Impact of Neuro-Psychological Factors on Smoking-Associated Lung Cancer

Abstract
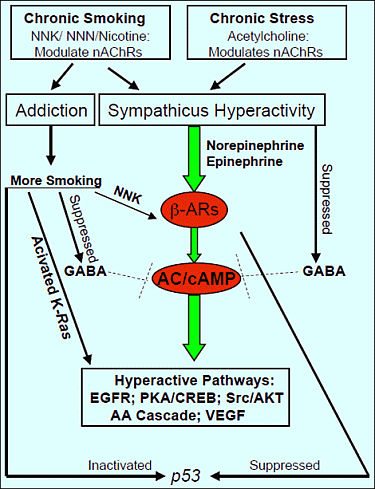
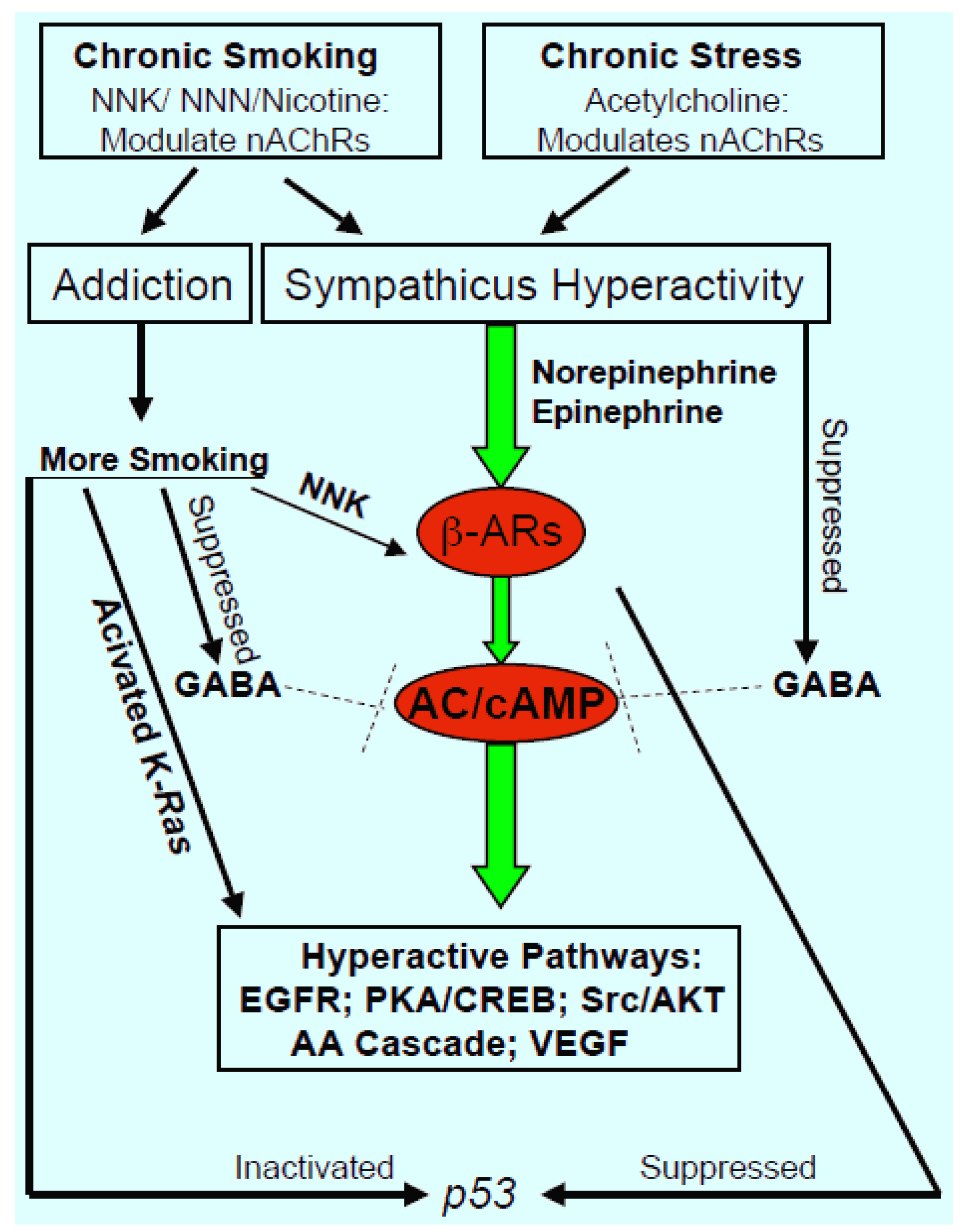
:
{kind=link}
{kind=link}
1. Introduction
2. Functions of nAChRs and ARs
3. Impact of “Nicotine Addiction” on Smoking Associated Lung Cancer
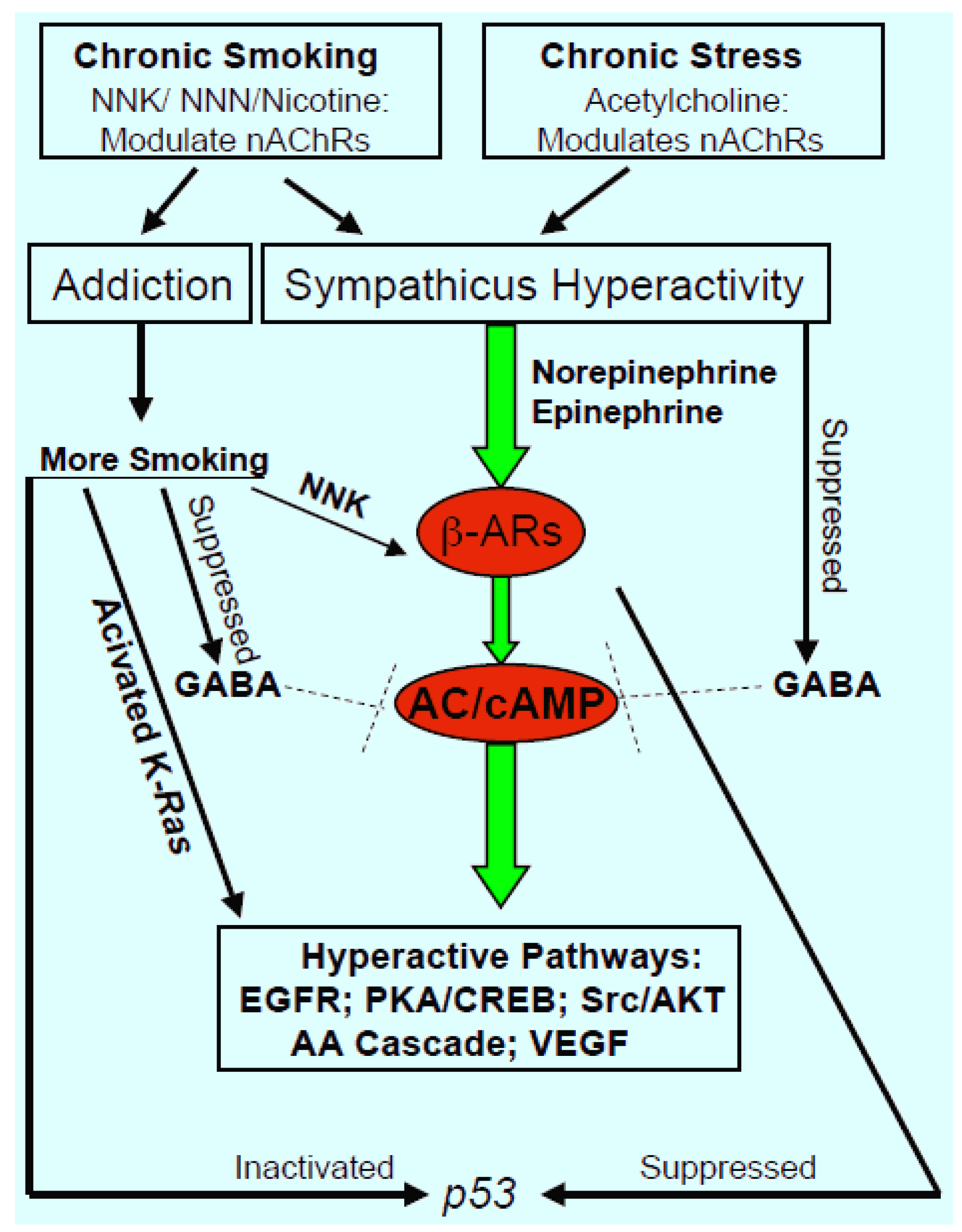
4. Impact of Chronic Psychological Stress on Lung Cancer
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Bunn, P.A., Jr.; Thatcher, N. Systemic treatment for advanced (stage IIIb/IV) non-small cell lung cancer: More treatment options; more things to consider. Conclusion. Oncologist 2008, 13, 37–46. [Google Scholar] [CrossRef]
- Holford, T.R.; Meza, R.; Warner, K.E.; Meernik, C.; Jeon, J.; Moolgavkar, S.H.; Levy, D.T. Tobacco control and the reduction in smoking-related premature deaths in the United States, 1964–2012. JAMA 2014, 311, 164–171. [Google Scholar] [CrossRef]
- Yano, T.; Haro, A.; Shikada, Y.; Maruyama, R.; Maehara, Y. Non-small cell lung cancer in never smokers as a representative “non-smoking-associated lung cancer”: Epidemiology and clinical features. Int. J. Clin. Oncol. 2011, 16, 287–293. [Google Scholar] [CrossRef]
- Devesa, S.S.; Bray, F.; Vizcaino, A.P.; Parkin, D.M. International lung cancer trends by histologic type: Male:female differences diminishing and adenocarcinoma rates rising. Int. J. Cancer 2005, 117, 294–299. [Google Scholar] [CrossRef]
- Toyoda, Y.; Nakayama, T.; Ioka, A.; Tsukuma, H. Trends in lung cancer incidence by histological type in Osaka, Japan. Jpn. J. Clin. Oncol. 2008, 38, 534–539. [Google Scholar] [CrossRef]
- Subramanian, J.; Govindan, R. Lung cancer in never smokers: A review. J. Clin. Oncol. 2007, 25, 561–570. [Google Scholar] [CrossRef]
- Flenaugh, E.L.; Henriques-Forsythe, M.N. Lung cancer disparities in African Americans: Health versus health care. Clin. Chest Med. 2006, 27, 431–439. [Google Scholar] [CrossRef]
- Hecht, S.S.; Chen, C.B.; Ohmori, T.; Hoffmann, D. Comparative carcinogenicity in F344 rats of the tobacco-specific nitrosamines, N'-nitrosonornicotine and 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1980, 40, 298–302. [Google Scholar]
- Gunning, W.T.; Castonguay, A.; Goldblatt, P.J.; Stoner, G.D. Strain A/J mouse lung adenoma growth patterns vary when induced by different carcinogens. Toxicol. Pathol. 1991, 19, 168–175. [Google Scholar] [CrossRef]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef]
- Hecht, S.S. Progress and challenges in selected areas of tobacco carcinogenesis. Chem. Res. Toxicol. 2008, 21, 160–171. [Google Scholar] [CrossRef]
- Schuller, H.M.; Orloff, M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem. Pharmacol. 1998, 55, 1377–1384. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol. Ther. 2006, 5, 511–517. [Google Scholar] [CrossRef]
