
Inhibition of Mitochondrial-Associated Protein MAGMAS Resensitizes Chemoresistant Prostate Cancer Cells to Docetaxel

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Chemicals
2.2. Antibodies
2.3. Immunoblotting
2.4. Quantitative Real-Time PCR
2.5. MTT Viability Assay and Determination of IC50 Values
2.6. RNA Interference
2.7. Clonogenic Assays
2.8. Kaplan–Meier Curve Data
3. Results
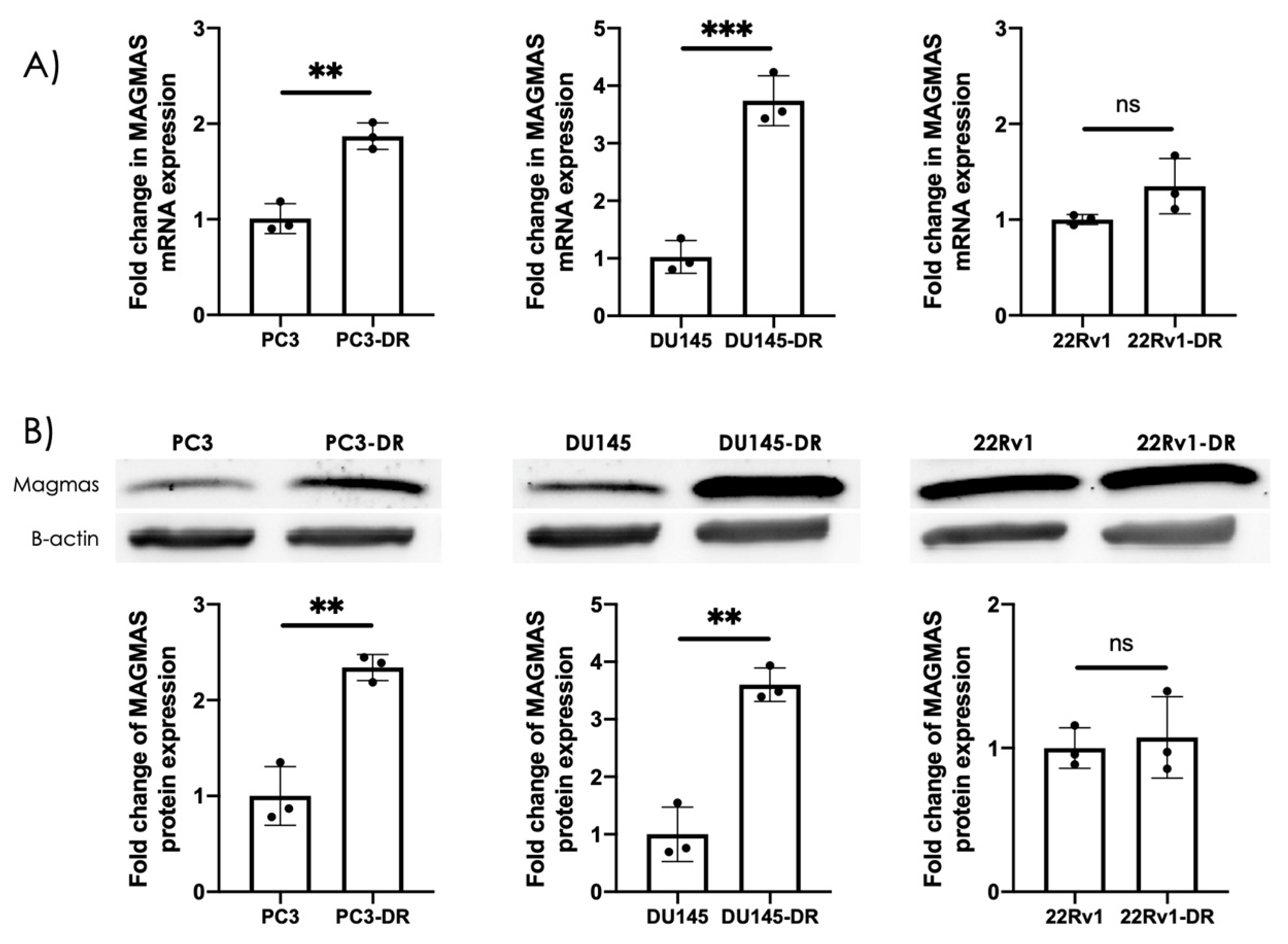
3.1. MAGMAS Is Endogenously Overexpressed in DTX-Resistant PCa Cells
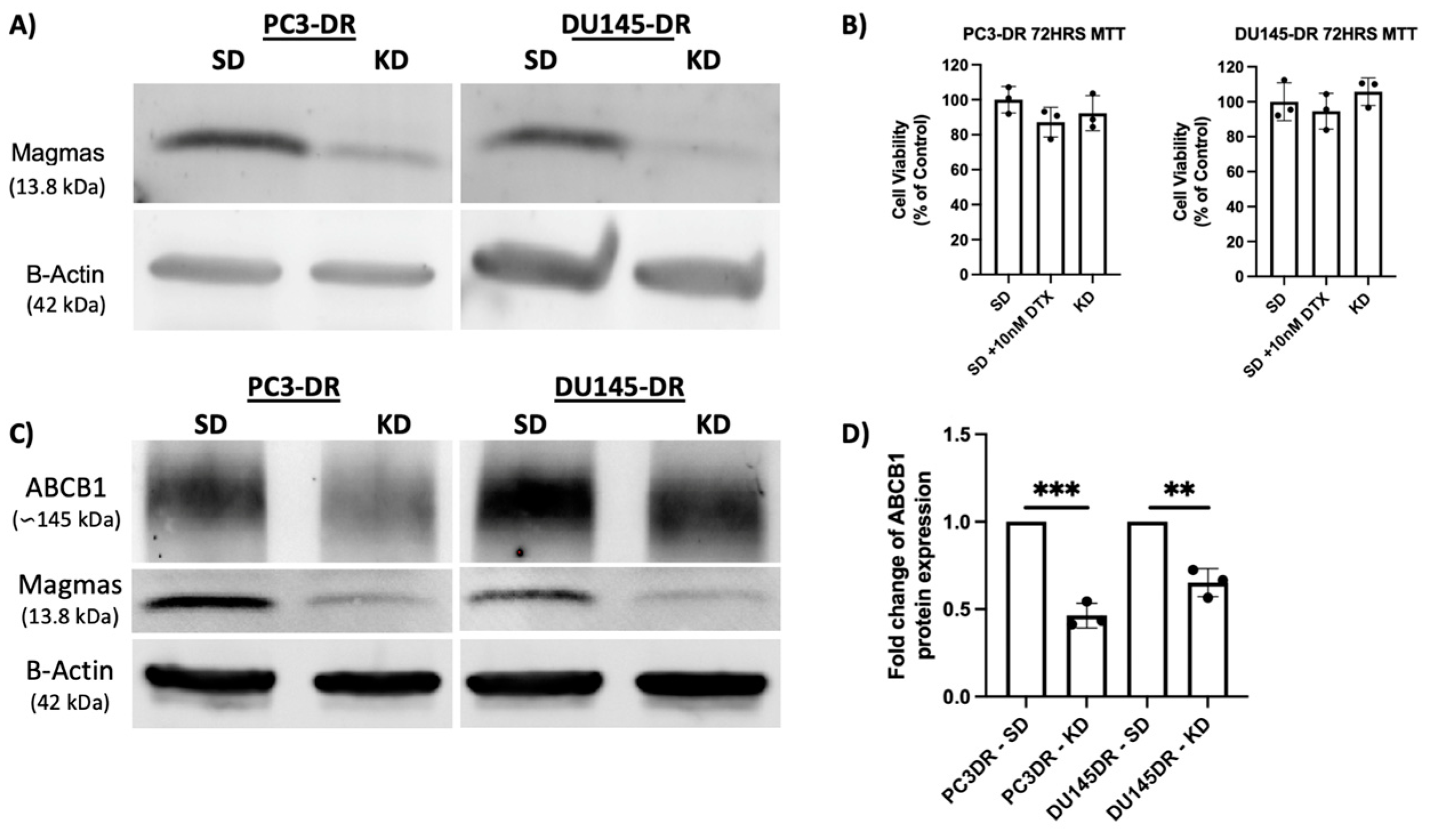
3.2. MAGMAS Depletion Leads to Decreased ABCB1 Abundance in DTX-Resistant PCa Cells
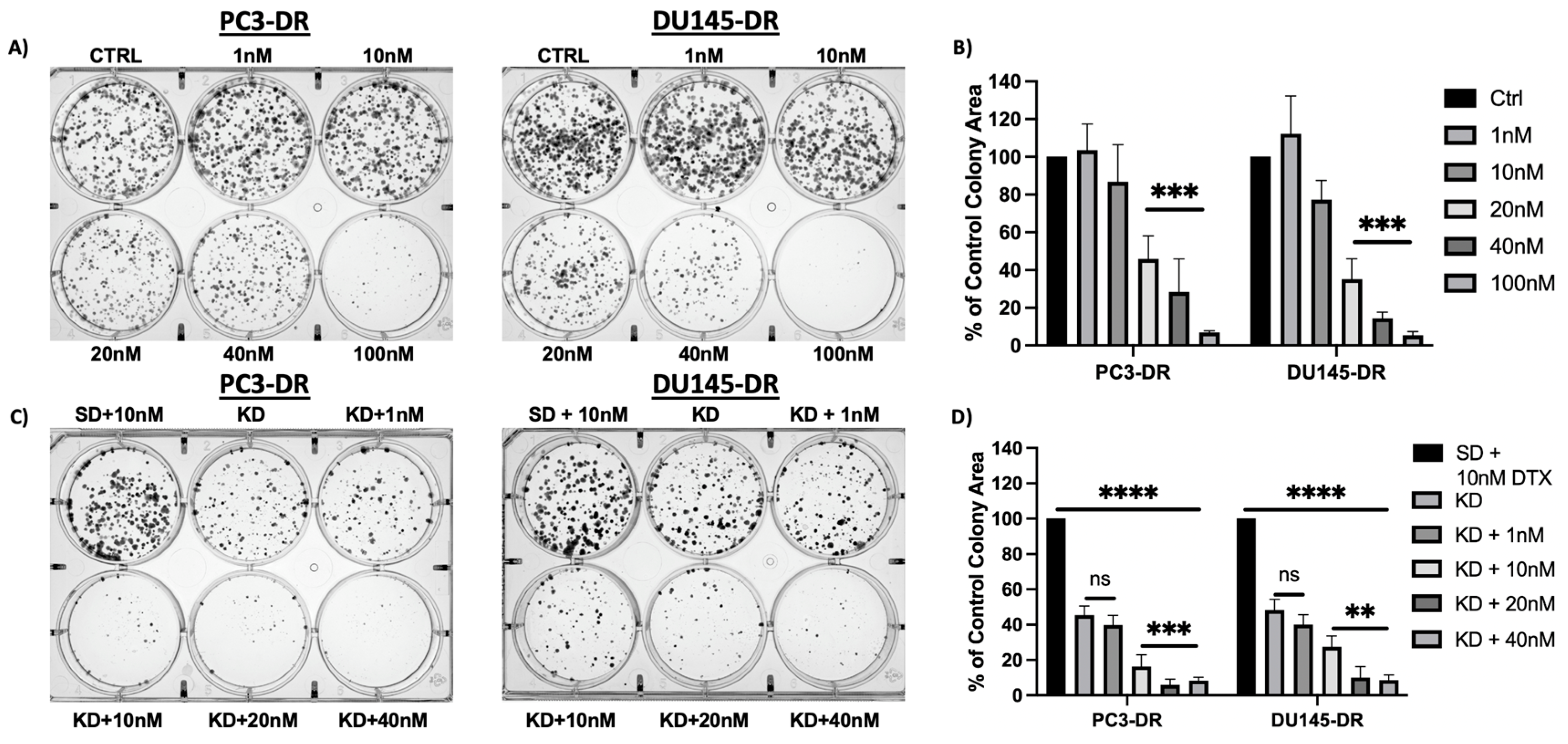
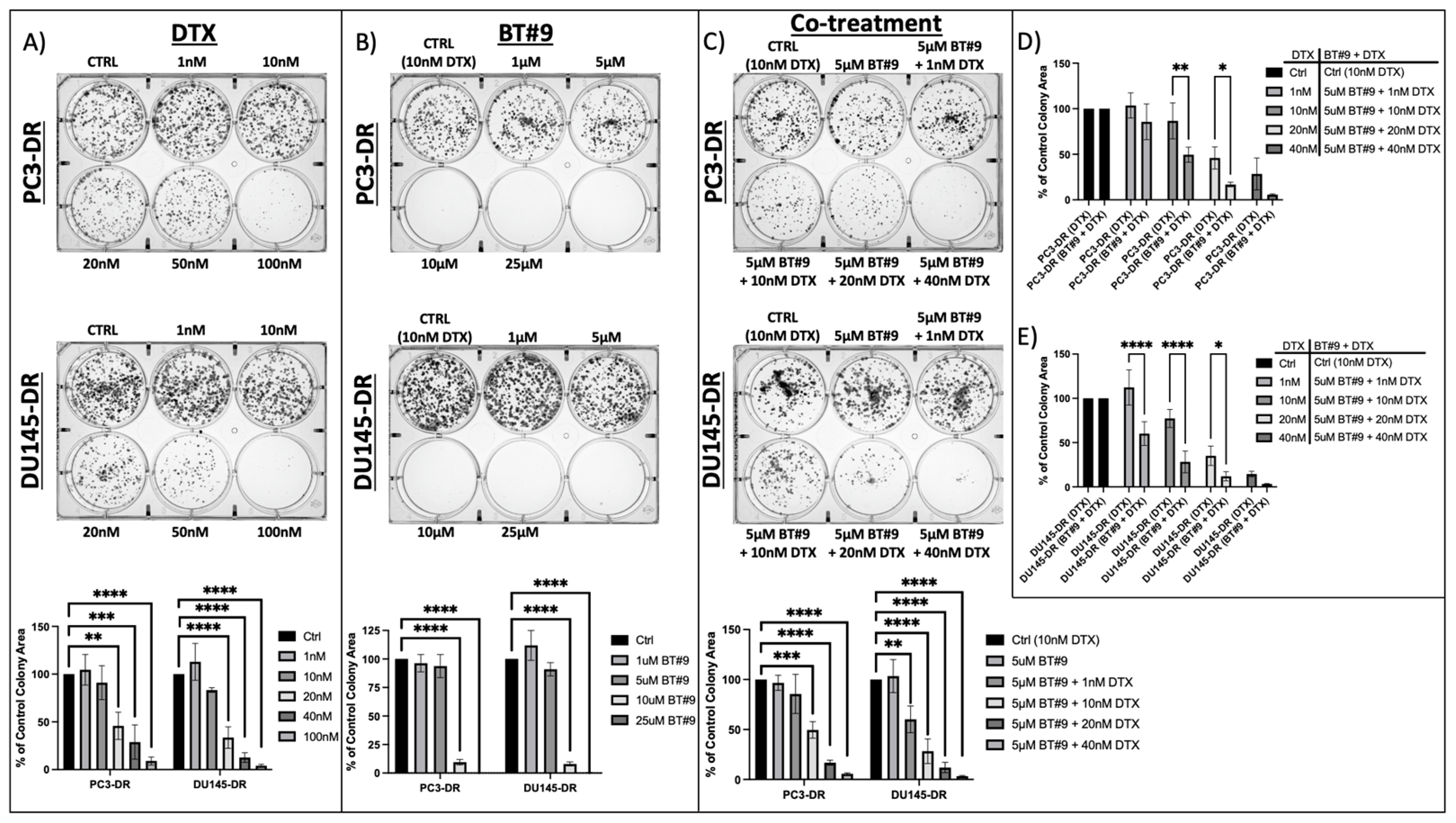
3.3. MAGMAS Depletion Inhibits the Clonogenicity Capacity of DTX-Resistant PCa Cells
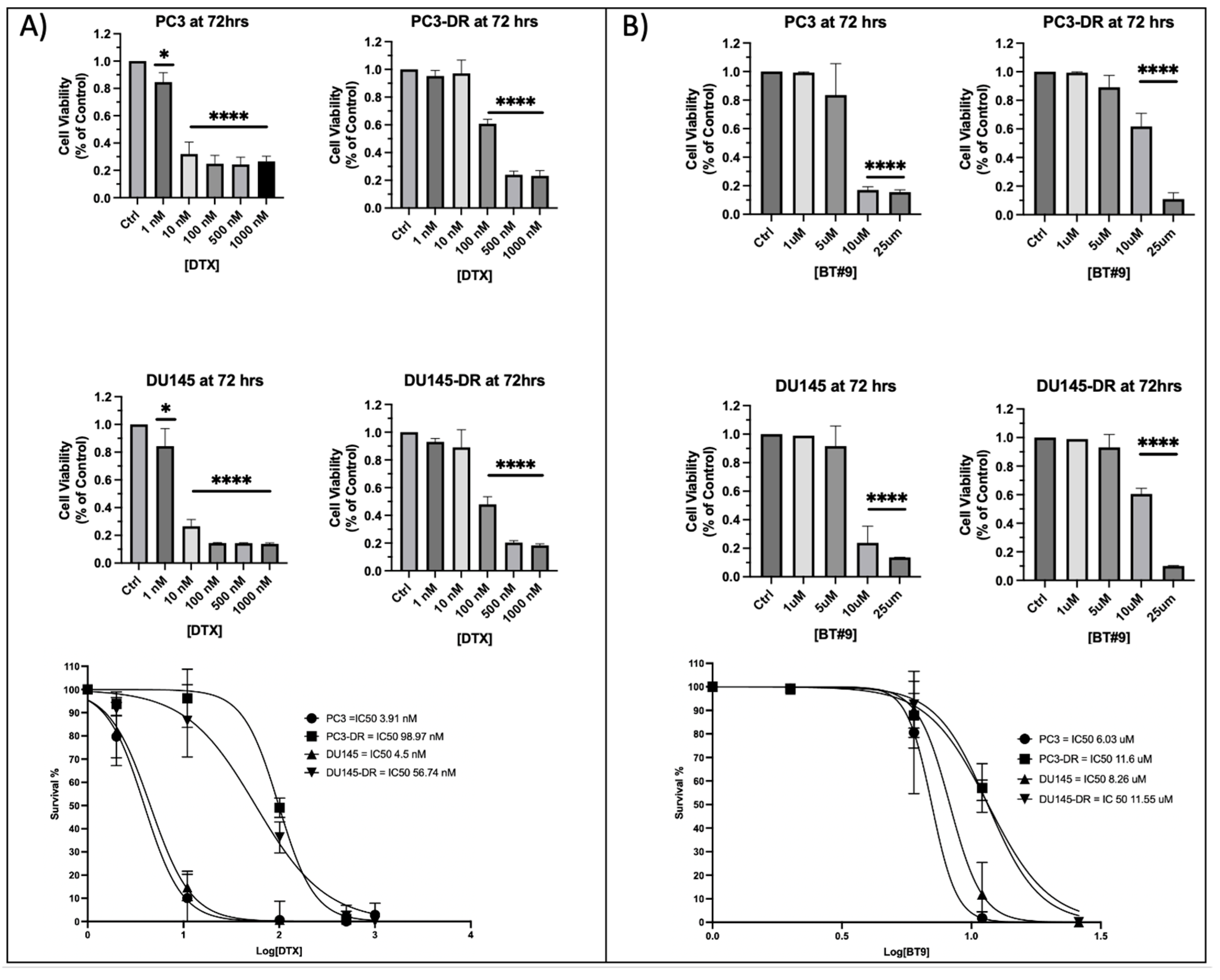
3.4. DTX-Resistant PCa Cells Exhibit Resistance to MAGMAS Inhibitor BT#9
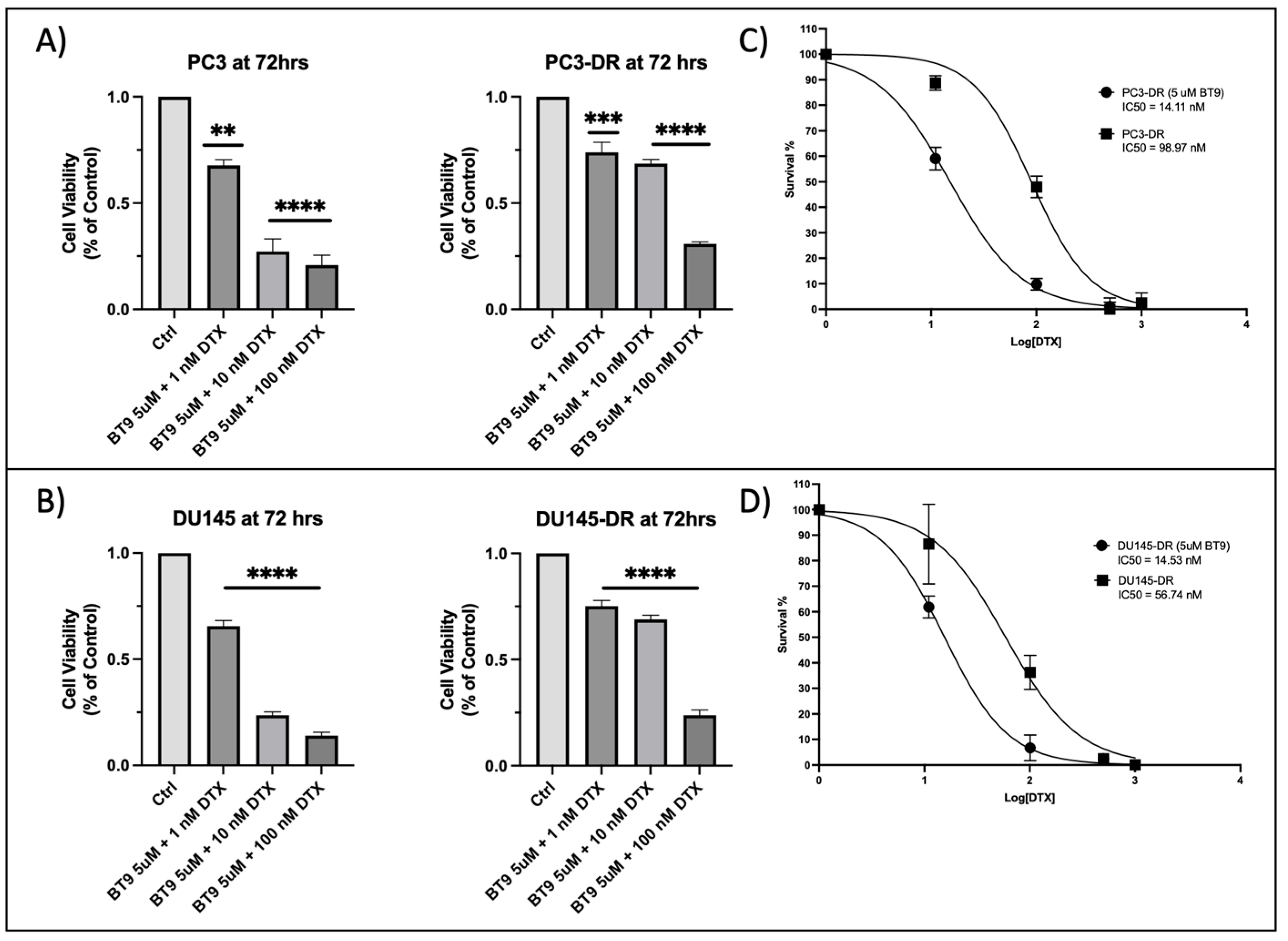
3.5. Sub-Therapeutic Inhibition of MAGMAS with BT#9 Sensitizes Chemoresistant PCa Cells to Docetaxel
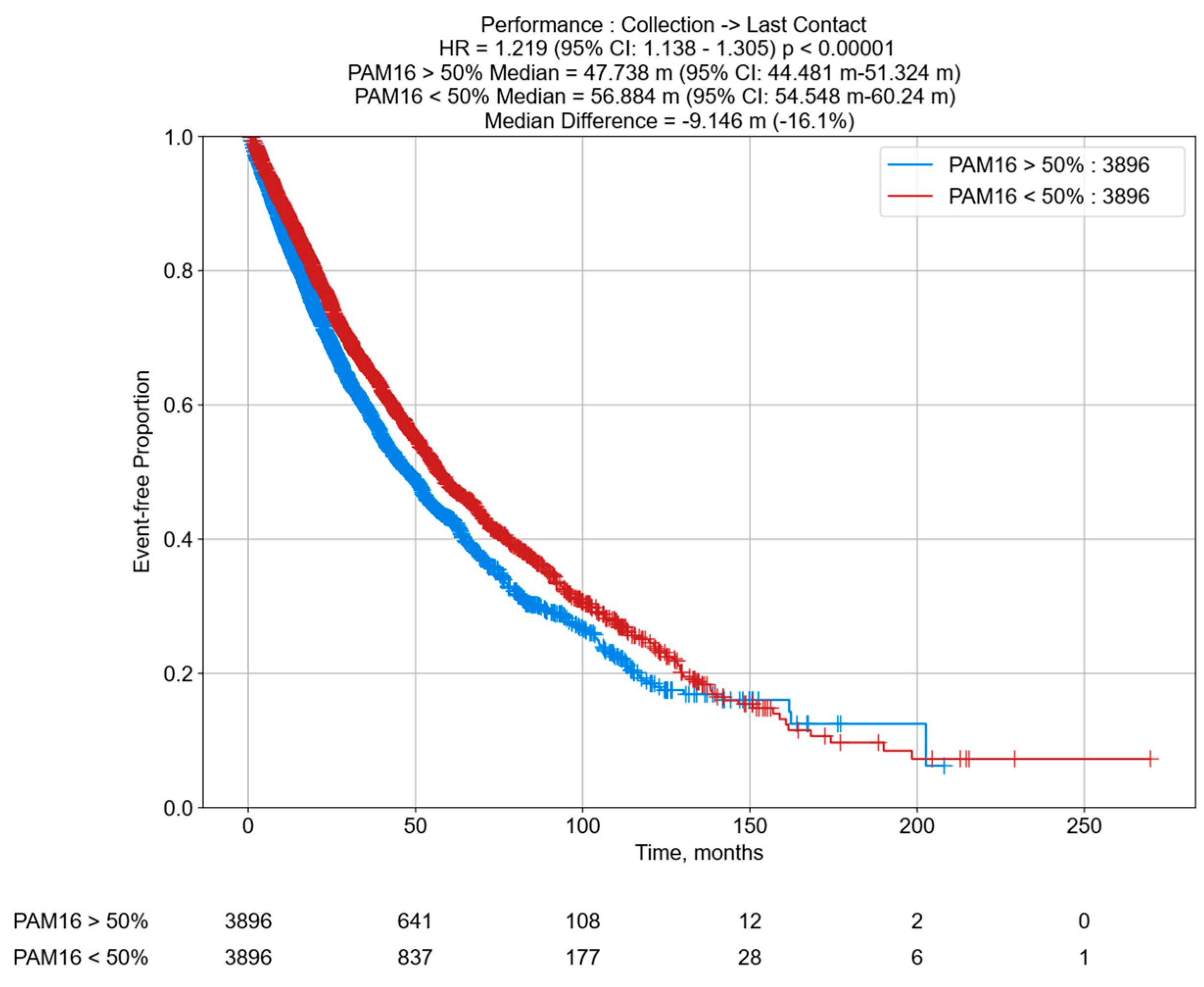
3.6. Decreased OS in PCa Patients According to PAM16 RNA Tumor Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Heyns, C.; Simonin, M.; Grosgurin, P.; Schall, R.; Porchet, H. Comparative efficacy of triptorelin pamoate and leuprolide acetate in men with advanced prostate cancer. BJU Int. 2003, 92, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Scher, H.I. Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res. 2009, 15, 4792–4798. