Desmoplasia and Chemoresistance in Pancreatic Cancer

{kind=link}
Abstract
:1. Introduction
2. Concepts of Therapy Resistance
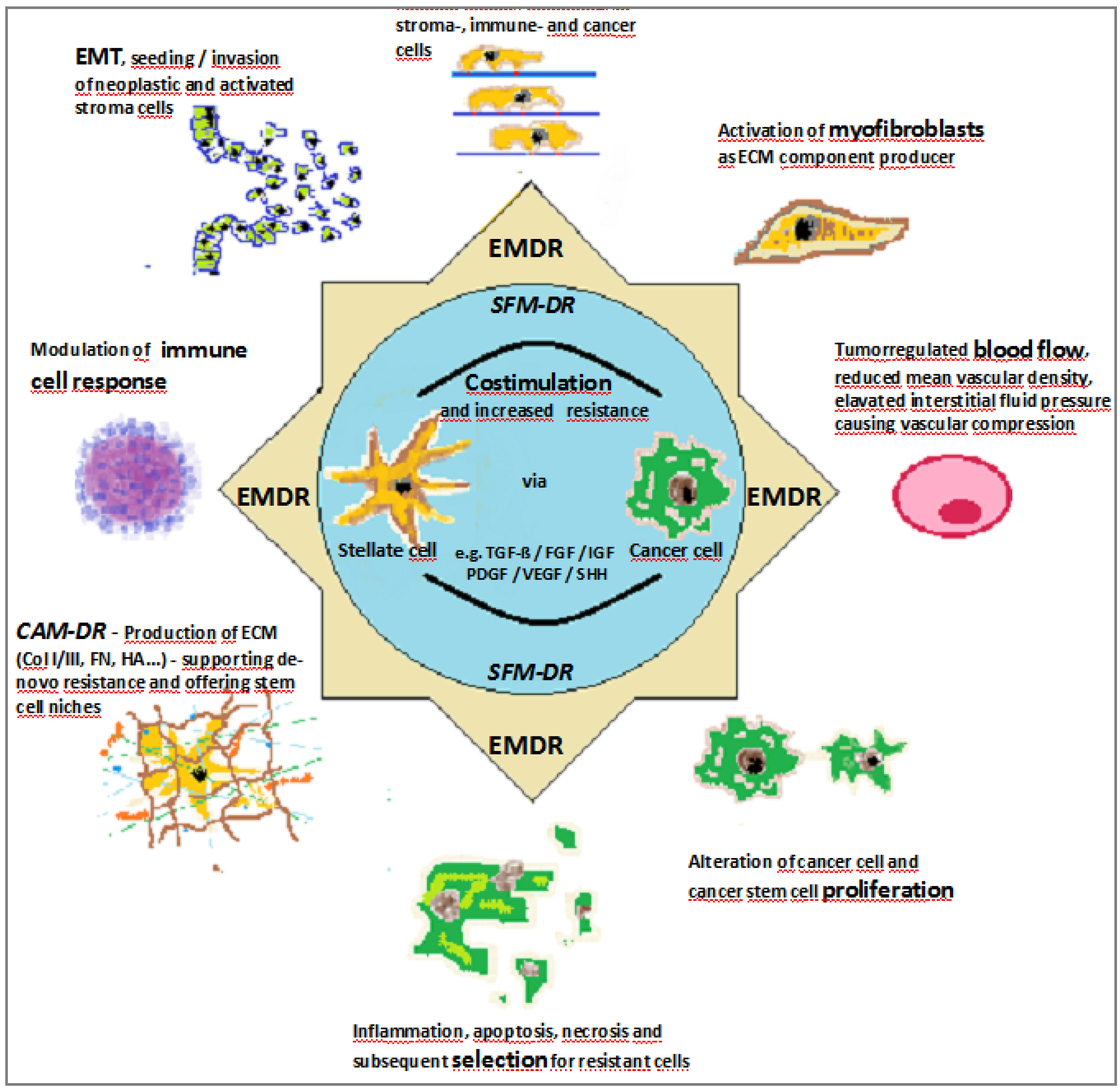
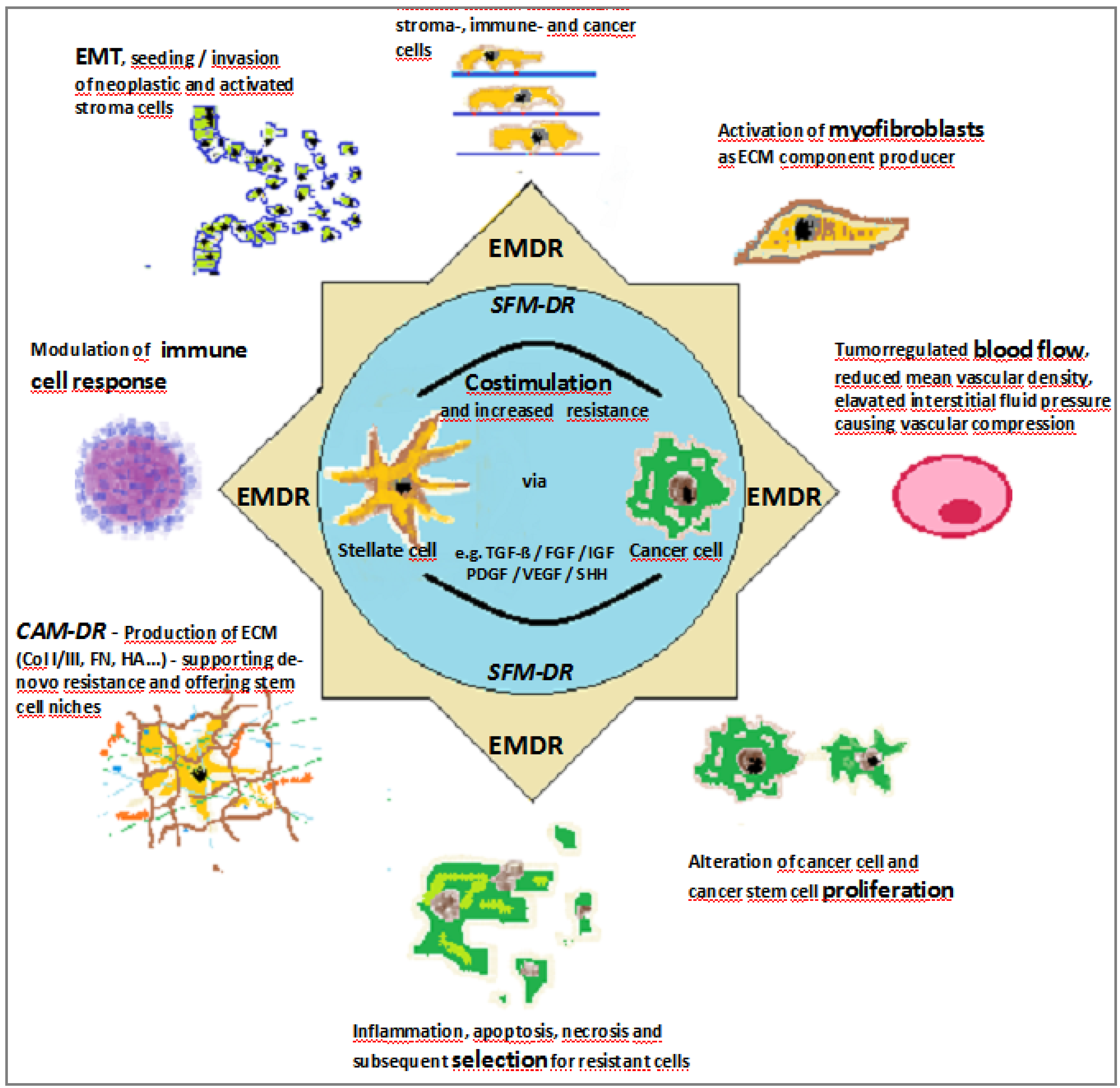
- Cancer cells or cancer stem cells (CSCs) promote resistance mechanisms
- Stroma cells promote resistance via establishment of the desmoplastic microenvironment
- Matrix components increase drug resistance
- High interstitial pressure prevents intra-tumoral drug deposition
2.1. Identifying Resistance Mechanisms of Cancer Cells or CSCs
2.2. Stromal Cells Promote Resistance via Establishment of the Desmoplastic Microenvironment
2.3. Matrix Components Increase Drug Resistance
2.4. Tumor Perfusion to Increase Drug Accumulation
3. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar]
- Kleeff, J.; Michalski, C.; Friess, H.; Buchler, M.W. Pancreatic cancer: From bench to 5-year survival. Pancreas 2006, 33, 111–118. [Google Scholar] [CrossRef]
- Löhr, M. Is it possible to survive pancreatic cancer? Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 236–237. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P. Risk factors for pancreatic cancer. J. Cell. Biochem. 2005, 95, 649–656. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Hwang, I.G.; Jang, J.S.; Oh, S.Y.; Lee, S.; Kwon, H.C.; Lee, G.W.; Go, S.; Kang, M.H.; Cha, Y.J.; Kang, J.H. A phase ii trial of erlotinib in combination with gemcitabine and cisplatin in advanced pancreatic cancer. Investig. New Drugs 2012, 30, 2371–2376. [Google Scholar] [CrossRef]
- Lim, K.H.; Chung, E.; Khan, A.; Cao, D.; Linehan, D.; Ben-Josef, E.; Wang-Gillam, A. Neoadjuvant therapy of pancreatic cancer: The emerging paradigm? Oncologist 2012, 17, 192–200. [Google Scholar] [CrossRef]
- Reni, M.; Balzano, G.; Aprile, G.; Cereda, S.; Passoni, P.; Zerbi, A.; Tronconi, M.C.; Milandri, C.; Saletti, P.; Rognone, A.; et al. Adjuvant pefg (cisplatin, epirubicin, 5-fluorouracil, gemcitabine) or gemcitabine followed by chemoradiation in pancreatic cancer: A randomized phase ii trial. Ann. Surg. Oncol. 2012, 19, 2256–2263. [Google Scholar] [CrossRef]
- Katz, M.H.; Fleming, J.B.; Lee, J.E.; Pisters, P.W. Current status of adjuvant therapy for pancreatic cancer. Oncologist 2010, 15, 1205–1213. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase iii trial of the national cancer institute of canada clinical trials group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2010, 60, 861–868. [Google Scholar] [CrossRef]
- Blaine, S.A.; Ray, K.C.; Branch, K.M.; Robinson, P.S.; Whitehead, R.H.; Means, A.L. Epidermal growth factor receptor regulates pancreatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G434–G441. [Google Scholar] [CrossRef]
