CDK11B | 38 simple coding mutation/2 nonsensense substitution/21 missense mutations/12 synonymous mutations/2 inframe insertions/2 deletion frameshifts | wide variety of cancer tissues | [135] |
| CDK12 | 189 simple coding mutation/17 nonsensense substitution/123 missense mutations/30 synonymous mutations/5 frameshift insertions/2 inframe deletions/11 deletion frameshifts/2 complex | wide variety of cancer tissues | [135] |
| CDK13 | 124 simple coding mutation/4 nonsensense substitution/96 missense mutations/22 synonymous mutations/1 inframe deletions/5 deletion frameshifts/1 complex | wide variety of cancer tissues | [135] |
| CDK14 | overexpression associated with increased cell migratory properties | hepatocellular carcinoma | [208] |
| overexpression associated with enhanced of chemoresistance | oesophageal squamous cell carcinoma | [209] | |
| 92 simple coding mutation/3 nonsensense substitution/62 missense mutations/20 synonymous mutations/1 inframe deletions/1 deletion frameshift | wide variety of cancer tissues | [135] | |
| CDK15 | 68 simple coding mutation/4 nonsensense substitution/42 missense mutations/14 synonymous mutations/3 deletion frameshifts | wide variety of cancer tissues | [135] |
| CDK16 | 35 simple coding mutation/1 nonsensense substituation/29 missense mutations/4 synonymous mutations | wide variety of cancer tissues | [135] |
| CDK17 | 76 simple coding mutation/7 nonsensense substituation/47 missense mutations/13 synonymous mutations/1 deletion frameshift/1 complex | wide variety of cancer tissues | [135] |
| CDK18 | 48 simple coding mutation/1 nonsensense substituation/28 missense mutations/19 synonymous mutations/1 deletion frameshift | wide variety of cancer tissues | [135] |
| CDK19 | 65 simple coding mutation/1 nonsensense substituation/45 missense mutations/16 synonymous mutations/1 deletion frameshift | wide variety of cancer tissues | [135] |
| CDK20 | 1 simple coding mutation/missense mutation (A165V) | malignant melanoma | [135] |
| Cyclin A | overexpression | esophageal squamous cell carcinoma, acute myeloid leukemia, soft tissue sarcoma, hepatocellular carcinoma, thyroid carcinoma, endometrial adenocarcinoma | [131,143,148,150,151,152] |
| overexpression | colorectal cancer | [147,159,160] | |
| amplification | breast cancer | [129] | |
| truncated form due to integration of hepatitis B virus DNA | hepatocellular carcinoma | [151,162] | |
| Cyclin B | Overexpression | breast cancers, esophageal squamous cell carcinoma, NSCLC, thyroid carcinoma | [139,140,141,142,143] |
| overexpression/nuclear localization | breast cancer | [132] | |
| Cyclin D | Overexpression | Follicular mantle cell lymphoma, lung cancer, breast cancer, head and neck, esophageal cancer | [133] |
| Overexpression | Colorectal adenocarcinomas | [210] | |
| Overexpression | lung cancer | [167] | |
| Overexpression | pancreatic cancer | [211] | |
| Overexpression | endometrial carcinoma | [212] | |
| Amplification/overexpression | head and neck carcinoma | [213,214,215] | |
| IGH translocation and overexpression | multiple myeloma | [216,217] | |
| IGH translocation and overexpression | mantle cell lymphoma | [218,219] | |
| Mutation that disrupts phosphorylation-dependent nuclear export | Esophageal cancer | [220] | |
| Truncated form (cyclin D1b) (A870G polymorphism)/Nuclear Accumulation | NSCLC | [221,222,223] | |
| Truncated form (cyclin D1b) (A870G polymorphism)/Nuclear Accumulation | esophageal and prostate cancer | [224,225] | |
| Truncated form (cyclin D1b) (A870G polymorphism)/Nuclear Accumulation | prostate cancer | [226] | |
| Truncated form (cyclin D1b) (A870G polymorphism)/Nuclear Accumulation | breast cancer | [227] | |
| Truncated form (cyclin D1b) and its co-expression with cyclin D1a | breast cancer | [228] | |
| cyclin D1a isoforms with truncated 3' UTRs, not alternatively spliced cyclin D1b mRNA isoforms/alterations of CCND1 3' UTR structure | mantle cell lymphoma | [229] | |
| Cyclin E | amplification | ovarian cancers | [154,155] |
| Overexpression | acute and chronic leukemias, Hodgkin’s and non-Hodgkin’s lymphomas | [230,231] | |
| Overexpression | osteosarcoma, NSCLC, pancreatic cancer | [156,157,158] | |
| Overexpression/amplification | colorectal cancer | [147,161] | |
| Overexpression/High nuclear expression | early development of breast cancers | [153] | |
| Overexpression of small isoforms | breast cancers | [145,232,233,234] | |
| Low mol weight (LMW) isoform (truncated) | breast cancer, melanoma, ovarian carcinoma tumors | [130,235,236,237] |
2.2. CDK5
2.3. Transcriptional CDKs
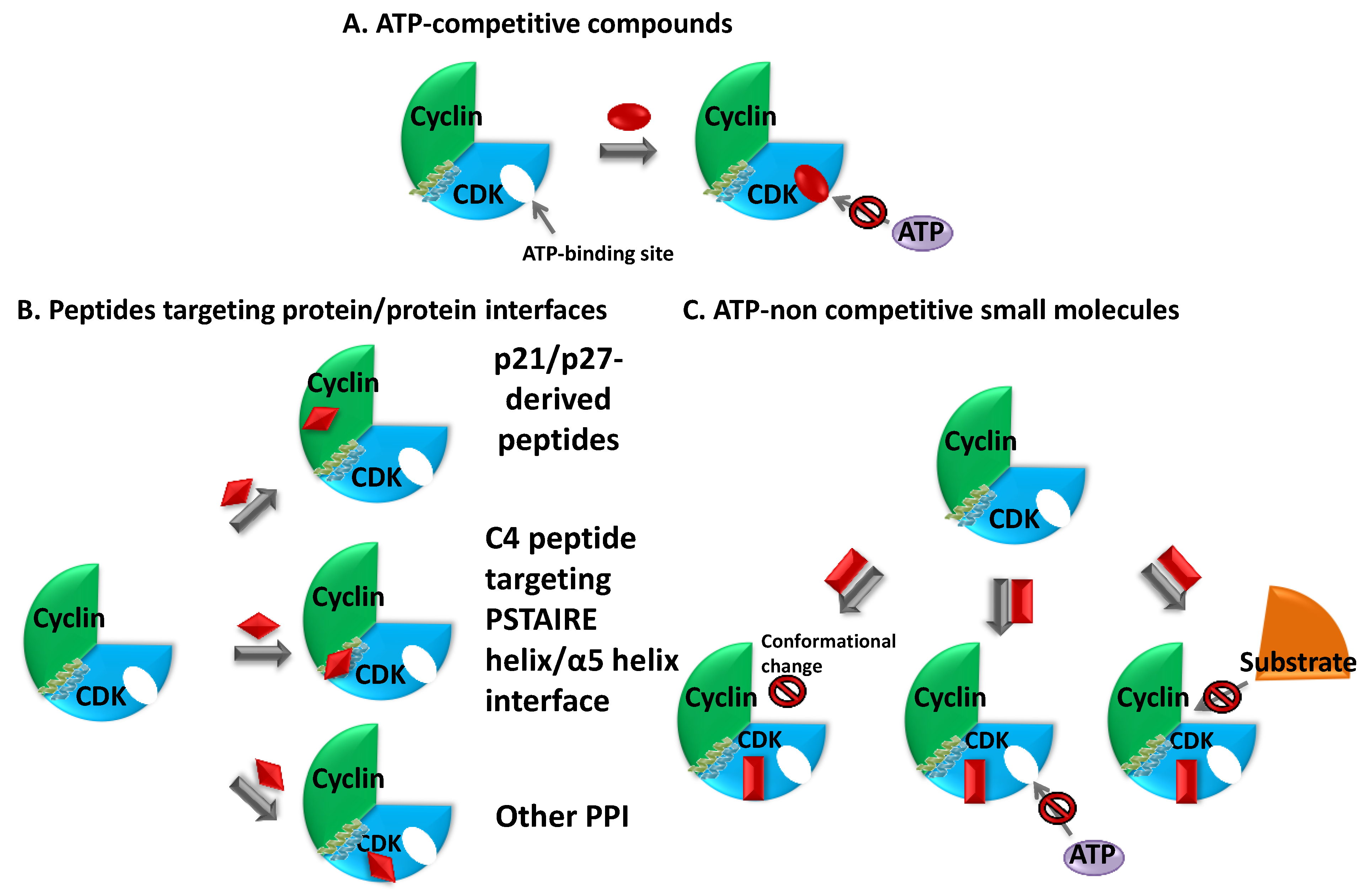
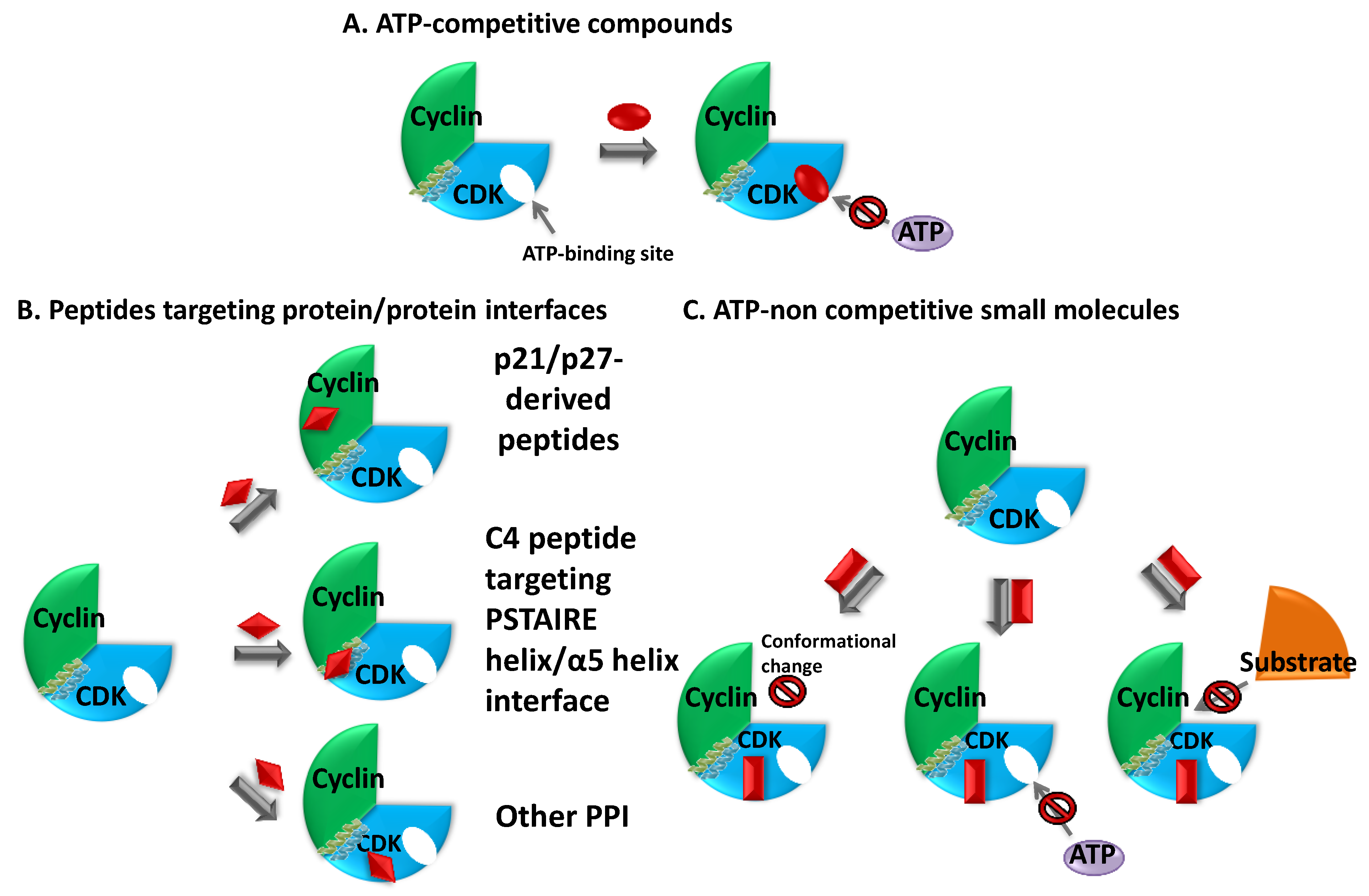
3. Targeting Cyclin-Dependent Kinases—Strategies and Inhibitors
| Inhibitor | Type/Nature/Class | Target | References |
|---|---|---|---|
| ATP-competitive compounds | |||
| Butyrolactone I | ATP-competitive/natural product | CDK1 > CDK2 | [290,291,292,293] |
| Staurosporine | ATP-competitive/alkaloid | CDK1, CDK2, CDK4 | [294,295] |
| 7-hydroxystaurosporine/UCN01 | ATP-competitive/alkaloid | CDK2, pRb, Chk1 | [270,296,297] |
| Flavopiridol/Alvocidib | ATP-competitive/flavonoid | CDK2, CDK4, CDK6, CDK9 | [270,298,299,300,301,302,303,304] |
| P276-00 | ATP-competitive/flavone | CDK1, CDK4, CDK9 | [305,306] |
| Hymenialdisine | ATP-competitive/natural product | CDK5, GSK3beta, CDK2, CDK1, Chk1 | [307] |
| Fascaplysine | ATP-competitive/natural product | CDK4 | [308] |
| Meriolins | ATP-competitive/aminopyrimidine indole | CDK1, CDK4, CDK9 | [309] |
| Roscovitine/CYC202/Seliciclib/CYC065 | ATP-competitive/trisubstituted purine | CDK5, CDK2, CDK1, CDK7, CDK9 | [310,311,312,313,314,315] |
| NU2058 & NU6027 | ATP-competitive/purine/pyrimidine | CDK1, CDK2 | [316,317] |
| Purvalanol-A | ATP-competitive/purine | CDK1,2, 5 | [318,319] |
| NU6140 | ATP-competitive/purine | [320] | |
| Olomoucine | ATP-competitive/purine | CDK1, CDK2, CDK5 | [321,322] |
| Indirubin-5 | ATP-competitive/indolinone | CDK1 > CDK2 > CDK5 | [323,324] |
| SU9516 | ATP-competitive/3-substituted indolinone | CDK2, CDK4 | [325,326] |
| Paullones | ATP-competitive/paullone | CDKs | [323,324,327] |
| R547/Ro-4584820 | ATP-competitive/Diaminopyrimidine | CDK1, CDK2, CDK4 | [328,329,330] |
| Dinaciclib (SCH 727965) | ATP-competitive/pyrimidine | CDK9, CDK1, CDK2, CDK5 | [331,332,333,334] |
| CDKI-73 | ATP-competitive/pyrimidine | CDK9 | [335] |
| PD-0183812 | ATP-competitive/pyridine | CDK4, CDK6 | [336] |
| PD-0322991/Palbociclib | ATP-competitive/pyrido-pyrimidine | CDK4, CDK6 | [337,338,339,340,341,342] |
| LEE011/LY2835219 | Small molecule | CDK1, CDK2, CDK4 | [343,344,345] |
| SNS-032/BMS-387032 | ATP-competitive/thiazole | CDK2, CDK7, CDK9 | [346,347] |
| RO-3306 | ATP-competitive/thiazolinone | CDK1 > CDK2 | [348] |
| AT7519 | ATP-competitive/pyrazole | CDK2, CDK9, CDK5, CDK4 | [349,350,351,352] |
| Peptides Targeting PPI | |||
| Spa310 and derivative from p130/pRb spacer domain | Peptide Competing with Substrate | CDK2/Cyclin A | [353,354] |
| CIP Peptide derived from p53, targeting CDK2/p53 | Peptide Competing with Substrate | CDK2/Cyclin A | [355] |
| C4 interface peptide derived from Cyclin A | Peptide Targeting CDK/Cyclin PPI | CDK2/CyclinA | [356] |
| NBI1 hexapeptide targeting Cyclin A surface pocket | Peptide Targeting CDK/Cyclin PPI | CDK2/Cyclin A | [357] |
| Interface Peptides derived from p35: CIP and p5 | Peptide Targeting CDK/Cyclin PPI | CDK5/p35 | [358,359,360,361,362,363] |
| RXL peptides | Peptide Targeting Cyclin-binding Groove | CDK2/CyclinA | [364] |
| C-terminal hexapeptide PRGPRP | Peptides Targeting CDK4 | CDK4/Cyclin D | [365] |
| Small peptides derived from E2F1 | Peptide Targeting Cyclin-binding Groove | CDK2 | [366] |
| Peptides derived from p21 | Peptide Targeting Cyclin-binding Groove | CDK2, CDK4 | [367,368,369,370,371,372] |
| Peptides derived from p27 | Peptide Targeting Cyclin-binding Groove | CDK2, CDK4 | [373] |
| Cyc103/cyclic peptide derived from p27 | Peptide Targeting Cyclin-binding Groove | CDK2 | [374] |
| Constrained peptidomimetic of p27 peptide | Peptide Targeting Cyclin-binding Groove | CDK2 | [375] |
| Peptide derived from P16INK4 | Peptide Targeting Cyclin-binding Groove | CDK4, CDK6 | [376,377] |
| ATP-Non Competitive Small molecules | |||
| SU9516 | ATP-competitive/3-substituted indolinone | CDK4 | [325,326] |
| Compound 1 | Small Molecule | CDK4 | [378] |
| 3-ATA: 3-amino thioacridone | Aminoacridines | CDK4 | [379] |
| CPD1—3alpha-amino-5alpha androstane | Small Molecule Non-ATP competitive | CDK5/p35 | [380,381] |
| Allosteric pocket in CDK2/CyclinA/p27 | Small Molecule Non-ATP competitive | CDK2/cyclinA/p27 | [382] |
| Chrysin-derivative/compound 69407 | Small Molecule Non-ATP competitive Allosteric | CDK2 & CDK4/CDK6 | [383] |
| ZK304709/MTGI/ZK-CDK | ATP-competitive | CDK1, CDK2,CDK4, CDK7, CDK9 | [384,385] |
| Cki-277 | ATP-competitive/thiazole urea | CDK1, CDK2 | [386] |
| JNJ-7706621 | ATP-competitive/acyl-substitutes triazole diamine | CDK1, CDK2/Aurora kinases | [387,388] |
| RGB-286199 | ATP-competitive/indenopyrazole | ||
| AG-024322 | Drug-like | CDK1, CDK2, CDK4 | [389] |
| AZD5438 | Drug-like | CDK1, CDK2, CDK9 | [390,391] |
| PHA-848125 | Drug like | CDK2 | |
| PHA-793887 | Drug like | CDK1, CDK2, CDK5, CDK7, CDK9 | |
| BAY-1000394 | Drug like | CDK1, CDK4, CDK9 | |
| CINK4 | ATP-competitive/triamino-pyrimidine | CDK4, CDK6 | [392] |
| 2-Aminoquinazoline inhibitors | Small molecule | CDK4 | [393] |
| 7X | Cyanopyridopyrimidine | CDK4 (ARK5) | [394] |
| Small molecule | CDK2, CDK4 | [395] | |
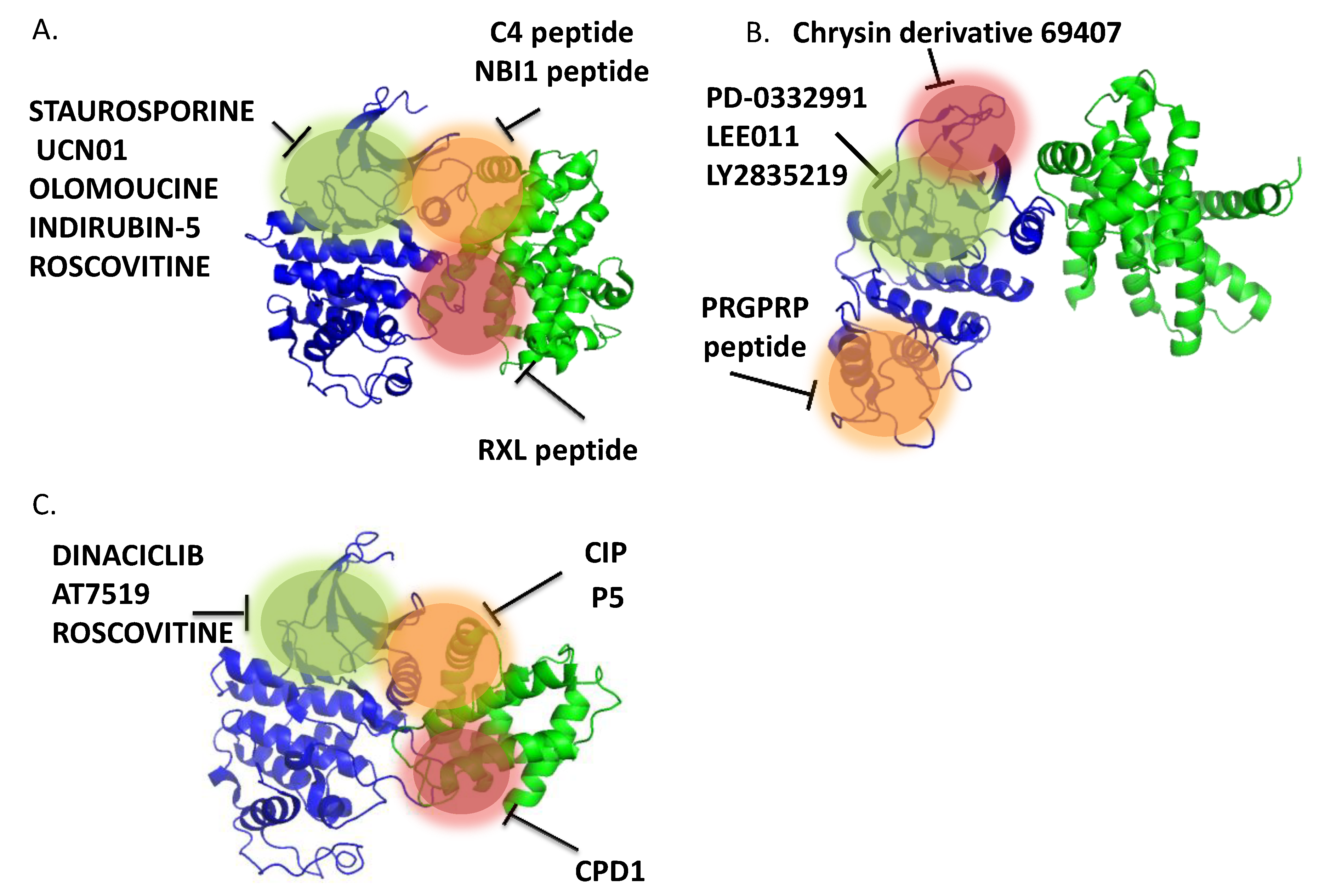
3.1. ATP-Competitive Inhibitors—From Natural Sources to SYNTHETIC Analogs
3.2. From First to Second Generation ATP-Competitive Inhibitors
3.3. ATP-Noncompetitive Inhibitors
3.3.1. Peptide Inhibitors of Protein/Protein Interactions
3.3.1.1. Substrate-Competitive Inhibitors of CDK2
3.3.1.2. Peptides Targeting the CDK2/CyclinA Interface
3.3.1.3. Targeting the Cyclin-Binding Groove of CDK2—Mimicking CKIs
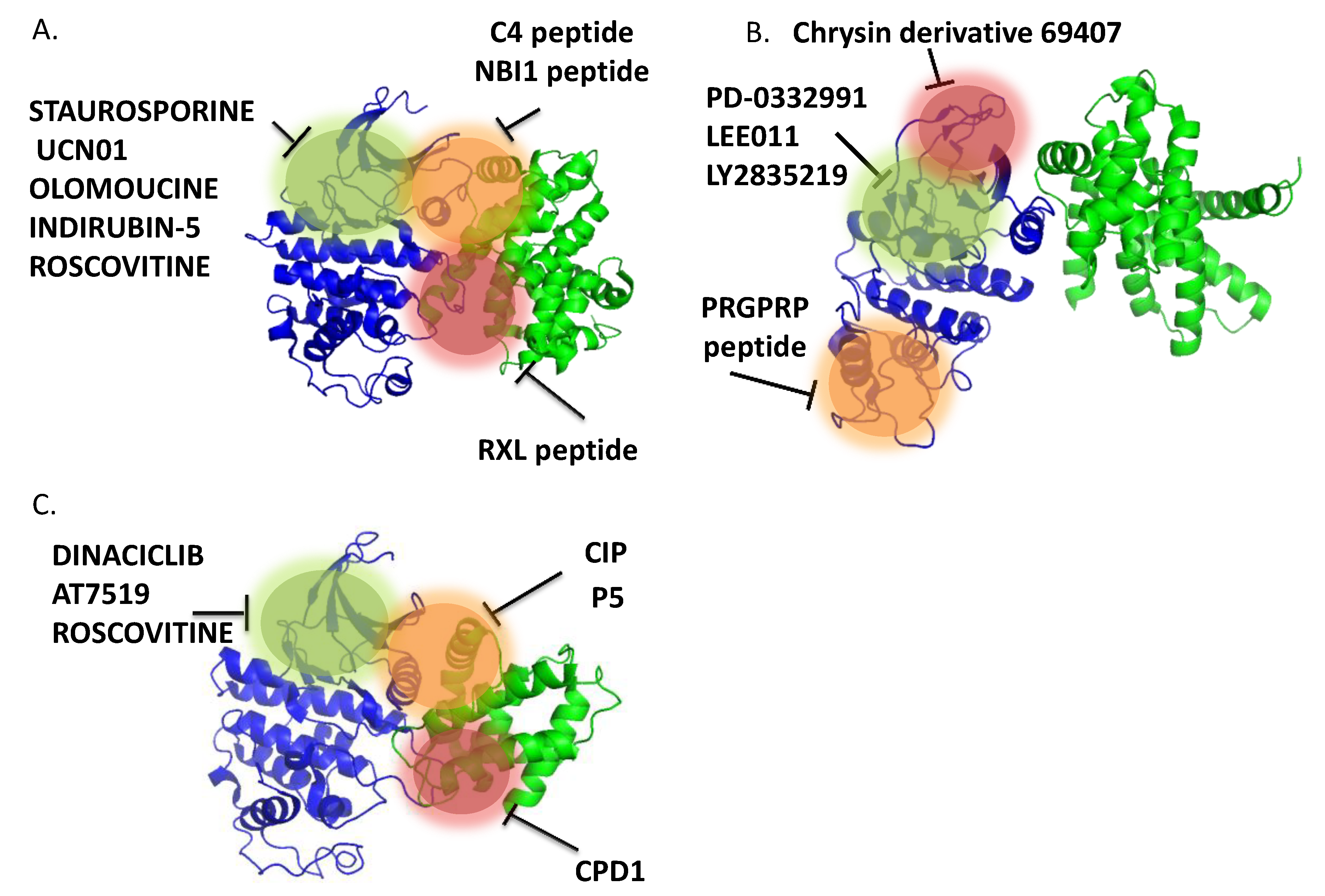
3.3.1.4. CDK4 Targeting Peptide
3.3.1.5. Peptide Inhibitors of CDK5/p25/p35
3.3.2. Small Molecule ATP-Noncompetitive Inhibitors—Allosteric Inhibitors [423,425,434,435]