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Aparicio, A.; Logothetis, C.J.; Maity, S.N. Understanding the lethal variant of prostate cancer: Power of examining extremes. Cancer Discov. 2011, 1, 466–468. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Beltran, H.; Tagawa, S.T.; Park, K.; MacDonald, T.; Milowsky, M.I.; Mosquera, J.M.; Rubin, M.A.; Nanus, D.M. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J. Clin. Oncol. 2012, 30, e386–e389. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef]
- Roubaud, G.; Liaw, B.C.; Oh, W.K.; Mulholland, D.J. Strategies to avoid treatment-induced lineage crisis in advanced prostate cancer. Nat. Rev. Clin. Oncol. 2017, 14, 269–283. [Google Scholar]
- de Kouchkovsky, I.; Chan, E.; Schloss, C.; Poehlein, C.; Aggarwal, R. Diagnosis and management of neuroendocrine prostate cancer. Prostate 2024, 84, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Culine, S.; El Demery, M.; Lamy, P.-J.; Iborra, F.; Avancès, C.; Pinguet, F. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J. Urol. 2007, 178 Pt 1, 844–848, discussion 848. [Google Scholar] [CrossRef]
- Papandreou, C.N.; Daliani, D.D.; Thall, P.F.; Tu, S.-M.; Wang, X.; Reyes, A.; Troncoso, P.; Logothetis, C.J. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J. Clin. Oncol. 2002, 20, 3072–3080. [Google Scholar] [CrossRef]
- Loriot, Y.; Massard, C.; Gross-Goupil, M.; Di Palma, M.; Escudier, B.; Bossi, A.; Fizazi, K. Combining carboplatin and etoposide in docetaxel-pretreated patients with castration-resistant prostate cancer: A prospective study evaluating also neuroendocrine features. Ann. Oncol. 2009, 20, 703–708. [Google Scholar] [CrossRef]
