
Extracellular Molecules Involved in Cancer Cell Invasion

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Molecule Name | Molecule Type | Molecules Co-involved in Cancer Cell Invasion |
|---|---|---|---|
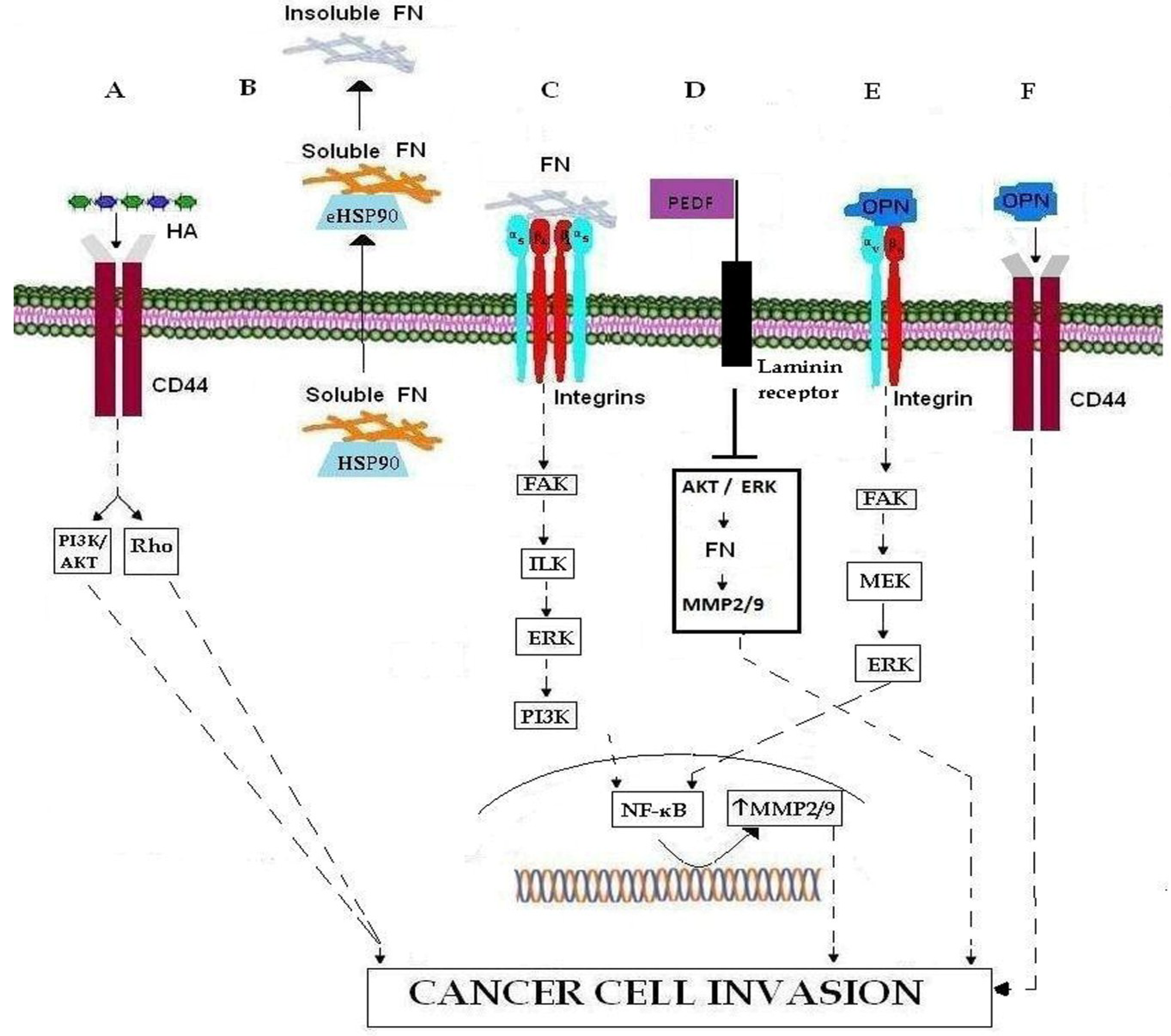
| ECM MOLECULES | Hyaluronan (HA) | glycosaminoglycan | CD44 |
| Fibronectin (FN) | glycoprotein | eHSP90, HSP90, MMP-9, MMP-9, FAK/PI3K/AKT/ERK/NF-κB, PEDF | |
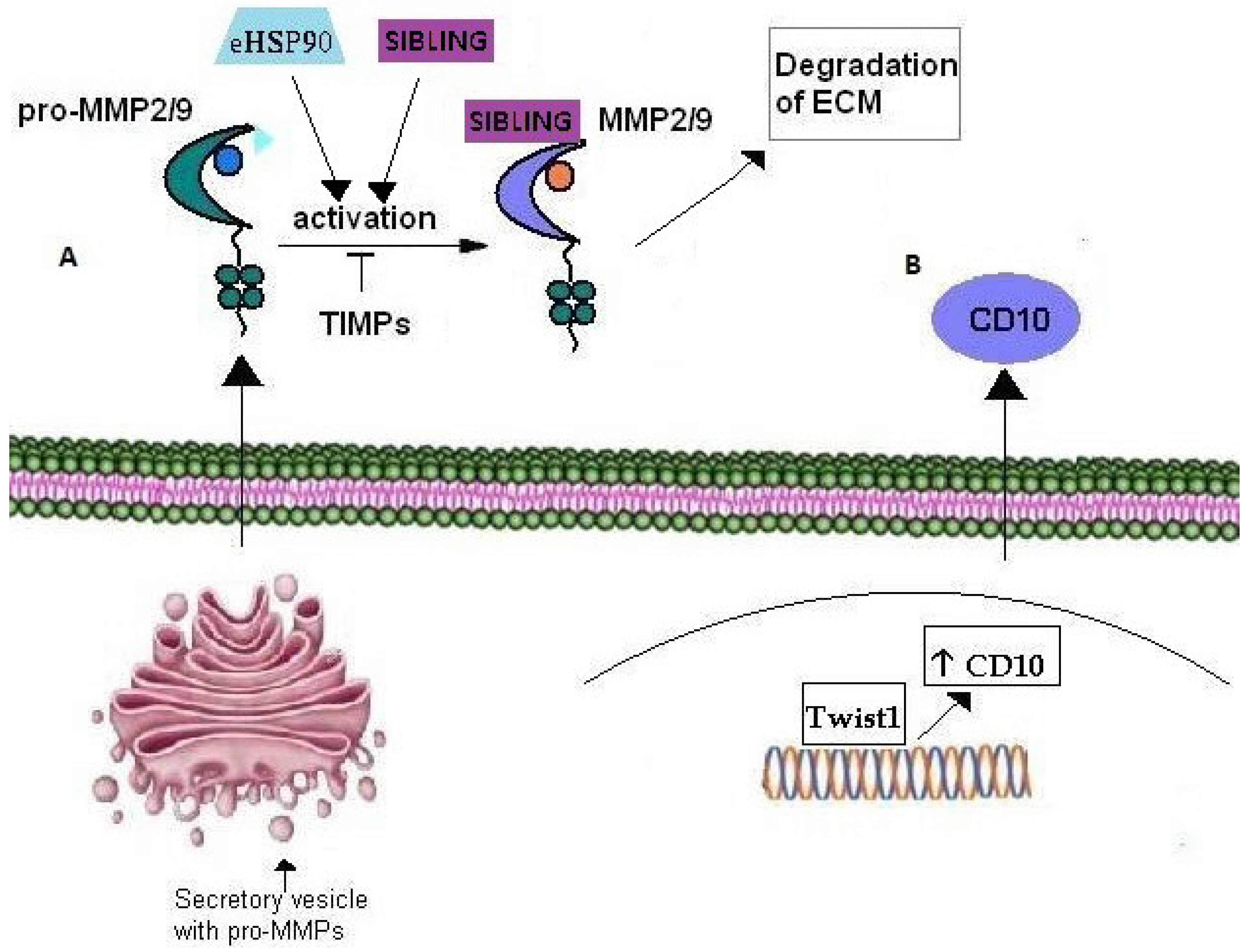
| SIBLING | Small Integrin-Binding Ligand, N-linked Glycoprotein | Pro-MMPs, MMP-2, MMP-9, MMP-3, αvβ3 integrin, FAK/MEK/ERK/NF-Κβ pathway, CD44v6 | |
| ECM RECEPTORS | Integrins | Cell surface receptors | Fibronectin, MMP-9, MMP-2, FAK/ILK/, ERK/, PI3K/NF-κB signaling cascades EGFR, osteopontin |
| CD44 | Cell surface receptors | Hyaluronan (HA), osteopontin | |
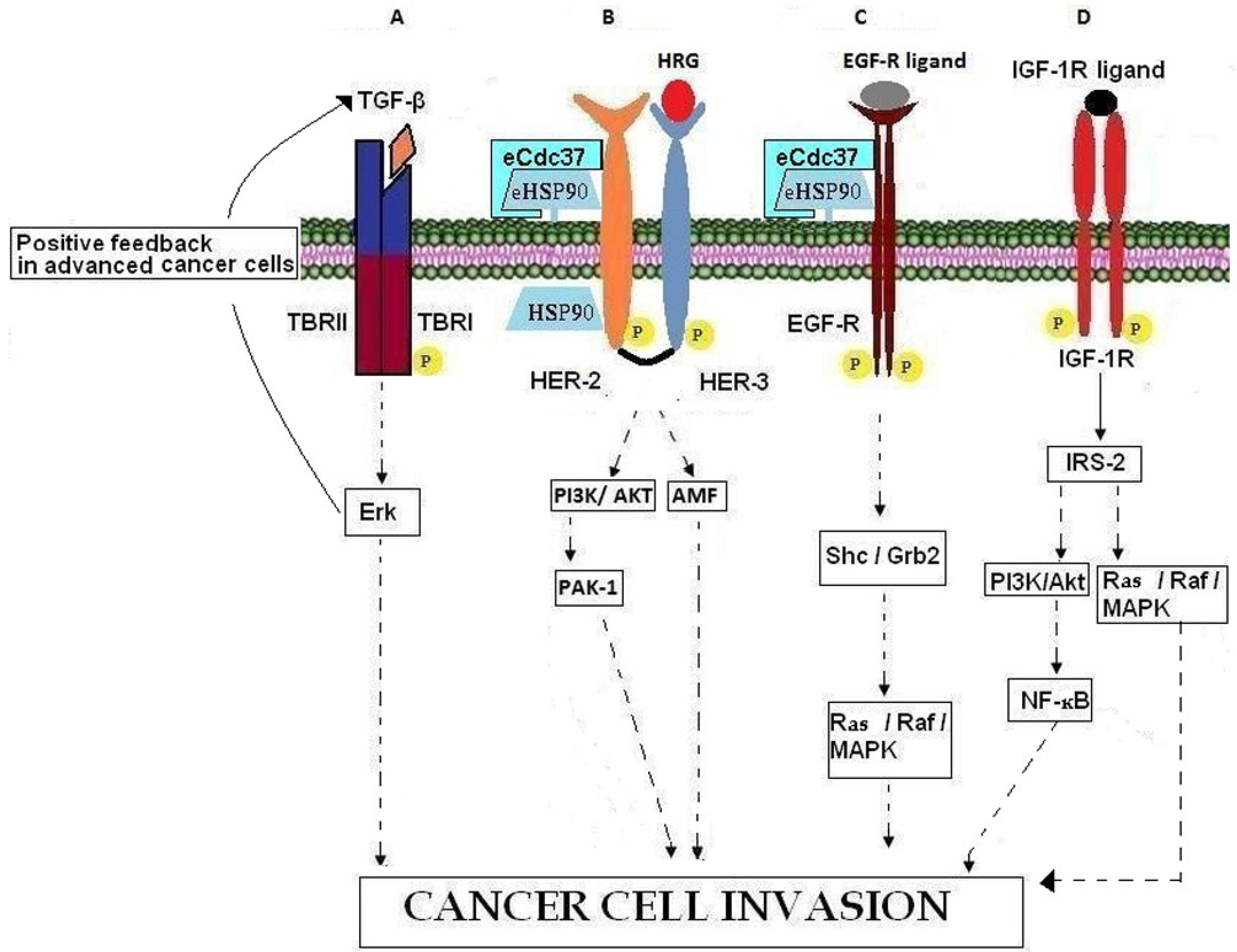
| GROWTH FACTORS | TGF-β | Growth factors | TBRI, TBRII, Erk, Ras |
| Heregulin | EGF-like growth and differentiation factor | ErbB3, ErbB4, PAK-1, AMF | |
| GROWTH FACTOR RECEPTORS | EGFR | Cell surface receptor | TGF-α, Grb2, Ras/Raf/MEK/MAPK |
| HER-2 | Cell surface co-receptor | HER-3, eHSP90, MAPK, PI3K/AKT | |
| IGF-R | Cell surface receptor | IGFs, IRS-2, PI3K/AKT, Ras/Raf/MAPK | |
| MATRIXMETALLO-PROTEINASES | Matrix Metalloproteinase (MMP)-9 | Zinc endopeptidase | eHSP90, HSP90, Rab40b, VAMP-4, gelatin type IV collagen, VEGF, bFGF |
| Matrix Metalloproteinase (MMP)-2 | Zinc endopeptidase | gelatine, type IV collagen, eHSP90, HSP90, Rab40b, VAMP-4, VEGF, bFGF | |
| CD10 | Zinc-dependent metalloproteinase | Twist1 | |
| CHAPERONES | eHSP90 | Chaperone | Cdc37, FN, HER-2, EGFR, pro-MMP-2, pro-MMP-9 |
| eCdc37 | Co-chaperone | HSP90, eHSP90, HER2, EGFR, Raf1, CDK4, EGFRvIII, Peuth-Jeghers cancer syndrome-associated kinase | |
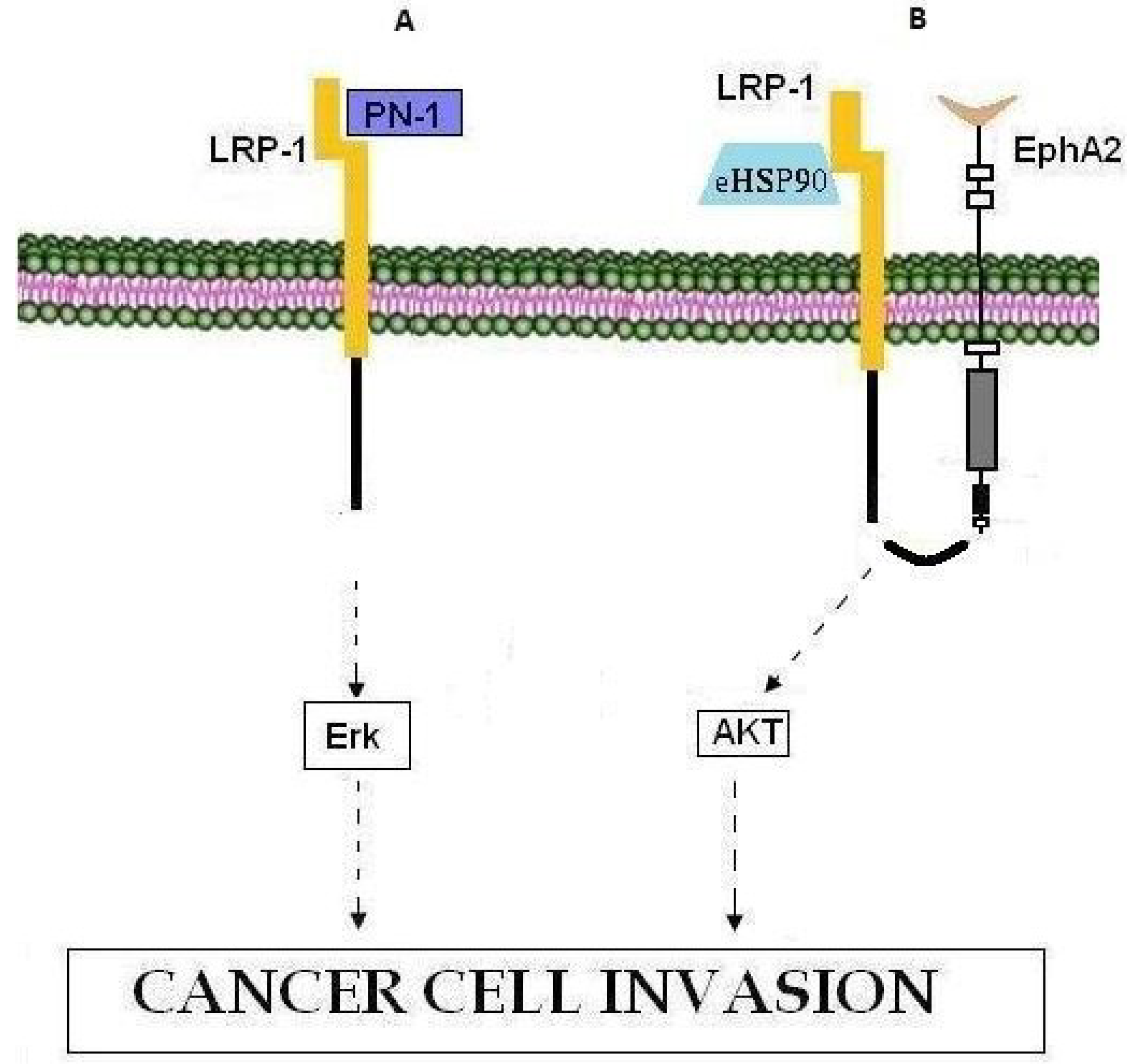
| LRP-1 | LRP-1 | Low-density lipoprotein (LDL) receptor | Nexin-1 (PN-1), Erk pathway, MMP-9, eHSP90, EphA2, AKT1, AKT2 |
2. ECM Molecules
2.1. Hyaluronan
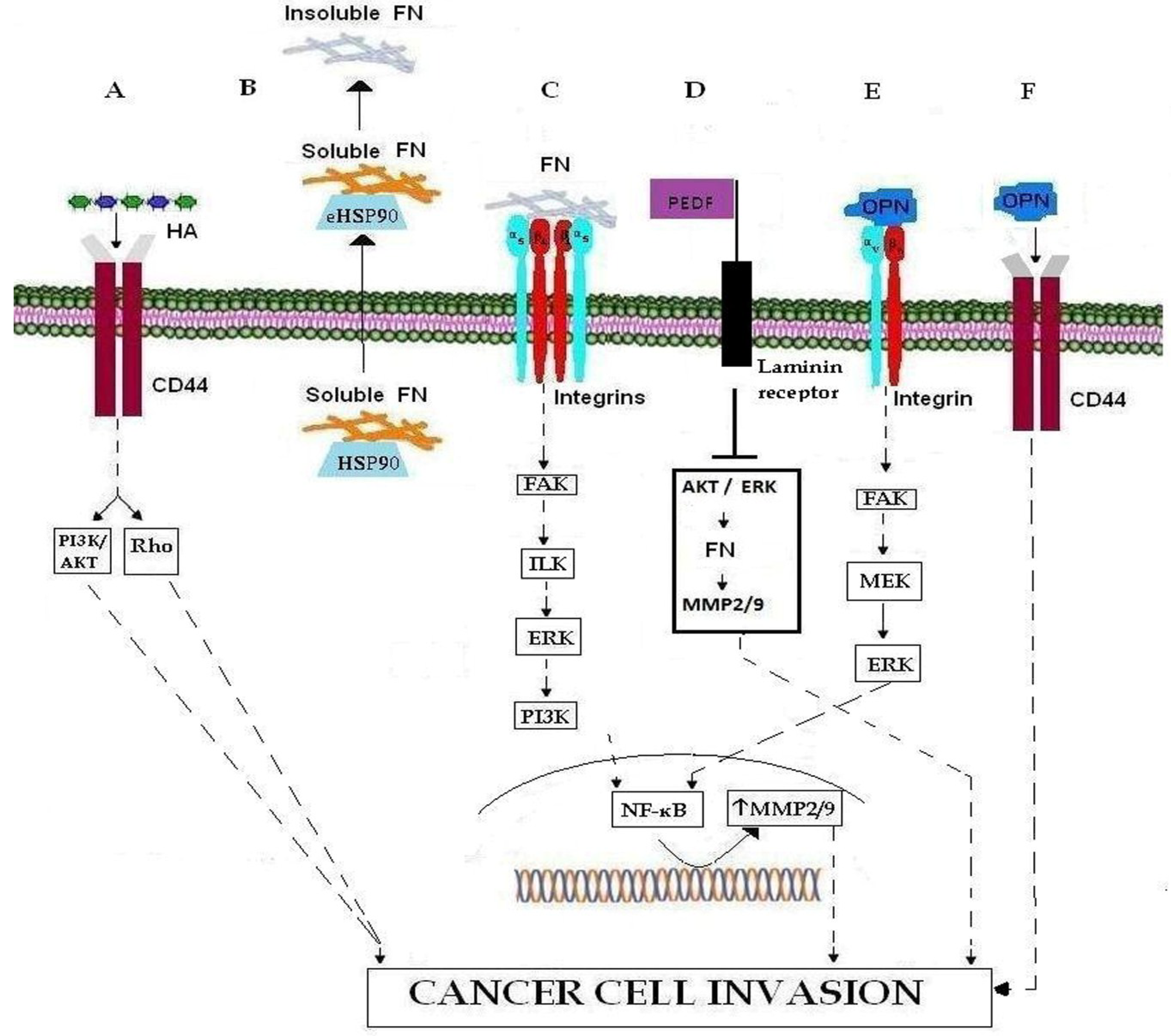
2.2. Fibronectin
2.3. SIBLING
3. ECM Receptors
3.1. Integrins
3.2. CD44
4. Growth Factors
4.1. TGF-β
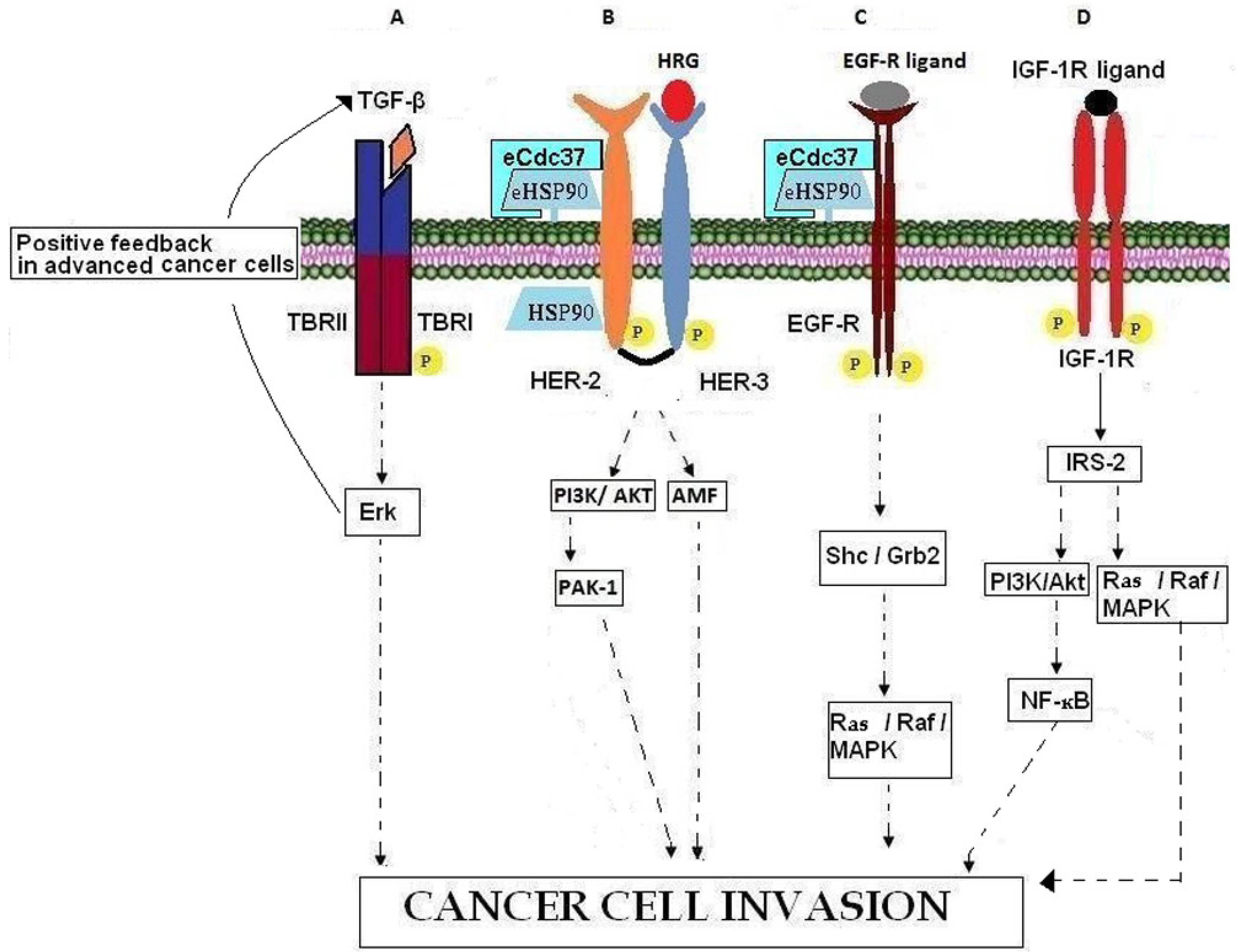
4.2. Heregulin
5. Growth Factor Receptors
5.1. ErbB Receptors
5.2. IGF-R
6. Matrix Metalloproteinases
6.1. MMPs
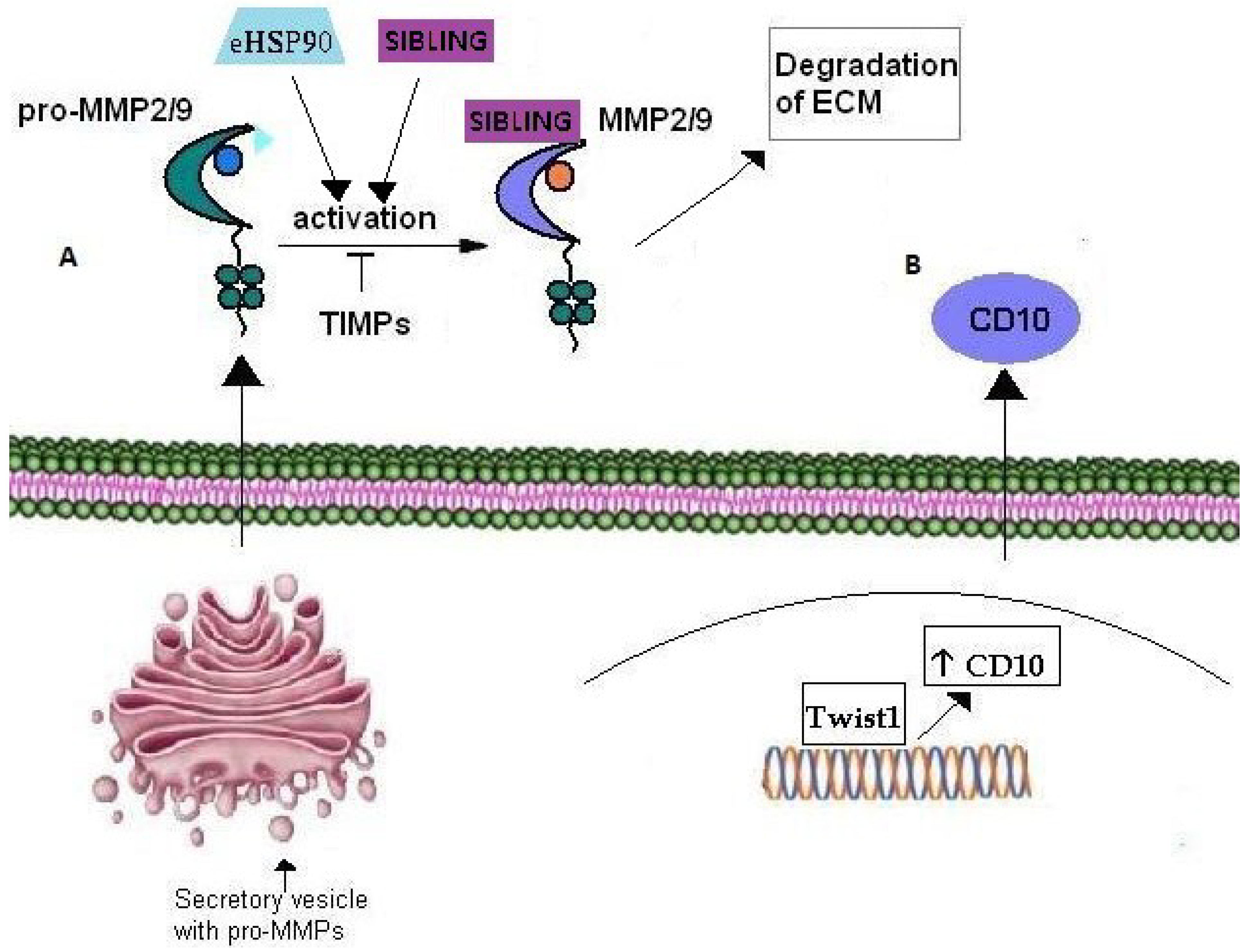
6.2. CD10
7. Chaperones
7.1. eHSP90
7.2. eCdc37
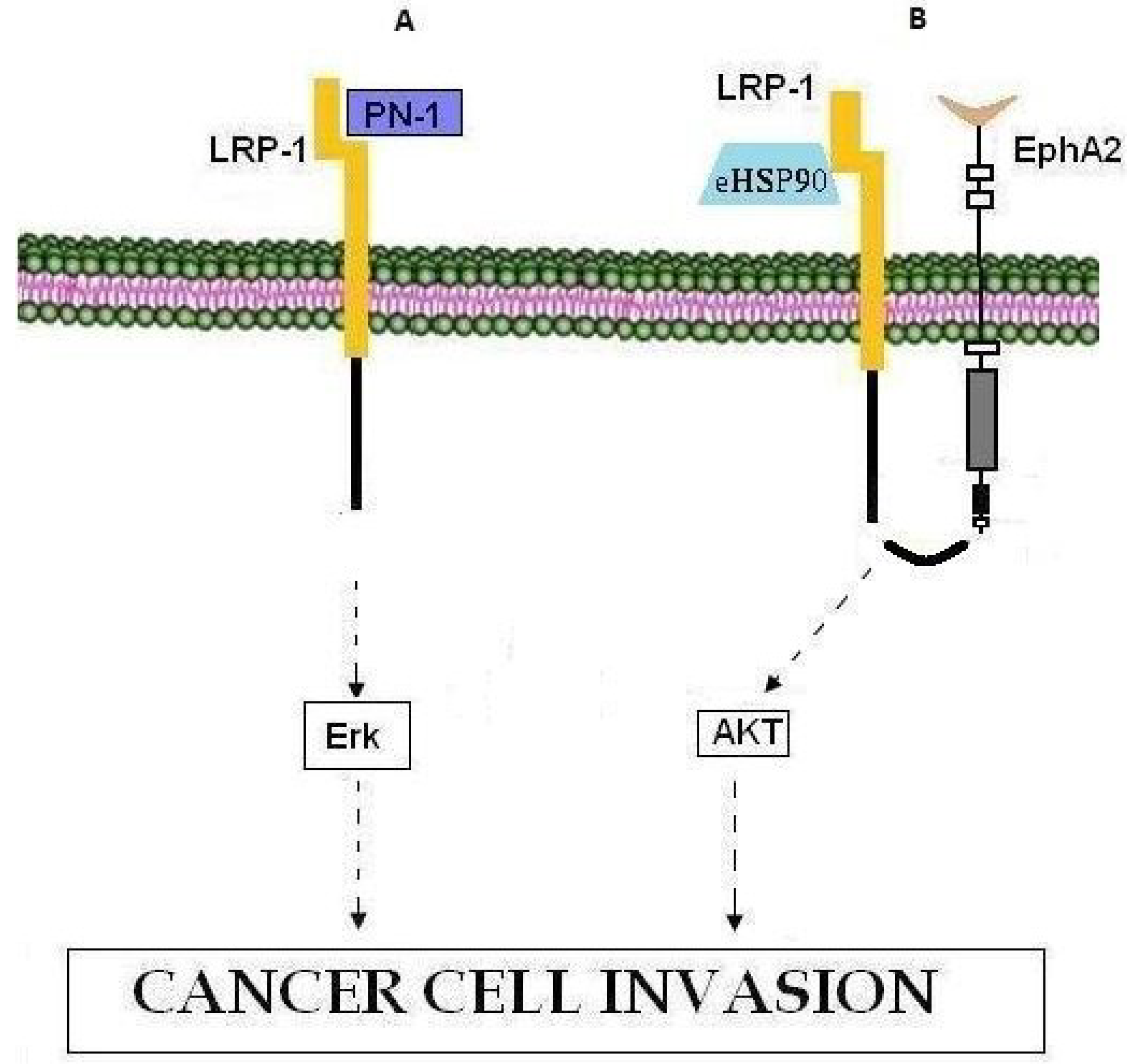
8. LRP-1
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.; Fodde, R. Cancer stemness and metastasis: Therapeutic consequences and perspectives. Eur J. Cancer 2010, 46, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.T.; Ye, Y.P.; Deng, Y.J.; Bian, X.W.; Ding, Y.Q. Metastatic cancer stem cells: From the concept to therapeutics. Am. J. Stem. Cells 2014, 3, 46–62. [Google Scholar] [PubMed]
- Spano, D.; Heck, C.; de Antonellis, P.; Christofori, G.; Zollo, M. Molecular networks that regulate cancer metastasis. Semin. Cancer Biol. 2012, 22, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, M.; Dello Sbarba, P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle 2008, 7, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Fabian, A.; Barok, M.; Vereb, G.; Szollosi, J. Die hard: Are cancer stem cells the bruce willises of tumor biology? Cytometry A 2009, 75, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem. Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Yip, G.W. Heparanase, hyaluronan, and CD44 in cancers: A breast carcinoma perspective. Cancer Res. 2006, 66, 10233–10237. [Google Scholar] [CrossRef] [PubMed]
- Udabage, L.; Brownlee, G.R.; Nilsson, S.K.; Brown, T.J. The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp. Cell Res. 2005, 310, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Gould, V.E.; Koukoulis, G.K.; Virtanen, I. Extracellular matrix proteins and their receptors in the normal, hyperplastic and neoplastic breast. Cell Differ. Dev. 1990, 32, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Zhou, T.; Fang, S.; Jia, M.; Xu, Z.; Dai, Z.; Li, C.; Li, S.; Li, L.; Zhang, T.; et al. Pigment epithelium-derived factor (PEDF) inhibits breast cancer metastasis by down-regulating fibronectin. Breast Cancer Res. Treat. 2014, 148, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.C.; O’Hagan, K.L.; Kenyon, A.; Dhanani, K.C.; Prinsloo, E.; Edkins, A.L. Hsp90 binds directly to fibronectin (FN) and inhibition reduces the extracellular fibronectin matrix in breast cancer cells. PLOS ONE 2014, 9, e86842. [Google Scholar] [CrossRef] [PubMed]