3.3.2.1. Small Molecule Inhibitors of CDK4 (Figure 6b)
3.3.2.2. Small Molecule Inhibitors of CDK5 (Figure 6C)
4. Concluding Remarks and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nurse, P. Genetic control of cell size at cell division in yeast. Nature 1975, 256, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic control of the cell division cycle in yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Nurse, P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 1987, 327, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Labbe, J.C.; Lee, M.G.; Nurse, P.; Picard, A.; Doree, M. Activation at M-phase of a protein kinase encoded by a starfish homologue of the cell cycle control gene cdc2+. Nature 1988, 335, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Lohka, M.J.; Hayes, M.K.; Maller, J.L. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA 1988, 85, 3009–3013. [Google Scholar] [CrossRef] [PubMed]
- Labbe, J.C.; Picard, A.; Peaucellier, G.; Cavadore, J.C.; Nurse, P.; Doree, M. Purification of MPF from starfish: Identification as the H1 histone kinase p34cdc2 and a possible mechanism for its periodic activation. Cell 1989, 57, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T. Cyclins and their partners: From a simple idea to complicated reality. Semin. Cell Biol. 1991, 2, 213–222. [Google Scholar] [PubMed]
- Dorée, M.; Hunt, T. From Cdc2 to Cdk1: When did the cell cycle kinase join its cyclin partner? J. Cell. Sci. 2002, 115, 2461–2464. [Google Scholar] [PubMed]
- Nurse, P. The incredible life and times of biological cells. Science 2000, 289, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Physiological relevance of cell cycle kinases. Physiol. Rev. 2011, 91, 973–1007. [Google Scholar] [CrossRef] [PubMed]
- Obaya, A.J.; Sedivy, J.M. Regulation of cyclin-Cdk activity in mammalian cells. Cell. Mol. Life Sci. 2002, 59, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.; Dowdy, S.F. Regulation of G(1) cell-cycle progression by oncogenes and tumor suppressor genes. Curr. Opin. Genet. Dev. 2002, 12, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J. Progression through the G1-phase of the on-going cell cycle. J. Cell. Biochem. 2003, 90, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Coudreuse, D.; Nurse, P. Driving the cell cycle with a minimal CDK control network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Jackman, M.; Lindon, C.; Nigg, E.A.; Pines, J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 2003, 5, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Dominguez-Brauer, C.; Wang, Z.; Asara, J.M.; Costa, R.H.; Tyner, A.L.; Lau, L.F.; Raychaudhuri, P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J. Biol. Chem. 2009, 284, 30695–30707. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Ji, Z.; Child, E.S.; Krause, E.; Mann, D.J.; Sharrocks, A.D. Cell cycle-dependent regulation of the forkhead transcription factor FOXK2 by CDK·cyclin complexes. J. Biol. Chem. 2010, 285, 35728–35739. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, J.; Hou, J.; Wu, Z.; Zhuang, Y.; Lu, M.; Zhang, Y.; Zhou, X.; Li, Z.; Xiao, W.; Zhang, W. Cdk1 interplays with Oct4 to repress differentiation of embryonic stem cells into trophectoderm. FEBS Lett. 2012, 586, 4100–4107. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Hindley, C.; McDowell, G.; Deibler, R.; Jones, A.; Kirschner, M.; Guillemot, F.; Philpott, A. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Development 2011, 138, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-S.; Lu, L.X.; Ohi, M.D.; Creamer, K.M.; English, C.; Partridge, J.F.; Ohi, R.; Gould, K.L. Cdk1 phosphorylation of the kinetochore protein Nsk1 prevents error-prone chromosome segregation. J. Cell Biol. 2011, 195, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Cortés-Ledesma, F.; Sartori, A.A.; Aguilera, A.; Jackson, S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 2008, 455, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bohrer, L.R.; Rai, A.N.; Pan, Y.; Gan, L.; Zhou, X.; Bagchi, A.; Simon, J.A.; Huang, H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell Biol. 2010, 12, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Li, G.; Son, J.; Xu, C.-F.; Margueron, R.; Neubert, T.A.; Reinberg, D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010, 24, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, Y.-H.; Li, L.-Y.; Lang, J.; Yeh, S.-P.; Shi, B.; Yang, C.-C.; Yang, J.-Y.; Lin, C.-Y.; Lai, C.-C.; et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011, 13, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Zhang, Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J. Biol. Chem. 2011, 286, 28511–28519. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, G.; St-Pierre, Y. Phosphorylation of human DNMT1: Implication of cyclin-dependent kinases. Biochem. Biophys. Res. Commun. 2011, 409, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.O.; Lukas, J.; Sørensen, C.S.; Bartek, J.; Helin, K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999, 18, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Coverley, D.; Pelizon, C.; Trewick, S.; Laskey, R.A. Chromatin-bound Cdc6 persists in S and G2 phases in human cells, while soluble Cdc6 is destroyed in a cyclin A-cdk2 dependent process. J. Cell. Sci. 2000, 113, 1929–1938. [Google Scholar] [PubMed]
- Ren, S.; Rollins, B.J. Cyclin C/Cdk3 Promotes Rb-Dependent G0 Exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Cho, Y.-Y.; Lau, A.T. Y.; Zhang, J.; Ma, W.-Y.; Bode, A.M.; Dong, Z. Cyclin-Dependent Kinase 3-Mediated Activating Transcription Factor 1 Phosphorylation Enhances Cell Transformation. Cancer Res. 2008, 68, 7650–7660. [Google Scholar] [CrossRef] [PubMed]
- Tomashevski, A.; Webster, D.R.; Grammas, P.; Gorospe, M.; Kruman, I.I. Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ. 2010, 17, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Vaites, L.P.; Kim, J.K.; Mellert, H.; Gurung, B.; Nakagawa, H.; Herlyn, M.; Hua, X.; Rustgi, A.K.; McMahon, S.B.; et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010, 18, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.S.; Greer, P.L.; Tsai, L.H. Cdk5 on the brain. Cell Growth Differ. 2001, 12, 277–283. [Google Scholar] [PubMed]
- Su, S.C.; Tsai, L.-H. Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, M.; Dudek, H.; Kwon, Y.T.; Ramos, Y.F.; Tsai, L.H. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996, 10, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Lilja, L.; Yang, S.-N.; Webb, D.-L.; Juntti-Berggren, L.; Berggren, P.-O.; Bark, C. Cyclin-dependent Kinase 5 Promotes Insulin Exocytosis. J. Biol. Chem. 2001, 276, 34199–34205. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Z.H.; Ip, N.Y. Cdk5: A multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012, 22, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.Y.; Fu, W.-Y.; Ng, A.K.Y.; Chien, W.W.Y.; Ng, Y.-P.; Wang, J.H.; Ip, N.Y. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc. Natl. Acad. Sci. USA 2004, 101, 6728–6733. [Google Scholar] [CrossRef] [PubMed]
- Tudhope, S.J.; Wang, C.-C.; Petrie, J.L.; Potts, L.; Malcomson, F.; Kieswich, J.; Yaqoob, M.M.; Arden, C.; Hampson, L.J.; Agius, L. A novel mechanism for regulating hepatic glycogen synthesis involving serotonin and cyclin-dependent kinase-5. Diabetes 2012, 61, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell. Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, Z.; Gustafsson, C.M. Emerging roles of Cdk8 in cell cycle control. Biochim. Biophys. Acta 2013, 1829, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Akoulitchev, S.; Chuikov, S.; Reinberg, D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000, 407, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Feng, D.; Wang, Q.; Abdulla, A.; Xie, X.-J.; Zhou, J.; Sun, Y.; Yang, E.S.; Liu, L.-P.; Vaitheesvaran, B.; et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J. Clin. Investig. 2012, 122, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fischer, P.M. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Zhao, R.; Hsu, E.L.; Cayer, J.; Ye, F.; Guo, Y.; Shyr, Y.; Cortez, D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010, 11, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; MacLachlan, T.K.; de Luca, A.; Claudio, P.P.; Condorelli, G.; Giordano, A. The cdc-2-related kinase, PISSLRE, is essential for cell growth and acts in G2 phase of the cell cycle. Cancer Res. 1995, 55, 3992–3995. [Google Scholar] [PubMed]
- Guen, V.J.; Gamble, C.; Flajolet, M.; Unger, S.; Thollet, A.; Ferandin, Y.; Superti-Furga, A.; Cohen, P.A.; Meijer, L.; Colas, P. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, 19525–19530. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Turner, N.C.; Elliott, R.; Syed, N.; Garrone, O.; Gasco, M.; Tutt, A.N.J.; Crook, T.; Lord, C.J.; Ashworth, A. Identification of CDK10 as an Important Determinant of Resistance to Endocrine Therapy for Breast Cancer. Cancer Cell 2008, 13, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Mayeda, A.; Trembley, J.H.; Lahti, J.M.; Kidd, V.J. CDK11 Complexes Promote Pre-mRNA Splicing. J. Biol. Chem. 2003, 278, 8623–8629. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Wang, Y.-C.; Fann, M.-J. Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol. Cell. Biol. 2006, 26, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-W.G.; Kuzyk, M.A.; Moradian, A.; Ichu, T.-A.; Chang, V.C.-D.; Tien, J.F.; Vollett, S.E.; Griffith, M.; Marra, M.A.; Morin, G.B. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol. Cell. Biol. 2012, 32, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Wong, Y.-H.; Geneviere, A.-M.; Fann, M.-J. CDK13/CDC2L5 interacts with L-type cyclins and regulates alternative splicing. Biochem. Biophys. Res. Commun. 2007, 354, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Shen, J.; Huang, Y.-L.; Su, Y.; Karaulanov, E.; Bartscherer, K.; Hassler, C.; Stannek, P.; Boutros, M.; Niehrs, C. Cell cycle control of wnt receptor activation. Dev. Cell 2009, 17, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.-Y.; Poon, V.Y.; Maeder, C.I.; Watanabe, S.; Lehrman, E.K.; Fu, A.K.Y.; Park, M.; Fu, W.-Y.; Jorgensen, E.M.; Ip, N.Y.; et al. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 2010, 141, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Watanabe, S.; Poon, V.Y.N.; Ou, C.-Y.; Jorgensen, E.M.; Shen, K. CYY-1/cyclin Y and CDK-5 differentially regulate synapse elimination and formation for rewiring neural circuits. Neuron 2011, 70, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Mikolcevic, P.; Sigl, R.; Rauch, V.; Hess, M.W.; Pfaller, K.; Barisic, M.; Pelliniemi, L.J.; Boesl, M.; Geley, S. Cyclin-dependent kinase 16/PCTAIRE kinase 1 is activated by cyclin Y and is essential for spermatogenesis. Mol. Cell. Biol. 2012, 32, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Nurse, P. Substrates for p34cdc2: In vivo veritas? Cell 1990, 61, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, P. The cdk-activating kinase (CAK): From yeast to mammals. Cell. Mol. Life Sci. 1999, 55, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Kasten, M.; de Falco, G.; Micheli, P.; Khalili, K.; Giordano, A. Regulatory functions of Cdk9 and of cyclin T1 in HIV tat transactivation pathway gene expression. J. Cell. Biochem. 1999, 75, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.H.; Takahashi, T.; Caviness, V.S.; Harlow, E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 1993, 119, 1029–1040. [Google Scholar] [PubMed]