- Papandreou, C.N.; Daliani, D.D.; Thall, P.F.; Tu, S.M.; Wang, X.; Reyes, A.; Troncoso, P.; Logothetis, C.J. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, Q.; Hicks, J.L.; Trabzonlu, L.; Ozbek, B.; Jones, T.; Vaghasia, A.M.; Larman, T.C.; Wang, R.; Markowski, M.C.; et al. MYC-driven increases in mitochondrial DNA copy number occur early and persist throughout prostatic cancer progression. JCI Insight 2023, 8, e169868. [Google Scholar] [CrossRef]
- Pujana-Vaquerizo, M.; Bozal-Basterra, L.; Carracedo, A. Metabolic adaptations in prostate cancer. Br. J. Cancer 2024, 131, 1250–1262. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef]
- Mizumachi, T.; Suzuki, S.; Naito, A.; Carcel-Trullols, J.; Evans, T.T.; Spring, P.M.; Oridate, N.; Furuta, Y.; Fukuda, S.; Higuchi, M. Increased mitochondrial DNA induces acquired docetaxel resistance in head and neck cancer cells. Oncogene 2007, 27, 831–838. [Google Scholar] [CrossRef]
- Haldar, S.; Mishra, R.; Billet, S.; Thiruvalluvan, M.; Placencio-Hickok, V.R.; Madhav, A.; Duong, F.; Angara, B.; Agarwal, P.; Tighiouart, M.; et al. Cancer epithelia-derived mitochondrial DNA is a targetable initiator of a paracrine signaling loop that confers taxane resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 8515–8523. [Google Scholar] [CrossRef]
- Peng, J.; Huang, C.-H.; Short, M.K.; Jubinsky, P.T. Magmas gene structure and evolution. Silico Biol. 2005, 5, 251–263. [Google Scholar] [CrossRef]