- Ioachim, E.; Charchanti, A.; Briasoulis, E.; Karavasilis, V.; Tsanou, H.; Arvanitis, D.L.; Agnantis, N.J.; Pavlidis, N. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: Their prognostic value and role in tumour invasion and progression. Eur. J. Cancer 2002, 38, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.E.; Foty, R.A.; Corbett, S.A. Fibronectin matrix assembly regulates α5β1-mediated cell cohesion. Mol. Biol. Cell 2004, 15, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka-Patynowski, I.; Schwarzbauer, J.E. The ins and outs of fibronectin matrix assembly. J. Cell Sci. 2003, 116, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Kim, A.; Kim, M.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum. Pathol. 2013, 44, 2028–2037. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Banerji, A.; Frei, E.; Chatterjee, A. Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci. 2008, 82, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Maity, G.; Choudhury, P.R.; Sen, T.; Ganguly, K.K.; Sil, H.; Chatterjee, A. Culture of human breast cancer cell line (MDA-MB-231) on fibronectin-coated surface induces pro-matrix metalloproteinase-9 expression and activity. Tumour Biol. 2011, 32, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.W.; Jain, A.; Tayback, M.; Fedarko, N.S. Small integrin binding ligand n-linked glycoprotein gene family expression in different cancers. Clin. Cancer Res. 2004, 10, 8501–8511. [Google Scholar] [CrossRef] [PubMed]
- Wai, P.Y.; Kuo, P.C. Osteopontin: Regulation in tumor metastasis. Cancer Metastasis Rev. 2008, 27, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Bellahcene, A.; Castronovo, V. Increased expression of osteonectin and osteopontin, two bone matrix proteins, in human breast cancer. Am. J. Pathol. 1995, 146, 95–100. [Google Scholar] [PubMed]
- Brown, L.F.; Papadopoulos-Sergiou, A.; Berse, B.; Manseau, E.J.; Tognazzi, K.; Perruzzi, C.A.; Dvorak, H.F.; Senger, D.R. Osteopontin expression and distribution in human carcinomas. Am. J. Pathol. 1994, 145, 610–623. [Google Scholar] [PubMed]
- Casson, A.G.; Wilson, S.M.; McCart, J.A.; O’Malley, F.P.; Ozcelik, H.; Tsao, M.S.; Chambers, A.F. Ras mutation and expression of the ras-regulated genes osteopontin and cathepsin l in human esophageal cancer. Int. J. Cancer 1997, 72, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, M.; Sakamoto, M.; Kanetaka, K.; Chuuma, M.; Hirohashi, S. Overexpression of osteopontin in hepatocellular carcinoma. Pathol. Int. 2002, 52, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Ito, A.; Nagoshi, J.; Takeda, M.; Kurata, A.; Takatsuka, Y.; Kohri, K.; Nomura, S.; Kitamura, Y. Expression of bone matrix protein messenger ribonucleic acids in human breast cancers. Possible involvement of osteopontin in development of calcifying foci. Lab. Invest. 1995, 72, 64–69. [Google Scholar] [PubMed]
- Senger, D.R.; Perruzzi, C.A.; Gracey, C.F.; Papadopoulos, A.; Tenen, D.G. Secreted phosphoproteins associated with neoplastic transformation: Close homology with plasma proteins cleaved during blood coagulation. Cancer Res. 1988, 48, 5770–5774. [Google Scholar] [PubMed]
- Singhal, H.; Bautista, D.S.; Tonkin, K.S.; O’Malley, F.P.; Tuck, A.B.; Chambers, A.F.; Harris, J.F. Elevated plasma osteopontin in metastatic breast cancer associated with increased tumor burden and decreased survival. Clin. Cancer Res. 1997, 3, 605–611. [Google Scholar] [PubMed]
- Tuck, A.B.; O’Malley, F.P.; Singhal, H.; Harris, J.F.; Tonkin, K.S.; Kerkvliet, N.; Saad, Z.; Doig, G.S.; Chambers, A.F. Osteopontin expression in a group of lymph node negative breast cancer patients. Int. J. Cancer 1998, 79, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Bramwell, V.H.; Doig, G.S.; Tuck, A.B.; Wilson, S.M.; Tonkin, K.S.; Tomiak, A.; Perera, F.; Vandenberg, T.A.; Chambers, A.F. Serial plasma osteopontin levels have prognostic value in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 3337–3343. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Kim, H.J.; Chang, J.; Ahn, C.M.; Kim, S.K. Elevated circulating level of osteopontin is associated with advanced disease state of non-small cell lung cancer. Lung Cancer 2007, 57, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, A.; Festuccia, C.; D’Andrea, G.; Teti, A.; Bologna, M. Osteopontin modulates prostate carcinoma invasive capacity through rgd-dependent upregulation of plasminogen activators. Biol Chem. 2002, 383, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Furger, K.A.; Allan, A.L.; Wilson, S.M.; Hota, C.; Vantyghem, S.A.; Postenka, C.O.; Al-Katib, W.; Chambers, A.F.; Tuck, A.B. Beta(3) integrin expression increases breast carcinoma cell responsiveness to the malignancy-enhancing effects of osteopontin. Mol. Cancer Res. 2003, 1, 810–819. [Google Scholar] [PubMed]
