Mutational Analysis of Merkel Cell Carcinoma

Abstract
:1. Introduction
2. The Role of Mutational Analysis in MCC
3. Merkel Cell Polyomavirus
4. Mutations in Tyrosine Kinase Signaling: KIT, PDGFRA, PIK3CA, AKT and PTEN

{kind=link}
| Generic Name | Trade/Code Name | Mechanism of Action | Trials in other Cancers | MCC Trial Phase | Trial Status | Additional |
|---|---|---|---|---|---|---|
| Pazopanib [46] | Votrient | Multi-targeted tyrosine kinase inhibitor | Renal cell, soft tissue sarcoma, lung, ovarian, breast, prostate, neuroendocrine, thyroid, cervical, cholangiocarcinoma, germ cell, urothelial and fallopian tube cancers | Phase 2 | Recruiting | |
| Cabozantinib [47] | Cometriq | Targeted inhibitor of c-Met and VEGFR2 | Thyroid, melanoma, breast, pancreatic, prostate, brain, bladder and ovarian cancers | Phase 2 | Recruiting | |
| Nelfinavir [48] | Viracept | Pancreatic, brain, cervical, head and neck, rectal, soft tissue sarcoma, and non-small cell lung cancers | Phase 1 | Unknown | ||
| Cixutumumab [49] | IMC-A12 | Monoclonal antibody targeting IGF-1R | Esophageal, soft tissue sarcoma, lung, liver, prostate, melanoma, breast, colorectal and thymoma cancers | Phase 1 | Ongoing, not recruiting | In combination with Everolimus |
| Everolimus [49,50] | Afinitor | Inhibitor of mTOR | Breast, brain, pancreatic, liver, colorectal, lung, head and neck, fallopian tube, gastric, thyroid, prostate, endometrial, renal cell, and cervical cancers | Phase 1 *, Phase 1 ** | Ongoing, not recruiting *, Ongoing, not recruiting ** | Separate trials in combination with Cixutumumab and Vatalanib |
| Vatalanib [50] | PTK787 | Inhibitor of VEGF-1 and 2, PDGFRβ and KIT | Hematologic, GIST, pancreatic, brain, colorectal, prostate, breast, melanoma, lung and mesothelioma cancers | Phase 1 | Ongoing, not recruiting | In combination with Everolimus |
| Temsirolimus [51] | Torisel | Inhibitor of mTOR | Thyroid, prostate, breast, liver, head and neck, endometrial, ovarian, neuroendocrine, gastric, cervical, pancreatic, renal, lung, colorectal, esophageal and brain cancers | Phase 1 | Ongoing, not recruiting | In combination with Vinorelbine |
| Imatinib [52] | Gleevec | Inhibitor of BCR-ABL | Hematologic, GIST, ovarian, breast, head and neck, lung, colorectal, thyroid, testicular, prostate, renal, gastric, brain, melanoma, pancreatic and sarcoma cancers | Phase 2 | Completed | No observed benefit |
5. Mutations in Tumor Suppressors: TP53 and RB1
6. Chromosomal Abnormalities
| Chromosome | Deletion/Amplification |
|---|---|
| 1 | Amplification of 1p34 in 9/23 (39%) tumor samples, contains L-Myc [67]. |
| Deletion of 1p32–33, 1p35 and 1p36 in 16/24 (73%) of MCC tumor samples, 1p36.33 contains p73 tumor suppressor [57]. | |
| Amplification of 1q11q31 in 32% of 19 primary MCC tumors analyzed, high-level amplification of 1q22q24 in 5% of samples [64]. | |
| Deletion of 1p35–36 in 7/10 (70%) of MCC samples [68]. | |
| Deletion of 1p arm in 3/3 (100%) of MCC samples [69]. | |
| 3 | 34 tumors samples from 24 patients revealed frequent loss for chromosomes 3p (46%), 5q (21%), 8p (21%), 10 (33%), 11q (17%), 13q (33%) and 17p (25%), and gains were seen for chromosomes 1 (63%), 3q (33%), 5p (38%), 8q (38%), 19 (63%), and X (41%) [70]. |
| 18/25 (69%) of tumor samples demonstrated 3p deletions ranging from 3p13–p21.1 [71]. | |
| 5 | Amplification of 5p in 32% and high-level amplification of 5p in 5% of 19 MCC samples [64]. |
| Deletion of 5q12–21 in 26% cases of 23 tumor samples [67]. | |
| 6 | Amplification of 6p in 8/19 cases (42%), most common 6pterqter [64]. |
| Trisomy in 8/17 cases (47%) [72]. | |
| Trisomy in 2/4 lymph nodes samples and 6/10 primary tumor samples [73]. | |
| Trisomy documented in a single patient case report of disease recurrence [74]. | |
| 7 | Case report document deletion of the long arm with break point at 7q31, as well as trisomy of chromosomes 6 and 11 [75]. |