- Srivastava, S.; Sinha, D.; Saha, P.P.; Marthala, H.; Silva, D. Magmas functions as a ROS regulator and provides cytoprotection against oxidative stress-mediated damages. Cell Death Dis. 2014, 5, e1394. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Das, B.C.; Aljitawi, O.; Kumar, A.; Das, S.; Veldhuizen, V.P. Magmas Inhibition in Prostate Cancer: A Novel Target for Treatment-Resistant Disease. Cancers 2022, 14, 2732. [Google Scholar] [CrossRef] [PubMed]
- Tagliati, F.; Gentilin, E.; Buratto, M.; Molè, D.; degli Uberti, E.C.; Zatelli, M.C. Magmas, a gene newly identified as overexpressed in human and mouse ACTH-secreting pituitary adenomas, protects pituitary cells from apoptotic stimuli. Endocrinology 2010, 151, 4635–4642. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Kadife, E.; Raza, A.; Short, M.; Jubinsky, P.T.; Kannourakis, G. Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods. Cells 2020, 9, 719. [Google Scholar] [CrossRef]
- Jubinsky, P.T.; Short, M.K.; Mutema, G.; Morris, R.E.; Ciraolo, G.M.; Li, M. Magmas expression in neoplastic human prostate. J. Mol. Histol. 2005, 36, 69–75. [Google Scholar] [CrossRef]
- Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Ochoa, P.T.; Elix, C.C.; Alkashgari, H.R.; McMullen, J.R.W.; Soto, U.; Martinez, S.R.; Osterman, C.J.D.; Mahler, M.; et al. The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells. Cells 2021, 10, 2723. [Google Scholar] [CrossRef]