- Senger, D.R.; Perruzzi, C.A. Cell migration promoted by a potent grgds-containing thrombin-cleavage fragment of osteopontin. Biochim. Biophys. Acta 1996, 1314, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wei, Y.Y.; Chen, H.T.; Fong, Y.C.; Hsu, C.J.; Tsai, C.H.; Hsu, H.C.; Liu, S.H.; Tang, C.H. Osteopontin increases migration and MMP-9 up-regulation via alphavbeta3 integrin, FAK, ERK, and NF-kappaB-dependent pathway in human chondrosarcoma cells. J. Cell Physiol. 2009, 221, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Bayless, K.J.; Salazar, R.; Davis, G.E. RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the αvβ3 and α5β1 integrins. Am. J. Pathol. 2000, 156, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-kappaB mediates αvβ3 integrin-induced endothelial cell survival. J. Cell Biol. 1998, 141, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Ledbetter, S.R.; Claffey, K.P.; Papadopoulos-Sergiou, A.; Peruzzi, C.A.; Detmar, M. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the αvβ3 integrin, osteopontin, and thrombin. Am. J. Pathol. 1996, 149, 293–305. [Google Scholar] [PubMed]
- Asosingh, K.; Gunthert, U.; Bakkus, M.H.; de Raeve, H.; Goes, E.; van Riet, I.; van Camp, B.; Vanderkerken, K. In vivo induction of insulin-like growth factor-I receptor and CD44V6 confers homing and adhesion to murine multiple myeloma cells. Cancer Res. 2000, 60, 3096–3104. [Google Scholar] [PubMed]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef]
- Gunthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zoller, M.; Haussmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Rudzki, Z.; Jothy, S. CD44 and the adhesion of neoplastic cells. Mol. Pathol. 1997, 50, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Takahashi, F.; Hirama, M.; Tanabe, K.K.; Fukuchi, Y. Restoration of CD44S in non-small cell lung cancer cells enhanced their susceptibility to the macrophage cytotoxicity. Lung Cancer 2003, 41, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Strokovich, K.; Springer, T.A.; Walz, T. Structure of integrin alpha5beta1 in complex with fibronectin. Embo J. 2003, 22, 4607–4615. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Keller, E.T.; Brown, J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J. Cell Biochem. 2004, 91, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adh. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Hosotani, R.; Kawaguchi, M.; Masui, T.; Koshiba, T.; Ida, J.; Fujimoto, K.; Wada, M.; Doi, R.; Imamura, M. Expression of integrin alphavbeta3 in pancreatic carcinoma: Relation to MMP-2 activation and lymph node metastasis. Pancreas 2002, 25, e30–e35. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin alpha(v)beta3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Ricono, J.M.; Huang, M.; Barnes, L.A.; Lau, S.K.; Weis, S.M.; Schlaepfer, D.D.; Hanks, S.K.; Cheresh, D.A. Specific cross-talk between epidermal growth factor receptor and integrin alphavbeta5 promotes carcinoma cell invasion and metastasis. Cancer Res. 2009, 69, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Berner, H.S.; Suo, Z.; Risberg, B.; Villman, K.; Karlsson, M.G.; Nesland, J.M. Clinicopathological associations of CD44 mrna and protein expression in primary breast carcinomas. Histopathology 2003, 42, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.K.; Zhou, X.; Wright, E.T.; Cristofanilli, M.; Smith, T.; Yang, Y.; Sneige, N.; Sahin, A.; Gilcrease, M.Z. CD44 expression is associated with increased survival in node-negative invasive breast carcinoma. Clin. Cancer Res. 2005, 11, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Rys, J.; Kruczak, A.; Lackowska, B.; Jaszcz-Gruchala, A.; Brandys, A.; Stelmach, A.; Reinfuss, M. The role of CD44V3 expression in female breast carcinomas. Pol. J. Pathol. 2003, 54, 243–247. [Google Scholar] [PubMed]
- Hiscox, S.; Baruha, B.; Smith, C.; Bellerby, R.; Goddard, L.; Jordan, N.; Poghosyan, Z.; Nicholson, R.I.; Barrett-Lee, P.; Gee, J. Overexpression of CD44 accompanies acquired tamoxifen resistance in MCF7 cells and augments their sensitivity to the stromal factors, heregulin and hyaluronan. BMC Cancer 2012. [Google Scholar] [CrossRef]
