Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tobacco
2.1. Tobacco Smoke
2.2. Secondhand Tobacco Smoke
2.3. Smokeless Tobacco
3. Tobacco-Specific Carcinogens
3.1. Carcinogens
3.2. Molecular Mechanisms of Nitrosamine-Induced Cancer
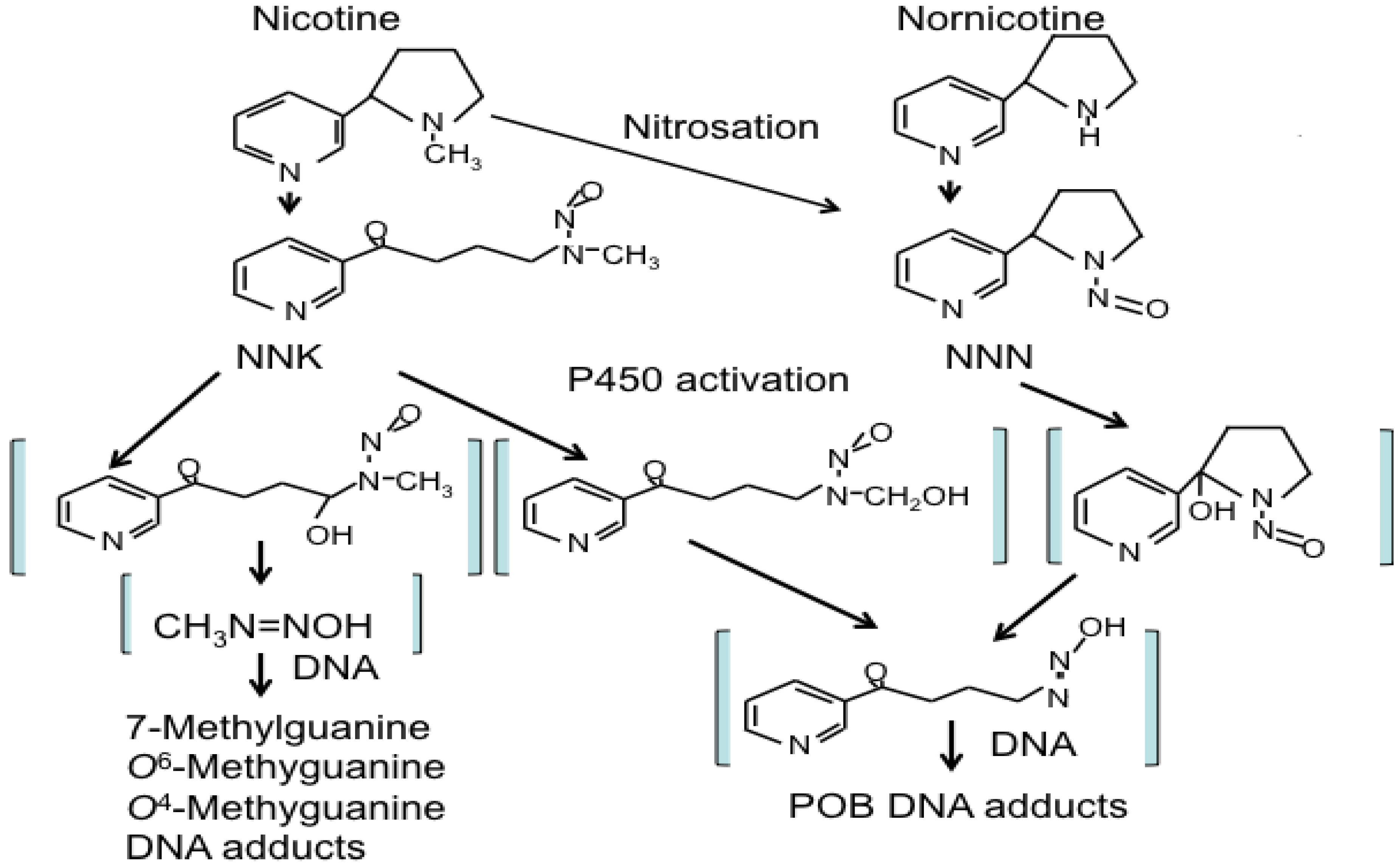
3.2.1. NNK and NNN Modulated Tumor Initiation: A Battle between DNA-Adducts Formation and Removal
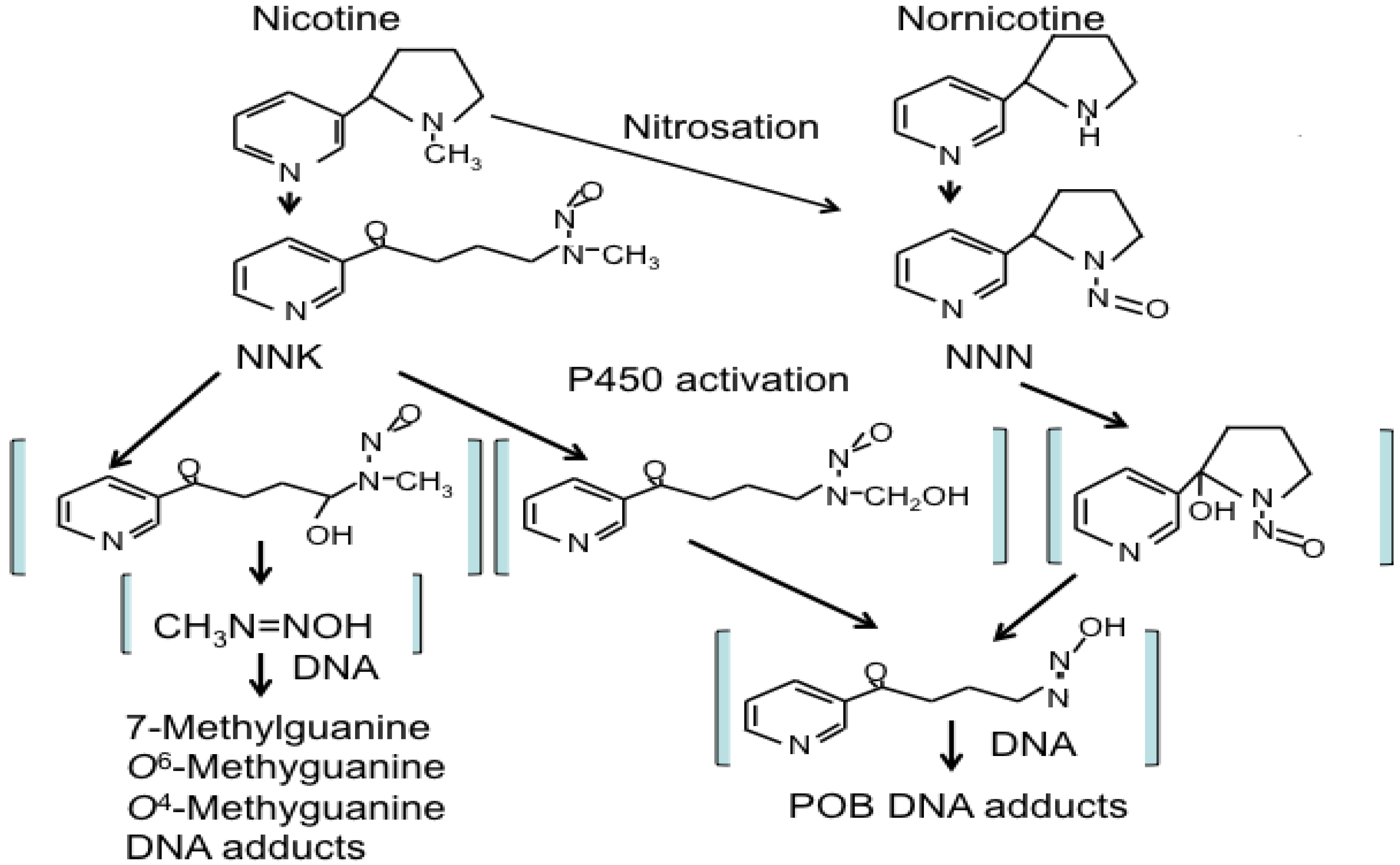
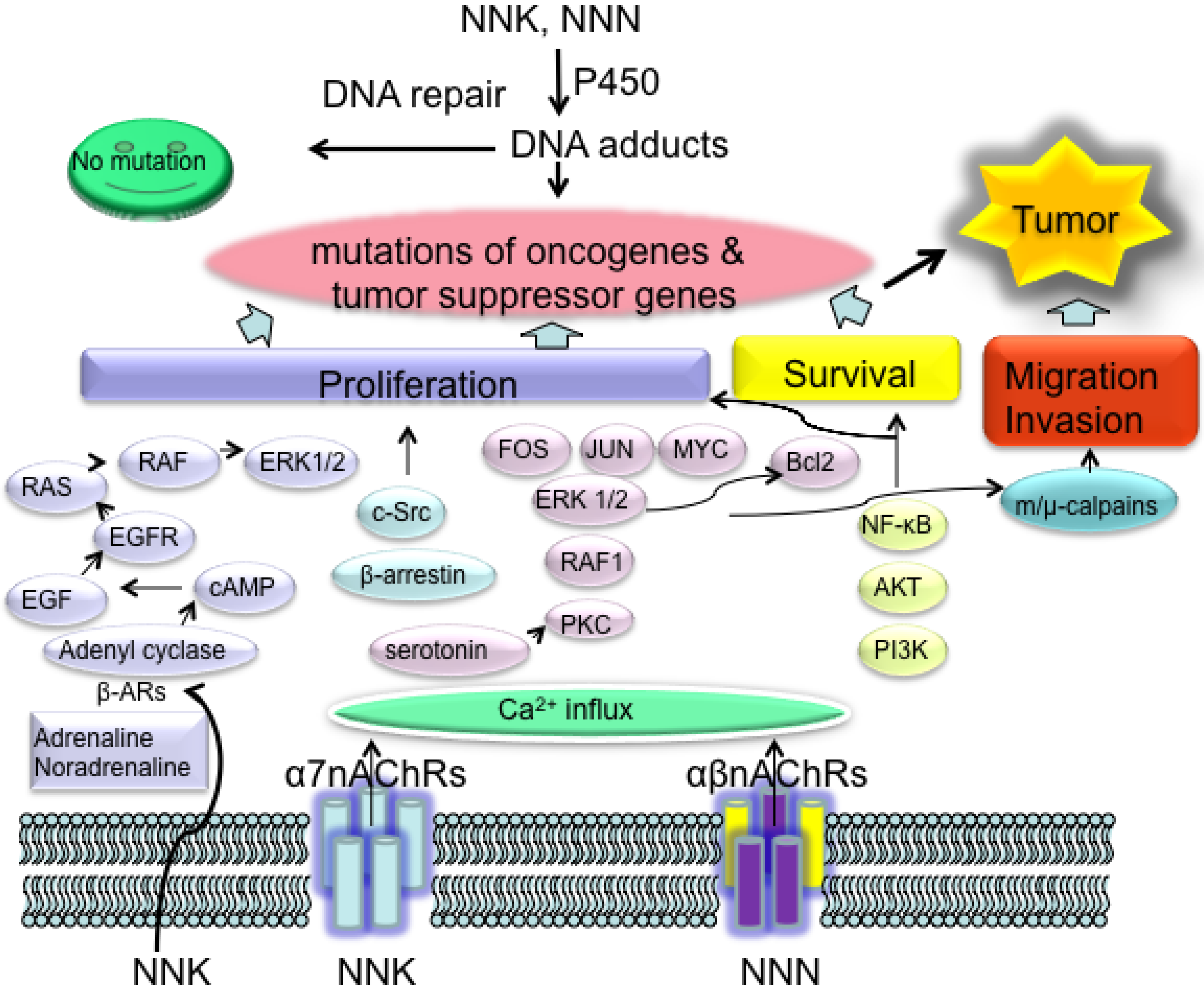
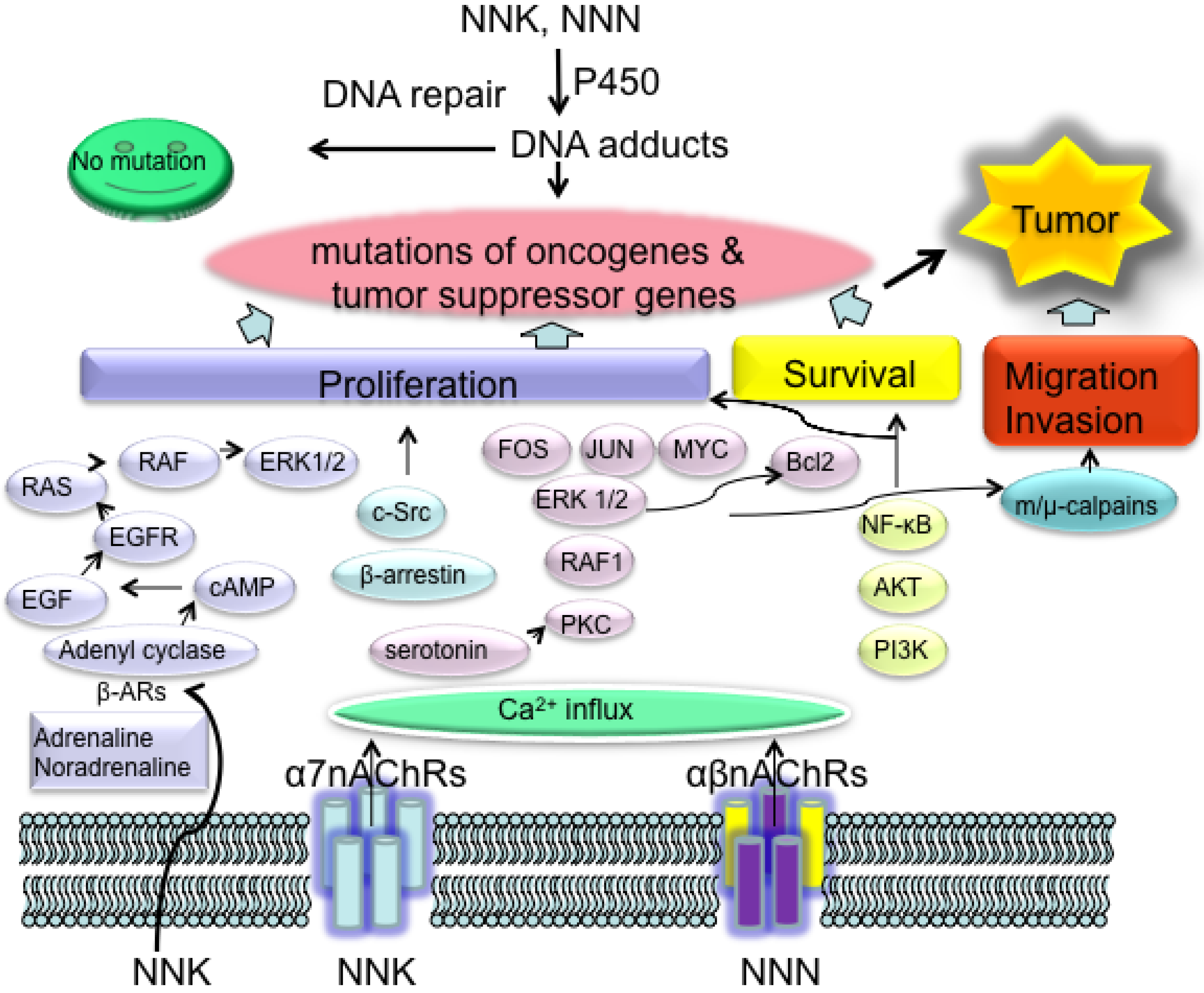
3.2.2. NNK and NNN Modulated Tumor Promotion and Progression: Creating a Microenvironment for Tumor Growth
4. Conclusions
Abbreviations
| NNK | 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone |
| NNN | N'-nitrosonornicotine |
| NNAL | 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butan-1-ol |
| PAH | polycyclic aromatic hydrocarbons |
| POB | pyridyloxobutyl |
| PHB | pyridylhydroxybutyl |
| O6mGua | O6-methylguanine |
| O4-mTh | O4-methylthymine |
| 7-mGua | 7-N-methylguanine |
| AGT | O6-alkylguanine DNA-alkyltransferase |
Acknowledgments
Conflicts of Interest
References
- Danaei, G.; Vander Hoorn, S.; Lopez, A.D.; Murray, C.J.; Ezzati, M.; Comparative Risk Assessment Collaborating Group. Causes of cancer in the world: Comparative risk assessment of nine behavioral and environmental risk factors. Lancet 2005, 366, 1784–1793. [Google Scholar] [CrossRef]