- Ross, C.K.C.-D.; Martinez, S.R.; Woods-Burnham, L.; Durán, A.M.; Roy, S.; Basu, A.; Ramirez, J.A.; Ortiz-Hernández, G.L.; Ríos-Colón, L.; Chirshev, E.; et al. RNA sequencing reveals upregulation of a transcriptomic program associated with stemness in metastatic prostate cancer cells selected for taxane resistance. Oncotarget 2018, 9, 30363–30384. [Google Scholar] [CrossRef]
- Ríos-Colón, L.; Ross, C.K.C.-D.; Basu, A.; Elix, C.; Alicea-Polanco, I.; Sanchez, T.W.; Radhakrishnan, V.; Chen, C.-S.; Casiano, C.A. Targeting the stress oncoprotein LEDGF/p75 to sensitize chemoresistant prostate cancer cells to taxanes. Oncotarget 2017, 8, 24915–24931. [Google Scholar] [CrossRef]
- Jubinsky, P.T.; Short, M.K.; Ghanem, M.; Das, B.C. Design, synthesis, and biological activity of novel Magmas inhibitors. Bioorganic Med. Chem. Lett. 2011, 21, 3479–3482. [Google Scholar] [CrossRef]
- Leoh, L.S.; Van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.R.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar] [CrossRef]
- Martinez, S.R.; Elix, C.C.; Ochoa, P.T.; Sanchez-Hernandez, E.S.; Alkashgari, H.R.; Ortiz-Hernandez, G.L.; Zhang, L.; Casiano, C.A. Glucocorticoid Receptor and beta-Catenin Interact in Prostate Cancer Cells and Their Co-Inhibition Attenuates Tumorsphere Formation, Stemness, and Docetaxel Resistance. Int. J. Mol. Sci. 2023, 24, 7130. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ Plugin to Automatically Quantify Colony Formation in Clonogenic Assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef] [PubMed]