- Bapat, S.A. Human ovarian cancer stem cells. Reproduction 2010, 140, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Wang, H.; He, L.; Zhang, J.; Ni, B.; Wang, X.; Jin, H.; Cahuzac, N.; Mehrpour, M.; Lu, Y.; Chen, Q. CD44 is of functional importance for colorectal cancer stem cells. Clin. Cancer Res. 2008, 14, 6751–6760. [Google Scholar] [CrossRef] [PubMed]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012. [Google Scholar] [CrossRef]
- Wang, R.; Lv, Q.; Meng, W.; Tan, Q.; Zhang, S.; Mo, X.; Yang, X. Comparison of mammosphere formation from breast cancer cell lines and primary breast tumors. J. Thorac. Dis. 2014, 6, 829–837. [Google Scholar] [PubMed]
- Buck, M.B.; Knabbe, C. TGF-beta signaling in breast cancer. Ann. N. Y. Acad. Sci. 2006, 1089, 119–126. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Vogelmann, R.; Nguyen-Tat, M.D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and pten. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B. Molecular and cell biology of TGF-beta. Miner. Electrolyte MeTable. 1998, 24, 111–119. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of Cancer. Proc. Jpn. Acad. B Phys. Biol. Sci. 2009, 85, 314–323. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- ten Dijke, P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008, 11, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; ten Dijke, P. TGF-beta signaling in breast cancer cell invasion and bone metastasis. J. Mammary Gland Biol. Neoplasia 2011, 16, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr. Opin. Genet. Dev. 2011, 21, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-beta: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014. [Google Scholar] [CrossRef]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-beta paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of TGF-beta paradox in benign and malignant cells. Front. Oncol. 2014. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-beta signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Garratt, A.N. “To erb-b or not to erb-b...” Neuregulin-1/erbb signaling in heart development and function. J. Mol. Cell Cardiol. 2006, 41, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Riese, D.J., 2nd; Gilbert, W.; Stern, D.F.; McMahan, U.J. Ligands for erbb-family receptors encoded by a neuregulin-like gene. Nature 1997, 387, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Harari, D.; Tzahar, E.; Romano, J.; Shelly, M.; Pierce, J.H.; Andrews, G.C.; Yarden, Y. Neuregulin-4: A novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene 1999, 18, 2681–2689. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.E.; Sliwkowski, M.X.; Akita, R.W.; Henzel, W.J.; Lee, J.; Park, J.W.; Yansura, D.; Abadi, N.; Raab, H.; Lewis, G.D.; et al. Identification of heregulin, a specific activator of p185erbb2. Science 1992, 256, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L., 3rd; Sliwkowski, M.X.; Akita, R.; Platko, J.V.; Guy, P.M.; Nuijens, A.; Diamonti, A.J.; Vandlen, R.L.; Cantley, L.C.; Cerione, R.A. The ErbB3 gene product is a receptor for heregulin. J. Biol. Chem. 1994, 269, 14303–14306. [Google Scholar] [PubMed]
- Stove, C.; Bracke, M. Roles for neuregulins in human Cancer. Clin. Exp. Metastasis. 2004, 21, 665–684. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.M.; Pankonin, M.S.; Loeb, J.A. Neuregulins: Versatile growth and differentiation factors in nervous system development and human disease. Brain Res. Rev. 2006, 51, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Falls, D.L. Neuregulins and the neuromuscular system: 10 years of answers and questions. J. Neurocytol. 2003, 32, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cleary, S.; Mandarano, M.A.; Long, W.; Birchmeier, C.; Jones, F.E. The breast proto-oncogene, hrgalpha regulates epithelial proliferation and lobuloalveolar development in the mouse mammary gland. Oncogene 2002, 21, 4900–4907. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Mehmi, I.; Lupu, R. Trastuzumab in combination with heregulin-activated Her-2 (ErbB-2) triggers a receptor-enhanced chemosensitivity effect in the absence of Her-2 overexpression. J. Clin. Oncol. 2006, 24, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Raj, E.H.; Skinner, A.; Mahji, U.; Nirmala, K.N.; Ravichandran, K.; Shanta, V.; Hurst, H.C.; Gullick, W.J.; Rajkumar, T. Neuregulin 1-alpha expression in locally advanced breast cancer. Breast (Edinburgh, Scotland) 2001, 10, 41–45. [Google Scholar] [CrossRef]