- Schuller, H.M.; Tithof, P.K.; Williams, M.; Plummer, H., 3rd. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999, 59, 4510–4515. [Google Scholar]
- Lefkowitz, R.J.; Hoffman, B.B.; Taylor, P. Neurohumoral transmission: The autonomic and somatic motor nervous systems. In the Pharmacological Basis of Therapeutics; Gilman, A.G., Rall, T.W., Nies, A.S., Taylor, P., Eds.; Pergamon Press: Oxford, UK, 1990; Volume 8, pp. 84–121. [Google Scholar]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef]
- McEwen, B.S. The neurobiology of stress: From serendipity to clinical relevance. Brain Res. 2000, 886, 172–189. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Plummer, H.K., 3rd; Ullah, M.F.; Unger, B.; Brody, J.R.; Schuller, H.M. Social stress promotes and gamma-aminobutyric acid inhibits tumor growth in mouse models of non-small cell lung cancer. Cancer Prev. Res. 2012, 5, 189–196. [Google Scholar] [CrossRef]
- Schuller, H.M. Effects of tobacco constituents and psychological stress on the beta-adrenergic regulation of non-small cell lung cancer and pancreatic cancer: Implications for intervention. Cancer Biomark. 2013, 13, 133–144. [Google Scholar]
- Schuller, H.M.; Al-Wadei, H.A. Beta-adrenergic signaling in the development and progression of pulmonary and pancreatic adenocarcinoma. Curr. Cancer Ther. Rev. 2012, 8, 116–127. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Lindstrom, J.; Anand, R.; Gerzanich, V.; Peng, X.; Wang, F.; Wells, G. Structure and function of neuronal nicotinic acetylcholine receptors. Prog. Brain Res. 1996, 109, 125–137. [Google Scholar] [CrossRef]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I.; Moretti, M.; Pedrazzi, P.; Pucci, L.; Zoli, M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef]
- Barik, J.; Wonnacott, S. Indirect modulation by alpha7 nicotinic acetylcholine receptors of noradrenaline release in rat hippocampal slices: Interaction with glutamate and GABA systems and effect of nicotine withdrawal. Mol. Pharmacol. 2006, 69, 618–628. [Google Scholar] [CrossRef]
- Gotti, C.; Moretti, M.; Gaimarri, A.; Zanardi, A.; Clementi, F.; Zoli, M. Heterogeneity and complexity of native brain nicotinic receptors. Biochem. Pharmacol. 2007, 74, 1102–1111. [Google Scholar] [CrossRef]
- Markou, A. Neurobiology of nicotine dependence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3159–3168. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Codignola, A.; Vicentini, L.M.; Clementi, F.; Sher, E. Nicotine stimulates a serotonergic autocrine loop in human small-cell lung carcinoma. Cancer Res. 1993, 53, 5566–5568. [Google Scholar]
- Jull, B.A.; Plummer, H.K., 3rd; Schuller, H.M. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 2001, 127, 707–717. [Google Scholar]