- Klarmann, G.J.; Hurt, E.M.; Mathews, L.A.; Zhang, X.; Duhagon, M.A.; Mistree, T.; Thomas, S.B.; Farrar, W.L. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin. Exp. Metastasis 2009, 26, 433–446. [Google Scholar] [CrossRef]
- Okasho, K.; Ogawa, O.; Akamatsu, S. Narrative review of challenges in the management of advanced neuroendocrine prostate cancer. Transl. Androl. Urol. 2021, 10, 3953–3962. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.-Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef]
- Li, W.; Cohen, A.; Sun, Y.; Squires, J.; Braas, D.; Graeber, T.G.; Du, L.; Li, G.; Li, Z.; Xu, X.; et al. The Role of CD44 in Glucose Metabolism in Prostatic Small Cell Neuroendocrine Carcinoma. Mol. Cancer Res. 2016, 14, 344–353. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Jamroze, A.; Chatta, G.; Tang, D.G. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021, 518, 1–9. [Google Scholar] [CrossRef]
- Butler, W.; Huang, J. Neuroendocrine cells of the prostate: Histology, biological functions, and molecular mechanisms. Precis. Clin. Med. 2021, 4, 25–34. [Google Scholar] [CrossRef]
- Clermont, P.-L.; Ci, X.; Pandha, H.; Wang, Y.; Crea, F. Treatment-Emergent Neuroendocrine Prostate Cancer: Molecularly Driven Clinical Guidelines. Int. J. Endocr. Oncol. 2019, 6, IJE20. [Google Scholar] [CrossRef]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.-W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef]