- Cheng, L.; Zha, Z.; Lang, B.; Liu, J.; Yao, X. Heregulin-beta1 promotes metastasis of breast cancer cell line SKBR3 through upregulation of snail and induction of epithelial-mesenchymal transition. Cancer Lett. 2009, 280, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jeong, H.; Lee, Y.; Kim, C.; Kim, H.; Kim, A. HRG-beta1-driven ErbB3 signaling induces epithelial-mesenchymal transition in breast cancer cells. BMC Cancer 2013, 13, 383. [Google Scholar] [CrossRef] [PubMed]
- Adam, L.; Vadlamudi, R.; Kondapaka, S.B.; Chernoff, J.; Mendelsohn, J.; Kumar, R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J. Biol. Chem. 1998, 273, 28238–28246. [Google Scholar] [CrossRef] [PubMed]
- Talukder, A.H.; Adam, L.; Raz, A.; Kumar, R. Heregulin regulation of autocrine motility factor expression in human tumor cells. Cancer Res. 2000, 60, 474–480. [Google Scholar] [PubMed]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell Physiol. 2000, 184, 1–16. [Google Scholar] [CrossRef]
- Loeb, J.A.; Fischbach, G.D. Aria can be released from extracellular matrix through cleavage of a heparin-binding domain. J. Cell Biol. 1995, 130, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L., 3rd; Weber, J.L.; Unger, M.J.; Ledesma, J.; Yu, N.; Gassmann, M.; Lai, C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature 1997, 387, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ErbB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.D.; Yarden, Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 2004, 23, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 2002, 3, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G. ErbB-4: Mechanism of action and biology. Exp. Cell Res. 2003, 284, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Skaria, K.B.; Yarden, Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Exp. Cell Res. 2003, 284, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.P.; Stellema, R.; Karrenbeld, A.; Koudstaal, J.; Gouw, A.S.; Sluiter, W.J.; Peeters, P.M.; Slooff, M.J.; de Vries, E.G. Clinical relevance of transforming growth factor alpha, epidermal growth factor receptor, p53, and ki67 in colorectal liver metastases and corresponding primary tumors. Hepatology 1998, 28, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Douziech, N.; Calvo, E.; Laine, J.; Morisset, J. Activation of map kinases in growth responsive pancreatic cancer cells. Cell Signal. 1999, 11, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J. The epidermal growth factor receptor as a target for cancer therapy. Endocr. Relat. Cancer 2001, 8, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol. 1996, 16, 5276–5287. [Google Scholar] [PubMed]
- Muss, H.B.; Thor, A.D.; Berry, D.A.; Kute, T.; Liu, E.T.; Koerner, F.; Cirrincione, C.T.; Budman, D.R.; Wood, W.C.; Barcos, M.; et al. C-ErbB-2 expression and response to adjuvant therapy in women with node-positive early breast Cancer. N. Engl. J. Med. 1994, 330, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the Her-2/NEU proto-oncogene in human breast and ovarian Cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Chavany, C.; Neckers, L. Polyubiquitination and proteasomal degradation of the p185c-ErbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J. Biol. Chem. 1996, 271, 22796–22801. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Mimnaugh, E.; Rosser, M.F.; Nicchitta, C.; Marcu, M.; Yarden, Y.; Neckers, L. Sensitivity of mature ErbB2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein hsp90. J. Biol. Chem. 2001, 276, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Kochupurakkal, B.S.; Yarden, Y. The achilles heel of ErbB-2/Her2: Regulation by the hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 2004, 3, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Gaitanou, M.; Stellas, D.; Matsas, R.; Patsavoudi, E. A critical role for hsp90 in cancer cell invasion involves interaction with the extracellular domain of Her-2. J. Biol. Chem. 2008, 283, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other erbb receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/Her receptor protein-tyrosine kinases and cancer. Biochem. Biophys. Res. Commun. 2004, 319, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D. Regulation of breast cancer metastasis by IGF signaling. J. Mammary Gland Biol. Neoplasia 2008, 13, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Zhang, X.; Matise, I.; Gaillard-Kelly, M.; Yee, D. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene 2010, 29, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Zhang, X.; Yoneda, T.; Yee, D. Regulation of breast cancer cell motility by insulin receptor substrate-2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene 2001, 20, 7318–7325. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Faria, T.N.; Stannard, B.; Roberts, C.T., Jr.; LeRoith, D. Essential role of tyrosine residues 1131, 1135, and 1136 of the insulin-like growth factor-I (IGF-I) receptor in IGF-I action. Mol. Endocrinol. 1994, 8, 40–50. [Google Scholar] [PubMed]