- Wong, H.P.; Yu, L.; Lam, E.K.; Tai, E.K.; Wu, W.K.; Cho, C.H. Nicotine promotes cell proliferation via α7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol. Appl. Pharmacol. 2007, 221, 261–267. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Masi, T.; Schuller, H.M. Chronic exposure to estrogen and the tobacco carcinogen NNK cooperatively modulates nicotinic receptors in small airway epithelial cells. Lung Cancer 2010, 69, 33–39. [Google Scholar] [CrossRef]
- Al-Wadei, M.H.; Al-Wadei, H.A.; Schuller, H.M. Pancreatic Cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors α3, α5, and α7. Mol. Cancer Res. 2012, 10, 239–249. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Schuller, H.M. Cooperative regulation of non-small cell lung carcinoma by nicotinic and beta-adrenergic receptors: A novel target for intervention. PLoS One 2012, 7, e29915. [Google Scholar]
- Cesario, A.; Russo, P.; Nastrucci, C.; Granone, P. Is alpha7-nAChR a possible target for lung cancer and malignant pleural mesothelioma treatment? Curr. Drug Targets 2012, 13, 688–694. [Google Scholar] [CrossRef]
- Schaal, C.; Chellappan, S.P. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef]
- Al-Wadei, M.H.; Al-Wadei, H.A.; Schuller, H.M. Effects of chronic nicotine on the autocrine regulation of pancreatic cancer cells and pancreatic duct epithelial cells by stimulatory and inhibitory neurotransmitters. Carcinogenesis 2012, 33, 1745–1753. [Google Scholar] [CrossRef]
- Drell, T.L., 4th; Joseph, J.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Effects of neurotransmitters on the chemokinesis and chemotaxis of MDA-MB-468 human breast carcinoma cells. Breast Cancer Res. Treat. 2003, 80, 63–70. [Google Scholar] [CrossRef]
- Masur, K.; Niggemann, B.; Zanker, K.S.; Entschladen, F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by β-blockers. Cancer Res. 2001, 61, 2866–2869. [Google Scholar]
- Palm, D.; Lang, K.; Niggemann, B.; Drell, T.L., 4th; Masur, K.; Zaenker, K.S.; Entschladen, F. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int. J. Cancer 2006, 118, 2744–2749. [Google Scholar] [CrossRef]
- Joseph, J.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. The neurotransmitter gamma-aminobutyric acid is an inhibitory regulator for the migration of SW 480 colon carcinoma cells. Cancer Res. 2002, 62, 6467–6469. [Google Scholar]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. GABA B receptor is a novel drug target for pancreatic cancer. Cancer 2008, 112, 767–778. [Google Scholar] [CrossRef]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 2008, 29, 1979–1985. [Google Scholar] [CrossRef]
- Carmella, S.G.; Borukhova, A.; Desai, D.; Hecht, S.S. Evidence for endogenous formation of tobacco-specific nitrosamines in rats treated with tobacco alkaloids and sodium nitrite. Carcinogenesis 1997, 18, 587–592. [Google Scholar] [CrossRef]