| 8 | Trisomy documented in a single patient case report of disease recurrence [74]. |
| Amplification of 8q21–q22 and loss of 4p15-pter [6]. | |
| 10 | Deletion of 10q23 in 9/21 (43%) cases, containing the PTEN locus [44]. |
| 13 | Deletion of 13q13q31 (21%), 4q (16%), and 16q (11%) in 19 MCC samples [64]. |
| Deletion of 13q14–21 in 26% of 23 tumor samples [67]. | |
| Deletion of 13p in 18/24 75% cases, most commonly deleted region was mapped close to the RB1 susceptibility gene 13p14.3 [62]. | |
| 22 | Case report documenting deletion of 22p in 100% and 22q in 85% of MCC cells from a patient sample [76]. |
7. MicroRNAs
8. Negative Mutational Findings
| Negative Study | Description |
|---|---|
| p14ARF, p16INK4, H-Ras, K-Ras, N-Ras, KIT | 1/20 (5%) p16INK4 mutations, no mutations in any of the other genes; hypermethylation of p14ARF suggesting downregulation of the tumor suppressor [56]. |
| p73 and TP53 | Missense mutation in p73 of unclear significance in 15 samples. One TP53 nonsense mutation [57]. |
| PTEN | Hemizygous mutations in 9/21 MCC samples, though remaining allele unmutated. Epigenetic silencing of remaining allele is possible though yet to be shown [44]. |
| PDGFA | Expression detected in 25/31 (81%) of cases, though no activating mutations [38]. |
| c-KIT | Expressed in 8/9 (89%) of cases, though no activating mutations [87]. |
| Wnt | Elevated β-catenin in only 1/12 (8%) cases, no mutations. Similarly no mutations in APC [88]. Lill et al. 2013 found no increased expression of β-catenin or cyclin D in MCC samples [89]. |
| BRAF | No mutations in exon 15 (commonly mutated region in melanoma) in 15 samples tested [90]. No. V600E mutations, which is found in 43% of melanomas, in 46 MCC samples [91]. |
| MAPK Pathway | High expression of Raf Kinase Inhibitor Protein (RKIP), which deactivates the pathway. Expression though lack of phosphorylated activation of ERK in 42/44 (95%) cases, representing lack of activation [91]. |
| ALK | Expressed in 26/32 (81%) of MCC samples, no rearrangement or other cytogenetic abnormality of the locus identified [92]. |
| HRAS, KRAS, NRAS, BRAF, cKIT | No mutations in exons 1 and 2 of all genes studied in 6 MCC cell lines [6]. |
| RON and MSP | No mutations, though transcription of both genes in 9/14 MCC samples and no controls [93]. |
| Notch-1 | Tumor suppressor downregulated in lung and gastrointestinal neuroendocrine tumors, found to be expressed in 30/31 (97%) of MCC samples, thus unlikely to play a role in oncogenesis. Mutational status no evaluated [94]. |
| APC, BRAF, β-catenin, EGFR, FLT3, JAK2, cKIT, KRas, NRas, Notch-1, PTEN | No mutations in hotspots of these genes in 60 MCC samples [39]. |
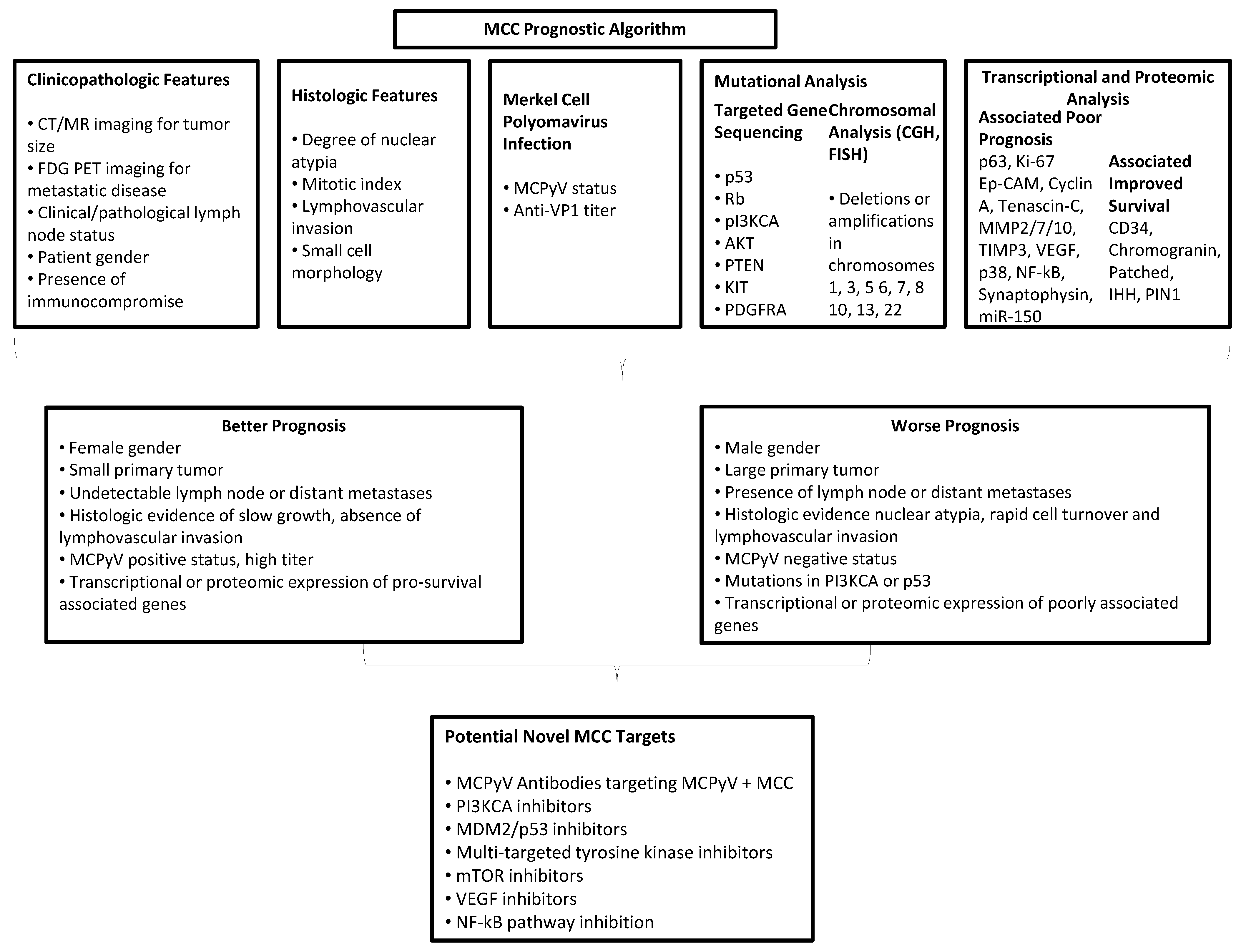
9. Molecular Prognostic Algorithm
| Expressed Marker | Association with MCC Prognosis |
|---|---|
| MCPyV | Associated with LT-Ag and RB1expression and absence of TP53 expression, and was associated with improved disease specific and overall survival (p < 0.01) on univariate analysis [95]. Polyomavirus-positive Merkel cell carcinomas showed better prognosis with one spontaneous regression case and significantly higher expression of retinoblastoma protein (p = 0.0003) and less TP53 expression (p = 0.0005) compared to MCPyV negative MCC [32]. |
| Intratumoral CD8 | Independent predictor of survival on multivariate analysis (p = 0.01) [31]. |
| Anti-LTAg | Associated with MCPyV infection, titer level correlated with disease progression. Rise in T-Ag titer preceded tumor recurrence, may have biomarker potential [96]. |
| Anti-VP1 | High anti-VP1 titers associated with improved progression free survival in MCC patients (p = 0.003) [97]. |
| p63 | p63 is expressed in more advanced disease, though its role as a prognostic tool is undetermined. In two different series, p63 expression was significantly associated with decreased survival [98,99]. However, a separate study of 95 patients found no correlation between p63 and prognosis [100]. |