- World Health Organization. Reducing risks, promoting healthy life. In The World Health Report; WHO: Geneva, Switzerland, 2002. [Google Scholar]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef]
- Jatoi, I.; Cummings, K.M.; Cazap, E. Global tobacco problem getting worse, not better. J. Oncol. Pract. 2009, 5, 21–23. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. Preventing Tobacco Use among Youth and Young Adults: A Report of the Surgeon General; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2012. [Google Scholar]
- World Health Organization. Global Cancer Rates Could Increase by 50% to 15 Million by 2020. Available online: http://www.who.int/mediacentre/news/releases/2003/pr27/en/ (accessed on 5 January 2014).
- Hukkanen, J.; Jacob, P., 3rd; Benowitz, N.L. Metabolism and disposition kinetics of nicotine. Pharmacol. Rev. 2005, 57, 79–115. [Google Scholar] [CrossRef]
- Warren, G.W.; Singh, A.K. Nicotine and lung cancer. J. Carcinog. 2013. [Google Scholar] [CrossRef]
- Doll, R.; Peto, R. The Causes of Cancer; Oxford Press: New York, NY, USA, 1981; pp. 1–144. [Google Scholar]
- US Department of Health and Human Services. Reducing the Health Consequences of Smoking: 25 Years of Progress. A Report of the Surgeon General; Centers for Disease Control and Prevention: Atlanta, GA, USA, 1989. [Google Scholar]