- Prokopczyk, B.; Hoffmann, D.; Bologna, M.; Cunningham, A.J.; Trushin, N.; Akerkar, S.; Boyiri, T.; Amin, S.; Desai, D.; Colosimo, S.; et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem. Res. Toxicol. 2002, 15, 677–685. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. The superfamily of heptahelical receptors. Nat. Cell. Biol. 2000, 2, E133–E136. [Google Scholar] [CrossRef]
- Frishman, W.H. β-Adrenergic blockade in cardiovascular disease. J. Cardiovasc Pharmacol. Ther. 2013, 18, 310–319. [Google Scholar] [CrossRef]
- Cazzola, M.; Page, C.P.; Rogliani, P.; Matera, M.G. β2-Agonist therapy in lung disease. Am. J. Respir. Crit. Care Med. 2013, 187, 690–696. [Google Scholar] [CrossRef]
- Hoffman, B.B.; Lefkowitz, R.J. Catecholamines and sympathomimetic drugs. In the Pharmacological Basis of Therapeutics, 8th ed.; Gilman, A.G., Rall, T.W., Nies, A.S., Taylor, P., Eds.; Pergamon Press: New York, NY, USA, 1990; pp. 187–220. [Google Scholar]
- Schuller, H.M. The neuro-psychological axis of pancreatic cancer as a novel target for intervention. Pancreat. Disor. Ther. 2013, 3. [Google Scholar] [CrossRef]
- Grau, M.; Soley, M.; Ramirez, I. Interaction between adrenaline and epidermal growth factor in the control of liver glycogenolysis in mouse. Endocrinology 1997, 138, 2601–2609. [Google Scholar]
- Shao, J.; Lee, S.B.; Guo, H.; Evers, B.M.; Sheng, H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003, 63, 5218–5223. [Google Scholar]
- Verhoeckx, K.C.; Doornbos, R.P.; Witkamp, R.F.; van der Greef, J.; Rodenburg, R.J. β-Adrenergic receptor agonists induce the release of granulocyte chemotactic protein-2, oncostatin M, and vascular endothelial growth factor from macrophages. Int. Immunopharmacol. 2006, 6, 1–7. [Google Scholar] [CrossRef]
- Weddle, D.L.; Tithoff, P.; Williams, M.; Schuller, H.M. β-Adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 2001, 22, 473–479. [Google Scholar] [CrossRef]
- Askari, M.D.; Tsao, M.S.; Schuller, H.M. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through β-adrenergic transactivation of EGF receptors. J. Cancer Res. Clin. Oncol. 2005, 131, 639–648. [Google Scholar] [CrossRef]
- Cooper, W.A.; Lam, D.C.; O’Toole, S.A.; Minna, J.D. Molecular biology of lung cancer. J. Thorac. Dis. 2013, 5, S479–S490. [Google Scholar]
- Govind, A.P.; Vezina, P.; Green, W.N. Nicotine-induced upregulation of nicotinic receptors: Underlying mechanisms and relevance to nicotine addiction. Biochem. Pharmacol. 2009, 78, 756–765. [Google Scholar] [CrossRef]
- Rahman, S. Nicotinic receptors as therapeutic targets for drug addictive disorders. CNS Neurol. Disord. Drug Targets 2013, 12, 633–640. [Google Scholar] [CrossRef]
- Esler, M.; Kaye, D. Measurement of sympathetic nervous system activity in heart failure: The role of norepinephrine kinetics. Heart Fail. Rev. 2000, 5, 17–25. [Google Scholar]