| Ki-67 | Ki-67 labeling index exceeding 50% correlated with poor prognosis [101]. |
| Ep-CAM | Highly expressed in metastasizing MCC [102]. |
| Cyclin A, Tenascin-C | Associated with worse prognosis [103]. |
| Patched and IHH | Sonic Hedgehog (SHH) pathway proteins were frequently and intensely over-expressed in the MCCs in this study (Sonic hedgehog, 93%; Indian hedgehog, 84%; Patched, 86%; Smoothened, 79%; Gli-1, 79%; Gli-2, 79%; Gli-3, 86%) compared with control samples. High levels of Patched and Indian hedgehog were significantly associated with an increase in patients overall (p = 0.015) and recurrence-free survival (p = 0.011), respectively [104]. |
| MMP2/7/10, TIMP3, VEGF, P38, NF-kappaB, and Synaptophysin | Expression correlated with metastatic tumor spread [105]. |
| PIN1 | Binds and stabilizes TP53, causing cell cycle arrest and growth inhibition. Found to be expressed in all 27 MCC samples studied to varying degrees. High expression was significantly associated with improved overall survival (50% 5-years survival vs. 14%; p = 0.03) [106]. |
| miR-150 | miR-150, mi-146, miR-630, miR-483-5p, and miR-142-3p associated with worse prognosis, though only miR-150 reached statistical significance [77]. |
| CD34 and Chromogranin | Trend towards favorable prognosis [107]. |
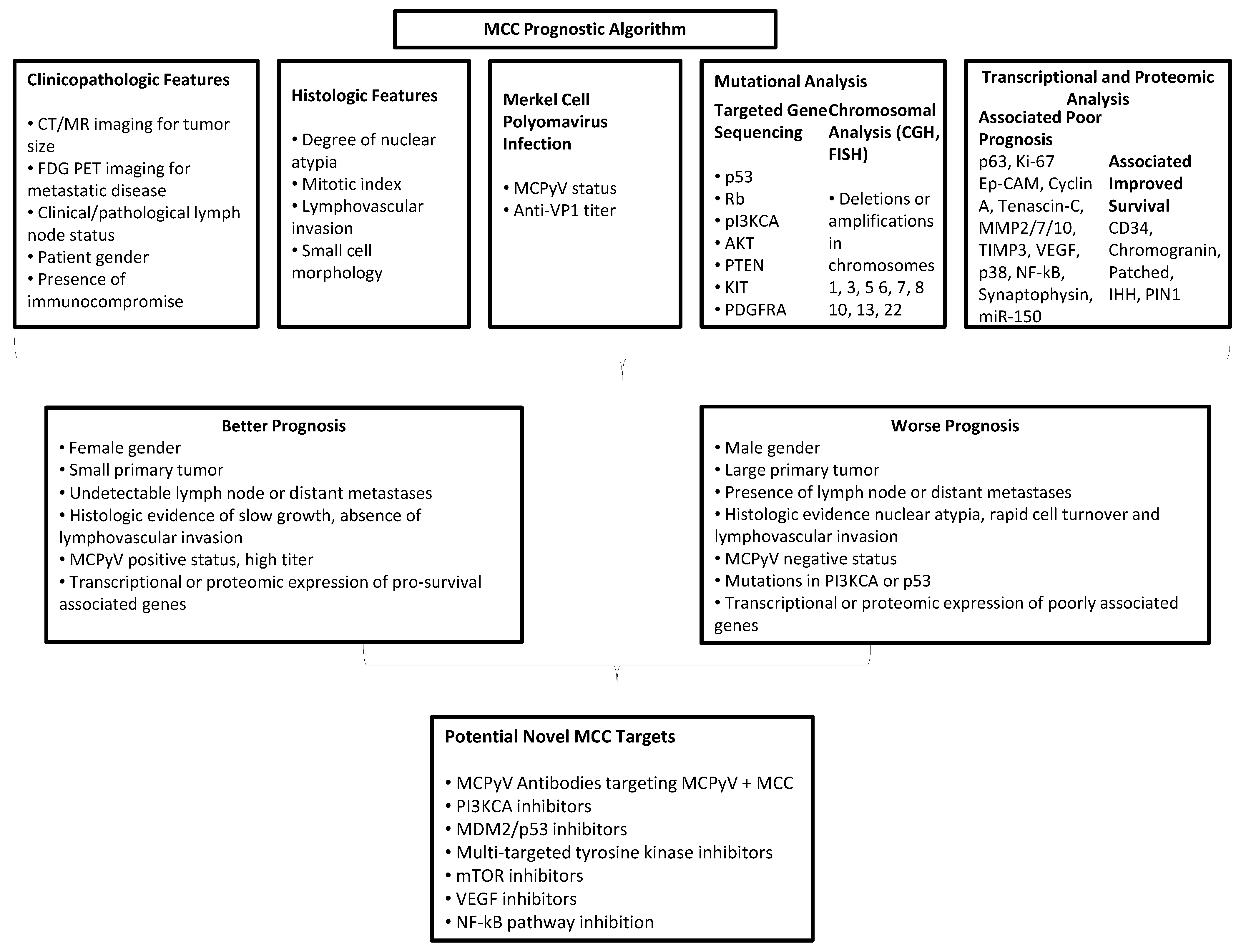
10. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Agelli, M.; Clegg, L.X.; Becker, J.C.; Rollison, D.E. The etiology and epidemiology of merkel cell carcinoma. Curr. Probl. Cancer 2010, 34, 14–37. [Google Scholar]
- Lunder, E.J.; Stern, R.S. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet a radiation. N. Engl. J. Med. 1998, 339, 1247–1248. [Google Scholar] [CrossRef]
- Howard, R.A.; Dores, G.M.; Curtis, R.E.; Anderson, W.F.; Travis, L.B. Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 1545–1549. [Google Scholar] [CrossRef]
- Laude, H.C.; Jonchere, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.F.; Dupin, N.; et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar]
- Sahi, H.; Savola, S.; Sihto, H.; Koljonen, V.; Bohling, T.; Knuutila, S. Rb1 gene in merkel cell carcinoma: Hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS 2014. [Google Scholar] [CrossRef]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. Uv-b-type mutations and chromosomal imbalances indicate common pathways for the development of merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 99, 352–360. [Google Scholar] [CrossRef]
- Albores-Saavedra, J.; Batich, K.; Chable-Montero, F.; Sagy, N.; Schwartz, A.M.; Henson, D.E. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: A population based study. J. Cutan. Pathol. 2010, 37, 20–27. [Google Scholar] [CrossRef]
- Andea, A.A.; Coit, D.G.; Amin, B.; Busam, K.J. Merkel cell carcinoma: Histologic features and prognosis. Cancer 2008, 113, 2549–2558. [Google Scholar]
- Skelton, H.G.; Smith, K.J.; Hitchcock, C.L.; McCarthy, W.F.; Lupton, G.P.; Graham, J.H. Merkel cell carcinoma: Analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J. Am. Acad. Dermatol. 1997, 37, 734–739. [Google Scholar] [CrossRef]
- Wong, H.H.; Wang, J. Merkel cell carcinoma. Arch. Pathol. Lab. Med. 2010, 134, 1711–1716. [Google Scholar]
- Hanly, A.J.; Elgart, G.W.; Jorda, M.; Smith, J.; Nadji, M. Analysis of thyroid transcription factor-1 and cytokeratin 20 separates merkel cell carcinoma from small cell carcinoma of lung. J. Cutan. Pathol. 2000, 27, 118–120. [Google Scholar]