- Yoneda, T.; Williams, P.J.; Hiraga, T.; Niewolna, M.; Nishimura, R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res. 2001, 16, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Myoui, A.; Hashimoto, N.; Sasaki, A.; Hata, K.; Morita, Y.; Yoshikawa, H.; Rosen, C.J.; Mundy, G.R.; Yoneda, T. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012, 72, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Saldana, S.M.; Lee, H.H.; Lowery, F.J.; Khotskaya, Y.B.; Xia, W.; Zhang, C.; Chang, S.S.; Chou, C.K.; Steeg, P.S.; Yu, D.; et al. Inhibition of type I insulin-like growth factor receptor signaling attenuates the development of breast cancer brain metastasis. PLOS ONE 2013, 8, e73406. [Google Scholar] [CrossRef] [PubMed]
- Min, K.W.; Kim, D.H.; Do, S.I.; Kim, K.; Lee, H.J.; Chae, S.W.; Sohn, J.H.; Pyo, J.S.; Oh, Y.H.; Kim, W.S.; et al. Expression patterns of stromal MMP-2 and tumoural MMP-2 and -9 are significant prognostic factors in invasive ductal carcinoma of the breast. Apmis 2014, 122, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Patsavoudi, E. Inhibiting matrix metalloproteinases, an old story with new potentials for cancer treatment. Anticancer Agents Med. Chem. 2012, 12, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [PubMed]
- Zuo, J.H.; Zhu, W.; Li, M.Y.; Li, X.H.; Yi, H.; Zeng, G.Q.; Wan, X.X.; He, Q.Y.; Li, J.H.; Qu, J.Q.; et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing emt-like phenotype change and MMP-9-mediated degradation of E-cadherin. J. Cell Biochem. 2011, 112, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Eustace, B.K.; Jay, D.G. Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 2004, 3, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; El Hamidieh, A.; Patsavoudi, E. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 2010. [Google Scholar] [CrossRef]
- Fedarko, N.S.; Jain, A.; Karadag, A.; Fisher, L.W. Three small integrin binding ligand n-linked glycoproteins (siblings) bind and activate specific matrix metalloproteinases. FASEB. J. 2004, 18, 734–736. [Google Scholar] [PubMed]
- Murphy, D.A.; Courtneidge, S.A. The “ins” and “outs” of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Jing, J.; Lee, J.; Schedin, P.; Gilbert, S.M.; Peden, A.A.; Junutula, J.R.; Prekeris, R. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J. Cell Sci. 2013, 126, 4647–4658. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Rab gtpases: Specifying and deciphering organelle identity and function. Trends Cell Biol. 2001, 11, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef]
- Campbell, N.E.; Kellenberger, L.; Greenaway, J.; Moorehead, R.A.; Linnerth-Petrik, N.M.; Petrik, J. Extracellular matrix proteins and tumor angiogenesis. J. Oncol. 2010. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Zuidwijk, K.; Verspaget, H.W.; de Jonge-Muller, E.S.; van Duijn, W.; Ferreira, V.; Fontijn, R.D.; David, G.; Hommes, D.W.; Lamers, C.B.; et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 2008, 44, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Stathopoulos, C.; Steffen, A.; Wiedemann, P.; Kohen, L.; Bringmann, A. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2007, 48, 4360–4367. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jilani, S.M.; Nikolova, G.V.; Carpizo, D.; Iruela-Arispe, M.L. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J. Cell Biol. 2005, 169, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Malfroy, B.; Kuang, W.J.; Seeburg, P.H.; Mason, A.J.; Schofield, P.R. Molecular cloning and amino acid sequence of human enkephalinase (neutral endopeptidase). FEBS Lett. 1988, 229, 206–210. [Google Scholar] [CrossRef]
- Ishimaru, F.; Mari, B.; Shipp, M.A. The type 2 CD10/neutral endopeptidase 24.11 promoter: Functional characterization and tissue-specific regulation by CBF/NF-Y isoforms. Blood 1997, 89, 4136–4145. [Google Scholar] [PubMed]