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: A new model of Takotsubo cardiomyopathy. Circulation 2012, 126, 697–706. [Google Scholar] [CrossRef]
- Haass, M.; Kubler, W. Nicotine and sympathetic neurotransmission. Cardiovasc. Drugs Ther. 1997, 10, 657–665. [Google Scholar] [CrossRef]
- Malpas, S.C. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol. Rev. 2010, 90, 513–557. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Schuller, H.M. Chronic nicotine stimulates lung adenocarcinoma in vivo and in vitro via modulation of nicotinic acetylcholine receptor regulated excitatory and inhibitory neurotransmission characteristic of nicotine addiction. In Proceedings of the AACR 101st Annual Meeting, Washington, DC, USA, 17–21 April 2010; AACR: Washington, DC, USA, 2010. [Google Scholar]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef]
- Davis, R.; Rizwani, W.; Banerjee, S.; Kovacs, M.; Haura, E.; Coppola, D.; Chellappan, S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One 2009, 4, e7524. [Google Scholar]
- Momi, N.; Ponnusamy, M.P.; Kaur, S.; Rachagani, S.; Kunigal, S.S.; Chellappan, S.; Ouellette, M.M.; Batra, S.K. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through α7nAChR-mediated MUC4 upregulation. Oncogene 2013, 32, 1384–1395. [Google Scholar] [CrossRef]
- Stepanov, I.; Carmella, S.G.; Han, S.; Pinto, A.; Strasser, A.A.; Lerman, C.; Hecht, S.S. Evidence for endogenous formation of N'-nitrosonornicotine in some long-term nicotine patch users. Nicotine Tob. Res. 2009, 11, 99–105. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Plummer, H.K., 3rd; Schuller, H.M. Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter gamma-aminobutyric acid. Carcinogenesis 2009, 30, 506–511. [Google Scholar] [CrossRef]
- Banerjee, J.; Al-Wadei, H.A.; Schuller, H.M. Chronic nicotine inhibits the therapeutic effects of gemcitabine on pancreatic cancer in vitro and in mouse xenografts. Eur. J. Cancer 2013, 49, 1152–1158. [Google Scholar] [CrossRef]
- Schuller, H.M. Cell type specific, receptor-mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem. Pharmacol. 1989, 38, 3439–3442. [Google Scholar] [CrossRef]
- Maneckjee, R.; Minna, J.D. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc. Natl. Acad. Sci. USA 1990, 87, 3294–3298. [Google Scholar] [CrossRef]
- Murphy, S.E.; von Weymarn, L.B.; Schutten, M.M.; Kassie, F.; Modiano, J.F. Chronic nicotine consumption does not influence 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis. Cancer Prev. Res. 2011, 4, 1752–1760. [Google Scholar] [CrossRef]
- Maier, C.R.; Hollander, M.C.; Hobbs, E.A.; Dogan, I.; Linnoila, R.I.; Dennis, P.A. Nicotine does not enhance tumorigenesis in mutant K-ras-driven mouse models of lung cancer. Cancer Prev. Res. 2011, 4, 1743–1751. [Google Scholar] [CrossRef]