- Tilling, T.; Moll, I. Which are the cells of origin in merkel cell carcinoma? J. Skin Cancer 2012. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Coursaget, P.; Samimi, M.; Nicol, J.T.; Gardair, C.; Touze, A. Human merkel cell polyomavirus: Virological background and clinical implications. APMIS 2013, 121, 755–769. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Penas, P.F.; Nghiem, P. Clinical characteristics of merkel cell carcinoma at diagnosis in 195 patients: The aeiou features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef]
- Tadmor, T.; Aviv, A.; Polliack, A. Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: An old bond with possible new viral ties. Ann. Oncol. 2011, 22, 250–256. [Google Scholar] [CrossRef]
- Harms, P.W.; Patel, R.M.; Verhaegen, M.E.; Giordano, T.J.; Nash, K.T.; Johnson, C.N.; Daignault, S.; Thomas, D.G.; Gudjonsson, J.E.; Elder, J.T.; et al. Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J. Investig. Dermatol. 2013, 133, 936–945. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4e-bp1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar]
- Shuda, M.; Chang, Y.; Moore, P.S. Merkel cell polyomavirus-positive merkel cell carcinoma requires viral small T-antigen for cell proliferation. J. Investig. Dermatol. 2014, 134, 1479–1481. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened merkel cell polyomavirus large T antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Brocker, E.B.; et al. An intact retinoblastoma protein-binding site in merkel cell polyomavirus large T antigen is required for promoting growth of merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [CrossRef]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T antigen locus of merkel cell polyomavirus downregulates human toll-like receptor 9 expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Santamaria-Barria, J.A.; Boland, G.M.; Yeap, B.Y.; Nardi, V.; Dias-Santagata, D.; Cusack, J.C., Jr. Merkel cell carcinoma: 30-year experience from a single institution. Ann. Surg. Oncol. 2013, 20, 1365–1373. [Google Scholar] [CrossRef]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Bohling, T.; Joensuu, H. Clinical factors associated with merkel cell polyomavirus infection in merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 938–945. [Google Scholar] [CrossRef]
- Bhatia, K.; Goedert, J.J.; Modali, R.; Preiss, L.; Ayers, L.W. Immunological detection of viral large T antigen identifies a subset of merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. Int. J. Cancer 2010, 127, 1493–1496. [Google Scholar] [CrossRef]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel cell polyomavirus-specific cd8(+) and cd4(+) T-cell responses identified in merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar]
- Paulson, K.G.; Iyer, J.G.; Tegeder, A.R.; Thibodeau, R.; Schelter, J.; Koba, S.; Schrama, D.; Simonson, W.T.; Lemos, B.D.; Byrd, D.R.; et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral cd8+ lymphocyte invasion as an independent predictor of survival. J. Clin. Oncol. 2011, 29, 1539–1546. [Google Scholar] [CrossRef]
- Higaki-Mori, H.; Kuwamoto, S.; Iwasaki, T.; Kato, M.; Murakami, I.; Nagata, K.; Sano, H.; Horie, Y.; Yoshida, Y.; Yamamoto, O.; et al. Association of merkel cell polyomavirus infection with clinicopathological differences in merkel cell carcinoma. Hum. Pathol. 2012, 43, 2282–2291. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Fernandez, A.P. The role of the immune response in merkel cell carcinoma. Cancers 2013, 5, 234–254. [Google Scholar] [CrossRef]
- Chompret, A.; Kannengiesser, C.; Barrois, M.; Terrier, P.; Dahan, P.; Tursz, T.; Lenoir, G.M.; Bressac-De Paillerets, B. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004, 126, 318–321. [Google Scholar] [CrossRef]
- Swick, B.L.; Ravdel, L.; Fitzpatrick, J.E.; Robinson, W.A. Platelet-derived growth factor receptor alpha mutational status and immunohistochemical expression in merkel cell carcinoma: Implications for treatment with imatinib mesylate. J. Cutan. Pathol. 2008, 35, 197–202. [Google Scholar]