- Löhr, J.M.; Haas, S.L.; Bechstein, W.O.; Bodoky, G.; Cwiertka, K.; Fischbach, W.; Folsch, U.R.; Jager, D.; Osinsky, D.; Prausova, J.; et al. Cationic liposomal paclitaxel plus gemcitabine or gemcitabine alone in patients with advanced pancreatic cancer: A randomized controlled phase II trial. Ann. Oncol. 2011, 23, 1214–1222. [Google Scholar] [CrossRef]
- Von Hoff, R.R.; Borad, M.; Laheru, D.; Smith, L.; Wood, T.; Korn, R.; Desai, N.; Iglesias, J.; Hidalgo, M. Sparc correlation with response to gemcitabine (g) plus nab-paclitaxel (nab-p) in patients with advanced metastatic pancreatic cancer: A phase I/II study. J. Clin. Oncol. 2009, 27. no. 15S 4525. [Google Scholar]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Neesse, A.; Frese, K.K.; Chan, D.S.; Bapiro, T.E.; Howat, W.J.; Richards, F.M.; Ellenrieder, V.; Jodrell, D.I.; Tuveson, D.A. Sparc independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut 2014, 63, 974–983. [Google Scholar]
- Kleeff, J.; Beckhove, P.; Esposito, I.; Herzig, S.; Huber, P.E.; Lohr, J.M.; Friess, H. Pancreatic cancer microenvironment. Int. J. Cancer 2007, 121, 699–705. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2012, 62, 112–120. [Google Scholar] [CrossRef]
- Michl, P.; Gress, T.M. Improving drug delivery to pancreatic cancer: Breaching the stromal fortress by targeting hyaluronic acid. Gut 2012, 61, 1377–1379. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Landowski, T.H.; Dalton, W.S. Role of the tumor microenvironment in mediating de novo resistance to drugs and physiological mediators of cell death. Oncogene 2003, 22, 7396–7402. [Google Scholar] [CrossRef]
- Jesenofsky, R.; Furst, D.; Ringel, J.; Chen, Y.; Schrodel, A.; Kleeff, J.; Kolb, A.; Schareck, W.D.; Lohr, M. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: Deactivation is induced by matrigel and n-acetylcysteine. Lab. Invest. 2005, 85, 1276–1291. [Google Scholar] [CrossRef]
- Löhr, J.M.; Jesenofsky, R. Pancreatic stellate cells and pancreatic carcinoma: An unholy alliance. JOP 2009, 10, 472–473. [Google Scholar]
- Borst, P.; Schinkel, A.H.; Smit, J.J.; Wagenaar, E.; Van Deemter, L.; Smith, A.J.; Eijdems, E.W.; Baas, F.; Zaman, G.J. Classical and novel forms of multidrug resistance and the physiological functions of p-glycoproteins in mammals. Pharmacol. Ther. 1993, 60, 289–299. [Google Scholar] [CrossRef]
- Burger, H.; Nooter, K.; Zaman, G.J.; Sonneveld, P.; van Wingerden, K.E.; Oostrum, R.G.; Stoter, G. Expression of the multidrug resistance-associated protein (mrp) in acute and chronic leukemias. Leukemia 1994, 8, 990–997. [Google Scholar]