- Long, D. Smoking as a coping strategy. Nurse Times 2003, 99, 50–53. [Google Scholar]
- Yong, H.H.; Borland, R. Functional beliefs about smoking and quitting activity among adult smokers in four countries: Findings from the International tobacco control four-country survey. Health Psychol. 2008, 27, S216–S223. [Google Scholar] [CrossRef]
- Lawrence, D.; Considine, J.; Mitrou, F.; Zubrick, S.R. Anxiety disorders and cigarette smoking: Results from the Australian survey of mental health and wellbeing. Aust. New Z. J. Psychiatry 2010, 44, 520–527. [Google Scholar]
- Fu, S.S.; McFall, M.; Saxon, A.J.; Beckham, J.C.; Carmody, T.P.; Baker, D.G.; Joseph, A.M. Post-traumatic stress disorder and smoking: A systematic review. Nicotine Tob. Res. 2007, 9, 1071–1084. [Google Scholar] [CrossRef]
- Mozayan, M.; Lee, T.J. Statins prevent cholinesterase inhibitor blockade of sympathetic α7-nAChR-mediated currents in rat superior cervical ganglion neurons. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1737–H1744. [Google Scholar] [CrossRef]
- Di Angelantonio, S.; Matteoni, C.; Fabbretti, E.; Nistri, A. Molecular biology and electrophysiology of neuronal nicotinic receptors of rat chromaffin cells. Eur. J. Neurosci. 2003, 17, 2313–2322. [Google Scholar] [CrossRef]
- Epperson, C.N.; O’Malley, S.; Czarkowski, K.A.; Gueorguieva, R.; Jatlow, P.; Sanacora, G.; Rothman, D.L.; Krystal, J.H.; Mason, G.F. Sex, GABA, and nicotine: The impact of smoking on cortical GABA levels across the menstrual cycle as measured with proton magnetic resonance spectroscopy. Biol. Psychiatry 2005, 57, 44–48. [Google Scholar] [CrossRef]
- D’Souza, M.S.; Markou, A. The “stop” and “go” of nicotine dependence: Role of GABA and glutamate. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, M.; Czeh, B.; Flugge, G.; Zhang, W. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 2010, 35, 1693–1707. [Google Scholar]
- Antoni, M.H.; Lutgendorf, S.K.; Cole, S.W.; Dhabhar, F.S.; Sephton, S.E.; McDonald, P.G.; Stefanek, M.; Sood, A.K. The influence of bio-behavioural factors on tumour biology: Pathways and mechanisms. Nat. Rev. Cancer 2006, 6, 240–248. [Google Scholar] [CrossRef]
- Park, P.G.; Merryman, J.; Orloff, M.; Schuller, H.M. β-Adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: Implications for individuals with preexisting chronic lung disease. Cancer Res. 1995, 55, 3504–3508. [Google Scholar]
- Laag, E.; Majidi, M.; Cekanova, M.; Masi, T.; Takahashi, T.; Schuller, H.M. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int. J. Cancer 2006, 119, 1547–1552. [Google Scholar] [CrossRef]
- Schuller, H.M.; Porter, B.; Riechert, A. Beta-adrenergic modulation of NNK-induced lung carcinogenesis in hamsters. J. Cancer Res. Clin. Oncol. 2000, 126, 624–630. [Google Scholar] [CrossRef]