- Andea, A.A.; Patel, R.; Ponnazhagan, S.; Kumar, S.; DeVilliers, P.; Jhala, D.; Eltoum, I.E.; Siegal, G.P. Merkel cell carcinoma: Correlation of kit expression with survival and evaluation of kit gene mutational status. Hum. Pathol. 2010, 41, 1405–1412. [Google Scholar]
- Swick, B.L.; Srikantha, R.; Messingham, K.N. Specific analysis of KIT and PDGFR-alpha expression and mutational status in merkel cell carcinoma. J. Cutan. Pathol. 2013, 40, 623–630. [Google Scholar] [CrossRef]
- Kartha, R.V.; Sundram, U.N. Silent mutations in kit and pdgfra and coexpression of receptors with SCF and PDGFA in merkel cell carcinoma: Implications for tyrosine kinase-based tumorigenesis. Mod. Pathol. 2008, 21, 96–104. [Google Scholar]
- Nardi, V.; Song, Y.; Santamaria-Barria, J.A.; Cosper, A.K.; Lam, Q.; Faber, A.C.; Boland, G.M.; Yeap, B.Y.; Bergethon, K.; Scialabba, V.L.; et al. Activation of PI3K signaling in merkel cell carcinoma. Clin. Cancer Res. 2012, 18, 1227–1236. [Google Scholar]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef]
- Lopez-Knowles, E.; Hernandez, S.; Malats, N.; Kogevinas, M.; Lloreta, J.; Carrato, A.; Tardon, A.; Serra, C.; Real, F.X. PIK3CA mutations are an early genetic alteration associated with FGFR3 mutations in superficial papillary bladder tumors. Cancer Res. 2006, 66, 7401–7404. [Google Scholar] [CrossRef]
- Hafner, C.; Hartmann, A.; Vogt, T. FGFR3 mutations in epidermal nevi and seborrheic keratoses: Lessons from urothelium and skin. J. Investig. Dermatol. 2007, 127, 1572–1573. [Google Scholar] [CrossRef]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT pathway in merkel cell carcinoma. PLoS One 2012, 7, e31255. [Google Scholar]
- Van Gele, M.; Leonard, J.H.; van Roy, N.; Cook, A.L.; de Paepe, A.; Speleman, F. Frequent allelic loss at 10q23 but low incidence of pten mutations in merkel cell carcinoma. Int. J. Cancer 2001, 92, 409–413. [Google Scholar] [CrossRef]
- Samlowski, W.E.; Moon, J.; Tuthill, R.J.; Heinrich, M.C.; Balzer-Haas, N.S.; Merl, S.A.; DeConti, R.C.; Thompson, J.A.; Witter, M.T.; Flaherty, L.E.; et al. A phase II trial of imatinib mesylate in merkel cell carcinoma (neuroendocrine carcinoma of the skin): A southwest oncology group study (s0331). Am. J. Clin. Oncol. 2010, 33, 495–499. [Google Scholar] [CrossRef]
- National Cancer Institute. Prospective randomized phase II trial of pazopanib (NSC # 737754) versus placebo in patients with progressive carcinoid tumors. Available online: http://clinicaltrials.gov/show/nct01841736 (accessed on 1 June 2014).
- Dana-farber Cancer Institute; Exelixis. Cabozantinib in recurrent/metastatic Merkel cell carcinoma. Available online: http://clinicaltrials.gov/show/nct02036476 (accessed on 1 June 2014).
- National Cancer Institute. Nelfinavir in treating patients with metastatic, refractory, or recurrent solid tumors. Available online: http://clinicaltrials.gov/show/nct00436735 (accessed on 1 June 2014).
- National Cancer Institute. Phase I study of anti-igf-1r monoclonal antibody, Imc-a12, and mtor inhibitor, everolimus, in advanced low to intermediate grade neuroendocrine carcinoma. Available online: http://clinicaltrials.gov/show/nct01204476 (accessed on 1 June 2014).
- National Cancer Institute. A Phase I trial of the mtor inhibitor Rad001 in combination with VEGF receptor tyrosine kinase inhibitor ptk787/zk 222584 in patients with advanced solid tumors. Available online: http://clinicaltrials.GOV/show/nct00655655 (accessed on 1 June 2014).
- Pfizer; University of Southern California. Phase I clinical trial of temsirolimus and vinorelbine in advanced solid tumors. Available online: http://clinicaltrials.gov/show/nct01155258 (accessed on 1 June 2014).
- National Cancer Institute. A phase II trial of sti-571/imatinib (Gleevec®) (Nsc-716051) in neuroendocrine carcinoma of the skin (Merkel Cell Carcinoma). Available online: http://clinicaltrials.gov/show/nct00068783 (accessed on 1 June 2014).