- Tsai, L.H.; Delalle, I.; Caviness, V.S.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal cdc2-like kinase: A cdc2-related protein kinase with predominantly neuronal expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Enders, G.H.; Wu, C.L.; Su, L.K.; Gorka, C.; Nelson, C.; Harlow, E.; Tsai, L.H. A family of human cdc2-related protein kinases. EMBO J. 1992, 11, 2909–2917. [Google Scholar] [PubMed]
- Lew, J.; Huang, Q.Q.; Qi, Z.; Winkfein, R.J.; Aebersold, R.; Hunt, T.; Wang, J.H. A brain-specific activator of cyclin-dependent kinase 5. Nature 1994, 371, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Qi, Z.; Yu, Y.P.; Wang, J.H. Neuronal Cdc2-like kinases: Neuron-specific forms of Cdk5. Int. J. Biochem. Cell Biol. 1997, 29, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Wang, J.H. Cyclin-dependent kinase 5 (Cdk5) and neuron-specific Cdk5 activators. Prog. Cell Cycle Res. 1996, 2, 205–216. [Google Scholar] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Lee, K.Y.; Qi, Z.; Matsuura, I.; Wang, J.H. Neuronal Cdc2-like kinase: From cell cycle to neuronal function. Biochem. Cell Biol. 1996, 74, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Dhariwala, F.A.; Rajadhyaksha, M.S. An unusual member of the Cdk family: Cdk5. Cell. Mol. Neurobiol. 2008, 28, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.L.; Lee, R.H.K.; Cheung, A.Y.; Yeung, P.K.; Chung, S.K.; Cheung, Z.H.; Ip, N.Y. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol. 2011, 13, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Rosales, J.L.; Lee, K.-Y. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays 2006, 28, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Vallejos, E.; Utreras, E.; Gonzalez-Billault, C. Going out of the brain: Non-nervous system physiological and pathological functions of Cdk5. Cell. Signal. 2012, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Ishiguro, K.; Ohnuma, J.; Takamatsu, M.; Yonekura, S.; Imahori, K. Precursor of cdk5 activator, the 23 kDa subunit of tau protein kinase II: Its sequence and developmental change in brain. FEBS Lett. 1994, 355, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kusakawa, G.; Saito, T.; Onuki, R.; Ishiguro, K.; Kishimoto, T.; Hisanaga, S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 2000, 275, 17166–17172. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chun, A.C.; Zhang, M.; Wang, J.H. Cyclin-dependent kinase 5 (Cdk5) activation domain of neuronal Cdk5 activator. Evidence of the existence of cyclin fold in neuronal Cdk5a activator. J. Biol. Chem. 1997, 272, 12318–12327. [Google Scholar] [CrossRef] [PubMed]
- Tarricone, C.; Dhavan, R.; Peng, J.; Areces, L.B.; Tsai, L.H.; Musacchio, A. Structure and regulation of the CDK5-p25(nck5a) complex. Mol. Cell 2001, 8, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.W.; Ando, D.M.; Taketa, D.A.; Zhou, H.; Dahlquist, F.W.; Lew, J. No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Szumlinski, K.K.; Lew, J. p10, the N-terminal domain of p35, protects against CDK5/p25-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 20041–20046. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, M.J.; Kushwaha, N.; Zhang, Y.; Aleyasin, H.; Callaghan, S.M.; Slack, R.S.; Albert, P.R.; Vincent, I.; Park, D.S. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J. Neurosci. 2005, 25, 8954–8966. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Yabut, O.; Fitzpatrick, H.; D’Arcangelo, G.; Herrup, K. Cdk5 suppresses the neuronal cell cycle by disrupting the E2F1-DP1 complex. J. Neurosci. 2010, 30, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, H.; Herrup, K. Cdk5 nuclear localization is p27-dependent in nerve cells: Implications for cell cycle suppression and caspase-3 activation. J. Biol. Chem. 2010, 285, 14052–14061. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Stinnakre, M.G.; Senamaud-Beaufort, C.; Winston, N.J.; Sweeney, C.; Kubelka, M.; Carrington, M.; Bréchot, C.; Sobczak-Thépot, J. Delayed early embryonic lethality following disruption of the murine cyclin A2 gene. Nat. Genet. 1997, 15, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Brandeis, M.; Rosewell, I.; Carrington, M.; Crompton, T.; Jacobs, M.A.; Kirk, J.; Gannon, J.; Hunt, T. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad. Sci. USA 1998, 95, 4344–4349. [Google Scholar] [CrossRef] [PubMed]
- De Bondt, H.L.; Rosenblatt, J.; Jancarik, J.; Jones, H.D.; Morgan, D.O.; Kim, S.H. Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massagué, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Lindstrom, D.L.; Harlow, E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. USA 1998, 95, 10453–10458. [Google Scholar] [CrossRef] [PubMed]
- Coleman, T.R.; Dunphy, W.G. Cdc2 regulatory factors. Curr. Opin. Cell Biol. 1994, 6, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Rosenblatt, J.; Morgan, D.O. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992, 11, 3995–4005. [Google Scholar] [PubMed]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Dozier, C.; Ducommun, B. The when and wheres of CDC25 phosphatases. Curr. Opin. Cell Biol. 2006, 18, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.D.; Tong, L.; Pavletich, N.P. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000, 14, 3115–3125. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massagué, J.; Pavletich, N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Lolli, G. Structural dissection of cyclin dependent kinases regulation and protein recognition properties. Cell Cycle 2010, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.R.; Noble, M.E.; Endicott, J.A.; Garman, E.F.; Wakatsuki, S.; Mitchell, E.; Rasmussen, B.; Hunt, T.; Johnson, L.N. The crystal structure of cyclin A. Structure 1995, 3, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Endicott, J.A.; Brown, N.R.; Johnson, L.N. The cyclin box fold: Protein recognition in cell-cycle and transcription control. Trends Biochem. Sci. 1997, 22, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Heitz, F.; Morris, M.C.; Fesquet, D.; Cavadore, J.C.; Dorée, M.; Divita, G. Interactions of cyclins with cyclin-dependent kinases: A common interactive mechanism. Biochemistry 1997, 36, 4995–5003. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Gondeau, C.; Tainer, J.A.; Divita, G. Kinetic mechanism of activation of the Cdk2/cyclin a complex. Key role of the C-lobe of the Cdk. J. Biol. Chem. 2002, 277, 23847–23853. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.R.; Noble, M.E.; Lawrie, A.M.; Morris, M.C.; Tunnah, P.; Divita, G.; Johnson, L.N.; Endicott, J.A. Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J. Biol. Chem. 1999, 274, 8746–8756. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, J.C.; Kirtley, M.P.; Stevenson, L.M.; Gergis, R.M.; Russo, A.A.; Pavletich, N.P.; Parsons, S.M.; Lew, J. Kinetic basis for activation of CDK2/cyclin A by phosphorylation. J. Biol. Chem. 2001, 276, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [PubMed]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Settleman, J. Cancer: Drivers and passengers. Nature 2007, 446, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Castro, I.; de Cárcer, G.; Malumbres, M. A census of mitotic cancer genes: New insights into tumor cell biology and cancer therapy. Carcinogenesis 2007, 28, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Van Dross, R.; Browning, P.J.; Pelling, J.C. Do truncated cyclins contribute to aberrant cyclin expression in cancer? Cell Cycle 2006, 5, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Husdal, A.; Bukholm, G.; Bukholm, I.R.K. The prognostic value and overexpression of cyclin A is correlated with gene amplification of both cyclin A and cyclin E in breast cancer patient. Cell. Oncol. 2006, 28, 107–116. [Google Scholar] [PubMed]
- Harwell, R.M.; Mull, B.B.; Porter, D.C.; Keyomarsi, K. Activation of cyclin-dependent kinase 2 by full length and low molecular weight forms of cyclin E in breast cancer cells. J. Biol. Chem. 2004, 279, 12695–12705. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Holm, C.; Jalili, S.; Richter, J.; Anagnostaki, L.; Landberg, G.; Persson, J.L. Expression of cyclin A1 and cell cycle proteins in hematopoietic cells and acute myeloid leukemia and links to patient outcome. Eur. J. Haematol. 2005, 75, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Urano, T.; Miki, Y.; Moriya, T.; Akahira, J.; Ishida, T.; Horie, K.; Inoue, S.; Sasano, H. Nuclear cyclin B1 in human breast carcinoma as a potent prognostic factor. Cancer Sci. 2007, 98, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Diehl, J.A. Nuclear cyclin D1: An oncogenic driver in human cancer. J. Cell. Physiol. 2009, 220, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- COSMIC Database. Available online: http://www.sanger.ac.uk/genetics/CGP/cosmic/ (accessed on 1 October 2014).