- Grant, C.E.; Valdimarsson, G.; Hipfner, D.R.; Almquist, K.C.; Cole, S.P.; Deeley, R.G. Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res. 1994, 54, 357–361. [Google Scholar]
- Bell, D.R.; Gerlach, J.H.; Kartner, N.; Buick, R.N.; Ling, V. Detection of P-glycoprotein in ovarian cancer: A molecular marker associated with multidrug resistance. J. Clin. Oncol. 1985, 3, 311–315. [Google Scholar]
- Kartner, N.; Riordan, J.R.; Ling, V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983, 221, 1285–1288. [Google Scholar] [CrossRef]
- Kartner, N.; Shales, M.; Riordan, J.R.; Ling, V. Daunorubicin-resistant chinese hamster ovary cells expressing multidrug resistance and a cell-surface p-glycoprotein. Cancer Res. 1983, 43, 4413–4419. [Google Scholar]
- Ling, V.; Kartner, N.; Sudo, T.; Siminovitch, L.; Riordan, J.R. Multidrug-resistance phenotype in chinese hamster ovary cells. Cancer Treat. Rep. 1983, 67, 869–874. [Google Scholar]
- Riordan, J.R.; Deuchars, K.; Kartner, N.; Alon, N.; Trent, J.; Ling, V. Amplification of p-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 1985, 316, 817–819. [Google Scholar] [CrossRef]
- Hagmann, W.; Jesnowski, R.; Faissner, R.; Guo, C.; Lohr, J.M. Atp-binding cassette c transporters in human pancreatic carcinoma cell lines. Upregulation in 5-fluorouracil-resistant cells. Pancreatology 2009, 9, 136–144. [Google Scholar] [CrossRef]
- Nambaru, P.K.; Hubner, T.; Kock, K.; Mews, S.; Grube, M.; Payen, L.; Guitton, J.; Sendler, M.; Jedlitschky, G.; Rimmbach, C.; et al. Drug efflux transporter multidrug resistance-associated protein 5 affects sensitivity of pancreatic cancer cell lines to the nucleoside anticancer drug 5-fluorouracil. Drug Metab. Dispos. 2010, 39, 132–139. [Google Scholar] [CrossRef]
- Hagmann, W.; Jesenofsky, R.; Lohr, J.M. Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia 2010, 12, 740–747. [Google Scholar]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Hagmann, W.; Faissner, R.; Schnölzer, M.; Löhr, J.-M.; Jesnowski, R. Membrane drug transporters and chemoresistance in human pancreatic carcinoma. Cancers 2011, 3, 106–125. [Google Scholar]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Drasin, D.J.; Robin, T.P.; Ford, H.L. Breast cancer epithelial-to-mesenchymal transition: Examining the functional consequences of plasticity. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef]
- Lonardo, E.; Hermann, P.C.; Mueller, M.T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef]
- Borovski, T.; de Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef]