- Schuller, H.M.; Al-Wadei, H.A.; Ullah, M.F.; Plummer, H.K., 3rd. Regulation of pancreatic cancer by neuropsychological stress responses: A novel target for intervention. Carcinogenesis 2012, 33, 191–196. [Google Scholar] [CrossRef]
- Sood, A.K.; Bhatty, R.; Kamat, A.A.; Landen, C.N.; Han, L.; Thaker, P.H.; Li, Y.; Gershenson, D.M.; Lutgendorf, S.; Cole, S.W. Stress hormone-mediated invasion of ovarian cancer cells. Clin. Cancer Res. 2006, 12, 369–375. [Google Scholar] [CrossRef]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A stress response pathway regulates DNA damage through β2-adrenoreceptors and beta-arrestin-1. Nature 2011, 477, 349–353. [Google Scholar] [CrossRef]
- Hara, M.R.; Sachs, B.D.; Caron, M.G.; Lefkowitz, R.J. Pharmacological blockade of a β(2)AR-β-arrestin-1 signaling cascade prevents the accumulation of DNA damage in a behavioral stress model. Cell Cycle 2013, 12, 219–224. [Google Scholar] [CrossRef]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O’Reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of β-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef]
- Melhem-Bertrandt, A.; Chavez-Macgregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef]
- Hamer, M.; Chida, Y.; Molloy, G.J. Psychological distress and cancer mortality. J. Psychosom. Res. 2009, 66, 255–258. [Google Scholar] [CrossRef]
- Brennan, P.; Crispo, A.; Zaridze, D.; Szeszenia-Dabrowska, N.; Rudnai, P.; Lissowska, J.; Fabianova, E.; Mates, D.; Bencko, V.; Foretova, L.; et al. High cumulative risk of lung cancer death among smokers and nonsmokers in central and eastern Europe. Am. J. Epidemiol. 2006, 164, 1233–1241. [Google Scholar] [CrossRef]
- Majidi, M.; Al-Wadei, H.A.; Takahashi, T.; Schuller, H.M. Nongenomic beta estrogen receptors enhance beta1 adrenergic signaling induced by the nicotine-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in human small airway epithelial cells. Cancer Res. 2007, 67, 6863–6871. [Google Scholar]
- Celli, B.R. Chronic obstructive pulmonary disease and lung cancer: Common pathogenesis, shared clinical challenges. Proc. Am. Thorac. Soc. 2012, 9, 74–79. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schuller, H.M. Impact of Neuro-Psychological Factors on Smoking-Associated Lung Cancer. Cancers 2014, 6, 580-594. https://doi.org/10.3390/cancers6010580
Schuller HM. Impact of Neuro-Psychological Factors on Smoking-Associated Lung Cancer. Cancers. 2014; 6(1):580-594. https://doi.org/10.3390/cancers6010580
Chicago/Turabian StyleSchuller, Hildegard M. 2014. "Impact of Neuro-Psychological Factors on Smoking-Associated Lung Cancer" Cancers 6, no. 1: 580-594. https://doi.org/10.3390/cancers6010580
APA StyleSchuller, H. M. (2014). Impact of Neuro-Psychological Factors on Smoking-Associated Lung Cancer. Cancers, 6(1), 580-594. https://doi.org/10.3390/cancers6010580