- Secretan, B.; Straif, K.; Baan, R.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part E: Tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009, 10, 1033–1034. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-attributable Disease. A Report of the Surgeon General; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010. [Google Scholar]
- International Agency for Research on Cancer. Smokeless tobacco and some tobacco-specific N-nitrosamines. IARC Monogr. Eval. Carcinog. Risks Hum. 2007, 89, 419–548. [Google Scholar]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef]
- International Union Against Cancer, 2007. Available online: http://www.deathsfromsmoking.net/ (accessed on 15 November 2013).
- International Agency for Research on Cancer. Betel-quid and areca-nut chewing and some arece-nut-derived nitrosamines. IARC Monogr. Eval. Carcinog. Risks Hum. 2004, 85, 44–300. [Google Scholar]
- Steward, S.L.; Cardinez, C.J.; Richardson, L.C.; Norman, L.; Kaufmann, R.; Pechacek, T.F.; Thompson, T.D.; Weir, H.K.; Sabatino, S.A. Surveillance for cancer associated with tobacco use—United States, 1999–2004. Morbid. Mortal. Wkly. Rep. 2008, 57, 1–33. [Google Scholar]
- International Agency for Research on Cancer. Tobacco smoke and involuntary smoking. IARC Monogr. Eval. Carcinog. Risks Hum. 2004, 83, 1–1438. [Google Scholar]
- Rodgman, A.; Perfetti, T.A. Alphabetical component index. In The Chemical Components of Tobacco and Tobacco Smoke; Rodgman, A., Perfetti, T.A., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 1483–1784. [Google Scholar]