- Topczewska, J.M.; Postovit, L.M.; Margaryan, N.V.; Sam, A.; Hess, A.R.; Wheaton, W.W.; Nickoloff, B.J.; Topczewski, J.; Hendrix, M.J. Embryonic and tumorigenic pathways converge via nodal signaling: Role in melanoma aggressiveness. Nat. Med. 2006, 12, 925–932. [Google Scholar] [CrossRef]
- Yoshinaga, K.; Inoue, H.; Utsunomiya, T.; Sonoda, H.; Masuda, T.; Mimori, K.; Tanaka, Y.; Mori, M. N-cadherin is regulated by activin a and associated with tumor aggressiveness in esophageal carcinoma. Clin. Cancer Res. 2004, 10, 5702–5707. [Google Scholar] [CrossRef]
- Yoshinaga, K.; Yamashita, K.; Mimori, K.; Tanaka, F.; Inoue, H.; Mori, M. Activin a causes cancer cell aggressiveness in esophageal squamous cell carcinoma cells. Ann. Surg. Oncol. 2008, 15, 96–103. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef]
- Shek, F.W.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Muller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Apte, M.V.; Wilson, J.S. Dangerous liaisons: Pancreatic stellate cells and pancreatic cancer cells. J. Gastroenterol. Hepatol. 2012, 27, S69–S74. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef]
- Yoshida, S.; Yokota, T.; Ujiki, M.; Ding, X.Z.; Pelham, C.; Adrian, T.E.; Talamonti, M.S.; Bell, R.H., Jr.; Denham, W. Pancreatic cancer stimulates pancreatic stellate cell proliferation and timp-1 production through the map kinase pathway. Biochem. Biophys. Res. Commun. 2004, 323, 1241–1245. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Neesse, A.; Wagner, M.; Ellenrieder, V.; Bachem, M.; Gress, T.M.; Buchholz, M. Pancreatic stellate cells potentiate proinvasive effects of serpine2 expression in pancreatic cancer xenograft tumors. Pancreatology 2007, 7, 380–385. [Google Scholar] [CrossRef]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (cam-dr): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar]
- Hazlehurst, L.A.; Damiano, J.S.; Buyuksal, I.; Pledger, W.J.; Dalton, W.S. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (cam-dr). Oncogene 2000, 19, 4319–4327. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Enkemann, S.A.; Beam, C.A.; Argilagos, R.F.; Painter, J.; Shain, K.H.; Saporta, S.; Boulware, D.; Moscinski, L.; Alsina, M.; et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res. 2003, 63, 7900–7906. [Google Scholar]
- Hall, P.A.; Coates, P.; Lemoine, N.R.; Horton, M.A. Characterization of integrin chains in normal and neoplastic human pancreas. J. Pathol. 1991, 165, 33–41. [Google Scholar] [CrossRef]
- Kornmann, M.; Beger, H.G.; Korc, M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas 1998, 17, 169–175. [Google Scholar] [CrossRef]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar]