- Zhao, M.Y.; Auerbach, A.; D’Costa, A.M.; Rapoport, A.P.; Burger, A.M.; Sausville, E.A.; Stass, S.A.; Jiang, F.; Sands, A.M.; Aguilera, N.; Zhao, X.F. Phospho-p70S6K/p85S6K and cdc2/cdk1 are novel targets for diffuse large B-cell lymphoma combination therapy. Clin. Cancer Res. 2009, 15, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, C.; Wang, X.; Becker, D. Expression analysis and molecular targeting of cyclin-dependent kinases in advanced melanoma. Cell Cycle 2011, 10, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Elkahloun, A.G.; Robertson, M.; Gills, J.J.; Tsurutani, J.; Shih, J.H.; Fukuoka, J.; Hollander, M.C.; Harris, C.C.; Travis, W.D.; et al. Loss of cytoplasmic CDK1 predicts poor survival in human lung cancer and confers chemotherapeutic resistance. PLoS One 2011, 6, e23849. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, K.; Amini, R.-M.; Heikkila, P.; Aittomaki, K.; Tamminen, A.; Nevanlinna, H.; Blomqvist, C. High cyclin B1 expression is associated with poor survival in breast cancer. Br. J. Cancer 2009, 100, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Begnami, M.D.; Fregnani, J.H.T.G.; Nonogaki, S.; Soares, F.A. Evaluation of cell cycle protein expression in gastric cancer: Cyclin B1 expression and its prognostic implication. Hum. Pathol. 2010, 41, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Furihata, M.; Ohtsuki, Y.; Ogoshi, S. Determination of the prognostic significance of cyclin B1 overexpression in patients with esophageal squamous cell carcinoma. Virchows Arch. 1999, 434, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Jang, S.J.; Khuri, F.R.; Hassan, K.; Liu, D.; Hong, W.K.; Mao, L. Overexpression of Cyclin B1 in Early-Stage Non-Small Cell Lung Cancer and Its Clinical Implication. Cancer Res. 2000, 60, 4000–4004. [Google Scholar] [PubMed]
- Nar, A.; Ozen, O.; Tutuncu, N.B.; Demirhan, B. Cyclin A and cyclin B1 overexpression in differentiated thyroid carcinoma. Med. Oncol. 2012, 29, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, J.; Singha, P.; Schadendorf, D. Expression of cyclins and cyclin dependent kinases in human benign and malignant melanocytic lesions. J. Clin. Pathol. 2001, 54, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Weroha, S.J.; Lingle, W.L.; Hong, Y.; Li, S.A.; Li, J.J. Specific overexpression of cyclin E·CDK2 in early preinvasive and primary breast tumors in female ACI rats induced by estrogen. Horm. Cancer 2010, 1, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Ma, H.-P.; Lu, X.-L.; Sun, S.-H.; Guo, X.; Li, F.-C. NF-κB induces abnormal centrosome amplification by upregulation of CDK2 in laryngeal squamous cell cancer. Int. J. Oncol. 2011, 39, 915–924. [Google Scholar] [PubMed]
- Wang, A.; Yoshimi, N.; Suzui, M.; Yamauchi, A.; Tarao, M.; Mori, H. Different expression patterns of cyclins A, D1 and E in human colorectal cancer. J. Cancer Res. Clin. Oncol. 1996, 122, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Furihata, M.; Ishikawa, T.; Inoue, A.; Yoshikawa, C.; Sonobe, H.; Ohtsuki, Y.; Araki, K.; Ogoshi, S. Determination of the prognostic significance of unscheduled cyclin A overexpression in patients with esophageal squamous cell carcinoma. Clin. Cancer Res. 1996, 2, 1781–1785. [Google Scholar] [PubMed]
- Donnellan, R.; Chetty, R. Cyclin E in human cancers. FASEB J. 1999, 13, 773–780. [Google Scholar] [PubMed]
- Huuhtanen, R.L.; Blomqvist, C.P.; Böhling, T.O.; Wiklund, T.A.; Tukiainen, E.J.; Virolainen, M.; Tribukait, B.; Andersson, L.C. Expression of Cyclin A in Soft Tissue Sarcomas Correlates with Tumor Aggressiveness. Cancer Res. 1999, 59, 2885–2890. [Google Scholar] [PubMed]
- Chao, Y.; Shih, Y.-L.; Chiu, J.-H.; Chau, G.-Y.; Lui, W.-Y.; Yang, W.K.; Lee, S.-D.; Huang, T.-S. Overexpression of Cyclin A but not Skp 2 Correlates with the Tumor Relapse of Human Hepatocellular Carcinoma. Cancer Res. 1998, 58, 985–990. [Google Scholar] [PubMed]
- Santala, S.; Talvensaari-Mattila, A.; Soini, Y.; Honkavuori-Toivola, M.; Santala, M. High expression of cyclin A is associated with poor prognosis in endometrial endometrioid adenocarcinoma. Tumor Biol. 2014, 35, 5395–5399. [Google Scholar] [CrossRef]
- Shaye, A.; Sahin, A.; Hao, Q.; Hunt, K.; Keyomarsi, K.; Bedrosian, I. Cyclin E deregulation is an early event in the development of breast cancer. Breast Cancer Res. Treat. 2009, 115, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010, 116, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.L.; Drapkin, R. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube derived high-grade serous ovarian cancers. Cancer Res. 2013, 74, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, W.W.; Stack, D.; Morris, T.; Grehan, D.; O’Keane, C.; Stewart, G.L.; Cumiskey, J.; Lam, W.L.; Squire, J.A.; Thomas, D.M.; et al. Cyclin E1 is amplified and overexpressed in osteosarcoma. J. Mol. Diagn. 2011, 13, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Koutsami, M.K.; Tsantoulis, P.K.; Kouloukoussa, M.; Apostolopoulou, K.; Pateras, I.S.; Spartinou, Z.; Drougou, A.; Evangelou, K.; Kittas, C.; Bartkova, J.; et al. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J. Pathol. 2006, 209, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Jiang, H.-Y. Expression of cell cycle regulator p57kip2, cyclinE protein and proliferating cell nuclear antigen in human pancreatic cancer: An immunohistochemical study. World J. Gastroenterol. 2005, 11, 5057–5060. [Google Scholar] [PubMed]
- Handa, K.; Yamakawa, M.; Takeda, H.; Kimura, S.; Takahashi, T. Expression of cell cycle markers in colorectal carcinoma: Superiority of cyclin A as an indicator of poor prognosis. Int. J. Cancer 1999, 84, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Q.; Miki, H.; Wu, F.; Saoo, K.; Nishioka, M.; Ohmori, M.; Imaida, K. Cyclin a correlates with carcinogenesis and metastasis, and p27kip1 correlates with lymphatic invasion, in colorectal neoplasms. Hum. Pathol. 2002, 33, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, K.; Yasui, W.; Kuniyasu, H.; Yokozaki, H.; Akama, Y.; Yunotani, S.; Hisatsugu, T.; Tahara, E. Concurrent amplification of cyclin E and CDK2 genes in colorectal carcinomas. Int. J. Cancer 1995, 62, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chenivesse, X.; Henglein, B.; Bréchot, C. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature 1990, 343, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, J.; Do, I.-G.; Jang, J.; Rho, K.; Ahn, S.; Maruja, L.; Kim, S.J.; Kim, K.-M.; Mao, M.; et al. Aberrant CDK4 Amplification in Refractory Rhabdomyosarcoma as Identified by Genomic Profiling. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- Wei, G.; Lonardo, F.; Ueda, T.; Kim, T.; Huvos, A.G.; Healey, J.H.; Ladanyi, M. CDK4 gene amplification in osteosarcoma: Reciprocal relationship with INK4A gene alterations and mapping of 12q13 amplicons. Int. J. Cancer 1999, 80, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.E.; Ichimura, K.; Reifenberger, G.; Collins, V.P. CDKN2 (p16/MTS1) Gene Deletion or CDK4 Amplification Occurs in the Majority of Glioblastomas. Cancer Res. 1994, 54, 6321–6324. [Google Scholar] [PubMed]
- Smalley, K.S.M.; Contractor, R.; Nguyen, T.K.; Xiao, M.; Edwards, R.; Muthusamy, V.; King, A.J.; Flaherty, K.T.; Bosenberg, M.; Herlyn, M.; et al. Identification of a novel subgroup of melanomas with KIT/cyclin-dependent kinase-4 overexpression. Cancer Res. 2008, 68, 5743–5752. [Google Scholar] [CrossRef] [PubMed]
- Dobashi, Y.; Goto, A.; Fukayama, M.; Abe, A.; Ooi, A. Overexpression of cdk4/cyclin D1, a possible mediator of apoptosis and an indicator of prognosis in human primary lung carcinoma. Int. J. Cancer 2004, 110, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Wunder, J.S.; Eppert, K.; Burrow, S.R.; Gokgoz, N.; Bell, R.S.; Andrulis, I.L.; Gogkoz, N. Co-amplification and overexpression of CDK4, SAS and MDM2 occurs frequently in human parosteal osteosarcomas. Oncogene 1999, 18, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Yu, M.M.; Lo, K.W.; Yim, S.F.; Chung, T.K.; Wong, Y.F. Alteration of cyclin D1 and CDK4 gene in carcinoma of uterine cervix. Cancer Lett. 2001, 166, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; de Plaen, E.; Hankeln, T.; Meyer zum Büschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Vidwans, S.J.; Flaherty, K.T.; Fisher, D.E.; Tenenbaum, J.M.; Travers, M.D.; Shrager, J. A melanoma molecular disease model. PLoS One 2011, 6, e18257. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Dubus, P.; Martín, J.; de la Cueva, E.; Ortega, S.; Malumbres, M.; Barbacid, M. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J. 2001, 20, 6637–6647. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Renner, O.; Dubus, P.; Ruiz-Cabello, J.; Martín-Caballero, J.; Barbacid, M.; Carnero, A.; Malumbres, M. Cooperation between Cdk4 and p27kip1 in tumor development: A preclinical model to evaluate cell cycle inhibitors with therapeutic activity. Cancer Res. 2005, 65, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Chawla, R.; Procknow, J.A.; Tantravahi, R.V.; Khurana, J.S.; Litvin, J.; Reddy, E.P. Cooperativity of Cdk4R24C and Ras in melanoma development. Cell Cycle 2010, 9, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.E.; McArthur, G.A. The cell-cycle regulator CDK4: An emerging therapeutic target in melanoma. Clin. Cancer Res. 2013, 19, 5320–5328. [Google Scholar] [CrossRef] [PubMed]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Fabert, C.; Fiancette, R.; Rouaud, P.; Baudet, C.; Truffinet, V.; Magnone, V.; Guillaudeau, A.; Cogné, M.; Dubus, P.; Denizot, Y. A defect of the INK4-Cdk4 checkpoint and Myc collaborate in blastoid mantle cell lymphoma-like lymphoma formation in mice. Am. J. Pathol. 2012, 180, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Eggers, J.P.; Grandgenett, P.M.; Collisson, E.C.; Lewallen, M.E.; Tremayne, J.; Singh, P.K.; Swanson, B.J.; Andersen, J.M.; Caffrey, T.C.; High, R.R.; et al. Cyclin-dependent kinase 5 is amplified and overexpressed in pancreatic cancer and activated by mutant K-Ras. Clin. Cancer Res. 2011, 17, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Li, L.; Zhang, J.; Lei, Y.; Wang, L.; Liu, D.-X.; Feng, J.; Hou, P.; Yao, R.; Zhang, Y.; Huang, B.; Lu, J. CDK5 is essential for TGF-β1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013. [Google Scholar] [CrossRef]
- Leshchenko, V.V.; Kuo, P.-Y.; Shaknovich, R.; Yang, D.T.; Gellen, T.; Petrich, A.; Yu, Y.; Remache, Y.; Weniger, M.A.; Rafiq, S.; et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood 2010, 116, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Lee, Y.; Park, K.H.; Sung, J.S.; Lee, J.-E.; Shin, E.-S.; Ryu, J.-S.; Kim, Y.H. Single-nucleotide polymorphisms in the promoter of the CDK5 gene and lung cancer risk in a Korean population. J. Hum. Genet. 2009, 54, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Strock, C.J.; Park, J.-I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-Dependent Kinase 5 Activity Controls Cell Motility and Metastatic Potential of Prostate Cancer Cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef] [PubMed]
- Levacque, Z.; Rosales, J.L.; Lee, K.-Y. Level of cdk5 expression predicts the survival of relapsed multiple myeloma patients. Cell Cycle 2012, 11, 4093–4095. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, M.M.; Mould, S.J.; Orchard, J.A.; Ibbotson, R.E.; Chapman, R.M.; Boright, A.P.; Platt, C.; Tsui, L.C.; Scherer, S.W.; Oscier, D.G. Dysregulation of cyclin dependent kinase 6 expression in splenic marginal zone lymphoma through chromosome 7q translocations. Oncogene 1999, 18, 6271–6277. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.F.; Plass, C.; Arap, W.; Chapman, V.M.; Held, W.A.; Berger, M.S.; Huang, H.-J.S.; Cavenee, W.K. Cyclin-dependent Kinase 6 (CDK6) Amplification in Human Gliomas Identified Using Two-dimensional Separation of Genomic DNA. Cancer Res. 1997, 57, 1250–1254. [Google Scholar] [PubMed]
- Chilosi, M.; Doglioni, C.; Yan, Z.; Lestani, M.; Menestrina, F.; Sorio, C.; Benedetti, A.; Vinante, F.; Pizzolo, G.; Inghirami, G. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am. J. Pathol. 1998, 152, 209–217. [Google Scholar] [PubMed]
- Easton, J.; Wei, T.; Lahti, J.M.; Kidd, V.J. Disruption of the Cyclin D/Cyclin-dependent Kinase/INK4/Retinoblastoma Protein Regulatory Pathway in Human Neuroblastoma. Cancer Res. 1998, 58, 2624–2632. [Google Scholar] [PubMed]
- Bellail, A.C.; Olson, J.J.; Hao, C. SUMO1 modification stabilizes CDK6 protein and drives the cell cycle and glioblastoma progression. Nat. Commun. 2014, 5, 4234. [Google Scholar] [CrossRef] [PubMed]
- Mendrzyk, F.; Radlwimmer, B.; Joos, S.; Kokocinski, F.; Benner, A.; Stange, D.E.; Neben, K.; Fiegler, H.; Carter, N.P.; Reifenberger, G.; et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J. Clin. Oncol. 2005, 23, 8853–8862. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; McCleland, M.L.; Truong, T.; Lau, S.; Modrusan, Z.; Soukup, T.M.; Roose-Girma, M.; Blackwood, E.M.; Firestein, R. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res. 2012, 72, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Shima, K.; Nosho, K.; Irahara, N.; Baba, Y.; Bojarski, E.; Giovannucci, E.L.; Hahn, W.C.; Fuchs, C.S.; Ogino, S. CDK8 expression in 470 colorectal cancers in relation to β-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 2010, 126, 2863–2873. [Google Scholar] [PubMed]
- Seo, J.-O.; Han, S.I.; Lim, S.-C. Role of CDK8 and beta-catenin in colorectal adenocarcinoma. Oncol. Rep. 2010, 24, 285–291. [Google Scholar] [PubMed]