- Tutka, P.; Mosiewicz, J.; Wielosz, M. Pharmacokinetics and metabolism of nicotine. Pharmacol. Rev. 2005, 57, 143–153. [Google Scholar]
- International Agency for Research on Cancer. Tobacco habits other than smoking: Betel quid and Areca nut chewing and some related nitrosamines. IARC Monogr. Eval. Carcinog. Risks Hum. 1985, 37, 37–202. [Google Scholar]
- Zu, K.; Giovannucci, E. Smoking and aggressive prostate cancer: A review of the epidemiologic evidence. Cancer Causes Control 2009, 20, 1799–1810. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. Report on Carcinogens, 12th ed.; US Department of Health and Human Services, Public Health Service, National Toxicology Program: Research Triangle Park, NC, USA, 2011. [Google Scholar]
- US Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke. A Report of the Surgeon General; US Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, USA, 2006. [Google Scholar]
- National Cancer Institutes. Health effects of exposure to environmental tobacco smoke. In Smoking and Tobacco Control Monograph; National Cancer Institute: Bethesda, MD, USA, 1999; Volume 10. [Google Scholar]
- National Cancer Institute. Cancer Progress Report-2003 Update; National Cancer Institute, NIH, DHHS: Bethesda, MD, USA, 2004. [Google Scholar]
- National Cancer Institute. Smokeless Tobacco and Cancer. In FactSheet. Available online: http://www.cancer.gov/cancertopics/factsheet/Tobacco/smokeless/ (accessed on 22 March 2014).