- Sumitomo, M.; Shen, R.; Walburg, M.; Dai, J.; Geng, Y.; Navarro, D.; Boileau, G.; Papandreou, C.N.; Giancotti, F.G.; Knudsen, B.; et al. Neutral endopeptidase inhibits prostate cancer cell migration by blocking focal adhesion kinase signaling. J. Clin. Invest. 2000, 106, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Shipp, M.A.; Stefano, G.B.; Switzer, S.N.; Griffin, J.D.; Reinherz, E.L. CD10 (CALLA)/neutral endopeptidase 24.11 modulates inflammatory peptide-induced changes in neutrophil morphology, migration, and adhesion proteins and is itself regulated by neutrophil activation. Blood 1991, 78, 1834–1841. [Google Scholar] [PubMed]
- Bachelard-Cascales, E.; Chapellier, M.; Delay, E.; Pochon, G.; Voeltzel, T.; Puisieux, A.; Caron de Fromentel, C.; Maguer-Satta, V. The CD10 enzyme is a key player to identify and regulate human mammary stem cells. Stem Cells 2010, 28, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Raouf, A.; Emerman, J.T.; Eaves, C.J. Epithelial progenitors in the normal human mammary gland. J. Mammary Gland Biol. Neoplasia 2005, 10, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Sunday, M.E.; Hua, J.; Torday, J.S.; Reyes, B.; Shipp, M.A. CD10/neutral endopeptidase 24.11 in developing human fetal lung. Patterns of expression and modulation of peptide-mediated proliferation. J. Clin. Invest. 1992, 90, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Buhring, H.J.; Battula, V.L.; Treml, S.; Schewe, B.; Kanz, L.; Vogel, W. Novel markers for the prospective isolation of human msc. Ann. N. Y. Acad. Sci. 2007, 1106, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Terauchi, M.; Kajiyama, H.; Shibata, K.; Ino, K.; Mizutani, S.; Kikkawa, F. Anti-progressive effect of neutral endopeptidase 24.11 (NEP/CD10) on cervical carcinoma in vitro and in vivo. Oncology 2005, 69, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Tokuhara, T.; Adachi, M.; Hashida, H.; Ishida, H.; Taki, T.; Higashiyama, M.; Kodama, K.; Tachibana, S.; Sasaki, S.; Miyake, M. Neutral endopeptidase/CD10 and aminopeptidase N/CD13 gene expression as a prognostic factor in non-small cell lung Cancer. Jpn. J. Thorac. Cardiovasc. Surg. 2001, 49, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology 2010, 139, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Maguer-Satta, V.; Besancon, R.; Bachelard-Cascales, E. Concise review: Neutral endopeptidase (CD10): A multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 2011, 29, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Omran, O.M. CD10 and E-cad expression in urinary bladder urothelial and squamous cell carcinoma. J. Environ. Pathol. Toxicol. Oncol. 2012, 31, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, G.Y.; Kim, Y.W.; Park, Y.K.; Song, J.Y.; Lim, S.J. Stromal CD10 expression and relationship to the E-cadherin/beta-catenin complex in breast carcinoma. Histopathology 2010, 56, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Makretsov, N.A.; Hayes, M.; Carter, B.A.; Dabiri, S.; Gilks, C.B.; Huntsman, D.G. Stromal CD10 expression in invasive breast carcinoma correlates with poor prognosis, estrogen receptor negativity, and high grade. Mod. Pathol. 2007, 20, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Kim, J.H.; Han, S.; Sung, C.O.; Do, I.G.; Ko, Y.H.; Um, S.H.; Kim, S.H. Twist1 is an independent prognostic factor of esophageal squamous cell carcinoma and associated with its epithelial-mesenchymal transition. Ann. Surg. Oncol. 2012, 19, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Sung, C.O.; Kim, J.H.; Kang, M.; Yoo, H.Y.; Kim, H.H.; Um, S.H.; Kim, S.H. CD10 expression is enhanced by twist1 and associated with poor prognosis in esophageal squamous cell carcinoma with facilitating tumorigenicity in vitro and in vivo. Int. J. Cancer 2015, 136, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Samiotaki, M.; Yfanti, E.; Panayotou, G.; Patsavoudi, E. Involvement of cell surface hsp90 in cell migration reveals a novel role in the developing nervous system. J. Biol. Chem. 2004, 279, 45379–45388. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Multhoff, G.; Farkas, B.; Wild, P.J.; Landthaler, M.; Stolz, W.; Vogt, T. Induction of hsp90 protein expression in malignant melanomas and melanoma metastases. Exp. Dermatol. 2004, 13, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Karameris, A.; Patsavoudi, E. Monoclonal antibody 4C5 immunostains human melanomas and inhibits melanoma cell invasion and metastasis. Clin. Cancer Res. 2007, 13, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Beebe, K.; Neckers, L. Impact of heat-shock protein 90 on cancer metastasis. Future Oncol. 2009, 5, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rao, R.; Shen, J.; Tang, Y.; Fiskus, W.; Nechtman, J.; Atadja, P.; Bhalla, K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008, 68, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sahu, D.; Tsen, F. Secreted heat shock protein-90 (hsp90) in wound healing and Cancer. Biochim. Biophys. Acta 2012, 1823, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Stivarou, T.; Vartzi, G.; Thomaidou, D.; Patsavoudi, E.; Hellenic Pasteur Institute, Athens, Greece. 2015; Personal observations.