- Kim, M.-Y.; Han, S.I.; Lim, S.-C. Roles of cyclin-dependent kinase 8 and β-catenin in the oncogenesis and progression of gastric adenocarcinoma. Int. J. Oncol. 2011, 38, 1375–1383. [Google Scholar] [PubMed]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; leRoy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Luo, Q.-F.; Wei, C.-K.; Li, D.-F.; Fang, L. siRNA-mediated silencing of CDK8 inhibits proliferation and growth in breast cancer cells. Int. J. Clin. Exp. Pathol. 2014, 7, 92–100. [Google Scholar] [PubMed]
- Gu, W.; Wang, C.; Li, W.; Hsu, F.-N.; Tian, L.; Zhou, J.; Yuan, C.; Xie, X.-J.; Jiang, T.; Addya, S.; et al. Tumor-suppressive effects of CDK8 in endometrial cancer cells. Cell Cycle 2013, 12, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Bellan, C.; de Falco, G.; Lazzi, S.; Micheli, P.; Vicidomini, S.; Schürfeld, K.; Amato, T.; Palumbo, A.; Bagella, L.; Sabattini, E.; et al. CDK9/CYCLIN T1 expression during normal lymphoid differentiation and malignant transformation. J. Pathol. 2004, 203, 946–952. [Google Scholar] [CrossRef] [PubMed]
- De Falco, G.; Bellan, C.; D’Amuri, A.; Angeloni, G.; Leucci, E.; Giordano, A.; Leoncini, L. Cdk9 regulates neural differentiation and its expression correlates with the differentiation grade of neuroblastoma and PNET tumors. Cancer Biol. Ther. 2005, 4, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-H.; Zhong, X.-Y.; Zhang, W.-G.; Wang, Z.-D.; Dong, Q.; Tai, S.; Li, H.; Cui, Y.-F. CDK10 functions as a tumor suppressor gene and regulates survivability of biliary tract cancer cells. Oncol. Rep. 2012, 27, 1266–1276. [Google Scholar] [PubMed]
- Zhong, X.; Xu, X.; Yu, J.; Jiang, G.; Yu, Y.; Tai, S.; Wang, Z.; Cui, Y. Clinical and biological significance of Cdk10 in hepatocellular carcinoma. Gene 2012, 498, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Lahti, J.M.; Valentine, M.; Xiang, J.; Jones, B.; Amann, J.; Grenet, J.; Richmond, G.; Look, A.T.; Kidd, V.J. Alterations in the PITSLRE protein kinase gene complex on chromosome 1p36 in childhood neuroblastoma. Nat. Genet. 1994, 7, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, A.; Shi, J.; Feng, Y.; Holubec, H.; M.Shanas, R.; Bhattacharyya, A.K.; Zheng, W.; Nelson, M.A. Haploinsufficiency of the cdc2l gene contributes to skin cancer development in mice. Carcinogenesis 2007, 28, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhang, J.; Choy, E.; Harmon, D.; Liu, X.; Nielsen, P.; Mankin, H.; Gray, N.S.; Hornicek, F.J. Systematic kinome shRNA screening identifies CDK11 (PITSLRE) kinase expression is critical for osteosarcoma cell growth and proliferation. Clin. Cancer Res. 2012, 18, 4580–4588. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Choy, E.; Cote, G.; Harmon, D.; Ye, S.; Kan, Q.; Mankin, H.; Hornicek, F.; Duan, Z. Cyclin-dependent kinase 11 (CDK11) is crucial in the growth of liposarcoma cells. Cancer Lett. 2014, 342, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.C.; Ching, A.K.K.; Chan, A.W.H.; Poon, T.C.W.; Mian, H.; Wong, A.S.T.; To, K.-F.; Wong, N. A novel interplay between oncogenic PFTK1 protein kinase and tumor suppressor TAGLN2 in the control of liver cancer cell motility. Oncogene 2011, 30, 4464–4475. [Google Scholar] [CrossRef] [PubMed]
- Miyagaki, H.; Yamasaki, M.; Miyata, H.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; Fujiwara, Y.; Ishii, H.; Tanaka, F.; et al. Overexpression of PFTK1 predicts resistance to chemotherapy in patients with oesophageal squamous cell carcinoma. Br. J. Cancer 2012, 106, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; Chung, E.J.; Kim, H.; Kim, G.E.; Kim, N.K.; Sohn, S.K.; Min, J.S.; Suh, C.O. Assessment of biomarkers in paired primary and recurrent colorectal adenocarcinomas. Int. J. Radiat. Oncol. Biol. Phys. 1999, 45, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Gansauge, S.; Gansauge, F.; Ramadani, M.; Stobbe, H.; Rau, B.; Harada, N.; Beger, H.G. Overexpression of Cyclin D1 in Human Pancreatic Carcinoma Is Associated with Poor Prognosis. Cancer Res. 1997, 57, 1634–1637. [Google Scholar] [PubMed]
- Moreno-Bueno, G.; Rodríguez-Perales, S.; Sánchez-Estévez, C.; Marcos, R.; Hardisson, D.; Cigudosa, J.C.; Palacios, J. Molecular alterations associated with cyclin d1 overexpression in endometrial cancer. Int. J. Cancer 2004, 110, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Akervall, J.A.; Michalides, R.J.; Mineta, H.; Balm, A.; Borg, A.; Dictor, M.R.; Jin, Y.; Loftus, B.; Mertens, F.; Wennerberg, J.P. Amplification of cyclin D1 in squamous cell carcinoma of the head and neck and the prognostic value of chromosomal abnormalities and cyclin D1 overexpression. Cancer 1997, 79, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Meredith, S.D.; Levine, P.A.; Burns, J.A.; Gaffey, M.J.; Boyd, J.C.; Weiss, L.M.; Erickson, N.L.; Williams, M.E. Chromosome 11q13 amplification in head and neck squamous cell carcinoma. Association with poor prognosis. Arch. Otolaryngol. Head Neck Surg. 1995, 121, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Michalides, R.J.; van Veelen, N.M.; Kristel, P.M.; Hart, A.A.; Loftus, B.M.; Hilgers, F.J.; Balm, A.J. Overexpression of cyclin D1 indicates a poor prognosis in squamous cell carcinoma of the head and neck. Arch. Otolaryngol. Head Neck Surg. 1997, 123, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M. Molecular Pathogenesis and a Consequent Classification of Multiple Myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L.; Brents, L.A.; Smith, C.M.; Gerhard, D.S.; Kuehl, W.M. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines [see comments]. Blood 1996, 88, 674–681. [Google Scholar] [PubMed]
- Bertoni, F.; Rinaldi, A.; Zucca, E.; Cavalli, F. Update on the molecular biology of mantle cell lymphoma. Hematol. Oncol. 2006, 24, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Gaillard, F.; Moreau, A.; Harousseau, J.-L.; Laboisse, C.; Milpied, N.; Bataille, R.; Avet-Loiseau, H. Detection of Translocation t(11;14)(q13;q32) in Mantle Cell Lymphoma by Fluorescence in Situ Hybridization. Am. J. Pathol. 1999, 154, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Benzeno, S.; Lu, F.; Guo, M.; Barbash, O.; Zhang, F.; Herman, J.G.; Klein, P.S.; Rustgi, A.; Diehl, J.A. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene 2006, 25, 6291–6303. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, O.; Ratschiller, D.; Gugger, M.; Betticher, D.C.; Heighway, J. Cyclin D1 in non-small cell lung cancer: A key driver of malignant transformation. Lung Cancer 2007, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Betticher, D.C.; Thatcher, N.; Altermatt, H.J.; Hoban, P.; Ryder, W.D.; Heighway, J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene 1995, 11, 1005–1011. [Google Scholar] [PubMed]
- Li, R.; An, S.-J.; Chen, Z.-H.; Zhang, G.-C.; Zhu, J.-Q.; Nie, Q.; Xie, Z.; Guo, A.-L.; Mok, T.S.; Wu, Y.-L. Expression of cyclin D1 splice variants is differentially associated with outcome in non-small cell lung cancer patients. Hum. Pathol. 2008, 39, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Diehl, J.A.; Haiman, C.A.; Knudsen, E.S. Cyclin D1: Polymorphism, aberrant splicing and cancer risk. Oncogene 2006, 25, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.J.; Petre, C.E.; Morey, L.M.; Wang, Y.; Revelo, M.P.; Haiman, C.A.; Lu, S.; Fenoglio-Preiser, C.M.; Li, J.; Knudsen, E.S.; et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Comstock, C.E.S.; Augello, M.A.; Benito, R.P.; Karch, J.; Tran, T.H.; Utama, F.E.; Tindall, E.A.; Wang, Y.; Burd, C.J.; Groh, E.M.; et al. Cyclin D1 splice variants: Polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin. Cancer Res. 2009, 15, 5338–5349. [Google Scholar] [CrossRef] [PubMed]
- Millar, E.K.A.; Dean, J.L.; McNeil, C.M.; O’Toole, S.A.; Henshall, S.M.; Tran, T.; Lin, J.; Quong, A.; Comstock, C.E.S.; Witkiewicz, A.; et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene 2009, 28, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Abramson, V.G.; Troxel, A.B.; Feldman, M.; Mies, C.; Wang, Y.; Sherman, L.; McNally, S.; Diehl, A.; Demichele, A. Cyclin D1b in human breast carcinoma and coexpression with cyclin D1a is associated with poor outcome. Anticancer Res. 2010, 30, 1279–1285. [Google Scholar] [PubMed]
- Wiestner, A.; Tehrani, M.; Chiorazzi, M.; Wright, G.; Gibellini, F.; Nakayama, K.; Liu, H.; Rosenwald, A.; Muller-Hermelink, H.K.; Ott, G.; et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007, 109, 4599–4606. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, M.; Portin, C.; Linderholm, B.; Lindh, J.; Roos, G.; Landberg, G. Expression of cyclin E and the cyclin-dependent kinase inhibitor p27 in malignant lymphomas-prognostic implications. Blood 1998, 92, 770–777. [Google Scholar] [PubMed]
- Wołowiec, D.; Benchaib, M.; Pernas, P.; Deviller, P.; Souchier, C.; Rimokh, R.; Felman, P.; Bryon, P.A.; Ffrench, M. Expression of cell cycle regulatory proteins in chronic lymphocytic leukemias. Comparison with non-Hodgkin’s lymphomas and non-neoplastic lymphoid tissue. Leukemia 1995, 9, 1382–1388. [Google Scholar] [PubMed]
- Keyomarsi, K.; Conte, D.; Toyofuku, W.; Fox, M.P. Deregulation of cyclin E in breast cancer. Oncogene 1995, 11, 941–950. [Google Scholar] [PubMed]
- Porter, D.C.; Keyomarsi, K. Novel splice variants of cyclin E with altered substrate specificity. Nucl. Acids Res. 2000, 28, E101. [Google Scholar] [CrossRef] [PubMed]
- Wingate, H.; Puskas, A.; Duong, M.; Bui, T.; Richardson, D.; Liu, Y.; Tucker, S.L.; van Pelt, C.; Meijer, L.; Hunt, K.; et al. Low molecular weight cyclin E is specific in breast cancer and is associated with mechanisms of tumor progression. Cell Cycle 2009, 8, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Akli, S.; Keyomarsi, K. Low-molecular-weight cyclin E: The missing link between biology and clinical outcome. Breast Cancer Res. 2004, 6, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Bedrosian, I.; Lu, K.H.; Verschraegen, C.; Keyomarsi, K. Cyclin E deregulation alters the biological properties of ovarian cancer cells. Oncogene 2004, 23, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Bales, E.; Mills, L.; Milam, N.; McGahren-Murray, M.; Bandyopadhyay, D.; Chen, D.; Reed, J.A.; Timchenko, N.; van den Oord, J.J.; Bar-Eli, M.; et al. The low molecular weight cyclin E isoforms augment angiogenesis and metastasis of human melanoma cells in vivo. Cancer Res. 2005, 65, 692–697. [Google Scholar] [PubMed]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar] [PubMed]
- Su, W.T.; Alaminos, M.; Mora, J.; Cheung, N.-K.; la Quaglia, M.P.; Gerald, W.L. Positional gene expression analysis identifies 12q overexpression and amplification in a subset of neuroblastomas. Cancer Genet. Cytogenet. 2004, 154, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Koster, J.; Ebus, M.E.; van Sluis, P.; Westerhout, E.M.; de Preter, K.; Gisselsson, D.; Øra, I.; Speleman, F.; Caron, H.N.; et al. Copy number defects of G1-cell cycle genes in neuroblastoma are frequent and correlate with high expression of E2F target genes and a poor prognosis. Genes Chromosomes Cancer 2012, 51, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Dei Tos, A.P.; Doglioni, C.; Piccinin, S.; Sciot, R.; Furlanetto, A.; Boiocchi, M.; dal Cin, P.; Maestro, R.; Fletcher, C.D.; Tallini, G. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J. Pathol. 2000, 190, 531–536. [Google Scholar] [CrossRef] [PubMed]
- An, H.X.; Beckmann, M.W.; Reifenberger, G.; Bender, H.G.; Niederacher, D. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am. J. Pathol. 1999, 154, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gillett, C.; Fantl, V.; Smith, R.; Fisher, C.; Bartek, J.; Dickson, C.; Barnes, D.; Peters, G. Amplification and Overexpression of Cyclin D1 in Breast Cancer Detected by Immunohistochemical Staining. Cancer Res. 1994, 54, 1812–1817. [Google Scholar] [PubMed]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.C.; Tseng, H.-C.; Goldman, J.A.; Shih, H.; Tsai, L.-H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 2003, 40, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Julien, J.-P. Cyclin-dependent kinase 5 in amyotrophic lateral sclerosis. Neurosignals 2003, 12, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.-F.; Ahlijanian, M.K. Role of cdk5 in the pathogenesis of Alzheimer’s disease. Neurosignals 2003, 12, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.D.; Crocker, S.J.; Jackson-Lewis, V.; Jordan-Sciutto, K.L.; Hayley, S.; Mount, M.P.; O’Hare, M.J.; Callaghan, S.; Slack, R.S.; Przedborski, S.; et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 13650–13655. [Google Scholar] [CrossRef] [PubMed]
- Bu, B.; Li, J.; Davies, P.; Vincent, I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J. Neurosci. 2002, 22, 6515–6525. [Google Scholar] [PubMed]
- Paudel, H.K.; Lew, J.; Ali, Z.; Wang, J.H. Brain proline-directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer’s paired helical filaments. J. Biol. Chem. 1993, 268, 23512–23518. [Google Scholar] [PubMed]
- Baumann, K.; Mandelkow, E.M.; Biernat, J.; Piwnica-Worms, H.; Mandelkow, E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993, 336, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P.; Couck, A.M. Cortical and brainstem-type Lewy bodies are immunoreactive for the cyclin-dependent kinase 5. Am. J. Pathol. 1995, 147, 1465–1476. [Google Scholar] [PubMed]
- Rubio de la Torre, E.; Luzón-Toro, B.; Forte-Lago, I.; Minguez-Castellanos, A.; Ferrer, I.; Hilfiker, S. Combined kinase inhibition modulates parkin inactivation. Hum. Mol. Genet. 2009, 18, 809–823. [Google Scholar] [PubMed]