- Hecht, S.S.; Carmella, S.G.; Murphy, S.E.; Riley, W.T.; Le, C.; Luo, X.; Mooney, M.; Hatsukami, D.K. Similar exposure to a tobacco-specific carcinogen in smokeless tobacco user and cigarette smokers. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1567–1567. [Google Scholar] [CrossRef]
- Henningfield, J.E.; Benowitz, N.L.; Slade, J.; Houston, T.P.; Davis, R.M.; Deitchman, S.D. Reducing the addictiveness of cigarettes. Tob. Control 1998, 7, 281–293. [Google Scholar] [CrossRef]
- Severson, H.H.; Hatsukami, D. Smokeless tobacco cessation. Prim. Care 1999, 26, 529–551. [Google Scholar] [CrossRef]
- Hoffmann, D.; Hoffmann, I.; El-Bayoumy, K. The less harmful cigarette: A controversial issue: A tribute to Ernst L. Wynder. Chem. Res. Toxicol. 2001, 14, 767–790. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef]
- Stepanov, I.; Carmella, S.G.; Briggs, A.; Hertsgaard, L.; Lindgren, B.; Hatsukami, D.; Hecht, S.S. Presence of the carcinogen N'-nitrosonornicotine in the urine of some users of oral nicotine replacement therapy products. Cancer Res. 2009, 69, 8236–8240. [Google Scholar] [CrossRef]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Hoffmann, D.; Hecht, S.S. Nicotine-derived N-nitrosamines and tobacco-related cancer: Current status and future directions. Cancer Res. 1985, 45, 935–944. [Google Scholar]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hoffmann, D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis 1988, 9, 875–884. [Google Scholar] [CrossRef]
- Hoffmann, D.; Brunnemann, K.D.; Prokopczyk, B.; Djordjevic, M.V. Tobacco-specific N-nitrosamines and Areca-derived N-nitrosamines: Chemistry, biochemistry, carcinogenicity, and relevance to humans. J. Toxicol. Environ. Health 1994, 41, 1–52. [Google Scholar] [CrossRef]
- Sturla, S.J.; Scott, J.; Lao, Y.; Hecht, S.S.; Villalta, P.W. Mass spectrometric analysis of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2005, 18, 1048–1055. [Google Scholar] [CrossRef]
- Kiyohara, C.; Yoshimasu, K.; Takayama, K.; Nakanishi, Y. EPHX1 polymorphisms and the risk of lung cancer: A HuGE review. Epidemiology 2006, 17, 89–99. [Google Scholar] [CrossRef]
- Kiyohara, C.; Yoshimasu, K.; Takayama, K.; Nakanishi, Y. NQO1, MPO, and the risk of lung cancer: A HuGE review. Genet. Med. 2005, 7, 463–478. [Google Scholar]
- Hecht, S.S.; Carmella, S.G.; Kenney, P.M.; Low, S.H.; Arakawa, K.; Yu, M.C. Effects of cruciferous vegetable consumption on urinary metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in Singapore Chinese. Cancer Epidemiol. Biomark. Prev. 2004, 13, 997–1004. [Google Scholar]
- Boyland, E.; Roe, F.J.; Gorrod, J.W. Induction of Pulmonary tumors in mice by nitrosonornicotine, a possible constituent of tobacco smoke. Nature 1964, 202, 1126. [Google Scholar] [CrossRef]
- Murphy, S.E.; Spina, D.A.; Nunes, M.G.; Pullo, D.A. Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem. Res. Toxicol. 1995, 8, 772–779. [Google Scholar] [CrossRef]
- Peterson, L.A. Formation, repair, and genotoxic properties of bulky DNA adducts formed from tobacco-specific nitrosamines. J. Nucleic. Acids 2010. [Google Scholar] [CrossRef]
- Maser, E.; Richter, E.; Friebertshauser, J. The identification of 11 beta-hydroxysteroid dehydrogenase as carbonyl reductase of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Eur. J. Biochem. 1996, 238, 484–489. [Google Scholar]
- Wong, H.L.; Murphy, S.E.; Hecht, S.S. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N'-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol. 2005, 18, 61–79. [Google Scholar] [CrossRef]
- Murphy, S.E.; Isaac, I.S.; Ding, X.; McIntee, E.J. Specificity of cytochrome P450 2A3-catalyzed alpha-hydroxylation of N′-nitrosonornicotine enantiomers. Drug Metab. Dispos. 2000, 28, 1263–1266. [Google Scholar]
- Yuan, J.M.; Knezevich, A.D.; Wang, R.; Gao, Y.T.; Hecht, S.S.; Stepanov, I. Urinary levels of the tobacco-specific carcinogen N'-nitrosonornicotine and its glucuronide are strongly associated with esophageal cancer risk in smokers. Carcinogenesis 2011, 32, 1366–1371. [Google Scholar] [CrossRef]
- Patten, C.J.; Smith, T.J.; Friesen, M.J.; Tynes, R.E.; Yang, C.S.; Murphy, S.E. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts for N'-nitrosonornicotine alpha-hydroxylation by human liver microsomes. Carcinogenesis 1997, 18, 1623–1630. [Google Scholar] [CrossRef]