- Löhr, M.; Trautmann, B.; Gottler, M.; Peters, S.; Zauner, I.; Maier, A.; Kloppel, G.; Liebe, S.; Kreuser, E.D. Expression and function of receptors for extracellular matrix proteins in human ductal adenocarcinomas of the pancreas. Pancreas 1996, 12, 248–259. [Google Scholar] [CrossRef]
- Löhr, M.; Trautmann, B.; Gottler, M.; Peters, S.; Zauner, I.; Maillet, B.; Klöppel, G. Human ductal adenocarcinomas of the pancreas express extracellular matrix proteins. Br. J. Cancer 1994, 69, 144–151. [Google Scholar] [CrossRef]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef]
- Uhm, J.H.; Dooley, N.P.; Kyritsis, A.P.; Rao, J.S.; Gladson, C.L. Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin. Cancer Res. 1999, 5, 1587–1594. [Google Scholar]
- Kouniavsky, G.; Khaikin, M.; Zvibel, I.; Zippel, D.; Brill, S.; Halpern, Z.; Papa, M. Stromal extracellular matrix reduces chemotherapy-induced apoptosis in colon cancer cell lines. Clin. Exp. Metastasis 2002, 19, 55–60. [Google Scholar] [CrossRef]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar]
- Grutzmann, R.; Boriss, H.; Ammerpohl, O.; Luttges, J.; Kalthoff, H.; Schackert, H.K.; Kloppel, G.; Saeger, H.D.; Pilarsky, C. Meta-analysis of microarray data on pancreatic cancer defines a set of commonly dysregulated genes. Oncogene 2005, 24, 5079–5088. [Google Scholar] [CrossRef]
- Wibe, E. Resistance to vincristine of human cells grown as multicellular spheroids. Br. J. Cancer 1980, 42, 937–941. [Google Scholar] [CrossRef]
- Sutherland, R.M. Cell and environment interactions in tumor microregions: The multicell spheroid model. Science 1988, 240, 177–184. [Google Scholar] [CrossRef]
- Sutherland, R.M.; Eddy, H.A.; Bareham, B.; Reich, K.; Vanantwerp, D. Resistance to adriamycin in multicellular spheroids. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1225–1230. [Google Scholar] [CrossRef]
- Desoize, B.; Jardillier, J. Multicellular resistance: A paradigm for clinical resistance? Crit. Rev. Oncol. Hematol. 2000, 36, 193–207. [Google Scholar] [CrossRef]
- Olive, P.L.; Durand, R.E. Drug and radiation resistance in spheroids: Cell contact and kinetics. Cancer Metastasis Rev. 1994, 13, 121–138. [Google Scholar] [CrossRef]
- Kerr, D.J.; Wheldon, T.E.; Hydns, S.; Kaye, S.B. Cytotoxic drug penetration studies in multicellular tumour spheroids. Xenobiotica 1988, 18, 641–648. [Google Scholar]
- He, J.M.; Wang, F.C.; Qi, H.B.; Li, Y.; Liang, H.J. Down-regulation of alphav integrin by retroviral delivery of small interfering rna reduces multicellular resistance of ht29. Cancer Lett. 2009, 284, 182–188. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Wang, Z.X.; Chang, P.A.; Li, J.J.; Pan, F.; Yang, L.; Bian, Z.H.; Zou, L.; He, J.M.; Liang, H.J. Knockdown of focal adhesion kinase reverses colon carcinoma multicellular resistance. Cancer Sci. 2009, 100, 1708–1713. [Google Scholar] [CrossRef]