- Nakamura, S.; Kawamoto, Y.; Nakano, S.; Akiguchi, I.; Kimura, J. p35nck5a and cyclin-dependent kinase 5 colocalize in Lewy bodies of brains with Parkinson’s disease. Acta Neuropathol. 1997, 94, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Urban, S.; Yan, E.; Hao, C.; Barron, G.; Allalunis-Turner, J. Expression and localization of cyclin-dependent kinase 5 in apoptotic human glioma cells. Neurooncology 2001, 3, 89–98. [Google Scholar]
- Kato, G.; Maeda, S. Neuron-specific Cdk5 kinase is responsible for mitosis-independent phosphorylation of c-Src at Ser75 in human Y79 retinoblastoma cells. J. Biochem. 1999, 126, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Wang, X.-Y.; Huang, B.-X.; Zhu, F.; Zhang, R.-G.; Wu, G. Expression of CDK5/p35 in resected patients with non-small cell lung cancer: Relation to prognosis. Med. Oncol. 2011, 28, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Mishra, A.; Hong, S.-M.; Bisht, S.; Strock, C.J.; Ball, D.W.; Goggins, M.; Maitra, A.; Nelkin, B.D. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010, 70, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Hahn, W.C. Revving the Throttle on an Oncogene: CDK8 Takes the Driver Seat. Cancer Res. 2009, 69, 7899–7901. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ji, J.-Y. Dysregulation of CDK8 and Cyclin C in tumorigenesis. J. Genet. Genomics 2011, 38, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, Z.; Zhang, W.; Qian, K.; Li, H.; Kong, D.; Li, Y.; Tang, Y. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/β-catenin signaling pathway. Cancer Lett. 2015, 356, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Romano, G. Deregulations in the cyclin-dependent kinase-9-related pathway in cancer: Implications for drug discovery and development. ISRN Oncol. 2013, 2013, 305371. [Google Scholar] [PubMed]
- Simone, C.; Giordano, A. Abrogation of signal-dependent activation of the cdk9/cyclin T2a complex in human RD rhabdomyosarcoma cells. Cell Death Differ. 2007, 14, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Duan, H.O.; Chang, C. Androgen receptor interacts with the positive elongation factor P-TEFb and enhances the efficiency of transcriptional elongation. J. Biol. Chem. 2001, 276, 9978–9984. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.D.; Fattaey, A. CDK inhibition and cancer therapy. Curr. Opin. Genet. Dev. 1999, 9, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.M.; Lane, D.P. Inhibitors of cyclin-dependent kinases as anti-cancer therapeutics. Curr. Med. Chem. 2000, 7, 1213–1245. [Google Scholar] [CrossRef] [PubMed]
- Rosania, G.R.; Chang, Y.-T. Targeting hyperproliferative disorders with cyclin dependent kinase inhibitors. Expert Opin. Ther. Patents 2000, 10, 215–230. [Google Scholar] [CrossRef]
- Senderowicz, A.M.; Sausville, E.A. Preclinical and clinical development of cyclin-dependent kinase modulators. J. Natl. Cancer Inst. 2000, 92, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, M.; Greengard, P.; Meijer, L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci. 2002, 23, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.M.; Endicott, J.; Meijer, L. Cyclin-dependent kinase inhibitors. Prog. Cell Cycle Res. 2003, 5, 235–248. [Google Scholar] [PubMed]
- Huwe, A.; Mazitschek, R.; Giannis, A. Small molecules as inhibitors of cyclin-dependent kinases. Angew. Chem. Int. Ed. Engl. 2003, 42, 2122–2138. [Google Scholar] [CrossRef]
- Vermeulen, K.; van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.; Kaye, S.; Workman, P.; Garrett, M.; Walton, M.; de Bono, J. Clinical anticancer drug development: Targeting the cyclin-dependent kinases. Br. J. Cancer 2005, 92, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Bruyère, C.; Meijer, L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr. Opin. Cell Biol. 2013, 25, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Krämer, A.; Matthess, Y.; Yan, R.; Spänkuch, B.; Gätje, R.; Knecht, R.; Kaufmann, M.; Strebhardt, K. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene 2006, 25, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Gros, E.; Aldrian-Herrada, G.; Choob, M.; Archdeacon, J.; Heitz, F.; Divita, G. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucl. Acids Res. 2007, 35, e49. [Google Scholar] [CrossRef] [PubMed]
- Androic, I.; Krämer, A.; Yan, R.; Rödel, F.; Gätje, R.; Kaufmann, M.; Strebhardt, K.; Yuan, J. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 2008, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Mc Master, G.; Coll, J.-L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucl. Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Schwartz, G.K. Development of cell-cycle inhibitors for cancer therapy. Curr. Oncol. 2009, 16, 36–43. [Google Scholar] [PubMed]
- Orzáez, M.; Gortat, A.; Mondragón, L.; Bachs, O.; Pérez-Payá, E. ATP-noncompetitive inhibitors of CDK-cyclin complexes. ChemMedChem 2009, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Valius, M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, D.; Pentimalli, F.; Giordano, A. Peptides or small molecules? Different approaches to develop more effective CDK inhibitors. Curr. Med. Chem. 2011, 18, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.A.; Pentimalli, F.; Esposito, L.; Giordano, A. ATP-noncompetitive CDK inhibitors for cancer therapy: An overview. Expert Opin. Investig. Drugs 2013, 22, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.; Indovina, P.; Magnotti, F.; Conti, D.; Giordano, A. Anticancer therapeutic strategies based on CDK inhibitors. Curr. Pharm. Des. 2013, 19, 5327–5332. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Okabe, T.; Ogino, H.; Matsumoto, H.; Suzuki-Takahashi, I.; Kokubo, T.; Higashi, H.; Saitoh, S.; Taya, Y.; Yasuda, H. Butyrolactone I, a selective inhibitor of cdk2 and cdc2 kinase. Oncogene 1993, 8, 2425–2432. [Google Scholar] [PubMed]
- Nishio, K.; Ishida, T.; Arioka, H.; Kurokawa, H.; Fukuoka, K.; Nomoto, T.; Fukumoto, H.; Yokote, H.; Saijo, N. Antitumor effects of butyrolactone I, a selective cdc2 kinase inhibitor, on human lung cancer cell lines. Anticancer Res. 1996, 16, 3387–3395. [Google Scholar] [PubMed]
- Wada, M.; Hosotani, R.; Lee, J.U.; Doi, R.; Koshiba, T.; Fujimoto, K.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Okuyama, A.; et al. An exogenous cdk inhibitor, butyrolactone-I, induces apoptosis with increased Bax/Bcl-2 ratio in p53-mutated pancreatic cancer cells. Anticancer Res. 1998, 18, 2559–2566. [Google Scholar] [PubMed]
- Yamamoto, H.; Monden, T.; Miyoshi, H.; Izawa, H.; Ikeda, K.; Tsujie, M.; Ohnishi, T.; Sekimoto, M.; Tomita, N.; Monden, M. Cdk2/cdc2 expression in colon carcinogenesis and effects of cdk2/cdc2 inhibitor in colon cancer cells. Int. J. Oncol. 1998, 13, 233–239. [Google Scholar] [PubMed]
- Gadbois, D.M.; Hamaguchi, J.R.; Swank, R.A.; Bradbury, E.M. Staurosporine is a potent inhibitor of p34cdc2 and p34cdc2-like kinases. Biochem. Biophys. Res. Commun. 1992, 184, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid Ca++ dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Yoshida, T.; Tsujita, T.; Shimizu, M.; Mizukami, T.; Okabe, M.; Akinaga, S. G1 phase accumulation induced by UCN-01 is associated with dephosphorylation of Rb and CDK2 proteins as well as induction of CDK inhibitor p21/Cip1/WAF1/Sdi1 in p53-mutated human epidermoid carcinoma A431 cells. Cancer Res. 1997, 57, 1495–1501. [Google Scholar] [PubMed]
- Bhonde, M.R.; Hanski, M.-L.; Magrini, R.; Moorthy, D.; Müller, A.; Sausville, E.A.; Kohno, K.; Wiegand, P.; Daniel, P.T.; Zeitz, M.; et al. The broad-range cyclin-dependent kinase inhibitor UCN-01 induces apoptosis in colon carcinoma cells through transcriptional suppression of the Bcl-x(L) protein. Oncogene 2005, 24, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, H.; Czech, J.; Naik, R.; Kaur, G.; Worland, P.; Losiewicz, M.; Parker, B.; Carlson, B.; Smith, A.; Senderowicz, A.; et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol. 1996, 9, 1143–1168. [Google Scholar] [PubMed]
- Losiewicz, M.D.; Carlson, B.A.; Kaur, G.; Sausville, E.A.; Worland, P.J. Potent inhibition of CDC2 kinase activity by the flavonoid L86–8275. Biochem. Biophys. Res. Commun. 1994, 201, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Senderowicz, A.M.; Pinto, D.; Igishi, T.; Raffeld, M.; Quintanilla-Martinez, L.; Ensley, J.F.; Sausville, E.A.; Gutkind, J.S. Flavopiridol, a novel cyclin-dependent kinase inhibitor, suppresses the growth of head and neck squamous cell carcinomas by inducing apoptosis. J. Clin. Investig. 1998, 102, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Schwartz, G.K.; Mohammad, R.M.; al-Katib, A.; Gabrilove, J.L. The novel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2 and induces growth arrest and apoptosis in chronic B-cell leukemia lines. Blood 1997, 90, 4307–4312. [Google Scholar] [PubMed]
- Christian, B.A.; Grever, M.R.; Byrd, J.C.; Lin, T.S. Flavopiridol in the treatment of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2007, 19, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Ruppert, A.S.; Johnson, A.J.; Fischer, B.; Heerema, N.A.; Andritsos, L.A.; Blum, K.A.; Flynn, J.M.; Jones, J.A.; Hu, W.; et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009, 27, 6012–6018. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Kasimis, B.S.; Cogswell, J.; Schwarzenberger, P.; Shapiro, G.I.; Fidias, P.; Bukowski, R.M. Phase I study of flavopiridol in combination with Paclitaxel and Carboplatin in patients with non-small-cell lung cancer. Clin. Lung Cancer 2008, 9, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.S.; Rathos, M.J.; Joshi, R.D.; Sivakumar, M.; Mascarenhas, M.; Kamble, S.; Lal, B.; Sharma, S. In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276–00. Mol. Cancer Ther. 2007, 6, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.S.; Rathos, M.J.; Mahajan, P.; Wagh, V.; Shenoy, S.; Bhatia, D.; Chile, S.; Sivakumar, M.; Maier, A.; Fiebig, H.-H.; et al. P276–00, a novel cyclin-dependent inhibitor induces G1-G2 arrest, shows antitumor activity on cisplatin-resistant cells and significant in vivo efficacy in tumor models. Mol. Cancer Ther. 2007, 6, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Thunnissen, A.-M.; White, A.; Garnier, M.; Nikolic, M.; Tsai, L.-H.; Walter, J.; Cleverley, K.; Salinas, P.; Wu, Y.-Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Tirado, O.M.; Marionneau-Lambot, S.; Ferandin, Y.; Lozach, O.; Morris, J.C.; Mateo-Lozano, S.; Drueckes, P.; Schächtele, C.; Kubbutat, M.H.G.; et al. Meriolins, a new class of cell death inducing kinase inhibitors with enhanced selectivity for cyclin-dependent kinases. Cancer Res. 2007, 67, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Mueller-Dieckmann, H.J.; Schulze-Gahmen, U.; Worland, P.J.; Sausville, E.; Kim, S.H. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Gray, N.S.; Rosania, G.R.; Sutherlin, D.P.; Kwon, S.; Norman, T.C.; Sarohia, R.; Leost, M.; Meijer, L.; Schultz, P.G. Synthesis and application of functionally diverse 2,6,9-trisubstituted purine libraries as CDK inhibitors. Chem. Biol. 1999, 6, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef] [PubMed]
- Demange, L.; Abdellah, F.N.; Lozach, O.; Ferandin, Y.; Gresh, N.; Meijer, L.; Galons, H. Potent inhibitors of CDK5 derived from roscovitine: Synthesis, biological evaluation and molecular modelling. Bioorg. Med. Chem. Lett. 2013, 23, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [PubMed]
- Berberich, N.; Uhl, B.; Joore, J.; Schmerwitz, U.K.; Mayer, B.A.; Reichel, C.A.; Krombach, F.; Zahler, S.; Vollmar, A.M.; Fürst, R. Roscovitine blocks leukocyte extravasation by inhibition of cyclin-dependent kinases 5 and 9. Br. J. Pharmacol. 2011, 163, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Arris, C.E.; Boyle, F.T.; Calvert, A.H.; Curtin, N.J.; Endicott, J.A.; Garman, E.F.; Gibson, A.E.; Golding, B.T.; Grant, S.; Griffin, R.J.; et al. Identification of novel purine and pyrimidine cyclin-dependent kinase inhibitors with distinct molecular interactions and tumor cell growth inhibition profiles. J. Med. Chem. 2000, 43, 2797–2804. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Bentley, J.; Arris, C.E.; Boyle, F.T.; Curtin, N.J.; Endicott, J.A.; Gibson, A.E.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; et al. Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Nat. Struct. Biol. 2002, 9, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Villerbu, N.; Gaben, A.-M.; Redeuilh, G.; Mester, J. Cellular effects of purvalanol A: A specific inhibitor of cyclin-dependent kinase activities. Int. J. Cancer 2002, 97, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.S.; Wodicka, L.; Thunnissen, A.M.; Norman, T.C.; Kwon, S.; Espinoza, F.H.; Morgan, D.O.; Barnes, G.; leClerc, S.; Meijer, L.; et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science 1998, 281, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Pennati, M.; Campbell, A.J.; Curto, M.; Binda, M.; Cheng, Y.; Wang, L.-Z.; Curtin, N.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; et al. Potentiation of paclitaxel-induced apoptosis by the novel cyclin-dependent kinase inhibitor NU6140: A possible role for survivin down-regulation. Mol. Cancer Ther. 2005, 4, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Veselý, J.; Havlicek, L.; Strnad, M.; Blow, J.J.; Donella-Deana, A.; Pinna, L.; Letham, D.S.; Kato, J.; Detivaud, L.; Leclerc, S. Inhibition of cyclin-dependent kinases by purine analogues. Eur. J. Biochem. 1994, 224, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Glab, N.; Labidi, B.; Qin, L.X.; Trehin, C.; Bergounioux, C.; Meijer, L. Olomoucine, an inhibitor of the cdc2/cdk2 kinases activity, blocks plant cells at the G1 to S and G2 to M cell cycle transitions. FEBS Lett. 1994, 353, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.E.; Yu, B.; Rice, A.; Lipson, K.E.; Liang, C.; Sun, L.; Tang, C.; McMahon, G.; Pestell, R.G.; Wadler, S. A novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res. 2001, 61, 6170–6177. [Google Scholar] [PubMed]