- Jansen, J.G.; de Groot, A.J.; van Teijlingen, C.M.; Tates, A.D.; Vrieling, H.; van Zeeland, A.A. Induction of hprt gene mutations in splenic T-lymphocytes from the rat exposed in vivo to DNA methylating agents is correlated with formation of O6-methylguanine in bone marrow and not in the spleen. Carcinogenesis 1996, 17, 2183–2191. [Google Scholar] [CrossRef]
- Margison, G.P.; Santibanez Koref, M.F.; Povey, A.C. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis 2002, 17, 483–487. [Google Scholar] [CrossRef]
- Povey, A.C.; Badawi, A.F.; Cooper, D.P.; Hall, C.N.; Harrison, K.L.; Jackson, P.E.; Lees, N.P.; O’Connor, P.J.; Margison, G.P. DNA alkylation and repair in the large bowel: Animal and human studies. J. Nutr. 2002, 132, 3518S–3521S. [Google Scholar]
- Plosky, B.; Samson, L.; Engelward, B.P.; Gold, B.; Schlaen, B.; Millas, T.; Magnotti, M.; Schor, J.; Scicchitano, D.A. Base excision repair and nucleotide excision repair contribute to the removal of N-methylpurines from active genes. DNA Repair 2002, 1, 683–696. [Google Scholar]
- Liu, L.; Castonguay, A.; Gerson, S.L. Lack of correlation between DNA methylation and hepatocarcinogenesis in rats and hamsters treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1992, 13, 2137–2140. [Google Scholar] [CrossRef]
- Wang, L.; Spratt, T.E.; Liu, X.K.; Hecht, S.S.; Pegg, A.E.; Peterson, L.A. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1997, 10, 562–567. [Google Scholar] [CrossRef]
- Thomson, N.M.; Kenney, P.M.; Peterson, L.A. The pyridyloxobutyl DNA adduct, O6-[4-oxo-4-(3-pyridyl)butyl]guanine, is detected in tissues from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-treated A/J mice. Chem. Res. Toxicol. 2003, 16, 1–6. [Google Scholar] [CrossRef]
- Pauly, G.T.; Peterson, L.A.; Moschel, R.C. Mutagenesis by O(6)-[4-oxo-4-(3-pyridyl)butyl]guanine in Escherichia coli and human cells. Chem. Res. Toxicol. 2002, 15, 165–169. [Google Scholar] [CrossRef]
- Brown, P.J.; Bedard, L.L.; Massey, T.E. Repair of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA pyridyloxobutylation by nucleotide excision repair. Cancer Lett. 2008, 260, 48–55. [Google Scholar] [CrossRef]
- Li, L.; Perdigao, J.; Pegg, A.E.; Lao, Y.; Hecht, S.S.; Lindgren, B.R.; Reardon, J.T.; Sancar, A.; Wattenberg, E.V.; Peterson, L.A. The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2009, 22, 1464–1472. [Google Scholar] [CrossRef]
- Fan, J.; Wilson, D.M. Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic. Biol. Med. 2005, 38, 1121–1138. [Google Scholar] [CrossRef]
- Petit, C.; Sancar, A. Nucleotide excision repair: From E. coli to man. Biochimie 1999, 81, 15–25. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Thompson, L.H.; Brookman, K.W.; Dillehay, L.E.; Mooney, C.L.; Carrano, A.V. Hypersensitivity to mutation and sister-chromatid-exchange induction in CHO cell mutants defective in incising DNA containing UV lesions. Somatic Cell Genet. 1982, 8, 759–573. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.Z.; El-Zein, R.A. The 399Gln polymorphism in the DNA repair gene XRCC1 modulates the genotoxic response induced in human lymphocytes by the tobacco-specific nitrosamine NNK. Cancer Lett. 2000, 159, 63–71. [Google Scholar] [CrossRef]
- Affatato, A.A.; Wolfe, K.J.; Lopez, M.S.; Hallberg, C.; Ammenheuser, M.M.; Abdel-Rahman, S.Z. Effect of XPD/ERCC2 polymorphisms on chromosome aberration frequencies in smokers and on sensitivity to the mutagenic tobacco-specific nitrosamine NNK. Environ. Mol. Mutagen 2004, 44, 65–73. [Google Scholar] [CrossRef]
- De Marco, F. Oxidative stress and HPV carcinogenesis. Viruses 2013, 5, 708–731. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Pryor, W.A.; Stone, K.; Zang, L.Y.; Bermudez, E. Fractionation of aqueous cigarette tar extracts: Fractions that contain the tar radical cause DNA damage. Chem. Res. Toxicol. 1998, 11, 441–448. [Google Scholar] [CrossRef]
- Rosa, J.G.; Prokopczyk, B.; Desai, D.H.; Amin, S.G.; El-Bayoumy, K. Elevated 8-hydroxy-2'-deoxyguanosine levels in lung DNA of A/J mice and F344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and inhibition by dietary 1,4-phenylene- bis(methylene)selenocyanate. Carcinogenesis 1998, 19, 1783–1788. [Google Scholar] [CrossRef]