- Sanchez-Hernandez, E.S.; Ochoa, P.T.; Suzuki, T.; Ortiz-Hernandez, G.L.; Unternaehrer, J.J.; Alkashgari, H.R.; Osterman, C.J.D.; Martinez, S.R.; Chen, Z.; Kremsky, I.; et al. Glucocorticoid Receptor Regulates and Interacts with LEDGF/p75 to Promote Docetaxel Resistance in Prostate Cancer Cells. Cells 2023, 12, 2046. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Di, K.; Lomeli, N.; Bota, D.A.; Das, B.C. Magmas inhibition as a potential treatment strategy in malignant glioma. J. Neuro-Oncol. 2019, 141, 267–276. [Google Scholar] [CrossRef]
- Bardelmeijer, H.A.; Ouwehand, M.; Buckle, T.; Huisman, M.T.; Schellens, J.H.; Beijnen, J.H.; van Tellingen, O. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res. 2002, 62, 6158–6164. [Google Scholar] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Giddings, E.L.; Champagne, D.P.; Wu, M.-H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Loda, M. Metabolic Vulnerabilities of Prostate Cancer: Diagnostic and Therapeutic Opportunities. Cold Spring Harb. Perspect. Med. 2018, 8, a030569. [Google Scholar] [CrossRef]
- Ahn, H.K.; Lee, Y.H.; Koo, K.C. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef]
- Basu, H.S.; Wilganowski, N.; Robertson, S.; Reuben, J.M.; Cohen, E.N.; Zurita, A.; Ramachandran, S.; Xiao, L.; Titus, M.; Wilding, G. Prostate cancer cells survive anti-androgen and mitochondrial metabolic inhibitors by modulating glycolysis and mitochondrial metabolic activities. Prostate 2021, 81, 799–811. [Google Scholar] [CrossRef]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef]