- Pearl, L.H. Hsp90 and cdc37—A chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 2005, 15, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Workman, P. Targeting cdc37: An alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle 2009, 8, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M.; Grammatikakis, N.; Cochran, B.H.; Chinkers, M.; Pratt, W.B. P50(cdc37) binds directly to the catalytic domain of raf as well as to a site on hsp90 that is topologically adjacent to the tetratricopeptide repeat binding site. J. Biol. Chem. 1998, 273, 20090–20095. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Rutherford, S.L.; Miyata, Y.; Yahara, I.; Freeman, B.C.; Yue, L.; Morimoto, R.I.; Lindquist, S. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997, 11, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Hartson, S.D.; Irwin, A.D.; Shao, J.; Scroggins, B.T.; Volk, L.; Huang, W.; Matts, R.L. P50(cdc37) is a nonexclusive hsp90 cohort which participates intimately in hsp90-mediated folding of immature kinase molecules. Biochemistry 2000, 39, 7631–7644. [Google Scholar] [CrossRef] [PubMed]
- El Hamidieh, A.; Grammatikakis, N.; Patsavoudi, E. Cell surface cdc37 participates in extracellular hsp90 mediated cancer cell invasion. PLOS ONE 2012, 7, e42722. [Google Scholar] [CrossRef] [PubMed]
- Fayard, B.; Bianchi, F.; Dey, J.; Moreno, E.; Djaffer, S.; Hynes, N.E.; Monard, D. The serine protease inhibitor protease nexin-1 controls mammary cancer metastasis through LRP-1-mediated MMP-9 expression. Cancer Res. 2009, 69, 5690–5698. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Li, Y.; Lee, J.; Schwartz, A.L.; Bu, G. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 2009, 69, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Staudt, N.D.; Jo, M.; Hu, J.; Bristow, J.M.; Pizzo, D.P.; Gaultier, A.; VandenBerg, S.R.; Gonias, S.L. Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. 2013, 73, 3902–3912. [Google Scholar] [CrossRef] [PubMed]
- Tsen, F.; Bhatia, A.; O’Brien, K.; Cheng, C.F.; Chen, M.; Hay, N.; Stiles, B.; Woodley, D.T.; Li, W. Extracellular heat shock protein 90 signals through subdomain II and the npvy motif of LRP-1 receptor to AKT1 and AKT2: A circuit essential for promoting skin cell migration in vitro and wound healing in vivo. Mol. Cell Biol. 2013, 33, 4947–4959. [Google Scholar] [CrossRef] [PubMed]
- Montel, V.; Gaultier, A.; Lester, R.D.; Campana, W.M.; Gonias, S.L. The low-density lipoprotein receptor-related protein regulates cancer cell survival and metastasis development. Cancer Res. 2007, 67, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Bohonowych, J.E.; Lema-Tome, C.; Liu, A.; Garrett-Mayer, E.; Wang, B.; Isaacs, J.S. A novel extracellular hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLOS ONE 2011, 6, e17649. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Debinski, W. The EphA2 receptor and ephrina1 ligand in solid tumors: Function and therapeutic targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Dedieu, S.; Langlois, B.; Devy, J.; Sid, B.; Henriet, P.; Sartelet, H.; Bellon, G.; Emonard, H.; Martiny, L. LRP-1 silencing prevents malignant cell invasion despite increased pericellular proteolytic activities. Mol. Cell Biol. 2008, 28, 2980–2995. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stivarou, T.; Patsavoudi, E. Extracellular Molecules Involved in Cancer Cell Invasion. Cancers 2015, 7, 238-265. https://doi.org/10.3390/cancers7010238
Stivarou T, Patsavoudi E. Extracellular Molecules Involved in Cancer Cell Invasion. Cancers. 2015; 7(1):238-265. https://doi.org/10.3390/cancers7010238
Chicago/Turabian StyleStivarou, Theodora, and Evangelia Patsavoudi. 2015. "Extracellular Molecules Involved in Cancer Cell Invasion" Cancers 7, no. 1: 238-265. https://doi.org/10.3390/cancers7010238
APA StyleStivarou, T., & Patsavoudi, E. (2015). Extracellular Molecules Involved in Cancer Cell Invasion. Cancers, 7(1), 238-265. https://doi.org/10.3390/cancers7010238