- Chung, F.L.; Xu, Y. Increased 8-oxodeoxyguanosine levels in lung DNA of A/J mice and F344 rats treated with the tobacco-specific nitrosamine 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1992, 13, 1269–1272. [Google Scholar] [CrossRef]
- Bilodeau, J.F.; Wang, M.; Chung, F.L.; Castonguay, A. Effects of nonsteroidal antiinflammatory drugs on oxidative pathways in A/J mice. Free Radic. Biol. Med. 1995, 18, 47–54. [Google Scholar] [CrossRef]
- Nishimura, S. Mammalian Ogg1/Mmh gene plays a major role in repair of the 8-hydroxyguanine lesion in DNA. Progr. Nucleic Acid Res. Mol. Biol. 2001, 68, 107–123. [Google Scholar] [CrossRef]
- Nishimura, S. Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA. Free Radic Biol. Med. 2002, 32, 813–821. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, J.H. Activation of the leukotriene B4 receptor 2-reactive oxygen species (BLT2-ROS) cascade following detachment confers anoikis resistance in prostate cancer cells. J. Biol. Chem. 2013, 288, 30054–30063. [Google Scholar] [CrossRef]
- Galzi, J.L.; Revah, F.; Bessis, A.; Changeux, J.P. Functional architecture of the nicotinic acetylcholine receptor: From electric organ to brain. Annu. Rev. Pharmacol. Toxicol. 1991, 31, 37–72. [Google Scholar] [CrossRef]
- Lindstrom, J. Neuronal nicotinic acetylcholine receptors. Ion Channels 1996, 4, 377–450. [Google Scholar] [CrossRef]
- Gotti, C.; Fornasari, D.; Clementi, F. Human neuronal nicotinic receptors. Progr. Neurobiol. 1997, 53, 199–237. [Google Scholar] [CrossRef]
- Singh, S.; Pillai, S.; Chellappan, S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J. Oncol. 2011. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Schuller, H.M.; Orloff, M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem. Pharmacol. 1998, 55, 1377–1384. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol. Ther. 2006, 5, 511–517. [Google Scholar] [CrossRef]
- Plummer, H.K.; Dhar, M.; Schuller, H.M. Expression of the alpha7 nicotinic acetylcholine receptor in human lung cells. Respir. Res. 2005, 6, 29. [Google Scholar] [CrossRef]
- Sartelet, H.; Maouche, K.; Totobenazara, J.L.; Petit, J.; Burlet, H.; Monteau, M.; Tournier, J.M.; Birembaut, P. Expression of nicotinic receptors in normal and tumoral pulmonary neuroendocrine cells (PNEC). Pathol. Res. Pract. 2008, 204, 891–898. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. Overexpression of SLURP-1 and -2 alleviates the tumorigenic action of tobacco-derived nitrosamine on immortalized oral epithelial cells. Biochem. Pharmacol. 2007, 74, 1315–1359. [Google Scholar] [CrossRef]
- Sheppard, B.J.; Williams, M.; Plummer, H.K.; Schuller, H.M. Activation of voltage-operated Ca2+-channels in human small cell lung carcinoma by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Int. J. Oncol. 2000, 16, 513–518. [Google Scholar]
- Jull, B.A.; Plummer, H.K.; Schuller, H.M. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of C-MYC in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 2001, 127, 707–717. [Google Scholar]
- Cattaneo, M.G.; Codignola, A.; Vicentini, L.M.; Clementi, F.; Sher, E. Nicotine stimulates a serotonergic autocrine loop in human small-cell lung carcinoma. Cancer Res. 1993, 53, 5566–5568. [Google Scholar]
- Jin, Z.; Gao, F.; Flagg, T.; Deng, X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and C-MYC through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 2004, 279, 40209–40219. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General; US Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, USA, 2014. [Google Scholar]
- Purdue, M.P.; Gold, L.; Jarvholm, B.; Alavanja, M.C.; Ward, M.H.; Vermeulen, R. Impaired lung function and lung cancer incidence in a cohort of Swedish construction workers. Thorax 2007, 62, 51–56. [Google Scholar]
- Mannino, D.M. Chronic obstructive pulmonary disease: Definition and epidemiology. Respir. Care 2003, 48, 1185–1191. [Google Scholar]
- Gwilt, C.R.; Donnelly, L.E.; Rogers, D.F. The non-neuronal cholinergic system in the airways: An unappreciated regulatory role in pulmonary inflammation? Pharmacol. Ther. 2007, 115, 208–222. [Google Scholar]
- Schuller, H.M. Carbon dioxide potentiates the mitogenic effects of nicotine and its carcinogenic derivative, NNK, in normal and neoplastic neuroendocrine lung cells via stimulation of autocrine and protein kinase C-dependent mitogenic pathways. Neurotoxicology 1994, 15, 877–886. [Google Scholar]