- Croix, B.S.; Rak, J.W.; Kapitain, S.; Sheehan, C.; Graham, C.H.; Kerbel, R.S. Reversal by hyaluronidase of adhesion-dependent multicellular drug resistance in mammary carcinoma cells. J. Natl. Cancer Inst. 1996, 88, 1285–1296. [Google Scholar] [CrossRef]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.R.; Rehnmark, S.; Verbeke, C.; Toftgard, R.; Lohr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef]
- Sofuni, A.; Iijima, H.; Moriyasu, F.; Nakayama, D.; Shimizu, M.; Nakamura, K.; Itokawa, F.; Itoi, T. Differential diagnosis of pancreatic tumors using ultrasound contrast imaging. J. Gastroenterol. 2005, 40, 518–525. [Google Scholar] [CrossRef]
- Sakamoto, H.; Kitano, M.; Suetomi, Y.; Maekawa, K.; Takeyama, Y.; Kudo, M. Utility of contrast-enhanced endoscopic ultrasonography for diagnosis of small pancreatic carcinomas. Ultrasound Med. Biol. 2008, 34, 525–532. [Google Scholar] [CrossRef]
- Komar, G.; Kauhanen, S.; Liukko, K.; Seppanen, M.; Kajander, S.; Ovaska, J.; Nuutila, P.; Minn, H. Decreased blood flow with increased metabolic activity: A novel sign of pancreatic tumor aggressiveness. Clin. Cancer Res. 2009, 15, 5511–5517. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53r172h and krasg12d cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef]
- Gerling, M.; Zhao, Y.; Nania, S.; Norberg, K.J.; Verbeke, C.S.; Englert, B.; Kuiper, R.V.; Bergström, A.; Hassan, M.; Neesse, A.; et al. Real-time assessment of tissue hypoxia in vivo with combined photoacoustics and high-frequency ultrasound. Theranostics 2014, 4, 604–613. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. Emt and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.-M. Desmoplasia and Chemoresistance in Pancreatic Cancer. Cancers 2014, 6, 2137-2154. https://doi.org/10.3390/cancers6042137
Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, Heuchel RL, Haas SL, Löhr J-M. Desmoplasia and Chemoresistance in Pancreatic Cancer. Cancers. 2014; 6(4):2137-2154. https://doi.org/10.3390/cancers6042137
Chicago/Turabian StyleSchober, Marvin, Ralf Jesenofsky, Ralf Faissner, Cornelius Weidenauer, Wolfgang Hagmann, Patrick Michl, Rainer L. Heuchel, Stephan L. Haas, and J.-Matthias Löhr. 2014. "Desmoplasia and Chemoresistance in Pancreatic Cancer" Cancers 6, no. 4: 2137-2154. https://doi.org/10.3390/cancers6042137
APA StyleSchober, M., Jesenofsky, R., Faissner, R., Weidenauer, C., Hagmann, W., Michl, P., Heuchel, R. L., Haas, S. L., & Löhr, J.-M. (2014). Desmoplasia and Chemoresistance in Pancreatic Cancer. Cancers, 6(4), 2137-2154. https://doi.org/10.3390/cancers6042137