- Carson, H.J.; Lueck, N.E.; Horten, B.C. Comparison of mutant and wild-type p53 proteins in merkel cell carcinoma. Clin. Diagn. Lab. Immunol. 2000, 7, 326. [Google Scholar]
- Lill, C.; Schneider, S.; Item, C.B.; Loewe, R.; Houben, R.; Halbauer, D.; Heiduschka, G.; Brunner, M.; Thurnher, D. P53 mutation is a rare event in merkel cell carcinoma of the head and neck. Eur. Arch. Otorhinolaryngol. 2011, 268, 1639–1646. [Google Scholar] [CrossRef]
- Waltari, M.; Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Bohling, T.; Joensuu, H. Association of merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in merkel cell carcinoma. Int. J. Cancer 2011, 129, 619–628. [Google Scholar]
- Lassacher, A.; Heitzer, E.; Kerl, H.; Wolf, P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in merkel cell carcinoma. J. Investig. Dermatol. 2008, 128, 1788–1796. [Google Scholar] [CrossRef]
- Van Gele, M.; Kaghad, M.; Leonard, J.H.; van Roy, N.; Naeyaert, J.M.; Geerts, M.L.; van Belle, S.; Cocquyt, V.; Bridge, J.; Sciot, R.; et al. Mutation analysis of p73 and tp53 in merkel cell carcinoma. Br. J. Cancer 2000, 82, 823–826. [Google Scholar] [CrossRef]
- Harris, C.C.; Hollstein, M. Clinical implications of the p53 tumor-suppressor gene. N. Engl. J. Med. 1993, 329, 1318–1327. [Google Scholar]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in merkel cell carcinoma cells are independent of the merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef]
- Leonard, J.H.; Hayard, N. Loss of heterozygosity of chromosome 13 in merkel cell carcinoma. Genes Chromosomes Cancer 1997, 20, 93–97. [Google Scholar] [CrossRef]
- Cimino, P.J.; Robirds, D.H.; Tripp, S.R.; Pfeifer, J.D.; Abel, H.J.; Duncavage, E.J. Retinoblastoma gene mutations detected by whole exome sequencing of merkel cell carcinoma. Mod. Pathol. 2014, 27, 1073–1087. [Google Scholar] [CrossRef]
- Larramendy, M.L.; Koljonen, V.; Bohling, T.; Tukiainen, E.; Knuutila, S. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary merkel cell carcinomas. Mod. Pathol. 2004, 17, 561–567. [Google Scholar] [CrossRef]
- Harle, M.; Arens, N.; Moll, I.; Back, W.; Schulz, T.; Scherthan, H. Comparative genomic hybridization (cgh) discloses chromosomal and subchromosomal copy number changes in merkel cell carcinomas. J. Cutan. Pathol. 1996, 23, 391–397. [Google Scholar]
- Van Gele, M.; Leonard, J.H.; van Roy, N.; van Limbergen, H.; van Belle, S.; Cocquyt, V.; Salwen, H.; de Paepe, A.; Speleman, F. Combined karyotyping, CGH and M-FISH analysis allows detailed characterization of unidentified chromosomal rearrangements in merkel cell carcinoma. Int. J. Cancer 2002, 101, 137–145. [Google Scholar]
- Paulson, K.G.; Lemos, B.D.; Feng, B.; Jaimes, N.; Penas, P.F.; Bi, X.; Maher, E.; Cohen, L.; Leonard, J.H.; Granter, S.R.; et al. Array-CGH reveals recurrent genomic changes in merkel cell carcinoma including amplification of L-Myc. J. Investig. Dermatol. 2009, 129, 1547–1555. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Merino, M.J.; Boni, R.; Liotta, L.A.; Cavazzana, A.; Zhuang, Z. Genetic changes associated with primary merkel cell carcinoma. Am. J. Clin. Pathol. 1998, 109, 565–570. [Google Scholar]
- Harnett, P.R.; Kearsley, J.H.; Hayward, N.K.; Dracopoli, N.C.; Kefford, R.F. Loss of allelic heterozygosity on distal chromosome 1p in merkel cell carcinoma. A marker of neural crest origins? Cancer Genet. Cytogenet. 1991, 54, 109–113. [Google Scholar] [CrossRef]
- Van Gele, M.; Speleman, F.; Vandesompele, J.; van Roy, N.; Leonard, J.H. Characteristic pattern of chromosomal gains and losses in merkel cell carcinoma detected by comparative genomic hybridization. Cancer Res. 1998, 58, 1503–1508. [Google Scholar]
- Leonard, J.H.; Williams, G.; Walters, M.K.; Nancarrow, D.J.; Rabbitts, P.H. Deletion mapping of the short arm of chromosome 3 in merkel cell carcinoma. Genes Chromosomes Cancer 1996, 15, 102–107. [Google Scholar] [CrossRef]
- Gancberg, D.; Feoli, F.; Hamels, J.; de Saint-Aubain, N.; Andre, J.; Rouas, G.; Verhest, A.; Larsimont, D. Trisomy 6 in merkel cell carcinoma: A recurrent chromosomal aberration. Histopathology 2000, 37, 445–451. [Google Scholar]
- Vasuri, F.; Magrini, E.; Foschini, M.P.; Eusebi, V. Trisomy of chromosome 6 in merkel cell carcinoma within lymph nodes. Virchows Arch. 2008, 452, 559–563. [Google Scholar]
- Suciu, V.; Botan, E.; Valent, A.; Chami, L.; Spatz, A.; Vielh, P. The potential contribution of fluorescent in situ hybridization analysis to the cytopathological diagnosis of merkel cell carcinoma. Cytopathology 2008, 19, 48–51. [Google Scholar]