- Moshinsky, D.J.; Bellamacina, C.R.; Boisvert, D.C.; Huang, P.; Hui, T.; Jancarik, J.; Kim, S.-H.; Rice, A.G. SU9516: Biochemical analysis of cdk inhibition and crystal structure in complex with cdk2. Biochem. Biophys. Res. Commun. 2003, 310, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Zaharevitz, D.W.; Gussio, R.; Leost, M.; Senderowicz, A.M.; Lahusen, T.; Kunick, C.; Meijer, L.; Sausville, E.A. Discovery and initial characterization of the paullones, a novel class of small-molecule inhibitors of cyclin-dependent kinases. Cancer Res. 1999, 59, 2566–2569. [Google Scholar] [PubMed]
- Diab, S.; Eckhardt, S.; Tan, A.; Frenette, G.; Gore, L.; Depinto, W.; Grippo, J.; DeMario, M.; Mikulski, S.; Papadimitrakopoulou, S. A phase I study of R547, a novel, selective inhibitor of cell cycle and transcriptional cyclin-dependent kinases (CDKs). J. Clin. Oncol. 2007, 25, 3528. [Google Scholar]
- DePinto, W.; Chu, X.-J.; Yin, X.; Smith, M.; Packman, K.; Goelzer, P.; Lovey, A.; Chen, Y.; Qian, H.; Hamid, R.; et al. In vitro and in vivo activity of R547: A potent and selective cyclin-dependent kinase inhibitor currently in phase I clinical trials. Mol. Cancer Ther. 2006, 5, 2644–2658. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-J.; DePinto, W.; Bartkovitz, D.; So, S.-S.; Vu, B.T.; Packman, K.; Lukacs, C.; Ding, Q.; Jiang, N.; Wang, K.; et al. Discovery of [4-Amino-2-(1-methanesulfonylpiperidin-4-ylamino)pyrimidin-5-yl](2,3-difluoro-6-methoxyphenyl)methanone (R547), a potent and selective cyclin-dependent kinase inhibitor with significant in vivo antitumor activity. J. Med. Chem. 2006, 49, 6549–6560. [Google Scholar] [CrossRef] [PubMed]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.-Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of Dinaciclib (SCH 727965): A Potent and Selective Inhibitor of Cyclin-Dependent Kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Mishra, A.; Bisht, S.; Karikari, C.; Garrido-Laguna, I.; Rasheed, Z.; Ottenhof, N.A.; Dadon, T.; Alvarez, H.; Fendrich, V.; et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. Ther. 2011, 12, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Bannerji, R.; Small, K.; Black, S.; Statkevich, P.; Abutarif, M.; Moseley, J.; Yao, S.; Takimoto, C.H.; Mita, M.M. A phase I dose-escalation study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the novel cyclin-dependent kinase inhibitor SCH 727965 administered every 3 weeks in subjects with advanced malignancies. ASCO Meeting Abstracts 2008, 26, 3532. [Google Scholar]
- Walsby, E.; Pratt, G.; Shao, H.; Abbas, A.Y.; Fischer, P.M.; Bradshaw, T.D.; Brennan, P.; Fegan, C.; Wang, S.; Pepper, C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget 2013, 5, 375–385. [Google Scholar]
- Fry, D.W.; Bedford, D.C.; Harvey, P.H.; Fritsch, A.; Keller, P.R.; Wu, Z.; Dobrusin, E.; Leopold, W.R.; Fattaey, A.; Garrett, M.D. Cell cycle and biochemical effects of PD 0183812. A potent inhibitor of the cyclin D-dependent kinases CDK4 and CDK6. J. Biol. Chem. 2001, 276, 16617–16623. [Google Scholar] [CrossRef] [PubMed]
- Guha, M. Blockbuster dreams for Pfizer’s CDK inhibitor. Nat. Biotechnol. 2013, 31, 187. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; Toogood, P.L. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [PubMed]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef] [PubMed]
- Cen, L.; Carlson, B.L.; Schroeder, M.A.; Ostrem, J.L.; Kitange, G.J.; Mladek, A.C.; Fink, S.R.; Decker, P.A.; Wu, W.; Kim, J.-S.; et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neurooncology 2012, 14, 870–881. [Google Scholar]
- Baughn, L.B.; di Liberto, M.; Wu, K.; Toogood, P.L.; Louie, T.; Gottschalk, R.; Niesvizky, R.; Cho, H.; Ely, S.; Moore, M.A.S.; et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006, 66, 7661–7667. [Google Scholar] [CrossRef] [PubMed]
- Menu, E.; Garcia, J.; Huang, X.; di Liberto, M.; Toogood, P.L.; Chen, I.; Vanderkerken, K.; Chen-Kiang, S. A novel therapeutic combination using PD 0332991 and bortezomib: Study in the 5T33MM myeloma model. Cancer Res. 2008, 68, 5519–5523. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A. Molecular Pathways: CDK4 Inhibitors for Cancer Therapy. Clin. Cancer Res. 2014, 20, 3379–3383. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Loo, A.; Chopra, R.; Caponigro, G.; Huang, A.; Vora, S.; Parasuraman, S.; Howard, S.; Keen, N.; Sellers, W.; et al. Abstract PR02: LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6-Reactivating Rb in cancer. Mol Cancer Ther 2013, 12, PR02. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Kreklau, E.; Wishart, G.N.; et al. Abstract B233: Identification and characterization of LY2835219: A potent oral inhibitor of the cyclin-dependent kinases 4 and 6 (CDK4/6) with broad in vivo antitumor activity. Mol. Cancer Ther. 2011, 10, B233. [Google Scholar] [CrossRef]
- Misra, R.N.; Xiao, H.; Kim, K.S.; Lu, S.; Han, W.-C.; Barbosa, S.A.; Hunt, J.T.; Rawlins, D.B.; Shan, W.; Ahmed, S.Z.; et al. N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J. Med. Chem. 2004, 47, 1719–1728. [Google Scholar]
- Kamath, A.V.; Chong, S.; Chang, M.; Marathe, P.H. P-glycoprotein plays a role in the oral absorption of BMS-387032, a potent cyclin-dependent kinase 2 inhibitor, in rats. Cancer Chemother. Pharmacol. 2005, 55, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, P.G.; Woodhead, A.J.; Berdini, V.; Boulstridge, J.A.; Carr, M.G.; Cross, D.M.; Davis, D.J.; Devine, L.A.; Early, T.R.; Feltell, R.E.; et al. Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. J. Med. Chem. 2008, 51, 4986–4999. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.-M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Cooke, L.; Lock, V.; Qi, W.; Lewis, E.J.; Thompson, N.T.; Lyons, J.F.; Mahadevan, D. AT7519, a cyclin-dependent kinase inhibitor, exerts its effects by transcriptional inhibition in leukemia cell lines and patient samples. Mol. Cancer Ther. 2010, 9, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Plummer, R.; Squires, M.S.; Rensvold, D.; Kurtin, S.; Pretzinger, C.; Dragovich, T.; Adams, J.; Lock, V.; Smith, D.M.; et al. A phase I pharmacokinetic and pharmacodynamic study of AT7519, a cyclin-dependent kinase inhibitor in patients with refractory solid tumors. Ann. Oncol. 2011, 22, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Bagella, L.; Sun, A.; Tonini, T.; Abbadessa, G.; Cottone, G.; Paggi, M.G.; de Luca, A.; Claudio, P.P.; Giordano, A. A small molecule based on the pRb2/p130 spacer domain leads to inhibition of cdk2 activity, cell cycle arrest and tumor growth reduction in vivo. Oncogene 2007, 26, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Bellacchio, E.; Bagella, L.; Paggi, M.G. Interaction between the Cdk2/cyclin A complex and a small molecule derived from the pRb2/p130 spacer domain: A theoretical model. Cell Cycle 2007, 6, 2591–2593. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.; Luciani, M.G.; Finlan, L.; Rankin, E.M.; Ibbotson, S.; Fersht, A.; Hupp, T.R. The development of a CDK2-docking site peptide that inhibits p53 and sensitizes cells to death. Cell Cycle 2004, 3, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gondeau, C.; Gerbal-Chaloin, S.; Bello, P.; Aldrian-Herrada, G.; Morris, M.C.; Divita, G. Design of a novel class of peptide inhibitors of cyclin-dependent kinase/cyclin activation. J. Biol. Chem. 2005, 280, 13793–13800. [Google Scholar] [CrossRef] [PubMed]
- Canela, N.; Orzáez, M.; Fucho, R.; Mateo, F.; Gutierrez, R.; Pineda-Lucena, A.; Bachs, O.; Pérez-Payá, E. Identification of an hexapeptide that binds to a surface pocket in cyclin A and inhibits the catalytic activity of the complex cyclin-dependent kinase 2-cyclin A. J. Biol. Chem. 2006, 281, 35942–35953. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.T.; Ohki, S.Y.; Tang, D.; Cheng, H.C.; Wang, J.H.; Zhang, M. Identification and structure characterization of a Cdk inhibitory peptide derived from neuronal-specific Cdk5 activator. J. Biol. Chem. 1999, 274, 7120–7127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-L.; Li, B.-S.; Amin, N.D.; Albers, W.; Pant, H.C. A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur. J. Biochem. 2002, 269, 4427–4434. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-L.; Kesavapany, S.; Gravell, M.; Hamilton, R.S.; Schubert, M.; Amin, N.; Albers, W.; Grant, P.; Pant, H.C. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005, 24, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-L.; Amin, N.D.; Hu, Y.-F.; Rudrabhatla, P.; Shukla, V.; Kanungo, J.; Kesavapany, S.; Grant, P.; Albers, W.; Pant, H.C. A 24-residue peptide (p5), derived from p35, the Cdk5 neuronal activator, specifically inhibits Cdk5-p25 hyperactivity and tau hyperphosphorylation. J. Biol. Chem. 2010, 285, 34202–34212. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Zheng, Y.-L.; Mishra, S.K.; Amin, N.D.; Steiner, J.; Grant, P.; Kesavapany, S.; Pant, H.C. A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J. 2013, 27, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, J.R.; Poore, C.P.; Sulaimee, N.H.B.; Pareek, T.; Asad, A.B.M.A.; Rajkumar, R.; Cheong, W.F.; Wenk, M.R.; Dawe, G.S.; Chuang, K.-H.; et al. Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J. Neurosci. 2013, 33, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, N.; Fong, S.; Marsters, J.; Koeppen, H.; Schwall, R.; Wickramasinghe, D. Selective cyclin-dependent kinase 2/cyclin A antagonists that differ from ATP site inhibitors block tumor growth. Cancer Res. 2003, 63, 1020–1024. [Google Scholar] [PubMed]
- Warenius, H.M.; Kilburn, J.D.; Essex, J.W.; Maurer, R.I.; Blaydes, J.P.; Agarwala, U.; Seabra, L.A. Selective anticancer activity of a hexapeptide with sequence homology to a non-kinase domain of Cyclin Dependent Kinase 4. Mol. Cancer 2011, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Sellers, W.R.; Sharma, S.K.; Wu, A.D.; Nalin, C.M.; Kaelin, W.G. Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol. Cell. Biol. 1996, 16, 6623–6633. [Google Scholar] [PubMed]
- Chen, I.T.; Akamatsu, M.; Smith, M.L.; Lung, F.D.; Duba, D.; Roller, P.P.; Fornace, A.J.; O’Connor, P.M. Characterization of p21Cip1/Waf1 peptide domains required for cyclin E/Cdk2 and PCNA interaction. Oncogene 1996, 12, 595–607. [Google Scholar] [PubMed]
- Bonfanti, M.; Taverna, S.; Salmona, M.; D’Incalci, M.; Broggini, M. p21WAF1-derived peptides linked to an internalization peptide inhibit human cancer cell growth. Cancer Res. 1997, 57, 1442–1446. [Google Scholar] [PubMed]
- Ball, K.L.; Lain, S.; Fâhraeus, R.; Smythe, C.; Lane, D.P. Cell-cycle arrest and inhibition of Cdk4 activity by small peptides based on the carboxy-terminal domain of p21WAF1. Curr. Biol. 1997, 7, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, M.; Lung, F.D.; Long, Y.Q.; Roller, P.P.; Sikorski, R.S.; O’Connor, P.M. A p21(Waf1/Cip1)carboxyl-terminal peptide exhibited cyclin-dependent kinase-inhibitory activity and cytotoxicity when introduced into human cells. Cancer Res. 1999, 59, 3480–3488. [Google Scholar] [PubMed]
- Zheleva, D.I.; McInnes, C.; Gavine, A.-L.; Zhelev, N.Z.; Fischer, P.M.; Lane, D.P. Highly potent p21(WAF1)-derived peptide inhibitors of CDK-mediated pRb phosphorylation: Delineation and structural insight into their interactions with cyclin A. J. Pept. Res. 2002, 60, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Kontopidis, G.; Andrews, M.J.; McInnes, C.; Plater, A.; Innes, L.; Renachowski, S.; Cowan, A.; Fischer, P.M. Truncation and Optimisation of Peptide Inhibitors of Cyclin-Dependent Kinase 2-Cyclin A Through Structure-Guided Design. ChemMedChem 2009, 4, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bolger, J.K.; Kirkland, L.O.; Premnath, P.N.; McInnes, C. Structural and functional analysis of cyclin D1 reveals p27 and substrate inhibitor binding requirements. ACS Chem. Biol. 2010, 5, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.R.; Ezhevsky, S.A.; Becker-Hapak, M.; Nagahara, H.; Wei, M.C.; Dowdy, S.F. Transduced p16INK4a Peptides Inhibit Hypophosphorylation of the Retinoblastoma Protein and Cell Cycle Progression Prior to Activation of Cdk2 Complexes in Late G1. Cancer Res. 1999, 59, 2577–2580. [Google Scholar] [PubMed]
- Dai, L.; Liu, Y.; Liu, J.; Wen, X.; Xu, Z.; Wang, Z.; Sun, H.; Tang, S.; Maguire, A.R.; Quan, J.; et al. A novel CyclinE/CyclinA-CDK Inhibitor targets p27Kip1 degradation, cell cycle progression and cell survival: Implications in cancer therapy. Cancer Lett. 2013, 333, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.J.I.; McInnes, C.; Kontopidis, G.; Innes, L.; Cowan, A.; Plater, A.; Fischer, P.M. Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes. Org. Biomol. Chem. 2004, 2, 2735–2741. [Google Scholar] [CrossRef] [PubMed]