- Vaz, C.V.; Marques, R.; Alves, M.G.; Oliveira, P.F.; Cavaco, J.E.; Maia, C.J.; Socorro, S. Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. J. Cancer Res. Clin. Oncol. 2016, 142, 5–16. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durán, A.M.; Whitley, K.; Santiago, K.; Yoo, C.; Valdez, G.; Cheng, K.W.; Ochoa, P.; de Semir, D.; Xiu, J.; Chokkalingam, P.; et al. Inhibition of Mitochondrial-Associated Protein MAGMAS Resensitizes Chemoresistant Prostate Cancer Cells to Docetaxel. Cancers 2025, 17, 1535. https://doi.org/10.3390/cancers17091535
Durán AM, Whitley K, Santiago K, Yoo C, Valdez G, Cheng KW, Ochoa P, de Semir D, Xiu J, Chokkalingam P, et al. Inhibition of Mitochondrial-Associated Protein MAGMAS Resensitizes Chemoresistant Prostate Cancer Cells to Docetaxel. Cancers. 2025; 17(9):1535. https://doi.org/10.3390/cancers17091535
Chicago/Turabian StyleDurán, Alfonso M., Kristen Whitley, Krystal Santiago, Christian Yoo, Giancarlo Valdez, Kai Wen Cheng, Pedro Ochoa, David de Semir, Joanne Xiu, Parthiban Chokkalingam, and et al. 2025. "Inhibition of Mitochondrial-Associated Protein MAGMAS Resensitizes Chemoresistant Prostate Cancer Cells to Docetaxel" Cancers 17, no. 9: 1535. https://doi.org/10.3390/cancers17091535
APA StyleDurán, A. M., Whitley, K., Santiago, K., Yoo, C., Valdez, G., Cheng, K. W., Ochoa, P., de Semir, D., Xiu, J., Chokkalingam, P., Das, S., Schaefer, E. S., Rowe, S. P., Das, B. C., Casiano, C. A., & Almaguel, F. (2025). Inhibition of Mitochondrial-Associated Protein MAGMAS Resensitizes Chemoresistant Prostate Cancer Cells to Docetaxel. Cancers, 17(9), 1535. https://doi.org/10.3390/cancers17091535