- Sandbrink, F.; Muller, L.; Fiebig, H.H.; Kovacs, G. Short communication: Deletion 7q, trisomy 6 and 11 in a case of merkel-cell carcinoma. Cancer Genet. Cytogenet. 1988, 33, 305–309. [Google Scholar]
- Shabtai, F.; Sternberg, A.; Klar, D.; Reiss, R.; Halbrecht, I. Involvement of chromosome 22 in a merkel cell carcinoma in a patient with a previous meningioma. Cancer Genet. Cytogenet. 1989, 38, 43–48. [Google Scholar] [CrossRef]
- Xie, H.; Lee, L.; Caramuta, S.; Hoog, A.; Browaldh, N.; Bjornhagen, V.; Larsson, C.; Lui, W.O. Microrna expression patterns related to merkel cell polyomavirus infection in human merkel cell carcinoma. J. Investig. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef]
- Boll, K.; Reiche, K.; Kasack, K.; Morbt, N.; Kretzschmar, A.K.; Tomm, J.M.; Verhaegh, G.; Schalken, J.; von Bergen, M.; Horn, F.; et al. Mir-130a, mir-203 and mir-205 jointly repress key oncogenic pathways and are downregulated in prostate carcinoma. Oncogene 2013, 32, 277–285. [Google Scholar]
- Chiang, Y.; Song, Y.; Wang, Z.; Chen, Y.; Yue, Z.; Xu, H.; Xing, C.; Liu, Z. Aberrant expression of mir-203 and its clinical significance in gastric and colorectal cancers. J. Gastrointest. Surg. 2011, 15, 63–70. [Google Scholar] [CrossRef]
- Jin, J.; Deng, J.; Wang, F.; Xia, X.; Qiu, T.; Lu, W.; Li, X.; Zhang, H.; Gu, X.; Liu, Y.; et al. The expression and function of microrna-203 in lung cancer. Tumour Biol. 2013, 34, 349–357. [Google Scholar] [CrossRef]
- Diao, Y.; Guo, X.; Jiang, L.; Wang, G.; Zhang, C.; Wan, J.; Jin, Y.; Wu, Z. Mir-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. J. Biol. Chem. 2014, 289, 529–539. [Google Scholar] [CrossRef]
- Knapp, C.F.; Sayegh, Z.; Schell, M.J.; Rawal, B.; Ochoa, T.; Sondak, V.K.; Messina, J.L. Expression of cxcr4, e-cadherin, bcl-2, and survivin in merkel cell carcinoma: An immunohistochemical study using a tissue microarray. Am. J. Dermatopathol. 2012, 34, 592–596. [Google Scholar] [CrossRef]
- Dresang, L.R.; Guastafierro, A.; Arora, R.; Normolle, D.; Chang, Y.; Moore, P.S. Response of merkel cell polyomavirus-positive merkel cell carcinoma xenografts to a survivin inhibitor. PLoS One 2013, 8, e80543. [Google Scholar]
- Ning, M.S.; Kim, A.S.; Prasad, N.; Levy, S.E.; Zhang, H.; Andl, T. Characterization of the merkel cell carcinoma mirnome. J. Skin Cancer 2014. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Edbauer, D. Good guy or bad guy: The opposing roles of microrna 125b in cancer. Cell Commun. Signal. 2014. [Google Scholar] [CrossRef]
- Segura, M.F.; Hanniford, D.; Menendez, S.; Reavie, L.; Zou, X.; Alvarez-Diaz, S.; Zakrzewski, J.; Blochin, E.; Rose, A.; Bogunovic, D.; et al. Aberrant mir-182 expression promotes melanoma metastasis by repressing foxo3 and microphthalmia-associated transcription factor. Proc. Natl. Acad. Sci. USA 2009, 106, 1814–1819. [Google Scholar] [CrossRef]
- Swick, B.L.; Ravdel, L.; Fitzpatrick, J.E.; Robinson, W.A. Merkel cell carcinoma: Evaluation of kit (cd117) expression and failure to demonstrate activating mutations in the c-kit proto-oncogene—Implications for treatment with imatinib mesylate. J. Cutan. Pathol. 2007, 34, 324–329. [Google Scholar] [CrossRef]
- Liu, S.; Daa, T.; Kashima, K.; Kondoh, Y.; Yokoyama, S. The wnt-signaling pathway is not implicated in tumorigenesis of merkel cell carcinoma. J. Cutan. Pathol. 2007, 34, 22–26. [Google Scholar]
- Lill, C.; Schneider, S.; Ghanim, B.; Brunner, M.; Heiduschka, G.; Loewe, R.; Thurnher, D. Expression of beta-catenin and cyclin d1 in merkel cell carcinomas of the head and neck. Wien. Klin. Wochenschr. 2013, 125, 501–507. [Google Scholar] [CrossRef]
- Worda, M.; Sreevidya, C.S.; Ananthaswamy, H.N.; Cerroni, L.; Kerl, H.; Wolf, P. T1796a braf mutation is absent in merkel cell carcinoma. Br. J. Dermatol. 2005, 153, 229–232. [Google Scholar] [CrossRef]
- Houben, R.; Michel, B.; Vetter-Kauczok, C.S.; Pfohler, C.; Laetsch, B.; Wolter, M.D.; Leonard, J.H.; Trefzer, U.; Ugurel, S.; Schrama, D.; et al. Absence of classical map kinase pathway signalling in merkel cell carcinoma. J. Investig. Dermatol. 2006, 126, 1135–1142. [Google Scholar] [CrossRef]
- Filtenborg-Barnkob, B.E.; Bzorek, M. Expression of anaplastic lymphoma kinase in merkel cell carcinomas. Hum. Pathol. 2013, 44, 1656–1664. [Google Scholar] [CrossRef]
- Nagahama, J.; Daa, T.; Yada, N.; Kashima, K.; Fujiwara, S.; Saikawa, T.; Yokoyama, S. Tyrosine kinase receptor ron and its ligand msp in merkel cell carcinoma. Pathol. Res. Pract. 2011, 207, 463–467. [Google Scholar] [CrossRef]