- Fåhraeus, R.; Paramio, J.M.; Ball, K.L.; Laín, S.; Lane, D.P. Inhibition of pRb phosphorylation and cell-cycle progression by a 20-residue peptide derived from p16CDKN2/INK4A. Curr. Biol. 1996, 6, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Prével, C.; Pellerano, M.; Van, T.N.N.; Morris, M.C. Fluorescent biosensors for high throughput screening of protein kinase inhibitors. Biotechnol. J. 2014, 9, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Prével, C.; Kurzawa, L.; Van, T.N.N.; Morris, M.C. Fluorescent biosensors for drug discovery new tools for old targets—Screening for inhibitors of cyclin-dependent kinases. Eur. J. Med. Chem. 2014, 88, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Corsino, P.; Horenstein, N.; Ostrov, D.; Rowe, T.; Law, M.; Barrett, A.; Aslanidi, G.; Cress, W.D.; Law, B. A novel class of cyclin-dependent kinase inhibitors identified by molecular docking act through a unique mechanism. J. Biol. Chem. 2009, 284, 29945–29955. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, K.; Huang, Z.; Park, C.-M.; Thimmegowda, N.R.; Jang, J.-H.; Ryoo, I.-J.; He, L.; Kim, S.-O.; Oi, N.; et al. A chrysin derivative suppresses skin cancer growth by inhibiting cyclin-dependent kinases. J. Biol. Chem. 2013, 288, 25924–25937. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-C.; Ngo, R.; Dai, K.; Li, C.; Liang, L.; Lee, J.; Emkey, R.; Eksterowicz, J.; Ventura, M.; Young, S.W.; et al. Development of a time-resolved fluorescence resonance energy transfer assay for cyclin-dependent kinase 4 and identification of its ATP-noncompetitive inhibitors. Anal. Biochem. 2012, 421, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Nakagawa, K.; Varma, R.K.; Conrad, N.K.; Cheng, J.Q.; Lee, W.-C.; Testa, J.R.; Johnson, B.E.; Kaye, F.J.; Kelley, M.J. The p16 Status of Tumor Cell Lines Identifies Small Molecule Inhibitors Specific for Cyclin-dependent Kinase 4. Clin. Cancer Res. 1999, 5, 4279–4286. [Google Scholar] [PubMed]
- Siemeister, G.; Luecking, U.; Wagner, C.; Detjen, K.; Mc Coy, C.; Bosslet, K. Molecular and pharmacodynamic characteristics of the novel multi-target tumor growth inhibitor ZK 304709. Biomed. Pharmacother. 2006, 60, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Wagner, K.; Welzel, M.; Remlinger, F.; Wiedenmann, B.; Siemeister, G.; Rosewicz, S.; Detjen, K.M. The oral multitarget tumour growth inhibitor, ZK 304709, inhibits growth of pancreatic neuroendocrine tumours in an orthotopic mouse model. Gut 2009, 58, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Payton, M.; Chung, G.; Yakowec, P.; Wong, A.; Powers, D.; Xiong, L.; Zhang, N.; Leal, J.; Bush, T.L.; Santora, V.; et al. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res. 2006, 66, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Connolly, P.J.; Lin, R.; Emanuel, S.; Middleton, S.A. Synthesis and evaluation of N-acyl sulfonamides as potential prodrugs of cyclin-dependent kinase inhibitor JNJ-7706621. Bioorg. Med. Chem. Lett. 2006, 16, 3639–3641. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, S.; Rugg, C.A.; Gruninger, R.H.; Lin, R.; Fuentes-Pesquera, A.; Connolly, P.J.; Wetter, S.K.; Hollister, B.; Kruger, W.W.; Napier, C.; Jolliffe, L.; Middleton, S.A. The in vitro and in vivo Effects of JNJ-7706621: A Dual Inhibitor of Cyclin-Dependent Kinases and Aurora Kinases. Cancer Res. 2005, 65, 9038–9046. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Courtney, C.L.; Criswell, K.A.; Holliman, C.L.; Evering, W.; Jessen, B.A. Toxicity and toxicokinetics of the cyclin-dependent kinase inhibitor AG-024322 in cynomolgus monkeys following intravenous infusion. Cancer Chemother. Pharmacol. 2008, 62, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Smethurst, D.; Growcott, J.; Barrass, N.C.; Foster, J.R.; Febbraro, S.; Swaisland, H.; Hughes, A. A first-in-man phase I tolerability and pharmacokinetic study of the cyclin-dependent kinase-inhibitor AZD5438 in healthy male volunteers. Cancer Chemother. Pharmacol. 2007, 60, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Byth, K.F.; Thomas, A.; Hughes, G.; Forder, C.; McGregor, A.; Geh, C.; Oakes, S.; Green, C.; Walker, M.; Newcombe, N.; et al. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol. Cancer Ther. 2009, 8, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; O’Reilly, T.; Furet, P.; Muller, L.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Fabbro, D.; Chaudhuri, B. Selective in vivo and in vitro effects of a small molecule inhibitor of cyclin-dependent kinase 4. J. Natl. Cancer Inst. 2001, 93, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Bathini, Y.; Singh, I.; Harvey, P.J.; Keller, P.R.; Singh, R.; Micetich, R.G.; Fry, D.W.; Dobrusin, E.M.; Toogood, P.L. 2-Aminoquinazoline inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. Lett. 2005, 15, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.R.; Akula, B.; Cosenza, S.C.; Athuluridivakar, S.; Mallireddigari, M.R.; Pallela, V.R.; Billa, V.K.; Subbaiah, D.R.C.V.; Bharathi, E.V.; Vasquez-del Carpio, R.; et al. Discovery of 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile (7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-related kinase 5 (ARK5). J. Med. Chem. 2014, 57, 578–599. [Google Scholar] [CrossRef] [PubMed]
- Barvian, M.; Boschelli, D.H.; Cossrow, J.; Dobrusin, E.; Fattaey, A.; Fritsch, A.; Fry, D.; Harvey, P.; Keller, P.; Garrett, M.; et al. Pyrido[2,3-d]pyrimidin-7-one inhibitors of cyclin-dependent kinases. J. Med. Chem. 2000, 43, 4606–4616. [Google Scholar] [CrossRef] [PubMed]
- Neant, I.; Guerrier, P. 6-Dimethylaminopurine blocks starfish oocyte maturation by inhibiting a relevant protein kinase activity. Exp. Cell Res. 1988, 176, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Havlíček, L.; Hanuš, J.; Veselý, J.; Leclerc, S.; Meijer, L.; Shaw, G.; Strnad, M. Cytokinin-Derived Cyclin-Dependent Kinase Inhibitors: Synthesis and cdc2 Inhibitory Activity of Olomoucine and Related Compounds. J. Med. Chem. 1997, 40, 408–412. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar]
- Lam, L.T.; Pickeral, O.K.; Peng, A.C.; Rosenwald, A.; Hurt, E.M.; Giltnane, J.M.; Averett, L.M.; Zhao, H.; Davis, R.E.; Sathyamoorthy, M.; et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Chao, S.H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; de la Fuente, C.; Deng, L.; Wang, L.; Zilberman, I.; Eadie, C.; Healey, M.; Stein, D.; Denny, T.; Harrison, L.E.; et al. Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors. J. Virol. 2001, 75, 7266–7279. [Google Scholar] [CrossRef] [PubMed]
- Gompel, M.; Leost, M.; de Kier Joffe, E.B.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Perry, N.B.; Ettouati, L.; Litaudon, M.; Blunt, J.W.; Munro, M.H.G.; Parkin, S.; Hope, H. Alkaloids from the Antarctic sponge Kirkpatrickia varialosa. Part1: Variolin B, a new antitumour and antiviral compound. Tetrahedron 1994, 50, 3987–3992. [Google Scholar] [CrossRef]
- Trimurtulu, G.; Faulkner, D.J.; Perry, N.B.; Ettouati, L.; Litaudon, M.; Blunt, J.W.; Munro, M.H.G.; Jameson, G.B. Alkaloids from the Antarctic sponge Kirkpatrickia varialosa. Part 2: Variolin A and N(3')-methyl tetrahydrovariolin B. Tetrahedron 1994, 50, 3993–4000. [Google Scholar] [CrossRef]
- Noble, M.; Barrett, P.; Endicott, J.; Johnson, L.; McDonnell, J.; Robertson, G.; Zawaira, A. Exploiting structural principles to design cyclin-dependent kinase inhibitors. Biochim. Biophys. Acta 2005, 1754, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Chemical genetics: Where genetics and pharmacology meet. Cell 2007, 128, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lopez, M.S.; Dar, A.C.; Ladow, E.; Finkbeiner, S.; Yun, C.-H.; Eck, M.J.; Shokat, K.M. Structure-guided inhibitor design expands the scope of analog-sensitive kinase technology. ACS Chem. Biol. 2013, 8, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.A. Complexities in the development of cyclin-dependent kinase inhibitor drugs. Trends Mol. Med. 2002, 8, S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.T.; Benson, B.G.; Bramson, H.N.; Chapman, D.E.; Dickerson, S.H.; Dold, K.M.; Eberwein, D.J.; Edelstein, M.; Frye, S.V.; Gampe, R.T.; et al. Prevention of Chemotherapy-Induced Alopecia in Rats by CDK Inhibitors. Science 2001, 291, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Jessen, B.A.; Lee, L.; Koudriakova, T.; Haines, M.; Lundgren, K.; Price, S.; Nonomiya, J.; Lewis, C.; Stevens, G.J. Peripheral white blood cell toxicity induced by broad spectrum cyclin-dependent kinase inhibitors. J. Appl. Toxicol. 2007, 27, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Pevarello, P.; Barbacid, M.; Bischoff, J.R. CDK inhibitors in cancer therapy: What is next? Trends Pharmacol. Sci. 2008, 29, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Echalier, A.; Hole, A.J.; Lolli, G.; Endicott, J.A.; Noble, M.E.M. An inhibitor’s-eye view of the ATP-binding site of CDKs in different regulatory states. ACS Chem. Biol. 2014, 9, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Németh, G.; Varga, Z.; Greff, Z.; Bencze, G.; Sipos, A.; Szántai-Kis, C.; Baska, F.; Gyuris, A.; Kelemenics, K.; Szathmáry, Z.; et al. Novel, selective CDK9 inhibitors for the treatment of HIV infection. Curr. Med. Chem. 2011, 18, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.; Abbas, A.Y.; Shao, H.; Teo, T.; Adams, J.; Li, P.; Bradshaw, T.D.; Fischer, P.M.; Walsby, E.; Pepper, C.; et al. Targeting RNA transcription and translation in ovarian cancer cells with pharmacological inhibitor CDKI-73. Oncotarget 2014, 5, 7691–7704. [Google Scholar] [PubMed]
- Santo, L.; Vallet, S.; Hideshima, T.; Cirstea, D.; Ikeda, H.; Pozzi, S.; Patel, K.; Okawa, Y.; Gorgun, G.; Perrone, G.; et al. AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene 2010, 29, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Walsby, E.; Lazenby, M.; Pepper, C.; Burnett, A.K. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine. Leukemia 2011, 25, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Grütter, C.; Rauh, D. Strategies for the selective regulation of kinases with allosteric modulators: Exploiting exclusive structural features. ACS Chem. Biol. 2013, 8, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Cherfils, J. Interfacial inhibition of macromolecular interactions: Nature’s paradigm for drug discovery. Trends Pharmacol. Sci. 2005, 26, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.A.; Thornton, J.M. Diversity of protein-protein interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, L.; Felding, J.; Audouze, K.; Nielsen, S.J.; Terry, R.B.; Krog-Jensen, C.; Butcher, S. Emerging classes of protein-protein interaction inhibitors and new tools for their development. Curr. Opin. Chem. Biol. 2004, 8, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Labbé, C.M.; Lagorce, D.; Laconde, G.; Sperandio, O. A leap into the chemical space of protein-protein interaction inhibitors. Curr. Pharm. Des. 2012, 18, 4648–4667. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Randal, M.; deLano, W.L.; Hyde, J.; Luong, T.N.; Oslob, J.D.; Raphael, D.R.; Taylor, L.; Wang, J.; McDowell, R.S.; et al. Binding of small molecules to an adaptive protein-protein interface. Proc. Natl. Acad. Sci. USA 2003, 100, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Barr, R.K.; Ketterman, A.J. Peptide inhibitors of protein kinases-discovery, characterisation and use. Biochim. Biophys. Acta 2005, 1754, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Ramsey, T.M.; Bair, K.W. Protein-protein interactions: Lessons learned. Curr. Med. Chem. Anticancer Agents 2002, 2, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Latham, P.W. Therapeutic peptides revisited. Nat. Biotechnol. 1999, 17, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Baines, I.C.; Colas, P. Peptide aptamers as guides for small-molecule drug discovery. Drug Discov. Today 2006, 11, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Ki Yeon, S.; Yang, X.-F.; Park, K.D.; Khanna, R. Differential regulation of collapsin response mediator protein 2 (CRMP2) phosphorylation by GSK3β and CDK5 following traumatic brain injury. Front. Cell. Neurosci. 2014, 8, 135. [Google Scholar] [PubMed]
- Warbrick, E.; Lane, D.P.; Glover, D.M.; Cox, L.S. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr. Biol. 1995, 5, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M.; Reisine, T. Human kinome drug discovery and the emerging importance of atypical allosteric inhibitors. Expert Opin. Drug Discov. 2010, 5, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Corbel, C.; Guéritte, F.; Couturier, C.; Bach, S.; Tan, V.B.C. An in silico approach for the discovery of CDK5/p25 interaction inhibitors. Biotechnol. J. 2011, 6, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Corbel, C.; Wang, Q.; Bousserouel, H.; Hamdi, A.; Zhang, B.; Lozach, O.; Ferandin, Y.; Tan, V.B.C.; Guéritte, F.; Colas, P.; et al. First BRET-based screening assay performed in budding yeast leads to the discovery of CDK5/p25 interaction inhibitors. Biotechnol. J. 2011, 6, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors. Cancers 2015, 7, 179-237. https://doi.org/10.3390/cancers7010179
Peyressatre M, Prével C, Pellerano M, Morris MC. Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors. Cancers. 2015; 7(1):179-237. https://doi.org/10.3390/cancers7010179
Chicago/Turabian StylePeyressatre, Marion, Camille Prével, Morgan Pellerano, and May C. Morris. 2015. "Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors" Cancers 7, no. 1: 179-237. https://doi.org/10.3390/cancers7010179
APA StylePeyressatre, M., Prével, C., Pellerano, M., & Morris, M. C. (2015). Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors. Cancers, 7(1), 179-237. https://doi.org/10.3390/cancers7010179