- Schuller, H.M. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007, 80, 2274–2280. [Google Scholar] [CrossRef]
- Schuller, H.M. Neurotransmission and cancer: Implications for prevention and therapy. Anticancer Drugs 2008, 19, 655–671. [Google Scholar] [CrossRef]
- Schuller, H.M.; Tithof, P.K.; Williams, M.; Plummer, H. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999, 59, 4510–4515. [Google Scholar]
- Schuller, H.M.; Porter, B.; Riechert, A. Beta-adrenergic modulation of NNK-induced lung carcinogenesis in hamsters. J. Cancer Res. Clin. Oncol. 2000, 126, 624–630. [Google Scholar] [CrossRef]
- Boswell-Smith, V.; Spina, D. PDE4 inhibitors as potential therapeutic agents in the treatment of COPD-focus on roflumilast. Int. J. Chron. Obstruct. Pulmon. Dis. 2007, 2, 121–129. [Google Scholar]
- Wang, D.; Cui, X. Evaluation of PDE4 inhibition for COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2006, 1, 373–379. [Google Scholar]
- West, K.A.; Brognard, J.; Clark, A.S.; Linnoila, I.R.; Yang, X.; Swain, S.M.; Harris, C.; Belinsky, S.; Dennis, P.A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Invest. 2003, 111, 81–90. [Google Scholar] [CrossRef]
- Tsurutani, J.; Castillo, S.S.; Brognard, J.; Granville, C.A.; Zhang, C.; Gills, J.J.; Sayyah, J.; Dennis, P.A. Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis 2005, 26, 1182–1195. [Google Scholar]
- Dasgupta, P.; Kinkade, R.; Joshi, B.; Decook, C.; Haura, E.; Chellappan, S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc. Natl. Acad. Sci. USA 2006, 103, 6332–6337. [Google Scholar] [CrossRef]
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Invest. 2006, 116, 2208–2217. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. The nicotinic receptor antagonists abolish pathobiologic effects of tobacco-derived nitrosamines on BEP2D cells. J. Cancer Res. Clin. Oncol. 2006, 32, 653–663. [Google Scholar] [CrossRef]
- Laag, E.; Majidi, M.; Cekanova, M.; Masi, T.; Takahashi, T.; Schuller, H.M. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int. J. Cancer 2006, 119, 1547–1552. [Google Scholar] [CrossRef]
- Majidi, M.; Al-Wadei, H.A.; Takahashi, T.; Schuller, H.M. Nongenomic beta estrogen receptors enhance beta1 adrenergic signaling induced by the nicotine-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in human small airway epithelial cells. Cancer Res. 2007, 67, 6863–6871. [Google Scholar]
- Lindstrom, J.; Anand, R.; Gerzanich, V.; Peng, X.; Wang, F.; Wells, G. Structure and function of neuronal nicotinic acetylcholine receptors. Progr. Brain Res. 1996, 109, 125–137. [Google Scholar] [CrossRef]
- Kawai, H.; Berg, D.K. Nicotinic acetylcholine receptors containing alpha 7 subunits on rat cortical neurons do not undergo long-lasting inactivation even when up-regulated by chronic nicotine exposure. J. Neurochem. 2001, 78, 1367–1378. [Google Scholar] [CrossRef]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 2008, 29, 1979–1985. [Google Scholar] [CrossRef]
- Nakazawa, K.; Ohno, Y. Block by phytoestrogens of recombinant human neuronal nicotinic receptors. J. Pharmacol. Sci. 2003, 93, 118–121. [Google Scholar] [CrossRef]
- Xu, L.; Deng, X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induces phosphorylation of mu- and m-calpain in association with increased secretion, cell migration, and invasion. J. Biol. Chem. 2004, 279, 53683–53690. [Google Scholar] [CrossRef]
- Sato, M.; Vaughan, M.B.; Girard, L.; Peyton, M.; Lee, W.; Shames, D.S.; Ramirez, R.D.; Sunaga, N.; Gazdar, A.F.; Shay, J.W.; et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006, 66, 2116–2128. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xue, J.; Yang, S.; Seng, S. Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN. Cancers 2014, 6, 1138-1156. https://doi.org/10.3390/cancers6021138
Xue J, Yang S, Seng S. Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN. Cancers. 2014; 6(2):1138-1156. https://doi.org/10.3390/cancers6021138
Chicago/Turabian StyleXue, Jiaping, Suping Yang, and Seyha Seng. 2014. "Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN" Cancers 6, no. 2: 1138-1156. https://doi.org/10.3390/cancers6021138
APA StyleXue, J., Yang, S., & Seng, S. (2014). Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN. Cancers, 6(2), 1138-1156. https://doi.org/10.3390/cancers6021138