- Panelos, J.; Batistatou, A.; Paglierani, M.; Zioga, A.; Maio, V.; Santi, R.; Pimpinelli, N.; de Giorgi, V.; Santucci, M.; Massi, D.; et al. Expression of notch-1 and alteration of the e-cadherin/beta-catenin cell adhesion complex are observed in primary cutaneous neuroendocrine carcinoma (merkel cell carcinoma). Mod. Pathol. 2009, 22, 959–968. [Google Scholar]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Bohling, T.; Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in merkel cell carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar]
- Paulson, K.G.; Carter, J.J.; Johnson, L.G.; Cahill, K.W.; Iyer, J.G.; Schrama, D.; Becker, J.C.; Madeleine, M.M.; Nghiem, P.; Galloway, D.A.; et al. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer Res. 2010, 70, 8388–8397. [Google Scholar] [CrossRef]
- Touze, A.; le Bidre, E.; Laude, H.; Fleury, M.J.; Cazal, R.; Arnold, F.; Carlotti, A.; Maubec, E.; Aubin, F.; Avril, M.F.; et al. High levels of antibodies against merkel cell polyomavirus identify a subset of patients with merkel cell carcinoma with better clinical outcome. J. Clin. Oncol. 2011, 29, 1612–1619. [Google Scholar] [CrossRef]
- Asioli, S.; Righi, A.; de Biase, D.; Morandi, L.; Caliendo, V.; Picciotto, F.; Macripo, G.; Maletta, F.; di Cantogno, L.V.; Chiusa, L.; et al. Expression of p63 is the sole independent marker of aggressiveness in localised (stage I-II) merkel cell carcinomas. Mod. Pathol. 2011, 24, 1451–1461. [Google Scholar] [CrossRef]
- Stetsenko, G.Y.; Malekirad, J.; Paulson, K.G.; Iyer, J.G.; Thibodeau, R.M.; Nagase, K.; Schmidt, M.; Storer, B.E.; Argenyi, Z.B.; Nghiem, P.; et al. P63 expression in merkel cell carcinoma predicts poorer survival yet may have limited clinical utility. Am. J. Clin. Pathol. 2013, 140, 838–844. [Google Scholar] [CrossRef]
- Lim, C.S.; Whalley, D.; Haydu, L.E.; Murali, R.; Tippett, J.; Thompson, J.F.; Hruby, G.; Scolyer, R.A. Increasing tumor thickness is associated with recurrence and poorer survival in patients with merkel cell carcinoma. Ann. Surg. Oncol. 2012, 19, 3325–3334. [Google Scholar]
- Llombart, B.; Monteagudo, C.; Lopez-Guerrero, J.A.; Carda, C.; Jorda, E.; Sanmartin, O.; Almenar, S.; Molina, I.; Martin, J.M.; Llombart-Bosch, A.; et al. Clinicopathological and immunohistochemical analysis of 20 cases of merkel cell carcinoma in search of prognostic markers. Histopathology 2005, 46, 622–634. [Google Scholar]
- Kurzen, H.; Kaul, S.; Egner, U.; Deichmann, M.; Hartschuh, W. Expression of muc 1 and ep-cam in merkel cell carcinomas: Implications for immunotherapy. Arch. Dermatol. Res. 2003, 295, 146–154. [Google Scholar]
- Koljonen, V.; Jahkola, T.; Tukiainen, E.; Granroth, G.; Haglund, C.; Bohling, T. Tenascin-c in primary merkel cell carcinoma. J. Clin. Pathol. 2005, 58, 297–300. [Google Scholar] [CrossRef]
- Brunner, M.; Thurnher, D.; Pammer, J.; Heiduschka, G.; Petzelbauer, P.; Schmid, C.; Schneider, S.; Erovic, B.M. Expression of hedgehog signaling molecules in merkel cell carcinoma. Head Neck 2010, 32, 333–340. [Google Scholar]
- Fernandez-Figueras, M.T.; Puig, L.; Musulen, E.; Gilaberte, M.; Lerma, E.; Serrano, S.; Ferrandiz, C.; Ariza, A. Expression profiles associated with aggressive behavior in merkel cell carcinoma. Mod. Pathol. 2007, 20, 90–101. [Google Scholar]
- Lill, C.; Schneider, S.; Pammer, J.; Loewe, R.; Gedlicka, W.; Houben, R.; Heiduschka, G.; Brunner, M.; Thurnher, D. Significant correlation of peptidyl-prolyl isomerase overexpression in merkel cell carcinoma with overall survival of patients. Head Neck 2011, 33, 1294–1300. [Google Scholar] [CrossRef]
- Koljonen, V.; Haglund, C.; Tukiainen, E.; Bohling, T. Neuroendocrine differentiation in primary merkel cell carcinoma—Possible prognostic significance. Anticancer Res. 2005, 25, 853–858. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erstad, D.J.; Cusack, J.C., Jr. Mutational Analysis of Merkel Cell Carcinoma. Cancers 2014, 6, 2116-2136. https://doi.org/10.3390/cancers6042116
Erstad DJ, Cusack JC Jr. Mutational Analysis of Merkel Cell Carcinoma. Cancers. 2014; 6(4):2116-2136. https://doi.org/10.3390/cancers6042116
Chicago/Turabian StyleErstad, Derek J., and James C. Cusack, Jr. 2014. "Mutational Analysis of Merkel Cell Carcinoma" Cancers 6, no. 4: 2116-2136. https://doi.org/10.3390/cancers6042116
APA StyleErstad, D. J., & Cusack, J. C., Jr. (2014). Mutational Analysis of Merkel Cell Carcinoma. Cancers, 6(4), 2116-2136. https://doi.org/10.3390/cancers6042116